

Abstract
Exome sequencing is increasingly being used to help diagnose pediatric neurology cases when clinical presentations are not specific. However, interpretation of equivocal results that include variants of uncertain significance remains a challenge. In those cases, follow-up testing and clinical correlation can help clarify the clinical relevance of the molecular findings. In this report, we describe the diagnostic odyssey of a 4-year-old girl who presented with global developmental delay and seizures, with leukodystrophy seen on MRI. Clinical evaluation, MRI, and comprehensive metabolic testing were performed, followed by whole-exome sequencing (WES), parental testing, follow-up testing, and retrospective detailed clinical evaluation. WES identified two candidate causative pathogenic variants in SAMHD1, a gene associated with the recessive condition Aicardi–Goutières syndrome (AGS) type 5 (OMIM 612952): a previously reported pathogenic variant NM_015474 c.602T>A (p.I201N), maternally inherited, and a rare missense variant of uncertain significance, c.1293A>T(p.L431F). Analysis of type I interferon-related biomarkers demonstrated that the patient has an interferon signature characteristic of AGS. Retrospective detailed clinical evaluation showed that the girl has a phenotype consistent with AGS5, a rare neurological condition. These results further define the phenotypic spectrum associated with specific SAMHD1 variants, including heterozygous variants in AGS carriers, and support the idea that autoinflammatory dysregulation is part of the disease pathophysiology. More broadly, this work highlights the issues and methodology involved in ascribing clinical relevance to interpretation of variants detected by WES.
Keywords: dysmyelinating leukodystrophy, immune dysregulation, upper limb spasticity
INTRODUCTION
Use of Whole-Exome Sequencing to Augment Diagnosis in Challenging Neurological Cases
Seizures or encephalopathies are presentations for a number of disorders with genetic, infectious, or inflammatory etiologies. Genetic encephalopathies, depending on the causative pathogenic variants, may have a characteristic age of onset, a set of described features, and rate of progression of symptoms. A genetic diagnosis can lead to more personalized management and facilitates risk assessment in family members. Although it is therefore important to pinpoint the molecular etiology, deciding which gene or set of genes to test can be difficult when based solely on clinical features, because of overlap of features and broad phenotypic variability in affected individuals. Accordingly, clinicians are increasingly turning to comprehensive genetic testing modalities such as whole-exome sequencing (WES) to complement existing diagnostic tools, including physical exams, radiologic imaging, biochemical testing, electromyography/nerve conduction study, and tissue biopsies. WES is particularly useful for cases in which monogenic conditions are suspected but lack of specific clinical clues forestall well-defined differential diagnoses. WES has provided a molecular diagnosis in challenging cases, especially in neurometabolic disorders with nonspecific presentation (Tarailo-Graovac et al. 2016). However, one of the major challenges posed by large-scale genomic sequencing modalities such as WES is simply the number of variants of uncertain significance (VUS) identified (Richards et al. 2015). VUS pose a challenge to the clinician in the sense that they raise doubts as to whether the diagnosis is likely, and whether these variants should be used for targeted testing in the future by family members to inform genetic risk assessment including prenatal diagnosis. In this report, we describe a case in which a follow-up laboratory and clinical assessment of a suspected pathogenic variant lead to a clinical diagnosis in a pediatric neurology case.
Aicardi–Goutières Syndrome
Aicardi–Goutières syndrome (AGS) was first described in 1984 as a familial progressive encephalopathy, and has since been reported in several cases from various ethnic backgrounds (Aicardi and Goutières 1984). The condition is considered a Mendelian mimic of congenital TORCH infection, and is characterized by spasticity, dystonia, delayed psychomotor development, chronic cerebrospinal fluid (CSF) leukocytosis, and leukodystrophy on brain imaging. Microcephaly, hepatosplenomegaly, cardiac arrhythmias, sensorineural hearing loss, glaucoma, and cortical blindness can be seen in severe cases (Crow et al. 2015; Tumienė et al. 2017). Clinically, children can present with nonspecific seizures and developmental delays, and CSF leukocytosis or abnormal brain MRI may go undetected unless a specific workup is undertaken. Chronic inflammatory skin conditions are commonly reported, including characteristic chilblains (red patches on the skin that occur in response to cold exposure) and recurrent mouth ulcers (Rice et al. 2007; Ravenscroft et al. 2011; Crow and Manel 2015). The disease presentation can be variable and may be severe depending on the involved gene and level of neurological sequelae associated with the initial disease episode (Rice et al. 2007).
