

Abstract
YES is a member of the SRC family kinase (SFK) group of non-receptor tyrosine kinases, which are implicated in multiple key cellular processes involved in oncogenesis. Antitubulin agents have been widely used as chemotherapeutics for cancer patients and these drugs arrest cells in mitosis, leading to subsequent cell death. In the present study, we define a mechanism for phospho-regulation of YES that is critical for its role in response to antitubulin agents. Specifically, we found that YES is phosphorylated at multiple sites on its N-terminal unique domain by the cell cycle kinase CDK1 during antitubulin drug-induced mitotic arrest. Phosphorylation of YES occurs during normal mitosis. Deletion of YES causes arrest in prometaphase and polyploidy in a p53-independent manner. We further show that YES regulates antitubulin chemosensitivity. Importantly, mitotic phosphorylation is essential for these effects. In support of our findings, we found that YES expression is high in recurrent ovarian cancer patients. Finally, through expression profiling, we documented that YES phosphorylation affects expression of multiple cell cycle regulators. Collectively, our results reveal a previously unrecognized mechanism for controlling the activity of YES during antitubulin chemotherapeutic treatment and suggest YES as a potential target for the treatment of antitubulin-resistant cancer.
Keywords: YES, mitotic phosphorylation, CDK1, taxol sensitivity, chemotherapy
1. Introduction
Antitubulin drugs, such as paclitaxel (taxol) and vinblastine, are widely used for various malignancies, including ovarian, breast, and non-small cell lung cancers [1–3]. These drugs induce mitotic arrest through activation of the spindle checkpoint and subsequent apoptotic cell death. However, despite their wide use in cancer treatment, patient response is highly variable, with drug resistance remaining a major clinical issue [3,4]. Thus, identification of new regulators and/or signaling pathways that are triggered by antitubulin agents may provide useful information on mechanisms underlying chemoresistance and lead to the development of novel prognostic and/or therapeutic approaches related to antitubulin chemotherapeutics.
The SFKs are implicated in multiple signaling processes in oncogenesis [5]. The SFKs include nine members: SRC, YES, LYN, FYN, FGR, HCK, LCK, BLK, and YRK [5]. SRC, YES, LYN, and FYN are expressed in various cell types, while the expression of other members is restricted to specific tissues [5,6]. The catalytic activity of SFKs is mainly regulated at two highly conserved tyrosine residues: the autophosphorylation site Y419 (numbering in SRC) in the activation loop and Y530 at the extreme C-terminus regulatory tail. While phosphorylation of Y419 promotes kinase activity, increased Y530 phosphorylation inhibits SRC catalytic ability Previous reports showed that YES is oncogenic in malignant mesothelioma [8], melanoma [9], and colorectal cancer cells [10]. A recent study also showed that aberrant YES expression affects cell cycle progression and apoptosis in ovarian cancer cells [11]. Despite the oncogenic function of YES, its role and regulation in response to antitubulin chemotherapeutics have not been defined.
Here we present that YES is hyperphosphorylated during antitubulin drug-induced mitotic arrest and dysregulation of YES influences antitubulin chemosensitivity. We further characterized the phosphorylation sites, the corresponding kinase, and the functional significance of the phosphorylation. Our data reveal a new layer of regulation for YES activity in response to antitubulin chemotherapeutics, suggesting YES as a potential target for the treatment of antitubulin drug-resistant cancer patients.
2. Materials and methods
2.1. Expression constructs, cell culture and transfection
Full-length human YES cDNA was a gift from William Hahn (Addgene 23938, pDONR223-YES1). To make the lentiviral-mediated Flag-tagged YES expression construct, the abovementioned cDNA was cloned into pSIN4-Flag-IRES vector [12]. Point mutations were generated by the QuikChange Site-Directed PCR Mutagenesis Kit (Stratagene, CA, USA) and verified by sequencing.
HEK293T, HeLa, U2OS, and OVCAR8 cell lines were purchased from American Type Culture Collection (ATCC, VA, USA) and were maintained in DMEM media supplemented with 10% FBS and 1% antibiotics. The HEK293T, HeLa, U2OS, and OVCAR8 cell lines were authenticated at ATCC and used at low passages. HCT116 and HCT116-p53−/− cell lines were kindly provided by Dr. Bert Vogelstein (Johns Hopkins University) and were cultured with McCoy’s 5A medium. Attractene (Qiagen, MD, USA) was used for transient overexpression following the manufacturer’s instructions. Nocodazole (100 ng/ml for 16–20 h) and taxol (0.1 µM for 16 h) were used to arrest cells in G2/M phase unless otherwise indicated. The VX680 (Aurora-A, -B, -C inhibitor), ZM447439 (Aurora-B, -C inhibitor), and BI2536 (Plk1 inhibitor) were from Selleck Chemicals (TX, USA). The MK5108 (Aurora-A inhibitor) was from Merck (NJ, USA), and RO3306 (CDK1 inhibitor) and Purvalanol A (CDK1/2/5 inhibitor) were from ENZO Life Sciences (NY, USA). All other chemicals were either from Sigma (MO, USA) or Thermo Fisher (MA, USA).
2.2. Cell line establishment
Stable overexpression and re-expression of YES (wild type and mutants) in YES-knockout (KO) cells were achieved by lentivirus-mediated infection. Lentivirus packaging, infection, and subsequent selection were done as we have described previously [13]. Gene knockout was achieved by a CRISPR-mediated method. CRISPR/double nickase knockout plasmids were purchased from Santa Cruz Biotechnology (SC-400261-NIC-2) (TX, USA) and transfection and clone selection were done as instructed by the manufacturer.
2.3. Phos-tag and Western blot analysis
Phos-tag™ was purchased from Wako Pure Chemical Industries, Ltd. (304–93521) (VA, USA) and used at 10 or 20 µM (with 100 µM MnCl2) in 8% SDS-acrylamide gels as we described previously [14]. Western blotting, immunoprecipitation, and lambda phosphatase treatment assays were done as described [13].
2.4. Recombinant protein purification and In vitro kinase assay
YES cDNA (encodes amino acids 1–300 of YES) was cloned into the pGEX-5X-1 vector. The glutathione S-transferase (GST)-tagged proteins were bacterially expressed and purified on GSTrap FF affinity columns (GE Healthcare, IL, USA) following the manufacturer’s instructions. GST-YES or GST-YES-5A (0.5–1 µg each) was incubated with 10 U recombinant CDK1/cyclin B complex (New England Biolabs, MA, USA) in kinase buffer [15] in the presence of 5 µCi γ−32P-ATP (3000 Ci/mmol, PerkinElmer, MA, USA). The samples were resolved by SDS-PAGE, transferred onto PVDF (Millipore, MA, USA) and visualized by autoradiography followed by Western blotting or detected by phospho-specific antibodies.
