

Abstract
Background
Chronic nicotine exposure produces neuroadaptations in brain reward systems and α4β2 nicotinic acetylcholine receptors (nAChRs) in the corticolimbic brain areas. We previously demonstrated opposite effects of nicotine exposure delivered by self-administration or pumps on brain reward thresholds that can be attributed to the different temporal pattern and contingency of nicotine exposure. We investigated the effects of these two factors on reward thresholds and somatic signs during nicotine withdrawal, and on nAChRs binding in corticolimbic brain areas.
Methods
The intracranial self-stimulation procedure was used to assess reward thresholds in rats prepared with pumps delivering various doses of nicotine continuously or intermittently. Separate group of rats were randomly exposed to nicotine via pumps (non-contingent) or nicotine self-administration (contingent) to determine [125I]-epibatidine binding at α4β2* nAChRs.
Results
Withdrawal from continuous non-contingent nicotine exposure led to significant elevations in thresholds and increases in somatic signs in rats, while there was no significant effect of withdrawal from intermittent non-contingent nicotine exposure at the same doses. nAChRs were upregulated during withdrawal from continuous non-contingent nicotine exposure. α4β2* nAChRs were upregulated in the ventral tegmental area and prelimbic cortex during withdrawal from non-contingent intermittent exposure and in the nucleus accumbens during withdrawal from contingent intermittent nicotine exposure to the same dose.
Conclusions
During non-contingent nicotine exposure, the temporal pattern of nicotine delivery differentially affected thresholds and somatic signs of withdrawal. Upregulation of α4β2* nAChRs was brain site-specific and depended on both temporal pattern and contingency of nicotine exposure.
Keywords: α4β2* nicotinic acetylcholine receptors, reward thresholds, somatic signs, intracranial self-stimulation, cotinine
1. Introduction
Chronic exposure to nicotine or other drugs of abuse produces changes in brain reward circuits resulting in the development of dependence (Koob and Volkow, 2010, Markou, 2008). It is well accepted that nicotine dependence is maintained by both the positive reinforcing and reward enhancing effects of nicotine, as well as the motivational effects of reward deficits associated with nicotine withdrawal (D'Souza and Markou, 2011). Extensive animal research in our laboratory demonstrated that both contingent exposure to nicotine self-administration (Kenny and Markou, 2006) and non-contingent acute (Harrison et al., 2002, Lindblom et al., 2005) or chronic (Cryan et al., 2003, Paterson et al., 2007, Skjei and Markou, 2003) nicotine administration via osmotic minipumps lowered brain reward thresholds reflecting reward enhancing effects of nicotine. By contrast, cessation of chronic subcutaneous non-contingent nicotine exposure resulted in elevations in brain reward thresholds reflecting a negative affective state, as well as somatic signs of withdrawal (Epping-Jordan et al., 1998, Semenova and Markou, 2003, Skjei and Markou, 2003). Similarly, the termination of extended 6 h access to nicotine self-administration produced somatic signs of nicotine withdrawal indicating the development of dependence on nicotine (Paterson and Markou, 2004). However, withdrawal from contingent 6 h exposure to nicotine self-administration resulted in an enhancement of brain reward function for at least 36 days after cessation of nicotine exposure (Kenny and Markou, 2006). This finding was unexpected and in contrast to the effects of nicotine withdrawal after non-contingent exposure via minipumps (Epping-Jordan et al., 1998, Semenova and Markou, 2003, Skjei and Markou, 2003). The temporal pattern and contingency of nicotine exposure may contribute to differential effects on brain reward function.
Differential adaptation in nicotinic acetylcholine receptor (nAChR) function may also be involved in the opposite effects of contingent vs non-contingent nicotine exposure on brain reward function. To date, 12 neuronal subunits of nAChRs have been identified with nine α-type (α2-α10) and three β-type (β2-β4) receptors that form structurally and functionally distinct hetero- and homo-pentametric receptors (Changeux, 2010, Collins et al., 2009). Nicotine dependence is believed to be mediated primarily via α4β2* (the asterisk indicates involvement of other subunits) nAChRs among others (Picciotto and Kenny, 2013). Chronic cigarette smoking in humans leads to nAChR upregulation of high affinity α4β2* nicotine binding sites in the brain (Benwell et al., 1988, Breese et al., 1997, Cosgrove et al., 2009, Gentry and Lukas, 2002, Perry et al., 1999, Staley et al., 2006). Studies in rodents showed an upregulation of high affinity nicotine binding sites after chronic non-contingent nicotine exposure via external or internal osmotic minipumps, repeated nicotine injections or intravenous infusions (Collins et al., 1990, Fasoli et al., 2016, Marks et al., 1983, Rowell and Li, 1997, Sanderson et al., 1993, Schwartz and Kellar, 1983, Ulrich et al., 1997). Similarly, exposure to chronic contingent nicotine administration also results in increased nAChR expression in rodents (Donny et al., 2000, Metaxas et al., 2010, Parker et al., 2004). The nicotine-induced upregulation of nAChR may contribute to increased sensitivity to nicotine (Dani and Heinemann, 1996, Wonnacott, 1990) and the long-lasting elevations in brain reward function after cessation of nicotine self-administration (Kenny and Markou, 2006).