AGS is genetically heterogeneous, and seven genes have been shown to cause AGS to date (TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1) (Rice et al. 2009; Dale et al. 2010; Crow et al. 2015). Biallelic pathogenic variants in SAMHD1 (20q11.23), which encodes a Sam domain and HD domain containing protein that functions as a deoxynucleoside triphosphate triphosphohydrolase, cause AGS5 (OMIM# 612952). AGS, most frequently transmitted in an autosomal recessive fashion with exception of AGS caused by TREX1, ADAR, and IFIH1, belongs to a group of monogenic diseases in which a constitutive up-regulation of type I IFN production leads to pathology (type I interferonopathy) (Crow and Manel 2015). All patients with confirmed pathogenic variants in one of the six AGS genes TREX1, RNASEH2A, RNASEH2C, SAMHD1, ADAR, or IFIH1 show sustained increased expression of type I interferon-stimulated gene transcripts in CSF and serum (interferon signature) (Rice et al. 2013; Crow and Manel 2015), reflecting overactivity of the innate immune system triggered by endogenous nucleic acid pathways. Thus, constitutive up-regulation of type 1 interferon activity directly underlies the disease pathophysiology and has important implications for understanding aspects of disease manifestation (such as development of lupus) and for designing potential treatment strategies (Rice et al. 2007; Crow et al. 2014).
We present a case report of a 4-year-old girl with truncal hypotonia and peripheral spasticity, seizures, and developmental delay, whose diagnostic odyssey culminated with WES and in which a combination of parental testing, targeted follow-up biomarker testing, and clinical correlation confirmed a diagnosis of AGS5 with associated autoimmune dysregulation.
Clinical Report
The female proband was born at 40-week gestational age via vaginal delivery to an unrelated couple (28-year-old mother and 24-year-old father) with no prior children. Birth history was uncomplicated, with birth weight of 6 pounds 9 ounces (27.8 percentile), birth length of 19 inches (26.8 percentile), and APGAR scores of 8 and 9 at 1 min and 5 min after birth, respectively. Her parents first noticed strabismus in the child at 4 months of age and noted that she was not reaching for objects. Motor delay persisted despite surgical correction of the congenital strabismus, and she was followed by neurology. She had significant central hypotonia and peripheral hypertonia and could not sit unsupported until 26 months of age. At around age 2.5 years, she developed epilepsy controlled by benzodiazepine and spasticity that required baclofen.
At age 4 years, the patient still had no ambulatory skills and required orthoses and an orthopedic walker, followed by a wheelchair. Her speech development was delayed and enunciation was difficult, but she showed progress without regression; she combined words at age 4. Hand slapping was exhibited accompanying various emotions, including nonagitated states. She had persistent erythema on her toes that would swell and exacerbate in the winter seasons (Fig. 1C).
Figure 1.

Digital photograph of the proband at 6 years of age, in her wheelchair (A) and in her orthoses (B). Throughout most of the year, and particularly in winter, chilblains were evident on the patient's toes (C). (D) Axial FLAIR image showing extensive and confluent abnormal FLAIR hyperintensity throughout the bilateral cerebral hemispheres (white arrows). (E) Coronal T2-weighted image showing extensive abnormal and confluent T2 hyperintensity throughout the bilateral cerebral hemisphere white matter (white arrow), with apparent sparing of the cerebellum. A single focus of T2 hypointensity is identified in the periventricular white matter of the left cerebral hemisphere (black arrow), which corresponds to susceptiblity artifact on the GRE sequence and is consistent with calcification. (F) Axial GRE image showing scattered punctate foci of magnetic susceptibility artifact (white arrows) consistent with calcification were noted throughout the cerebral white matter.