2.5. Antibodies
Anti-YES antibodies from BD Biosciences (Cat. No. 610375, CA, USA) were used for Western blotting throughout the study. Rabbit polyclonal phospho-specific antibodies against YES S11, T21, S40, T60, and T69 were generated and purified by AbMart (Shanghai, China). The peptides for generating the phospho-antibodies are as follows: SKENK-pS-PAIKY (pS11); VRPEN-pT-PEPVS (pT21); EPTTV-pS-PCPSS (pS40); SSLSM-pT-PFGGS (pT60); and GSSGV-pT-PFGGA (pT69). The corresponding non-phospho-peptides were also synthesized for antibody purification. Anti-β-actin, anti-cyclin B, and anti-CDC27 antibodies were from Santa Cruz Biotechnology. Anti-GST, -Aurora-A, -Lats1, and -Lats2 antibodies were from Bethyl Laboratories (TX, USA). Anti-p-T288/T232/T198 Aurora-A/B/C, - p-S10 H3, -p-S642 Wee1, -Wee1, -p-Y15 CDK1, -CDK1, -p-T160 CDK2, -CDK2, -CDC20, - p21, -p53, -p-S127 YAP, -p-S397 YAP, -YAP, -p-Lats1/2 T1079/T1041, -p-Y419 SRC, -p-Y530 SRC, -LYN, -FYN, -SRC, and -HCK antibodies were from Cell Signaling Technology (MA,USA). Anti-Flag and anti-β-tubulin (for immunofluorescence staining) antibodies were from Sigma.
2.6. Immunofluorescence staining and confocal microscopy
Cell fixation, permeabilization, fluorescence staining, and microscopy were done as previously described [16]. The stained cells were mounted with Fluoromount (Vector Laboratories, CA, USA) and visualized on an LSM710 or LSM 800 Zeiss fluorescence microscope (Carl Zeiss, NY, USA). The Slidebook 4.2 software (Intelligent Imaging Innovations, CO, USA) was used for analyzing and processing all immunofluorescence images.
2.7. Quantitative real time PCR
Total RNA isolation, RNA reverse transcription and quantitative real time-PCR were done as we have described previously [13]. The primer sequences were as follows: 5’-AAGACCATGTGGACCTGTCACTG (p21-Forward), 5’-AGGGCTTCCTCTTGGAGAAGATC (p21-Reverse), 5’-GAACCACCAAAGTTACCACGA (RBL1-Forward), and 5’-ATTAAACAGATCCTTAACACTGCAAG (RBL1-Reverse). The RT2 cell cycle arrays (Qiagen) were used, and experiments and data analysis were performed following manufacturer’s instructions.
2.8. Flow cytometry
DNA contents among cell lines were quantified via propidium iodide (PI) staining followed by flow cytometry analysis. Briefly, 1 × 106 cells were suspended in 100 µl PBS buffer and fixed by adding 1 ml 70% ethanol in −20 °C for an overnight. The cells were then mixed with 0.5 ml FxCycle™ PI/RNase Staining Solution (Invitrogen, F10797, MA, USA) and incubated for 30 minutes at room temperature in dark. The samples were analyzed by a FACSCalibur flow cytometer (Excitation at 488 nm/Emission at 530 nm). Profiles of cell cycle and polyploidy were examined by a standard method.
2.9. Annexin V/PI staining
The Dead Cell Apoptosis Kit with Annexin V Alexa Fluor™ 488 and propidium iodide (Invitrogen, V13241) was used for examination of apoptosis. Cells were seeded in 6-well plates at a density of 70–80% in duplicate. HeLa cells were treated with taxol at 50 nM for 24 h and OVCAR8 cells were treated with taxol at 20 nM for 24 h. Cell suspension preparation and staining was followed by procedures described in the manufacturer’s manual. Stained cells were analyzed by flow cytometry with fluorescence emission at 530 and 575 nm. Both early (Annexin V positive/PI negative) and late (Annexin V positive/PI positive) apoptotic proportions for each population were included.
2.10. Statistical analysis
Statistical significance was performed using a two-tailed, unpaired Student’s t-test.
3. Results
3.1. YES is phosphorylated during antitubulin drug-induced mitotic arrest
To explore how YES and other SFKs are regulated during antitubulin agent treatment, we treated HeLa cells with paclitaxel (taxol) or nocodazole (which both interfere with microtubules and arrest cells in prometaphase) and examined their responses during mitotic arrest using a Phos-tag approach. As shown in Figure 1A, B, the most prominent change we observed was the dramatic mobility up-shift of YES and, to a lesser extent, of SRC (Fig. 1A, B). Lambda phosphatase treatment largely converted slow-migrating bands to fast-migrating bands, indicating that the mobility shift of YES during mitotic arrest is caused by phosphorylation (Fig. 1C). Phosphorylation of YES was also evident in U2OS and HCT116 cells, suggesting this phospho-regulation is not cell type-specific (Fig. 1D). YES is a tyrosine kinase and its catalytic activity is regulated by phosphorylation at Y430 (main autophosphorylation site, Y419 in SRC) and Y537 (Y530 in SRC) [5,7]. We further explored whether the kinase activity is required for YES mitotic phosphorylation. Transfected YES-KD (kinase dead, K305M) and YES-Y537F (nonphosphorylatable mutant) were still shifted during taxol-induced mitotic arrest (Fig. 1E), suggesting that the mobility shift/phosphorylation of YES was likely not due to the phosphorylation at Y430 and or Y537. These observations suggest that YES is phosphorylated on novel sites other than Y430 and Y537 in response to antitubulin drugs.
Figure 1. YES is phosphorylated during antitubulin-induced mitotic arrest.
