

Abstract
Codon usage bias is universal to all genomes. Hepatitis A virus (HAV) codon usage is highly biased and deoptimized with respect to its host. Accordingly, HAV is unable to induce cellular translational shutoff and its internal ribosome entry site (IRES) is inefficient. Codon usage deoptimization may be seen as a hawk (host cell) versus dove (HAV) game strategy for accessing transfer RNA (tRNA). HAV avoids use of abundant host cell codons and thereby eludes competition for the corresponding tRNAs. Instead, codons that are abundant or rare in cellular messenger RNAs (mRNAs) are used relatively rarely in its genome, although intermediately abundant host cell codons are abundant in the viral genome. Rare codons in the capsid coding region slow down the translation elongation rate, and in doing so intrinsically modulate capsid folding, which is critical to the stability of a virus transmitted through the fecal–oral route. HAV is a paradigmatic example of what has been proposed as a codon usage “code” for protein structure.
Nonrandom usage of synonymous codons, known as codon bias, is common to most species in all domains of life, and each organism has a specific codon usage signature that reflects the evolutionary forces that have acted on its genome (Grantham et al. 1980). Although mutational bias is the main driving force for codon bias, selection may also play a role in shaping the codon usage of a genome (Sharp et al. 1993; Hershberg and Petrov 2008). Among selective forces, translation selection or adaptation to available transfer RNA (tRNA) pools to ensure the efficiency and accuracy of translation have been proposed in many organisms from prokaryotes to eukaryotes (Ikemura 1981; Bennetzen and Hall 1982; Stenico et al. 1994; Moriyama and Powell 1997; Musto et al. 2001; Shah and Gilchrist 2010; Gingold and Pilpel 2011). In particular, for viruses, because of their inability to synthesize tRNAs, translation selection demands an adaptation of their codon usage to that of their cellular hosts. Additionally, selection for specific translation kinetics has also been suggested to shape the codon usage of a genome. Translation kinetics depends on the rate of ribosome traffic on the messenger RNA (mRNA). Generally speaking, tRNAs translating abundant, or preferred codons, are abundant in the tRNA pool, whereas tRNAs translating nonpreferred, or rare codons, are scarce. Consequently, abundant codons speed up translation elongation, although rare codons slow down the speed of translation owing to ribosome stalls induced by the longer time required for incorporating scarce tRNAs into the ribosome A site (Tuller et al. 2010a; Plotkin and Kudla 2011; Wohlgemuth et al. 2013). The right combination of codons can modulate the local rate of translation elongation, temporally separating protein-folding events, and ensuring “beneficial” and avoiding “unwanted” interactions within the growing peptide (Yang and Nielsen 2008). Thus, the local rate of translation elongation can regulate cotranslational protein folding (Plotkin and Kudla 2011; Zhang and Ignatova 2011; Pechmann and Frydman 2013; Chaney and Clark 2015; Yu et al. 2015). This phenomenon has been defined as a codon usage “code” for protein structure (Yu et al. 2015).
Yet another selection pressure that may contribute to shaping the codon usage of viruses is the need to evade cell defense mechanisms. CpG and UpA dinucleotides may be sensed as pathogen-associated molecular patterns (PAMPs) by host cells (Takeshita et al. 2004; Sugiyama et al. 2005; Atkinson et al. 2014; Lester and Li 2014), and viruses thus tend to decrease the frequency of these dinucleotides in their genomes.
Hepatitis A virus (HAV) shows highly biased and deoptimized codon usage, the driving force for which is selection for finely tuned translation kinetics (Sánchez et al. 2003; Aragonès et al. 2010) that results in exceptional capsid stability (Costafreda et al. 2014). Remarkably, this codon deoptimization is very well conserved in the recently described seal HAV-like virus and small mammal hepatoviruses (Anthony et al. 2015; Drexler et al. 2015). This contrasts with other picornaviruses in which there is no such level of codon deoptimization. In addition, the CpG frequency in the HAV genome likely contributes to its codon bias. In this review, we explore the causes and consequences of the unique codon usage of HAV with a special emphasis on translation.
HAV GENOME COMPOSITION AND BEYOND
Codon usage bias is a universal feature of all genomes (Plotkin and Kudla 2011), and it may be measured through the effective number of codons (ENCs) statistic (Wright 1990), which ranges from 20 (extreme bias: only one codon per amino acid is used) to 61 (no bias: all codons are equally used).
The underlying causes of codon usage bias are mostly related to genome composition (mutation pressure), including GC content, GC content at the third position (GC3), mononucleotide composition, and dinucleotide composition. The HAV genome shows an ENC of around 39, among the lowest known of RNA viruses, a low GC content (37%), an even lower GC3 content (26%), and an extremely low dinucleotide CpG frequency (0.39%) (Table 1) (Jenkins and Holmes 2003; Sánchez et al. 2003; Bosch et al. 2010; Belalov and Lukashev 2013). The GC3 content and nucleotide composition at the third position do not explain the highly biased codon usage of HAV (Jenkins and Holmes 2003; D’Andrea et al. 2011; Pintó et al. 2012; Belalov and Lukashev 2013), although uneven base composition and dinucleotide composition may partially contribute to it (Jenkins and Holmes 2003; Bosch et al. 2010; Belalov and Lukashev 2013). Nevertheless, the exceptionally low dinucleotide CpG frequency of HAV, by far the lowest in the Picornaviridae family and among the lowest in the entire virus world, cannot be explained by the overall low GC content of its genome, because the dinucleotide GpC frequency is 2.93%, which is only minimally below that of other picornaviruses (Table 1) (Bosch et al. 2010). Consequently, it must result from other factors.