Brain MRI
The patient had a series of brain MRIs at ages 6, 18, and 30 months. All of these studies demonstrated diffuse and confluent abnormal T2/FLAIR hyperintensity within the bilateral cerebral white matter (Fig. 1D). Although all lobes were affected, there was frontal and parietal lobe predominance. Signal abnormality involved both the periventricular white matter as well as the subcortical U fibers. Of interest, there was clear sparing of the cerebellum (Fig. 1E). Several small foci of magnetic susceptibility artifact were identified within the lobar white matter bilaterally on the GRE sequences, considered consistent with calcification (Fig. 1F).
Biochemical and Metabolic Testing
Urine organic acids, plasma amino acids, plasma lactate, and plasma acylcarnitine profile were all normal. A lysosomal enzyme panel was normal, ruling out GM1 and GM2 gangliosidoses (including hexosaminidase A for Tay–Sachs disease), α- and β-mannosidoses, fucosidosis, metachromatic leukodystrophy, and Krabbe disease. Normal 7-dehydrocholesterol level ruled out Smith–Lemli Opitz syndrome. Very long chain fatty acids showed normal pattern ruling out peroxisomal disorders. N-glycan testing was normal ruling out congenital disorders of N-glycosylation.
Genetic Testing
Routine karyotype was normal. Single-nucleotide polymorphism chromosomal microarray showed numerous regions of homozygosity (ROH) of >3 Mb, totaling 3.8% of the genome. Fifty-two genes within these ROH were noted to be associated with recessive conditions, although none were a good phenotypic fit; the microarray did not show any regions of imbalance. Both parents are Caucasian and deny any consanguinity. Given the long list of genes that could be considered to underlie the patient's condition, she was enrolled into the North Carolina Genomic Evaluation by Next-Generation Exome Sequencing (NCGENES) research study, with full approval by the UNC IRB and parental consent. WES was performed, and any variants in genes associated with intellectual disability/autism and leukodystrophy were examined, as well as any variants from a broader set of genes associated with neuromuscular disorders (NMDs). These NMD diagnostic genes were curated from commercially available panels, as well as Online Mendelian Inheritance in Man, and PubMed. More than 500 genes were examined in total.
The WES identified two sequence variants in the SAMHD1 gene: SAMHD1 c.602T>A p.I201N, a previously reported pathogenic variant (Rice et al. 2009; Goncalves et al. 2012), and SAMHD1 c.1293A>T p.L431F, a VUS (Table 1). No other variants were reported. Compound heterozygous pathogenic variants in SAMHD1 are associated with autosomal recessive AGS5, which was believed to be a good phenotypic fit. The p.I201N variant had previously been reported in a compound heterozygous state in a family with AGS5 (Rice et al. 2009), and in the heterozygous state in a mother and son affected with familial autosomal dominant chilblain lupus-2 (Ravenscroft et al. 2011), now considered a forme fruste of AGS (Yarbrough et al. 2016). The SAMHD1 p.I201N is a missense pathogenic variant located within the HD (histidine-aspartate) domain of the encoded protein, which interferes with protein function, and is rare in the gnomAD population database (Lek et al. 2016). The SAMHD1 p.L431F variant is a rare missense variant that changes a single conserved amino acid from leucine to phenylalanine and is not within either of the known functional protein domains; the nearest missense variant reported to be associated with AGS5 is p.I448T (White et al. 2017), so no hotspot or domain information could be used to further classify the variant. The variant is predicted to be damaging by Condel (González-Pérez and López-Bigas 2011), is rare in the gnomAD database (2/277,126 alleles), and has not been published previously. The variant had not previously been reported in an affected individual, and no functional impact on protein function had been published. Accordingly, the p.L431F variant was considered a VUS.
Table 1.