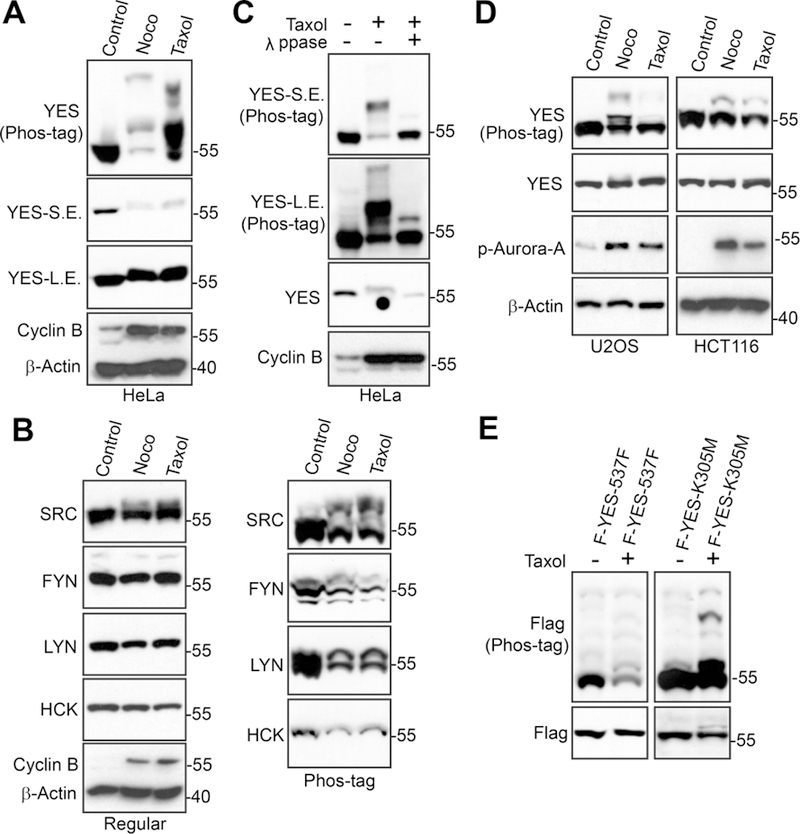
A, HeLa cells were treated with DMSO (control), taxol (0.1 µM for 16 h) or nocodazole (Noco, 100 ng/ml for 16 h). Total cell lysates were probed with the indicated antibodies. S.E.: short exposure; L.E.: long exposure. Increased cyclin B levels mark cells in mitotic arrest. B, HeLa cells were treated as in A and total cell lysates were probed with other members of SFKs. C, HeLa cells were treated with taxol as indicated and cell lysates were further treated with (+) or without (−) λ phosphatase (ppase). Total cell lysates were probed with anti-YES antibody. S.E.: short exposure; L.E.: long exposure. D, U2OS and HCT116 cancer cell lines were treated as indicated. Total cell lysates were analyzed as in A with the indicated antibodies. Increased p-Aurora-A levels mark cells in mitosis. E, HEK293T cells were transfected with Flag-YES-Y537F or Flag-YES-K305M. Thirty-six hours post-transfection, cells were treated with or without taxol for an additional 20 h. Total cell lysates were probed with Flag on both Phos-tag and regular gels.
3.2. Identification of the corresponding kinase for YES phosphorylation
Upon treatment of antitubulin agents, several mitotic kinases, including CDK1 (Cyclin-dependent kinase 1), Aurora, and Plk1 (Polo-like kinase 1), are activated [17][17]. Inhibition of Aurora (with VX-680 for Aurora-A, B, C, and MK5108 for Aurora-A) and Plk1 (with BI2536) kinases did not alter the YES phosphorylation (data not shown). Interestingly, addition of RO3306 (a CDK1 inhibitor) or Purvalanol A (inhibits CDK1, 2, 5) largely reverted the mobility shift induced by taxol in both HeLa and U2OS cells (Fig. 2A), suggesting that CDK1 is likely the kinase for YES phosphorylation during antitubulin agent-induced mitotic arrest.
Figure 2. CDK1 phosphorylates YES in vitro.
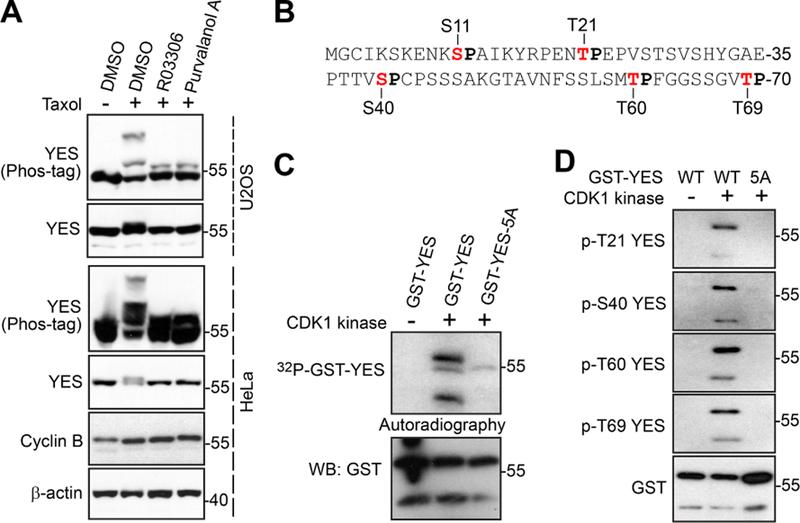
A, HeLa and U2OS cells were treated with taxol. RO3306 (CDK1 inhibitor) or Purvalanol A (CDK1/2/5 inhibitor) was added (with MG132 to block mitotic exit by stabilizing cyclin B) into the cells 1.5 h before harvesting the cells. Total cell lysates were subjected to Western blotting with the indicated antibodies. B, A diagram shows the N-terminal sequence of YES with 5 S/TP consensus. C, In vitro kinase assays with purified CDK1/cyclin B complex (New England Biolab). D, In vitro kinase assays were done as in C except anti-phospho-YES antibodies were used.
3.3. CDK1 phosphorylates YES in vitro
CDK1 recognizes a minimal S/T-P consensus sequence [18]. YES contains five S/TPs (S11, T21, S40, T60, and T69) (Fig. 2B). All five sites are located in the N-terminus (which is not conserved within the SRC family) and are unique to YES, consistent with our observations that among the SRC family proteins, YES was the most heavily phosphorylated in response to antitubulin drugs.
To determine whether CDK1 kinase can directly phosphorylate YES, we performed in vitro kinase assays with GST-tagged YES as substrates. Purified CDK1/cyclin B complex readily phosphorylated GST-YES in vitro (Fig. 2C). Importantly, mutating these five sites to alanines (GST-YES-5A) almost completely abolished the 32P incorporation, suggesting that this N-terminal S/TP cluster is the major phosphorylation region of YES (Fig. 2C). We generated phospho-specific antibodies against S11, T21, S40, T60, and T69 and in vitro kinase assays confirmed that all these sites (except S11) were phosphorylated in the presence of CDK1/cyclin B complex (Fig. 2D), suggesting that CDK1 directly phosphorylates YES in vitro.
3.4. CDK1 phosphorylates YES in cells
To explore whether T21, S40, T60, and T69 are also phosphorylated within cells during taxol-induced mitotic arrest, we transfected Flag-tagged YES or a corresponding non-phosphorylatable mutant (YES-5A) into cells, treated the cells with taxol, and determined levels of phosphorylation by phospho-antibodies. Taxol treatment significantly increased the phosphorylation of T21, S40, T60, and T69. The signal was abolished by mutating these sites to alanines (Fig. 3A), confirming the specificity of these phospho-specific antibodies. Addition of RO3306 or Purvalanol A abolished the phosphorylation induced by taxol (Fig. 3B), suggesting that phosphorylation of YES is CDK1 kinase dependent.
Figure 3. CDK1 phosphorylates YES in cells.