Table 1.
Genomic parameters of hepatitis A virus (HAV) compared to poliovirus (PV)
| GCa | GC3b | CpGc | GpCd | ENCe | RCDIf | AA coded by rare codonsg | Total rare codonsh | |
|---|---|---|---|---|---|---|---|---|
| HAV | 37 | 26 | 0.36 | 2.93 | 39 | 1.70 | 15 | 27 |
| PV | 46 | 47 | 2.68 | 4.64 | 54 | 1.14 | 5 | 8 |
aGC: % GC.
bGC3: % GC at the third position.
cCpG: % CpG dinucleotide.
dGpC: % GpC dinucleotide.
eENC: Effective number of codons.
fRCDI: Relative codon deoptimization index.
gNumber of different amino acids (AAs) coded by rare codons.
hTotal number of different rare codons used.
In addition to genome composition, other selective pressures may play a role in shaping the codon usage of an organism. For example, although the dinucleotide composition of a virus genome may contribute to its codon bias, the dinucleotide composition itself may be shaped evolutionarily by the need to elude cellular antiviral responses, although the mechanism by which RNA composition is sensed by cellular PAMP receptors is mostly still unknown (Sugiyama et al. 2005; Greenbaum et al. 2009; Atkinson et al. 2014). Hence, the scarcity of CpG dinucleotides in the HAV genome may result from evolutionary pressure to avoid antiviral responses that indirectly influence codon usage. CpG frequency has been proposed to correlate inversely with the replicative capacity of a virus owing to an inability of PAMP receptors to sense low CpG viral genomes (Atkinson et al. 2014). In light of this, it is intriguing that cell-adapted strains of HAV, which retain an extremely low CpG frequency, show instead very low replicative capacities.
For HAV, the need for a highly stable capsid structure that allows for both intrahost (low pH, high levels of biliary salts) and interhost (environmental resistance) phases of its biological cycle, may require extremely accurate cotranslational folding of the capsid proteins. This can be achieved through the use of an appropriate combination of abundant and rare codons in selected regions of the genome (Sánchez et al. 2003; Aragonès et al. 2010), thereby contributing further to the codon usage bias. Fifteen different amino acids are encoded by rare codons in the HAV genome, contributing to a total of 27 rare codons; in contrast, these numbers are 5 and 8, respectively, for poliovirus (Table 1) (Sánchez et al. 2003; Pintó et al. 2007). This increase in the use of rare codons comes from the fact that HAV uses, as rare codons, not only those that are rare in cellular mRNAs but also those that are abundantly used by the cell. HAV uses then, as preferred codons, those that are neither abundant nor rare in the cell. HAV codon usage thus can be considered to be opposite or deoptimized with respect to that of human cells. The relative codon deoptimization index (RCDI) measures how deoptimized a gene is with respect to a reference data set (Mueller et al. 2006). In our case, the RCDI indicates how deoptimized the HAV codon usage is with respect to its human host. An RCDI value of 1 indicates a perfect match between the virus and host codon usage, whereas a higher value signifies greater deviation from the host. The RCDI of HAV approximates 1.70, indicating that HAV has highly deoptimized codon usage compared to poliovirus, which has an RCDI of 1.14 (Table 1) (Aragonès et al. 2010).
In summary, the HAV genome has a low GC content, a low GC3 content, an extremely low CpG frequency, and highly biased and deoptimized codon usage (Table 1).
INTERRELATED MOLECULAR FEATURES PROMOTING SLOW RNA TRANSLATION
Viruses depend on the host translation machinery and resources for their own protein synthesis. Many viruses have developed strategies to hijack the cell translation apparatus, including modifications of key eukaryotic translation factors, the induction of so-called cellular shutoff, and the evolution of specialized and efficient cis-acting elements that recruit ribosomes, that is, the internal ribosome entry site (IRES) of picornaviruses (Walsh et al. 2013). HAV is relatively unique among pathogenic mammalian picornaviruses in not inducing cellular translational shutoff. The formation of translation initiation complexes on the HAV IRES requires both eIF4G and eIF4E, translation initiation factors that are proteolytically cleaved or suppressed, respectively, by many other picornaviruses to shut off translation of cellular mRNAs (Ali et al. 2001). Additionally, HAV has a very inefficient IRES (Brown et al. 1991; Whetter et al. 1994) and, as described above, highly deoptimized codon usage downstream from its IRES.
These three unique features of HAV are interconnected and, consequently, like the chicken and the egg, it is difficult to define which came first. Rather, they are the result of the coevolution of different parts of the genome and their epistatic interactions. The inability of HAV to shut down host cell protein synthesis can be envisioned as a sort of “hawk” versus “dove” game in the quest for resources such as ribosomes and tRNAs (Fig 1). The deoptimized HAV codon usage may be considered a “dove”-like strategy to tackle unfair competition for tRNAs within the cell, while keeping a certain proportion of codons pairing with incidentally very scarce tRNAs and hence behaving as rare codons during translation (Fig. 1). Under this scheme, translation is anticipated to be very slow overall, and locally extremely slow owing to ribosome stalls (Tuller et al. 2010a). If combined with a very active IRES, this could prompt the arrest of too many ribosomes on a single translating RNA molecule. An inefficient IRES would be instead preferable to avoid ribosome traffic jams.