SAMHD1 variants identified in the proband
Follow-up testing on the proband's mother showed that the p.I201N variant was maternally inherited but the p.L431F was not, suggesting that the two variants are in trans (Fig. 2), although the possibility remains the variant occurred de novo on the maternally inherited allele. The father was not available for testing.
Figure 2.
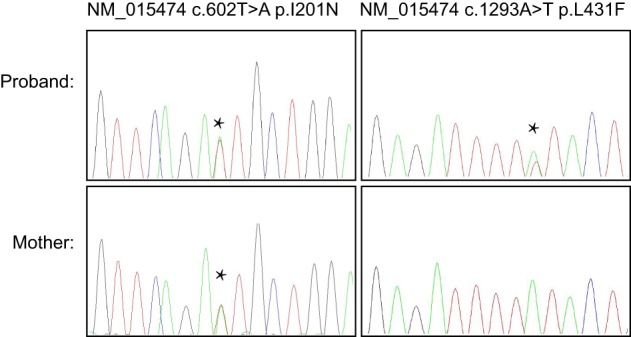
Sanger sequencing of WES-identified variants in the proband and her mother. The I201N variant was identified in the patient's mother, who per report does not experience chilblains.
Elevated levels of type I interferon have been demonstrated to play a role in Aicardi–Goutières syndrome (Rice et al. 2009, 2013). To further adjudicate the novel variant identified by WES, which implicated AGS5 as the diagnosis, peripheral blood was examined for the previously published “interferon signature” that is present in AGS patients. The testing showed the patient's blood had significantly increased levels of all six interferon-stimulated genes tested, consistent with a SAMHD1-mediated process, and consistent with the diagnosis of AGS5 (Fig. 3).
Figure 3.

Immune signature testing shows elevated levels of all analytes tested, six interferon-stimulated genes, in the patient sample (green) compared to control (blue). The decimalized age at sampling (4.81) and the interferon score (6.567) are shown.
Retrospective Clinical Evaluation
Given the rarity in the general population (PM2), the results of parental genetic testing (PM3), the in silico predictions (PP3), and the interferon signature (not counted as strong evidence because the interferon signature is not specific to the variant or gene), the SAMHD1 p.L431F variant met the ACMG classification criteria for likely pathogenic (Richards et al. 2015). Although we decided to use PM3 in this case, we cannot rule out the possibility that the variant arose as a de novo event on the maternally inherited copy. These results strongly implicated AGS5 as the diagnosis in the patient. However, with the variant not being definitively pathogenic and the interferon signature being selective for AGS but not pathognomonic, the diagnosis of AGS5 required further clinical correlation. Retrospective review of the patient's medical record and follow-up physical exam demonstrated features consistent with AGS5, including seizures, developmental delay, and leukodystrophy, with possible calcifications in the basal ganglia. The proband also displayed chilblains on her toes, the patches of small blood vessel inflammation that often occur in response to cold exposure that can be a characteristic feature of AGS5. Although chilblains are not specific to AGS5, this skin finding of swelling and redness that flares up during cold weather was specifically noted in the patient's chart and is seen in ∼40% of AGS cases. Examination of the proband's mother showed that she does not have chilblains, unlike the previous published report of a heterozygous SAMHD1 p.I201N carrier (Ravenscroft et al. 2011), raising the possibility that not all heterozygous SAMHD1 p.I201N carriers will manifest any clinical signs. This report is the first to demonstrate apparent incomplete penetrance of the SAMHD1 p.I201N variant with autosomal dominant chilblains.
DISCUSSION
AGS manifests as an early-onset encephalopathy that usually results in epilepsy, intellectual disability, and regression of motor skills. Affected infants or children present at variable times, frequently after a period of apparently normal development, typically with irritability, developmental regression, and spasticity. The disease is characterized by chilblain lesions seen in peripheral skin such as fingers, and toes. Milder cases have been reported with only cutaneous manifestation (Yarbrough et al. 2016). It is possible that the milder end of the clinical spectrum is underdiagnosed. SAMHD1-associated AGS5 confers high risk for glaucoma as well as cerebrovascular events compared to other AGS (Crow et al. 2015).