A, HEK293T cells were transfected with Flag-tagged YES and non-phosphorylatable mutant (YES-5A). Cells were treated with taxol at 48 h post-transfection. Total cell lysates were probed with the indicated antibodies. Increased p-Aurora-A,-B mark cells in mitosis. B, HEK293T cells were transfected with Flag-YES. At 30 h post-transfection, the cells were treated with taxol for 16 h. RO3306 (5 µM) or Purvalanol A (10 µM) was added to cells 1.5 h before harvesting as indicated. Proteasome inhibitor MG132 was also added (together with inhibitors) to prevent cyclin B from degradation and cells from exiting from mitosis. Total cell lysates were probed with anti-phospho-YES and subsequent anti-Flag antibodies. C, YES knockout (KO) HeLa cells were stably transduced with YES-WT or YES-5A. Total cell lysates from these cell lines were treated with taxol and probed with the indicated antibodies. S.E.: short exposure; LE: long exposure. D, HEK293T cells were transfected with Flag-tagged YES and non-phosphorylatable mutant (YES-5A). Cells were treated with taxol at 48 h post-transfection and YES proteins were immunoprecipitated (with Flag) antibody and analyzed with the indicated antibodies.
We next examined whether these sites affect the mobility of YES during taxol treatment. For this purpose, we deleted endogenous YES and re-expressed wild type (WT) YES or YES-5A mutant and compared their mobility shift in the presence of taxol. Mutation of all five sites largely abolished the mobility shift of YES, suggesting that these N-terminal five sites are the main phosphorylation region responsible for mobility shift of YES upon taxol treatment (Fig. 3C). Together, our data identified novel phosphorylation of YES and phosphorylation occurs in a CDK1-dependent manner during taxol-induced mitotic arrest. We further showed that the nonphosphorylatable mutant (YES-5A) completely lost its Y430 and Y537 phosphorylation, suggesting that CDK1-mediated mitotic phosphorylation is required for YES kinase activity (Fig. 3D).
3.5. Phosphorylation of YES is essential for proper mitotic progression
We next examined whether YES or its phosphorylation is involved in mitotic progression. To do so, we generated YES knockout (KO) HeLa cells using CRISPR/Cas9-nickase (Cas9n) (Fig. 4A). Knockout of YES resulted in moderate but significant mitotic arrest in HeLa cells (Fig. 4B). Adding back YES-WT completely rescued the effects, however, re-expression of YES-5A failed to do so, indicating that mitotic phosphorylation is required for mitotic progression (Fig. 4B). We further used a double thymidine block and release approach in these cell lines to determine which mitotic phase is affected. Figure 4C shows that a significantly higher percentage of cells was observed in prometaphase in YES-KO cells when compared with parental cells (Fig. 4C, D). Again, this phenotype was rescued by re-expression of YES-WT, but not the YES-5A mutant (Fig. 4D). Together, these observations suggest that YES and its mitotic phosphorylation are required for prometaphase to metaphase progression.
Figure 4. Generation and characterization of YES knockout (KO) HeLa cell line.
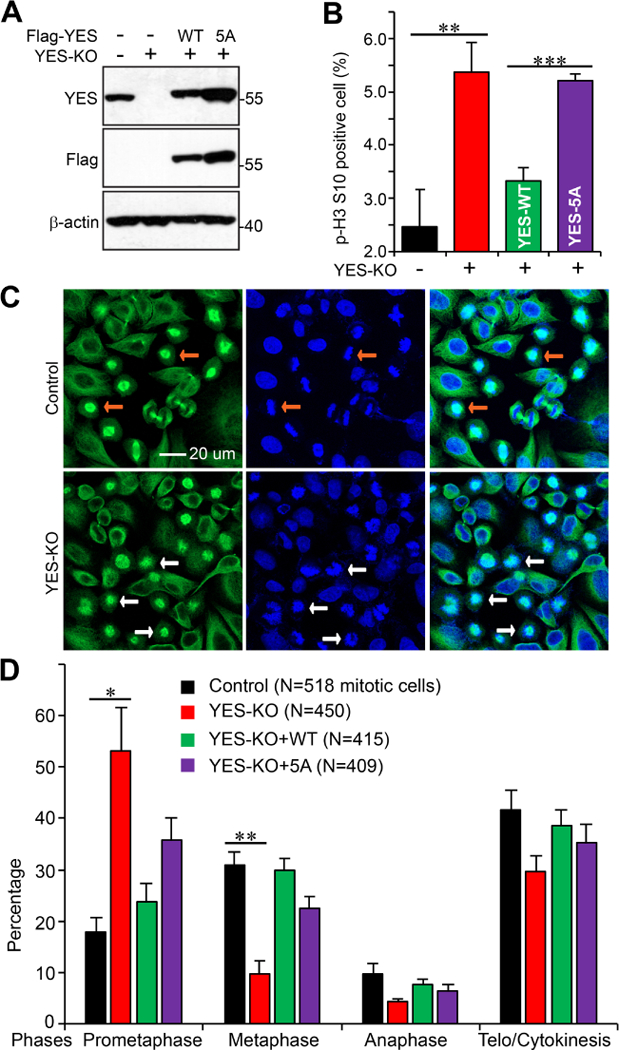
A, Western blot analysis of parental, YES KO, KO with exogenous YES-WT or YES-5A HeLa cell lines. B, HeLa cell lines established in A were used to quantify the percentage of cells with p-H3 S10 positive through flow cytometry. Data are from three independent experiments and were expressed as mean ± s.e.m. **: p<0.01; ***: p<0.001 (t-test). C, Representative immunofluorescence confocal images showing the difference of mitotic phases between the HeLa control and YES knockout cell lines. Cells were synchronized with a double thymidine block and release method [15] and cells were stained at 10 h post release. β-tubulin is shown in green and nuclei are stained with DAPI (blue). The white arrows point to cells in metaphase (chromosomes are condensed and aligned at the plate) and yellow arrows indicate cells in prometaphase (condensed but not aligned). D, The indicated HeLa cell lines were used to quantify the percentage of cells in different mitotic phases. Cells were treated as in C. Data are from three independent experiments and were expressed as mean ± s.e.m. *:p<0.05; **:p<0.01 (t-test).
3.6. YES knockout leads to tetraploidy independent of p53, but is phosphorylation-dependent
We further examined YES localization during cell cycle progression. In both interphase (YES is not phosphorylated) and mitotic cells (YES is phosphorylated), YES is mainly localized in cell membrane area and does not have obvious colocalization with microtubule (Fig. 5A).
Figure 5. YES knockout resulted in tetraploidy in HeLa and HCT116 cells.