Figure 1.
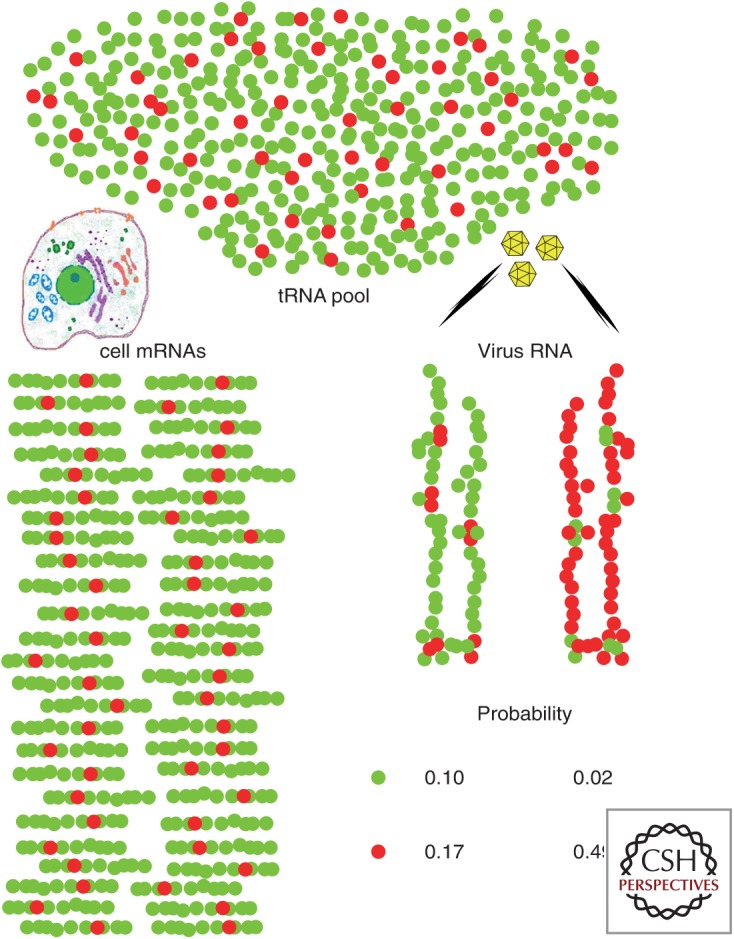
Probability that hepatitis A virus (HAV) RNA will pair successfully with the abundant or nonabundant transfer RNAs (tRNAs) required for its translation under two different codon usage simulations, considered within the framework of a very simplistic model for cellular codon usage involving only a single amino acid encoded by either a major abundant codon (green balls) or a minor rare codon (red balls). Three assumptions are made: (1) HAV is a very slowly replicating virus (inefficient internal ribosome entry site [IRES]) with few, but very lengthy translating RNAs compared to cellular messenger RNAs (mRNAs) (the model assumes one translating HAV RNA for every 50 cellular mRNAs per cell, and that it is 6× longer than the average cellular mRNA), (2) HAV is unable to shut down cellular protein synthesis, and (3) tRNA pools are limiting and adapted to the cellular mRNAs. In a hypothetical situation in which HAV adopts the same codon usage as the cell (optimized: viral RNA on the left), the probabilities of tRNA pairing with the most abundant codon and tRNA pairing with the less abundant codon are 0.1 and 0.17, respectively. In contrast, with codon usage opposite with respect to the cell (deoptimized: viral RNA on the right), these probabilities are 0.02 and 0.49. The latter scenario provides an overall advantageous outcome for the virus, with a higher probability of getting tRNAs pairing with its most abundant codons, while accomplishing the goal of using incidentally very low abundance tRNAs to slow down translation at certain positions.
A clue to elucidate such intricate genomic connections comes from the ripple effect that adaptation to replication in conditions of artificially induced shutoff has had on the codon usage and IRES activity of the cell-adapted HM175-43c variant of HAV (Lemon et al. 1991), henceforth termed L0 (initial lineage). An analysis of the RCDI along the capsid-coding region of the L0 genome reveals local differences in the level of codon optimization (Fig. 2A), with three highly deoptimized regions (higher peaks) and two more optimized regions (low valleys). We adapted the L0 virus to conditions in which cellular transcription is inhibited by actinomycin D. This was a lengthy process involving an initial loss of fitness followed by fitness recovery, driven by changes in codon usage within the capsid-coding region (Aragonès et al. 2010). The overall dynamics of the adaptive changes in codon usage were characterized first by re-deoptimization, followed subsequently by optimization (Costafreda et al. 2014). Virus populations that were well adapted to transcriptional shutoff were comprised of haplotypes with different degrees of codon optimization (Pérez-Rodríguez et al. 2016), as revealed by deep sequencing of a VP1 segment aligning with the second valley in the RCDI plot (Fig. 2A). One of these haplotypes (HM175-HP) has been extensively examined (Pérez-Rodríguez et al. 2016). The most evident changes in codon usage in HP virus compared to its ancestor, L0, were toward optimization and located in the VP1-coding region referred to above (Fig. 2A, green plot). The genetic analysis of quasispecies variation in this VP1 segment showed how critical the frequency of codons is with respect to cellular codon usage. In the L0 population, the number of nonsynonymous mutations per nonsynonymous site (Ka) was much higher than the number of synonymous mutations per synonymous site (Ks). This unexpected behavior is likely related to a need to maintain a basal codon bias among new codons generated through mutation, which is facilitated by nonsynonymous mutations. In contrast, compared to L0, the HP population showed a similar Ks but a significantly lower Ka. This different pattern may be related to the need to change codon frequencies under conditions of transcriptional shutoff, which is more likely to occur through synonymous mutations.