The brain MRI findings in our patient were generally consistent with previous reports of AGS, including confluent diffuse bilateral white matter involvement, involvement of subcortical U fibers, sparing of optic radiations and cerebellum, and findings consistent with calcifications. Nevertheless, we noted MRI findings in our patient distinct from those reported in prior AGS patients, including no calcifications in the lentiform nuclei, extensive periventricular white matter involvement, and overall lack of cerebral atrophy (Uggetti et al. 2009; Ramantani et al. 2010). Although the proband did not have a CT scan, which would be the gold standard for calcifications, and acknowledging that some of these features may develop over time, these results further delineate and expand the phenotypic spectrum of brain findings associated with AGS5.
Patients with AGS display a characteristic interferon signature in CSF or serum (Crow and Manel 2015). Accordingly, we were able to use this follow-up testing to help adjudicate a WES-identified VUS. Although elevated levels of interferon genes are seen in AGS, and are not seen in many other pediatric neurological conditions, it is not specific to AGS5. The characteristic AGS interferon signature, coupled with a detailed retrospective clinical analysis of the patient's features, enabled the diagnosis of SAMHD1-associated AGS5 in the current case. Although the genetic testing results correlate well with a clinical phenotype of AGS5, because the WES analysis was limited to neurological genes, the possibility of additional pathogenic variants in the proband's genome cannot be excluded; although the p.L431F variant has been upgraded to likely pathogenic, its causality as an AGS5 pathogenic variant is not yet definitive. Given the list of features that have been associated with biallelic SAMHD1 pathogenic variants, this patient is followed for signs of arthropathy and joint contractures, mouth ulcers, glaucoma, and intracranial vasculopathy (Rice et al. 2009). She is also monitored for insulin-dependent diabetes mellitus and other autoimmune disorders such as hypothyroidism. Knowing the full spectrum of features associated with AGS5 allows for optimal screening and intervention as needed in the child.
Although there is no definitive treatment at this time, clinical trials investigating the use of reverse transcriptase inhibitors in children with AGS are being developed (clinicaltrials.gov; NCT02363452). However, the clear pathophysiological etiology in this disease as a disorder of interferon-mediated autoinflammatory dysfunction presents potential avenues for the development of targeted therapeutics. As such, early diagnosis of children with AGS with the use of WES and other comprehensive genomic technologies could potentially lead to beneficial treatments being developed. Given the fact that the disease, for some, tends not to manifest until after a period of postnatal normality, there may be a therapeutic window of opportunity. If so, obtaining a precise diagnosis early on is of paramount importance.
METHODS
Sequencing Analysis
With approval from the UNC Institutional Review Board, genomic DNA was isolated from 10 ml of whole blood from the proband and family members using PureGene chemistry in the UNC Biospecimen Processing Facility. Sample quality and quantity were measured using an Agilent Bioanalyzer or Tape Station. Molecular barcoding, WES capture, and library preparation was done using the Agilent SureSelect ALL Exon Kit version 4 and carried out according to manufacturer's guidelines. Libraries were sequenced on a HiSeq2500 at the UNC High Throughput Sequencing Facility with paired-end sequencing at an average depth of 50×. Table 2 provides mean sample level exome coverage and exon-level coverage specifically for the SAMHD1 gene. Mapping (hg19), alignment, and variant calling were done according to the Broad Institute's best practices using BWA and GATK (McKenna et al. 2010). Variants were computationally annotated and filtered using an in-house bioinformatics pipeline based upon genomic position, minor allele frequency (MAF) within the 1000 Genomes database and the Exome Aggregation Consortium database (n = 61,486 exomes), and predicted effect on the resulting protein (Reilly et al. 2015). Variants were confirmed by bidirectional Sanger dideoxy sequencing in the UNC Hospitals CLIA-certified McLendon laboratory.
Table 2.