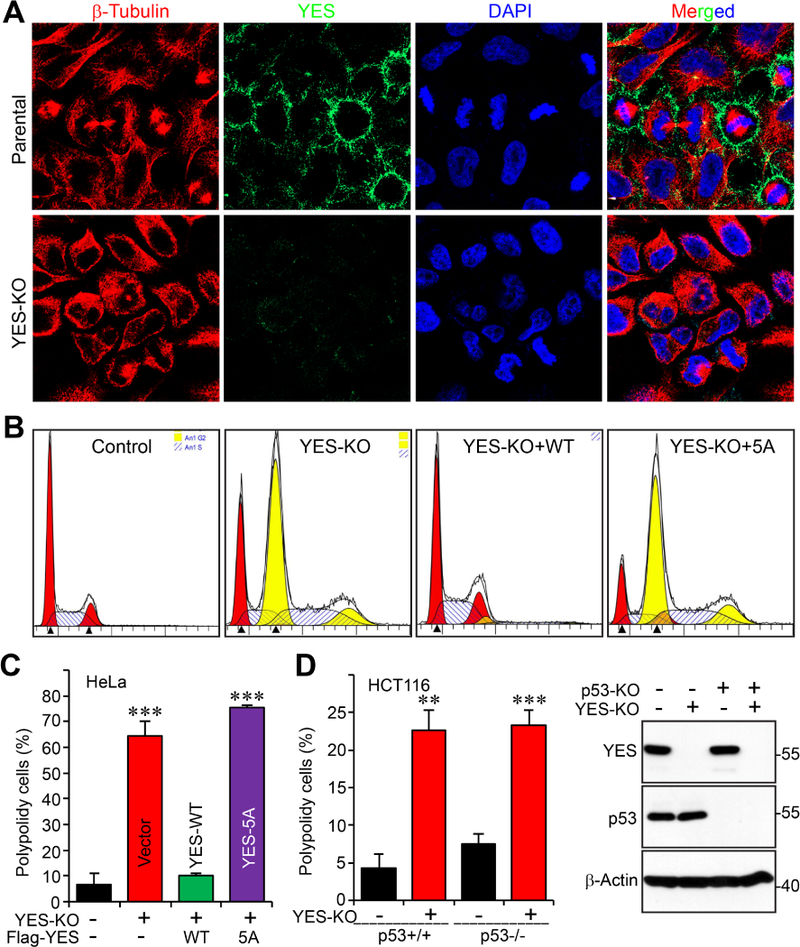
A, Immunofluorescence staining of YES (green) during cell cycle progression in HeLa parental and YES-KO cells. β-tubulin is shown in red and nuclei are stained with DAPI (blue). YES-KO cells serve as a negative control for antibody validation.
B, C, The specified HeLa cell lines were labeled with propidium iodide (PI) and flow cytometry analysis was performed to determine the percentage of tetraploid cells in the whole population. Representative flow diagrams are shown from each cell line (B). D, The indicated HCT116 cell lines were similarly analyzed as in B, C. Data are expressed as the mean ± s.e.m. of at least three independent experiments. **: p<0.01; ***: p<0.001 (t-test).
Mitotic defects often lead to genomic instability. We quantified the ploidy changes resulting from knockout of YES. Utilizing flow cytometry, we discovered that the number of tetraploid cells significantly increased in YES KO HeLa cells (Fig. 5B, C). Similar results, to a lesser extent, were observed in HCT116 cells (Fig. 5D). This phenotype was independent of p53 status since p53 KO did not affect YES KO-driven tetraploidy (Fig. 5D). WT YES, but not YES-5A mutant, reconstitution completely restored these tetraploid cells to diploid cells (Fig. 5B, C), indicating that mitotic phosphorylation of YES is required for maintaining genome integrity.
3.7. YES regulates expression of multiple cell cycle regulators
Given the sequence similarity and functional conservation among the SFKs, we determined whether YES deletion affects expression of other SFKs. Figure 6A shows that FYN levels were increased in YES-KO HeLa cells but not in HCT116 cells (Fig. 6A). LYN and SRC levels were not altered upon YES inactivation. Interestingly, LYN, but not YES, SRC, or FYN, expression levels were reduced in p53-KO HCT116 cells (Fig. 6A).
Figure 6. YES controls multiple cell cycle regulators.
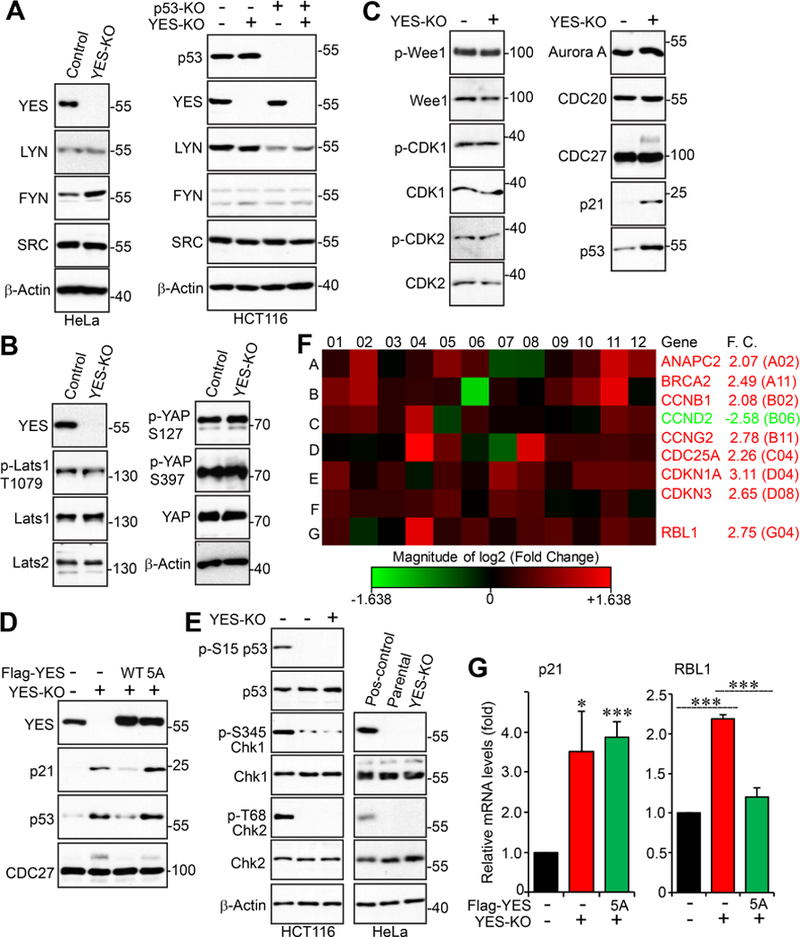
A, Western blot analysis of SFK members in the indicated cell lines. B, Western blot analysis of Lats-YAP signaling in parental and YES-KO HeLa cells. C, Total cell lysates from parental and YES-KO HeLa cells were subjected to Western blotting analysis with various cell cycle regulators. D, Total cell lysates from the indicated HeLa cells were subjected to Western blotting analysis. E, YES deletion resulted in no significant DNA damage. The positive controls (first lane) are HeLa cells treated with 5-FU (Fluorouracil) or Doxorubicin. F, Expression profiling of cell cycle regulators identified multiple genes that are regulated by YES. The RT2 cell cycle arrays (Qiagen) were used and experiments were performed following the manufacturer’s instructions. F.C.: fold change. G, Quantitative RT-PCR for p21 and RBL1 in indicated HeLa cell lines. Data are expressed as the mean ± s.e.m. of three independent experiments. ***: p< 0.001; *: p< 0.05 (t-test).
An earlier study identified YAP, a critical effector of the Hippo-YAP signaling [19–21], as a YES-associated protein [22], so we also examined total and phospho-YAP levels in parental and YES KO cells to determine if loss of YES would affect the status of YAP. We detected no noticeable changes to the total YAP protein level and phosphorylation levels at S127 and S397 (two major Hippo-mediated inactivating phosphorylation sites [23–25]) (Fig. 6B).