Figure 2.
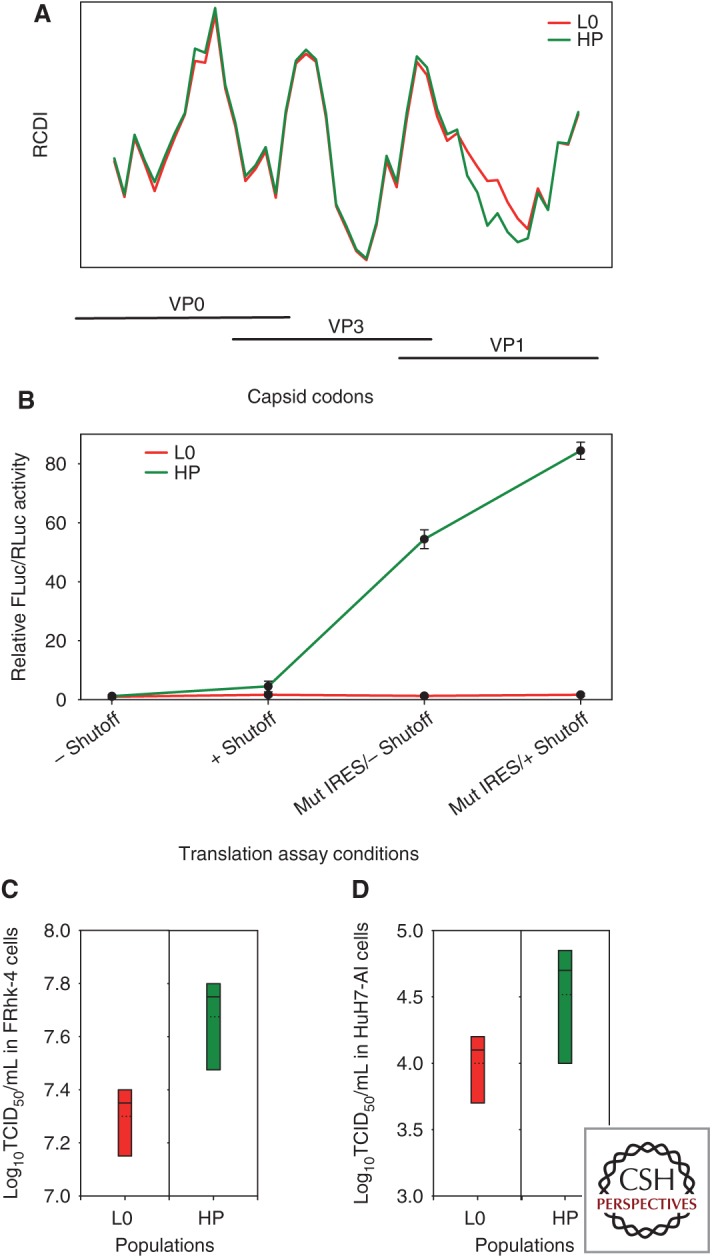
Codon usage and its relationship to the rate of translation and replication capacities of two variants derived from the HM175 strain of hepatitis A virus (HAV) (HM175-43c: L0 parental type; HM175-HP: HP fast-growing type) that differ in their level of codon optimization. (A) Relative codon deoptimization index (RCDI) of the L0 and HP strains with respect to the human codon usage. RCDI values shown correspond to overlapping 100 codon segments of the capsid-coding RNA with 15 codon-sliding windows. The higher the RCDI value, the higher the deviation of viral codon usage from host cell codon usage. The HP strain shows a remarkable decrease in RCDI values in a region extending 50% of the VP1 length, compared to the L0 strain, which denotes more optimized codon usage relative to the cell. (B) Rate of translation elongation of the VP1 fragment showing differences in the RCDI between L0 and HP strains. The elongation rate is measured as the relative FLuc/RLuc activity, using a bicistronic vector in which Renilla luciferase (RLuc) translation is cap dependent and Firefly luciferase (FLuc) translation is HAV internal ribosome entry site (IRES) dependent. The VP1 fragment is cloned under control of the IRES just upstream of and in frame with FLuc. Translation elongation was assayed using two bicistronic vectors representing the parental-type IRES (L0-type IRES: two first points of the kinetics) and the mutated-active-type internal ribosome entry site (IRES) (HP-type IRES: two last points of the kinetics), under conditions of no shutoff (–) or shutoff (+). Results are expressed as fold change of the elongation rate relative to the L0 population with the parental-type IRES and in the absence of shutoff. (C) Box plots of the virus yields obtained in FRhK-4 cells. (D) Box plots of virus yields obtained in HuH7-AI cells. In both C and D, the multiplicity of infection was 1. Dotted lines represent mean titer.