Sequencing coverage metrics
| Mean coverage (sample) | 83.6× | ||||
|---|---|---|---|---|---|
| Fraction >8× | Fraction >15× | Fraction >30× | Fraction >50× | ||
| Transcript coverage (NM_015474.3) | Transcript | 1.000 | 1.000 | 0.992 | 0.825 |
| Fraction >8× | Fraction >15× | Fraction >30× | Fraction >50× | ||
| Coverage (per exon; NM_015474.3) | Exon | ||||
| 1 | 1.000 | 1.000 | 0.953 | 0.797 | |
| 2 | 1.000 | 1.000 | 1.000 | 1.000 | |
| 3 | 1.000 | 1.000 | 1.000 | 0.753 | |
| 4 | 1.000 | 1.000 | 1.000 | 1.000 | |
| 5 | 1.000 | 1.000 | 1.000 | 1.000 | |
| 6 | 1.000 | 1.000 | 1.000 | 1.000 | |
| 7 | 1.000 | 1.000 | 0.969 | 0.456 | |
| 8 | 1.000 | 1.000 | 1.000 | 0.400 | |
| 9 | 1.000 | 1.000 | 1.000 | 1.000 | |
| 10 | 1.000 | 1.000 | 1.000 | 1.000 | |
| 11 | 1.000 | 1.000 | 1.000 | 1.000 | |
| 12 | 1.000 | 1.000 | 1.000 | 0.840 | |
| 13 | 1.000 | 1.000 | 1.000 | 1.000 | |
| 14 | 1.000 | 1.000 | 1.000 | 0.037 | |
| 15 | 1.000 | 1.000 | 1.000 | 1.000 | |
| 16 | 1.000 | 1.000 | 1.000 | 1.000 | |
Analysis of Expression of Interferon-Stimulated Genes
Blood from the proband was collected in a PaxGene tube and total RNA was extracted with a PaxGene isolation kit. Quantitative reverse transcription PCR was performed using the TaqMan Universal PCR Master mix (Applied Biosystems) and cDNA derived from 40 ng RNA. TaqMan probes for each specific target were used to quantify relative expression levels of IFI27, IFI44L, IFIT1, ISG15, RSAD2, and SIGLEC1 normalized to HPRT1 and 18S. The median fold change of the six interferon-stimulated genes, when compared to the median of 29 previously collected healthy controls, was used to calculate the interferon score for the patient (Rice et al. 2013). Scores higher than the mean of controls plus two standard deviations are considered positive.
ADDITIONAL INFORMATION
Data Deposition and Access
The variants described in this study were deposited at ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) under accession numbers SCV000803284.1 and SCV000803285.1. Raw sequencing data has been submitted to dbGaP (https://www.ncbi.nlm.nih.gov/gap) and can be found under accession number phs000827.v3.p1.
Ethics Statement
The patient described in this study was enrolled in the North Carolina Genomic Evaluation by Next-Generation Exome Sequencing (NCGENES) research study, with full approval by the University of North Carolina at Chapel Hill Institutional review board as well as parental consent.
Acknowledgments
We thank the family that participated in this study and Manyu Li for Sanger sequencing analysis. This work was supported by the National Human Genome Research Institute of the National Institutes of Health under award number U01HG006487 to J.P.E., J.S.B., and K.E.W.
Competing Interest Statement
The authors have declared no competing interest.