We next explored the downstream effectors/signaling regulated by YES using two approaches. First, we screened several well-known cell-cycle regulators in parental and YES-KO HeLa cells to attempt to identify proteins with altered expression or phosphorylation levels between the two cell lines (Fig. 6C). Interestingly, both p21 and p53 protein levels were significantly increased upon loss of YES and their expression was restored by re-expression of YES-WT (Fig. 6C, D). Again, YES-5A failed to rescue p21 and p53 levels in YES-KO cells, suggesting that the mitotic phosphorylation of YES is required for controlling p21 and p53 expression (Fig. 6D). Increased p53 expression is unlikely due to DNA damage since YES deletion failed to cause significant DNA damage revealed by phospho-Chk1/2 and phospho-p53 (Fig. 6E). We also found a small portion of CDC27 (Cell Division Cycle 27), a mitotic regulator, was phosphorylated in YES-KO cells (Fig. 6C, D). Second, we performed an mRNA array analysis involving 84 cell cycle regulators in control and YES-KO cells. Eight genes were upregulated and one gene was downregulated in YES-KO cells compared with parental cells (Fig. 6F). Of note, the mRNA levels of p53 were only modestly increased (1.5 fold, G11 in the array). We confirmed by qRT-PCR that p21 (CDKN1A: cyclin-dependent kinase inhibitor) mRNA levels were increased (Fig. 6F, G). Consistent with the results in Figure 6D, re-expression of YES-5A was not able to restore p21 expression (Fig. 6G). We also confirmed that RB-like protein 1 (RBL1) mRNA levels were increased (Fig. 6F, G). The YES-5A mutant has similar activity to WT YES in rescuing RBL1 mRNA levels, suggesting that YES phosphorylation is dispensable in controlling RBL1 expression (Fig. 6G). Together, these observations suggest that YES regulates expression of multiple cell cycle regulators.
3.8. YES and its phosphorylation controls taxol chemosensitivity
Antitubulin drugs are widely used in cancer patients and drug resistance is a major barrier in clinical treatment of cancer. Given the response of YES to antitubulin agents, we examined whether YES and its phosphorylation are involved in antitubulin drug-driven cytotoxicity. Deletion of YES did not cause significant apoptosis under normal conditions (Fig. 7A). Apoptosis was greatly increased (as revealed by cleaved PARP and Annexin-V staining) under taxol treatment in YES knockout cells compared to that in parental cells (Fig. 7A, B). Re-expression of YES-WT, but not YES-5A mutant, largely rescued the taxol sensitivity, suggesting that YES and its mitotic phosphorylation confer taxol resistance (Fig. 7A, B). In line with this loss-of-function observation, overexpression of YES-WT, but not the YES-5A mutant, in OVCAR8 (a high-grade serous ovarian cancer cell line) resulted in fewer apoptotic cells compared with controls under taxol treatment (Fig. 7C-E).
Figure 7. YES and its mitotic phosphorylation regulates taxol sensitivity.
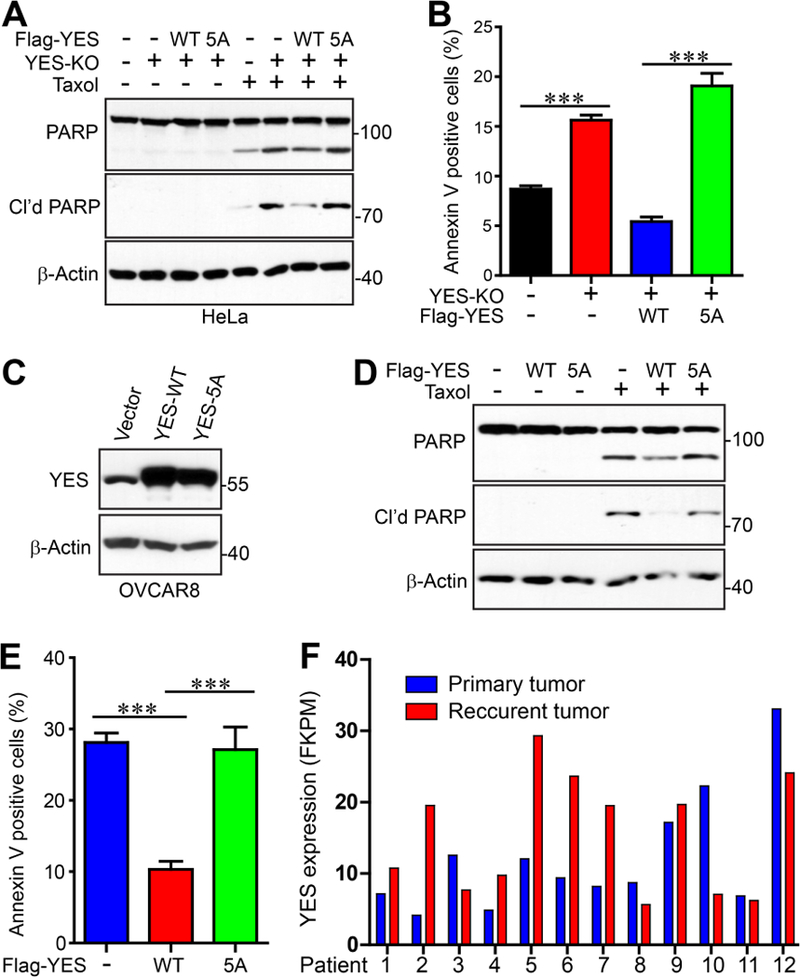
A, The specified HeLa cell lines were treated with DMSO or taxol (50 nM for 24 h). Total cell lysates were probed with the indicated antibodies. Cleaved PARP serves as an apoptotic marker. B, The cells were treated as in A and were then labeled with Annexin-V and PI. Flow cytometry analysis was performed to determine the percentage of apoptotic cells in the whole population. Data were expressed as the mean ± s.e.m. of three independent experiments. ***: p<0.001 (t-test). C, Establishment of OVCAR8 cell lines expressing vector (control), YES-WT, or YES-5A. D, The cell lines from C were treated with DMSO or taxol (20 nM for 24 h). Total cell lysates were probed with the indicated antibodies. E, Cells were treated as in D and apoptotic cells were identified and quantified by Annexin-V and PI staining. Data are expressed as the mean ± s.e.m. of three independent experiments. ***: p<0.001 (t-test). F, Clinical relevance of YES expression and chemoresistance. RNA-seq data were extracted from Patch et al., 2015 [26] and YES mRNA levels were analyzed in primary and recurrent high-grade serous ovarian tumors (paired, before and post-chemotherapy)
We analyzed the mRNA levels of YES in paired, recurrent post-chemotherapy high grade serous ovarian cancer and their primary untreated tumors, using RNA-seq data published by Patch et al.[26]. Of interest, expression of YES was higher (>1.5 fold) in 5 cases of 12 recurrent tumors compared to matched primary tumors (Fig. 7F). Increased expression with chemoresistance was much more common than decreased expression. These data support a potential association between YES upregulation with taxol chemoresistance.