More remarkably, we found that the HP strain harbors a more efficient IRES, caused by the acquisition of three mutations (U359C, U590C, and U726C), which may potentially affect the IRES RNA secondary structure and its interaction with 18S ribosomal RNA (rRNA) (Le et al. 1993; Scheper et al. 1994; Pérez-Rodríguez et al. 2016). The combined effect of codon optimization, coupled with the more active IRES, on the translation elongation rate of the VP1 segment was extraordinary (Fig. 2B). Whereas in the L0 population there was no substantial change in the translation elongation rate under shutoff conditions, or in the presence or absence of the IRES mutations (Fig. 2B; red plot), in the HP population, the IRES mutations caused a significant increase in the elongation rate, which was further increased with transcriptional shutoff (Fig. 2B; green plot). Ultimately, all of these changes resulted in a more rapidly replicating phenotype for the HP strain in different cell types, including the fetal rhesus kidney cells (FRhK-4) used for shutoff adaptation (Fig. 2C) and HuH7-AI human hepatocytes (Fig. 2D).
Overall, the efficiency with which HAV RNA is translated is determined by the efficiency of translation initiation, which depends on the RNA structure near the initiator AUG (i.e., the IRES) and by regulation of the rate of translation elongation driven by codon usage, as previously suggested in cell systems (Kudla et al. 2009; Tuller et al. 2010b). However, contrary to what has been suggested recently (Pop et al. 2014), it is also determined by the available tRNA pools, which in turn are likely to be affected by the abundance of translationally active cellular mRNAs (Fig. 3), as previously proposed (Sorensen et al. 1989).
Figure 3.

Cellular shutoff induces a ripple effect on codon usage optimization and the acquisition of internal ribosome entry site (IRES) mutations. In the absence of cellular shutoff with high cellular messenger RNA (mRNA) synthesis (top part of the figure), hepatitis A virus (HAV) shows a very inefficient IRES, a deoptimized codon usage, and very low yields of highly stable capsids. Under conditions of cellular shutoff with a decreased cellular mRNA synthesis (bottom part of the figure), HAV shows an optimized codon usage, a more active IRES, and produces high yields of capsids with subtle folding changes.
TRANSLATION ELONGATION RATE CONTRIBUTES TO AN EXCEPTIONALLY STABLE CAPSID PHENOTYPE
Despite the increasing importance accorded to the role of codon usage in regulating cotranslational folding of polypeptides, there are few clear examples that illustrate such an effect (Kimchi-Sarfaty et al. 2007; Yu et al. 2015).
We have proposed that the highly deoptimized codon usage of HAV has resulted from selection based on the fine-tuning of translation that contributes through a “chaperone-like” function to a highly stable capsid (Aragonès et al. 2010). However, for HAV, which is a fastidious virus to propagate in cell culture, subtle changes in capsid folding that might be expected to be associated with codon usage variation are not easily measured by directly studying capsid structure. Instead, we have assessed indirect measures of capsid integrity, such as the kinetics of RNA uncoating, which is dependent on the flexibility of the capsid. HAV has been described as requiring a very long time to uncoat in cell culture (Bishop and Anderson 2000). The L0 parental virus described above requires ∼18 h for 50% of the virions to be uncoated (Costafreda et al. 2014). In contrast, the HP strain requires significantly less time, ∼14 h. This more efficient uncoating may contribute to its fast-growing phenotype. Thus, the L0 strain is likely to have a more rigid capsid than HP virus, which is largely caused by differences in codon usage. Although the amino acid sequences of the L0 and HP capsid proteins differ at three positions, the positive relationship between translation elongation efficiency and transcriptional shutoff (Fig. 2B, green plot) is indicative of a major influence of codon frequency on translation speed. Indeed, the capsid folding of different HAV populations can be made to differ by simply changing the growth conditions, that is, by inhibiting cellular protein synthesis, without a single amino acid replacement (Costafreda et al. 2014). Additionally, subtle changes in capsid folding can be inferred from a slight loss of recognition of HP compared to L0 capsids by several monoclonal antibodies (mAbs), despite similar recognition by polyclonal Ab (Pérez-Rodríguez et al. 2016). Altogether, differences in folding of the capsid of HP versus L0 virus are likely caused by the sum of the effects of changes in translation elongation and the amino acid substitutions.
Another remarkable point is the high antigenic stability of the HAV capsid and the fact that there is only a single serotype. Although mAb-resistant mutants can be isolated in vitro (Nainan et al. 1992; Ping and Lemon 1992; Aragonès et al. 2008), only a few natural antigenic variants have been detected (Sánchez et al. 2002; Pérez-Sautu et al. 2011). This suggests that there are severe capsid structural constraints preventing the emergence of new serotypes. A relationship between the impact of codon usage on capsid folding and these structural constraints may contribute to this antigenically stable phenotype (Aragonès et al. 2008). An analysis of HAV quasispecies emerging under the pressure of mAbs revealed that 86% and 66% of amino acid replacements detected in VP1 and VP3, respectively, were transition mutations that maintained basal codon bias. Similarly, 7% and 12%, respectively, were transversions maintaining the codon frequency. In contrast, only 5% and 16%, and 2% and 6%, were transitions and transversions that resulted in a change of codon frequency. These data not only reflect the dominance of transitions over transversions, but also highlight the importance of codon frequency. Only 31% and 26% of all potential amino acid replacements that maintain the frequency of rare codons specifying VP3 and VP1 residues on the capsid surface, respectively, were induced by transition mutations. This may explain the lack of coincidence between residues encoded by rare codons (Fig. 4, white residues) and those at which replacements were detected in the L0 quasispecies under selective pressure of two mAbs (yellow residues); only a few matches are evident (clear blue residues) in a merged projection image of the HAV protomer (Wang et al. 2015).