Referees
Gholson Lyon
Anonymous
Anonymous
REFERENCES
- Aicardi J, Goutières F. 1984. A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann Neurol 15: 49–54. [DOI] [PubMed] [Google Scholar]
- Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL, Oojageer A, Anderson B, Pizzino A, Helman G, et al. 2015. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet Part A 167: 296–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, Manel N. 2015. Aicardi–Goutières syndrome and the type I interferonopathies. Nat Rev Immunol 15: 429–440. [DOI] [PubMed] [Google Scholar]
- Crow YJ, Vanderver A, Orcesi S, Kuijpers TW, Rice GI. 2014. Therapies in Aicardi–Goutières syndrome. Clin Exp Immunol 175: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale RC, Gornall H, Singh-Grewal D, Alcausin M, Rice GI, Crow YJ. 2010. Familial Aicardi–Goutières syndrome due to SAMHD1 mutations is associated with chronic arthropathy and contractures. Am J Med Genet A 152A: 938–942. [DOI] [PubMed] [Google Scholar]
- Goncalves A, Karayel E, Rice GI, Bennett KL, Crow YJ, Superti-Furga G, Bürckstümmer T. 2012. SAMHD1 is a nucleic-acid binding protein that is mislocalized due to Aicardi–Goutières syndrome–associated mutations. Hum Mutat 33: 1116–1122. [Google Scholar]
- González-Pérez A, López-Bigas N. 2011. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet 88: 440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB. 2016. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536: 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramantani G, Niggemann P, Bast T, Lee-Kirsch MA. 2010. Reconciling neuroimaging and clinical findings in Aicardi–Goutières syndrome: an autoimmune-mediated encephalopathy. AJNR Am J Neuroradiol 31: E62–E63; author reply E64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravenscroft JC, Suri M, Rice GI, Szynkiewicz M, Crow YJ. 2011. Autosomal dominant inheritance of a heterozygous mutation in SAMHD1 causing familial chilblain lupus. Am J Med Genet A 155A: 235–237. [DOI] [PubMed] [Google Scholar]
- Reilly J, Stanley A, McGee J, Owen P, Schmitt C, Wilhelmsen K. 2015. MaPSeq, a service-oriented architecture for genomics research within an academic biomedical research institution. Informatics 2: 20–30. [Google Scholar]
- Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, Fuller JC, Jackson RM, Lamb T, Briggs TA, et al. 2009. Mutations involved in Aicardi–Goutières syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet 41: 829–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Forte GM, Szynkiewicz M, Chase DS, Aeby A, Abdel-Hamid MS, Ackroyd S, Allcock R, Bailey KM, Balottin U, et al. 2013. Assessment of interferon-related biomarkers in Aicardi–Goutières syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case-control study. Lancet Neurol 12: 1159–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice G, Kitabayashi N, Barth M, Briggs TA, Burton ACE, Carpanelli ML, Cerisola AM, Colson C, Dale RC, Danti FR, et al. 2007. Clinical and molecular phenotype of Aicardi–Goutières syndrome. Am J Hum Genet 81: 713–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarailo-Graovac M, Shyr C, Ross CJ, Horvath GA, Salvarinova R, Ye XC, Zhang LH, Bhavsar AP, Lee JJ, Drögemöller BI, et al. 2016. Exome sequencing and the management of neurometabolic disorders. N Engl J Med 374: 2246–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumienė B, Voisin N, Preikšaitienė E, Petroška D, Grikinienė J, Samaitienė R, Utkus A, Reymond A, Kučinskas V. 2017. Inflammatory myopathy in a patient with Aicardi–Goutières syndrome. Eur J Med Genet 60: 154–158. [DOI] [PubMed] [Google Scholar]
- Uggetti C, La Piana R, Orcesi S, Egitto MG, Crow YJ, Fazzi E. 2009. Aicardi–Goutières syndrome: neuroradiologic findings and follow-up. AJNR Am J Neuroradiol 30: 1971–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White TE, Brandariz-Nuñez A, Martinez-Lopez A, Knowlton C, Lenzi G, Kim B, Ivanov D, Diaz-Griffero F. 2017. A SAMHD1 mutation associated with Aicardi–Goutières syndrome uncouples the ability of SAMHD1 to restrict HIV-1 from its ability to downmodulate type 1 interferon in humans. Hum Mutat 38: 658–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarbrough K, Danko C, Krol A, Zonana J, Leitenberger S. 2016. The importance of chilblains as a diagnostic clue for mild Aicardi–Goutières syndrome. Am J Med Genet A 170: 3308–3312. [DOI] [PubMed] [Google Scholar]