4. Discussion
Antitubulin agents are some of the most clinically successful chemotherapeutics for cancer [2,3,27]. Antitubulin compounds arrest cells in G2/M and induce cell death by unclear mechanisms. Some previous studies demonstrated that CDK1-mediated Bcl-xL (B-cell lymphoma-extra large) phosphorylation at S62 is essential for Bcl-xL’s inactivation, resulting in apoptosis in response to antitubulin agents [28,29]. Furthermore, the destruction of Mcl1 (myeloid cell leukemia sequence 1) was recently identified as having a critical role in mediating the apoptotic effect elicited by antitubulin chemotherapeutics. Mcl1 is phosphorylated by multiple kinases (including CDK1) and degraded through the proteasome pathway during mitotic arrest [30,31]. Thus, CDK1-Bcl2-Mcl1 signaling implicates a proapoptotic pathway that is important in how cells respond to antitubulin drugs [17]. Interestingly, recent studies showed that the Hippo-YAP signaling pathway may also function as an important regulator in antitubulin chemoresistance in breast and ovarian cancer [32–36]. This study identified YES as an alternative mediator that links mitotic arrest and cell death in response to antitubulin chemotherapeutics. Given the observation that YES is overexpressed in tumor samples from breast and ovarian cancer patients and in recurrent/resistant ovarian cancer patients [5] (Fig. 7E), our study implies YES is a potential therapeutic target and suggests that combining kinase inhibitors for YES with antitubulin agents will have enhanced efficacy in treatment of antitubulin-resistant and/or recurrent patients.
Activation of SFKs (e.g. SRC, YES, and LYN) has been well documented in antitubulin agent-arrested mitotic cells and in normal mitosis [6,37]. Furthermore, activation of SFKs is essential for mitotic progression since inhibition of SFKs blocks metaphase progression [38,39] and cytokinesis [40,41]. Phosphorylation and regulation on serine/threonine in the unique N-terminal region of SFKs are much less studied. In response to antitubulin agents, the most significant mobility change/phosphorylation was observed in YES among these SFKs examined (Fig. 1) and this mobility retardation is not due to tyrosine phosphorylation and activation. The current study identified novel phosphorylation of YES during antitubulin agent-induced mitotic arrest. Phosphorylation occurs at multiple sites on its N-terminal variable region (S11, T21, S40, T60, and T69) (Figs. 2, 3). Interestingly, SRC has only two S/TP sites (T47 and S75), and FYN (T45) and LCK (S59) contains only one in this region, supporting the observation that YES has the most mobility retardation due to phosphorylation. LYN does not have any S/TP consensus sequences in this domain. Of note, SRC (T47 and S75) and LCK (S59) have been shown to be phosphorylated by CDK1 during mitosis, but it has remained elusive whether their phosphorylation is involved in antitubulin agent-induced cell death. Future studies are also required to elucidate the overlapping and distinct roles among the SFKs in mediating antitubulin chemosensitivity in different cell types.
While the YES kinase activity is not required for its N-terminal serine/threonine phosphorylation in response to antitubulin agents (Fig. 1E), the phosphorylation is essential for YES kinase activity and regulates taxol sensitivity (Figs. 3, 7). Therefore, these observations have also identified a new layer of regulation for YES kinase activity. It is currently not known how this N-terminal phosphorylation affects YES tyrosine phosphorylation (Y430 and Y537) and activation. Interestingly, all SFKs have acylation signals in their N-terminal regions allowing for myristoylation and palmitoylation. This region links SFKs to the plasma membrane and maintains them in the active conformation [5]. Thus, it is possible that this phosphorylation regulates YES kinase activity through controlling its conformation and membrane translocation.
5. Conclusions:
In summary, we found that YES is phosphorylated at multiple sites on its N-terminal unique domain by CDK1 during antitubulin drug-induced mitotic arrest. Phosphorylation of YES occurs during normal mitosis. We further show that YES regulates antitubulin chemosensitivity. Importantly, mitotic phosphorylation is essential for these effects. Collectively, our results reveal a previously unrecognized mechanism for controlling the activity of YES during antitubulin chemotherapeutic treatment. Our study suggests YES as a potential target for the treatment of antitubulin drug-resistant cancer patients
Highlights.
YES kinase is phosphorylated during antitubulin agent-induced and normal mitosis
CDK1 phosphorylates YES at S11, T21, S40, T60, and T69 in vitro and in vivo
Mitotic phosphorylation is involved in mitotic progression
YES and its phosphorylation regulate antitubulin chemosensitivity.
Acknowledgements:
All fluorescence images were acquired by Zeiss LSM 710 or LSM 800 confocal microscopes at the Advanced Microscopy Core at the University of Nebraska Medical Center. The core is supported in part by grant P30 GM106397 from the National Institutes of Health (NIH). Research in the Dong laboratory is supported by Fred & Pamela Buffett Cancer Center Support Grant (P30 CA036727), grants P30 GM106397 and R01 GM109066 from the NIH, and W81XWH-14–1-0150 from the Department of Defense Health Program. Zhan Wang is supported by fellowships from Chinese Scholarship Council, China. This work is also supported by Natural Science Foundation of Shandong Province, China (ZR2016HQ03 to S.Y.) and National Natural Science Foundation of China (81602332 to S.Y.). We thank Dr. Joyce Solheim for critical reading and comments on the manuscript.