Figure 4.

Hepatitis A virus (HAV) protomer (Wang et al. 2015) showing residues encoded by rare codons (white), and residues replaced in VP3 and VP1 in quasispecies placed under the immune pressure of monoclonal antibodies (mAbs) (yellow). Although residues undergoing replacement are located very close to residues encoded by rare codons, there is a general lack of coincidence with only few matching positions (clear blue). (A) Selection with H7C27 mAb. (B) Selection with K34C8 mAb.
In summary, these data emphasize the role of codon usage, particularly the use of rare codons, in shaping the folding of the HAV capsid (Fig. 3), which turns out to be particularly resistant to low pH, high temperature, and desiccation (Abad et al. 1994; Wang et al. 2015). HAV thus may be a paradigmatic example of the proposed codon usage “code” for protein structure (Yu et al. 2015).
CONCLUDING REMARKS
HAV is a very unique virus with an extreme genomic composition characterized by very low GC, GC3, and CpG content, which results in highly biased codon usage. Multiple selective forces may contribute to this bias, including selection to escape PAMP receptor sensing, and selection for an optimal balance between accurate and efficient translation and a controlled rate of translation elongation. The most striking feature of HAV codon usage is its extreme deoptimization with regard to host cell codon usage, which is a paradox because viruses generally adapt to cellular tRNA pools. However, unlike many viruses that have evolved mechanisms to shut down cellular protein synthesis to gain a competitive advantage for translation resources, HAV fails to shut down cellular translation and instead adopts a dove-like behavior, settling for those tRNAs that are in less demand by the cell. Accordingly, its IRES is very inefficient. This situation results in an overall slow rate of translation that is slower still at certain genome locations as a result of the use of many different rare codons. There is a growing body of evidence that codon usage impacts local translational dynamics, and that variations in the translation elongation rate may facilitate cotranslational folding. The HAV capsid shows an exceptional physical stability, which can be, at least partially, related to the pace of translation elongation imposed by its very special codon usage. Similarly, the fact that only a single serotype of HAV exists may result from the need to maintain rare codon frequencies in and around key epitopes. HAV mutants with changes in capsid codon usage show differences in the local rate of translation, which in turn influences folding as revealed by the formation of physically and antigenically different capsids. In summary, the influence of HAV codon usage on the local translation elongation rate and its subsequent impact on capsid conformation is a prime example of how codon usage “encodes” protein folding.
ACKNOWLEDGMENTS
Work in our laboratory is supported by Grants BIO2011-23461 and BIO2014-53285-R from the Spanish Ministry of Economy, the last cofinanced by the European Regional Development Fund (ERDF), and by the Generalitat de Catalunya Biotechnology Reference Network (XRB). F.-J.P.-R. and M.dC. are recipients of fellowships from the Spanish Ministry of Science and Innovation and Ministry of Economy, respectively. L.D'A. is a recipient of a fellowship from the Spanish Ministry of External Affairs and Cooperation.
Footnotes
Editors: Stanley M. Lemon and Christopher Walker
Additional Perspectives on Enteric Hepatitis Viruses available at www.perspectivesinmedicine.org
REFERENCES
- Abad FX, Pintó RM, Bosch A. 1994. Survival of enteric viruses on environmental fomites. Appl Environ Microbiol 60: 3704–3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali IK, McKendrick L, Morley SJ, Jackson RJ. 2001. Activity of the hepatitis A virus IRES requires association between the cap-binding translation initiation factor (eIF4E) and eIF4G. J Virol 75: 7854–7863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony SJ, St Leger JA, Liang E, Hicks AL, Sanchez-Leon MD, Jain K, Lefkowitch JH, Navarrete-Macias I, Knowles N, Goldstein T, et al. 2015. Discovery of a novel hepatovirus (Phopivirus of seals) related to human hepatitis A virus. mBio 6: e01180–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragonès L, Bosch A, Pintó RM. 2008. Hepatitis A virus mutant spectra under the selective pressure of monoclonal antibodies: Codon usage constraints limit capsid variability. J Virol 82: 1688–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragonès L, Guix S, Ribes E, Bosch A, Pintó RM. 2010. Fine-tuning translation kinetics selection as the driving force of codon usage bias in the hepatitis A virus capsid. PLoS Pathog 6: e1000797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson NJ, Witteveldt J, Evans DJ, Simmonds P. 2014. The influence of CpG and UpA dinucleotide frequencies on RNA virus replication and characterization of the innate cellular pathways underlying virus attenuation and enhanced replication. Nucleic Acids Res 42: 4527–4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belalov IS, Lukashev AN. 2013. Causes and implications of codon usage bias in RNA viruses. PLoS ONE 8: e56642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennetzen JL, Hall BD. 1982. Codon selection in yeast. J Biol Chem 257: 3026–3031. [PubMed] [Google Scholar]
- Bishop NE, Anderson DA. 2000. Uncoating kinetics of hepatitis A virus virions and provirions. J Virol 74: 3423–3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch A, Mueller S, Pintó RM. 2010. Codon biases and viral fitness. In The picornaviruses (ed. Ehrenfeld E , Domingo E , Roos R ), pp. 271–283. American Society for Microbiology, Washington, DC. [Google Scholar]
- Brown EA, Day SP, Jansen RW, Lemon SM. 1991. The 5′ nontranslated region of hepatitis A virus RNA: Secondary structure and elements required for translation in vitro. J Virol 65: 5828–5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaney JL, Clark PL. 2015. Roles for synonymous codon usage in protein biogenesis. Annu Rev Biophys 44: 143–166. [DOI] [PubMed] [Google Scholar]
- Costafreda MI, Pérez-Rodríguez FJ, D’Andrea L, Guix S, Ribes E, Bosch A, Pintó RM. 2014. Hepatitis a virus adaptation to cellular shut off is driven by dynamic adjustments of codon usage and results in the selection of populations with altered capsids. J Virol 88: 5029–5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Andrea LA, Pintó RM, Bosch A, Musto HC, Cristina J. 2011. A detailed comparative analysis on the overall codon usage patterns in hepatitis A virus. Virus Res 157: 19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drexler JF, Corman VM, Lukashev AN, van den Brand JM, Gmyl AP, Brunink S, Rasche A, Seggewibeta N, Feng H, Leijten LM, et al. 2015. Evolutionary origins of hepatitis A virus in small mammals. Proc Natl Acad Sci 112: 15190–15195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingold H, Pilpel Y. 2011. Determinants of translation efficiency and accuracy. Mol Syst Biol 7: 481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grantham R, Gautier C, Gouy M, Mercier R, Pave A. 1980. Codon catalog usage and the genome hypothesis. Nucleic Acids Res 8: r49–r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenbaum BD, Rabadan R, Levine AJ. 2009. Patterns of oligonucleotide sequences in viral and host cell RNA identify mediators of the host innate immune system. PLoS ONE 4: e5969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberg R, Petrov DA. 2008. Selection on codon bias. Annu Rev Genet 42: 287–299. [DOI] [PubMed] [Google Scholar]
- Ikemura T. 1981. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes. J Mol Biol 146: 1–21. [DOI] [PubMed] [Google Scholar]
- Jenkins GM, Holmes EC. 2003. The extent of codon usage bias in human RNA viruses and its evolutionary origin. Virus Res 92: 1–7. [DOI] [PubMed] [Google Scholar]
- Kimchi-Sarfaty C, Oh JM, Kim IW, Sauna ZE, Calcagno AM, Ambudkar SV, Gottesman MM. 2007. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science 315: 525–528. [DOI] [PubMed] [Google Scholar]
- Kudla G, Murray AW, Tollervey D, Plotkin JB. 2009. Coding-sequence determinants of gene expression in Escherichia coli. Science 324: 255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le SY, Chen JH, Sonenberg N, Maizel JV Jr. 1993. Conserved tertiary structural elements in the 5′ nontranslated region of cardiovirus, aphthovirus and hepatitis A virus RNAs. Nucleic Acids Res 21: 2445–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemon SM, Murphy PC, Shields PA, Ping LH, Feinstone SM, Cromeans T, Jansen RW. 1991. Antigenic and genetic variation in cytopathic hepatitis A virus variants arising during persistent infection: Evidence for genetic recombination. J Virol 65: 2056–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester SN, Li K. 2014. Toll-like receptors in antiviral innate immunity. J Mol Biol 426: 1246–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama EN, Powell JR. 1997. Codon usage bias and tRNA abundance in Drosophila. J Mol Evol 45: 514–523. [DOI] [PubMed] [Google Scholar]
- Mueller S, Papamichail D, Coleman JR, Skiena S, Wimmer E. 2006. Reduction of the rate of poliovirus protein synthesis through large-scale codon deoptimization causes attenuation of viral virulence by lowering specific infectivity. J Virol 80: 9687–9696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musto H, Cruveiller S, D’Onofrio G, Romero H, Bernardi G. 2001. Translational selection on codon usage in Xenopus laevis. Mol Biol Evol 18: 1703–1707. [DOI] [PubMed] [Google Scholar]
- Nainan OV, Brinton MA, Margolis HS. 1992. Identification of amino acids located in the antibody-binding sites of human hepatitis A virus. Virology 191: 984–987. [DOI] [PubMed] [Google Scholar]