Abbreviations:
- CDK1
cyclin-dependent kinase 1
- CDKN1A (p21)
cyclin-dependent kinase inhibitor 1A
- CRISPR
clustered regularly-interspaced short palindromic repeats
- KO
knockout
- Plk1
Polo-like kinase 1
- RBL1
RB-like protein 1
- SFK
Src family non-receptor tyrosine kinase
- YAP
Yes-associated protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- [1].Jackson JR, Patrick DR, Dar MM, Huang PS. Nat. Rev. Cancer 7 (2007) 107–117. [DOI] [PubMed] [Google Scholar]
- [2].Janssen A, Medema RH. Oncogene 30 (2011) 2799–2809. [DOI] [PubMed] [Google Scholar]
- [3].Dominguez-Brauer C, Thu KL, Mason JM, Blaser H, Bray MR, Mak TW. Mol. Cell 60 (2015) 524–536. [DOI] [PubMed] [Google Scholar]
- [4].Gascoigne KE, Taylor SS. J. Cell. Sci 122 (2009) 2579–2585. [DOI] [PubMed] [Google Scholar]
- [5].Espada J, Martin-Perez J. Int. Rev. Cell. Mol. Biol 331 (2017) 83–122. [DOI] [PubMed] [Google Scholar]
- [6].Kuga T, Nakayama Y, Hoshino M, Higashiyama Y, Obata Y, Matsuda D, Kasahara K, Fukumoto Y, Yamaguchi N. Arch. Biochem. Biophys 466 (2007) 116–124. [DOI] [PubMed] [Google Scholar]
- [7].Nunes-Xavier CE, Martin-Perez J, Elson A, Pulido R. Biochim. Biophys. Acta 1836 (2013) 211–226. [DOI] [PubMed] [Google Scholar]
- [8].Sato A, Sekine M, Virgona N, Ota M, Yano T. Oncol. Rep 28 (2012) 1889–1893. [DOI] [PubMed] [Google Scholar]
- [9].Hamamura K, Tsuji M, Hotta H, Ohkawa Y, Takahashi M, Shibuya H, Nakashima H, Yamauchi Y, Hashimoto N, Hattori H, Ueda M, Furukawa K, Furukawa K. J. Biol. Chem 286 (2011) 18526–18537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Barraclough J, Hodgkinson C, Hogg A, Dive C, Welman A. Neoplasia 9 (2007) 745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Li L, He L, Zhao JL, Xiao J, Liu M, Li X, Tang H. J. Cell. Biochem 116 (2015) 1050–1059. [DOI] [PubMed] [Google Scholar]
- [12].Stauffer S, Zeng Y, Zhou J, Chen X, Chen Y, J. Dong. Cell. Signal 39 (2017) 74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Xiao L, Chen Y, Ji M, Dong J. J. Biol. Chem 286 (2011) 7788–7796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chen X, Stauffer S, Chen Y, Dong J. J. Biol. Chem 291 (2016) 14761–14772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Xiao L, Chen Y, Ji M, Volle DJ, Lewis RE, Tsai MY, Dong J. J. Biol. Chem 286 (2011) 36304–36315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhang L, Iyer J, Chowdhury A, Ji M, Xiao L, Yang S, Chen Y, Tsai MY, Dong J. J. Biol. Chem 287 (2012) 34069–34077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Matson DR, Stukenberg PT. Mol. Interv 11 (2011) 141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Nigg EA. Trends Cell Biol 3 (1993) 296–301. [DOI] [PubMed] [Google Scholar]
- [19].Fulford A, Tapon N, Ribeiro PS. Curr. Opin. Cell Biol 51 (2017) 22–32. [DOI] [PubMed] [Google Scholar]
- [20].Fu V, Plouffe SW, Guan KL. Curr. Opin. Cell Biol 49 (2018) 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yu FX, Zhao B, Guan KL. Cell 163 (2015) 811–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sudol M. Oncogene 9 (1994) 2145–2152. [PubMed] [Google Scholar]
- [23].Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, D. Pan. Cell 130 (2007) 1120–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, Zheng P, Ye K, Chinnaiyan A, Halder G, Lai ZC, Guan KL. Genes Dev 21 (2007) 2747–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. Genes Dev 24 (2010) 72–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Patch AM, Christie EL, Etemadmoghadam D, Garsed DW, George J, Fereday S, Nones K, Cowin P, Alsop K, Bailey PJ, Kassahn KS, Newell F, Quinn MC, Kazakoff S, Quek K, Wilhelm-Benartzi C, Curry E, Leong HS, Australian Ovarian Cancer Study Group, Hamilton A, Mileshkin L, Au-Yeung G, Kennedy C, Hung J, Chiew YE, Upreti PM, Galitovskaya EN, Chu R, Tackett AJ, Terrano DT, Granell S, editschke TC, Young G, Strachan K, Waring P, Azar W, Mitchell C, Traficante N, Hendley J, Thorne H, Shackleton M, Miller DK, Arnau GM, Tothill RW, Holloway TP, Semple T, Harliwong I, Nourse C, Nourbakhsh E, Manning S, Idrisoglu S, Bruxner TJ, Christ AN, Poudel B, Holmes O, Anderson M, Leonard C, Lonie A, Hall N, Wood S, Taylor DF, Xu Q, Fink JL, Waddell N, Drapkin R, Stronach E, Gabra H, Brown R, Jewell A, Nagaraj SH, Markham E, Wilson PJ, Ellul J, McNally O, Doyle MA, Vedururu R, Stewart C, Lengyel E, Pearson JV, Waddell N, deFazio A, Grimmond SM, Bowtell DD. Nature 521 (2015) 489–494. [DOI] [PubMed] [Google Scholar]
- [27].Jordan MA, Wilson L. Nat. Rev. Cancer 4 (2004) 253–265 [DOI] [PubMed] [Google Scholar]
- [28].Upreti M, Galitovskaya EN, Chu R, Tackett AJ, Terrano DT, Granell S, Chambers TC. J. Biol. Chem 283 (2008) 35517–35525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Terrano DT, Upreti M, Chambers TC. Mol. Cell. Biol 30 (2010) 640–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Harley ME, Allan LA, Sanderson HS, Clarke PR. EMBO J 29 (2010) 2407–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, Helgason E, Ernst JA, Eby M, Liu J, Belmont LD, Kaminker JS, O’Rourke KM, Pujara K, Kohli PB, Johnson AR, Chiu ML, Lill JR, Jackson PK, Fairbrother WJ, Seshagiri S, Ludlam MJ, Leong KG, Dueber EC, Maecker H, Huang DC, Dixit VM. Nature 471 (2011) 110–114. [DOI] [PubMed] [Google Scholar]
- [32].Aylon Y, Michael D, Shmueli A, Yabuta N, Nojima H, Oren M. Genes Dev 20 (2006)2687–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lai D, Ho KC, Hao Y, Yang X. Cancer Res 71 (2011) 2728–2738. [DOI] [PubMed] [Google Scholar]
- [34].Zhang X, George J, Deb S, Degoutin JL, Takano EA, Fox SB, AOCS Study group, Bowtell DD, Harvey KF. Oncogene 30 (2011) 2810–2822. [DOI] [PubMed] [Google Scholar]
- [35].Zhao Y, Yang X. Oncotarget 6 (2015) 21906–21917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zhao Y, Khanal P, Savage P, She YM, Cyr TD, Yang X. Cancer Res 74 (2014) 4493–4503. [DOI] [PubMed] [Google Scholar]
- [37].Chackalaparampil I, Shalloway D. Cell 52 (1988) 801–810. [DOI] [PubMed] [Google Scholar]
- [38].Moasser MM, Srethapakdi M, Sachar KS, Kraker AJ, Rosen N. Cancer Res 59 (1999) 6145–6152. [PubMed] [Google Scholar]
- [39].Thery M, Racine V, Pepin A, Piel M, Chen Y, Sibarita JB, Bornens M. Nat. Cell Biol 7 (2005) 947–953. [DOI] [PubMed] [Google Scholar]
- [40].Ng MM, Chang F, Burgess DR. Dev. Cell 9 (2005) 781–790. [DOI] [PubMed] [Google Scholar]
- [41].Kasahara K, Nakayama Y, Nakazato Y, Ikeda K, Kuga T, Yamaguchi N. J. Biol. Chem 282 (2007) 5327–5339. [DOI] [PubMed] [Google Scholar]