- Pechmann S, Frydman J. 2013. Evolutionary conservation of codon optimality reveals hidden signatures of cotranslational folding. Nat Struct Mol Biol 20: 237–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Rodríguez FJ, D’Andrea L, de Castellarnau M, Costafreda MI, Guix S, Ribes E, Quer J, Gregori J, Bosch A, Pintó RM. 2016. Improving virus production through quasispecies genomic selection and molecular breeding. Sci Rep 6: 35962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Sautu U, Costafreda MI, Cayla J, Tortajada C, Lite J, Bosch A, Pintó RM. 2011. Hepatitis A virus vaccine escape variants and potential new serotype emergence. Emerg Infect Dis 17: 734–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping LH, Lemon SM. 1992. Antigenic structure of human hepatitis A virus defined by analysis of escape mutants selected against murine monoclonal antibodies. J Virol 66: 2208–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pintó RM, Aragonès L, Costafreda MI, Ribes E, Bosch A. 2007. Codon usage and replicative strategies of hepatitis A virus. Virus Res 127: 158–163. [DOI] [PubMed] [Google Scholar]
- Pintó RM, D’Andrea L, Pérez-Rodríguez FJ, Costafreda MI, Ribes E, Guix S, Bosch A. 2012. Hepatitis A virus evolution and the potential emergence of new variants escaping the presently available vaccines. Future Microbiol 7: 331–346. [DOI] [PubMed] [Google Scholar]
- Plotkin JB, Kudla G. 2011. Synonymous but not the same: The causes and consequences of codon bias. Nat Rev Genet 12: 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop C, Rouskin S, Ingolia NT, Han L, Phizicky EM, Weissman JS, Koller D. 2014. Causal signals between codon bias, mRNA structure, and the efficiency of translation and elongation. Mol Syst Biol 10: 770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez G, Pintó RM, Vanaclocha H, Bosch A. 2002. Molecular characterization of hepatitis a virus isolates from a transcontinental shellfish-borne outbreak. J Clin Microbiol 40: 4148–4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez G, Bosch A, Pintó RM. 2003. Genome variability and capsid structural constraints of hepatitis A virus. J Virol 77: 452–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheper GC, Voorma HO, Thomas AA. 1994. Basepairing with 18S ribosomal RNA in internal initiation of translation. FEBS Lett 352: 271–275. [DOI] [PubMed] [Google Scholar]
- Shah P, Gilchrist MA. 2010. Effect of correlated tRNA abundances on translation errors and evolution of codon usage bias. PLoS Genet 6: e1001128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM, Stenico M, Peden JF, Lloyd AT. 1993. Codon usage: Mutational bias, translational selection, or both? Biochem Soc Trans 21: 835–841. [DOI] [PubMed] [Google Scholar]
- Sorensen MA, Kurland CG, Pedersen S. 1989. Codon usage determines translation rate in Escherichia coli. J Mol Biol 207: 365–377. [DOI] [PubMed] [Google Scholar]
- Stenico M, Lloyd AT, Sharp PM. 1994. Codon usage in Caenorhabditis elegans: Delineation of translational selection and mutational biases. Nucleic Acids Res 22: 2437–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama T, Gursel M, Takeshita F, Coban C, Conover J, Kaisho T, Akira S, Klinman DM, Ishii KJ. 2005. CpG RNA: Identification of novel single-stranded RNA that stimulates human CD14+CD11c+ monocytes. J Immunol 174: 2273–2279. [DOI] [PubMed] [Google Scholar]
- Takeshita F, Gursel I, Ishii KJ, Suzuki K, Gursel M, Klinman DM. 2004. Signal transduction pathways mediated by the interaction of CpG DNA with Toll-like receptor 9. Semin Immunol 16: 17–22. [DOI] [PubMed] [Google Scholar]
- Tuller T, Carmi A, Vestsigian K, Navon S, Dorfan Y, Zaborske J, Pan T, Dahan O, Furman I, Pilpel Y. 2010a. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell 141: 344–354. [DOI] [PubMed] [Google Scholar]
- Tuller T, Waldman YY, Kupiec M, Ruppin E. 2010b. Translation efficiency is determined by both codon bias and folding energy. Proc Natl Acad Sci 107: 3645–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D, Mathews MB, Mohr I. 2013. Tinkering with translation: Protein synthesis in virus-infected cells. Cold Spring Harb Perspect Biol 5: a012351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Ren J, Gao Q, Hu Z, Sun Y, Li X, Rowlands DJ, Yin W, Wang J, Stuart DI, et al. 2015. Hepatitis A virus and the origins of picornaviruses. Nature 517: 85–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whetter LE, Day SP, Elroystein O, Brown EA, Lemon SM. 1994. Low efficiency of the 5′ nontranslated region of hepatitis A virus RNA in directing cap-independent translation in permissive monkey kidney cells. J Virol 68: 5253–5263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlgemuth SE, Gorochowski TE, Roubos JA. 2013. Translational sensitivity of the Escherichia coli genome to fluctuating tRNA availability. Nucleic Acids Res 41: 8021–8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright F. 1990. The effective number of codons used in a gene. Gene 87: 23–29. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R. 2008. Mutation-selection models of codon substitution and their use to estimate selective strengths on codon usage. Mol Biol Evol 25: 568–579. [DOI] [PubMed] [Google Scholar]
- Yu CH, Dang Y, Zhou Z, Wu C, Zhao F, Sachs MS, Liu Y. 2015. Codon usage influences the local rate of translation elongation to regulate co-translational protein folding. Mol Cell 59: 744–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Ignatova Z. 2011. Folding at the birth of the nascent chain: Coordinating translation with co-translational folding. Curr Opin Struct Biol 21: 25–31. [DOI] [PubMed] [Google Scholar]