In the present study we compared the effects of nicotine dose and temporal pattern (intermittent vs continuous) of non-contingent nicotine exposure via osmotic minipumps on brain reward function and somatic signs during nicotine withdrawal in rats. We used intermittent nicotine exposure because the delivery method using continuous nicotine infusion to induce dependence does not closely mimic the intermittent pattern of nicotine intake of human smokers (Brynildsen et al., 2016). That is, a chronic smoker uses tobacco every 20 to 30 min with intermittent inhalation of mainstream smoke, with an extended period of withdrawal during sleep (Ghosheh et al., 2001). The rats were exposed to nicotine at doses equal to both contingently self-administered (Kenny and Markou, 2006, Paterson et al., 2008) and non-contingently administered via minipumps (Skjei and Markou, 2003). Further, we compared nAChR expression in corticolimbic brain areas in rats self-administering nicotine intravenously for 12 h/day (i.e., contingent intermittent exposure), and rats treated non-contingently with nicotine via minipumps (continuously or intermittently). Finally, nicotine and cotinine levels in plasma were compared after contingent intermittent, non-contingent intermittent and non-contingent continuous nicotine exposures for 21–22 days.
2. Materials and Methods
2.1. Subjects
Male Wistar rats (Charles River, Raleigh, NC) weighing 320–360 g at the beginning of the experiments) were housed in groups of two in a humidity- and temperature-controlled vivarium on a reverse 12 h light/dark cycle (lights off at 7 am). Rats had ad libitum access to food and water until behavioral training commenced. Training and testing occurred during the dark cycle. All experiments were in accordance with the guidelines of the American Association for the Accreditation of Laboratory Animal Care and the National Research Council’s Guide for Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee.
2.2. Drugs
(–)Nicotine hydrogen tartrate (Sigma, St. Louis, MO) was dissolved in sterile 0.9% saline solution and infused through 28-day subcutaneous osmotic minipumps (1.2, 3.16 or 6.32 mg/kg nicotine base per 12h or 24h for 20 days). For the 12 h nicotine exposure, nicotine concentrations were doubled to match the nicotine dose delivered during 24 h exposure. For intravenous self-administration, nicotine was dissolved in sterile saline (pH adjusted to ~7).
2.3. Intracranial self-stimulation (ICSS) apparatus, surgery, and procedure
The ICSS apparatus, surgery, and procedure were identical to those described previously (Harrison et al., 2001, Semenova and Markou, 2003). Briefly, training and testing occurred in 16 sound-attenuated (San Diego Instruments, San Diego, CA) Plexiglas chambers (Med Associates, St. Albans, VT) that contained a metal wheel manipulandum. Brain stimulation was delivered by constant current stimulators (San Diego Instruments, San Diego, CA). Under isoflurane/oxygen vapor mixture (1–1.5% isoflurane) anesthesia, subjects were prepared with bipolar stainless steel electrodes (Plastics One, Roanoke, VA) in the posterior lateral hypothalamus (anterior/posterior, −0.5 mm from bregma; lateral, ±1.7 mm; dorsal/ventral, −8.3 mm from dura) (Pellegrino et al., 1986) with the incisor bar elevated 5.0 mm above the interaural line. Subjects were trained to respond for electrical stimulation under a discrete-trial current-threshold intracranial self-stimulation procedure, modified from the original procedure developed by Kornetsky and colleagues (Kornetsky et al., 1979). Each test session lasted 30–40 min and provided two dependent variables for behavioral assessment: reward threshold and response latency (Harrison et al., 2001, Markou and Koob, 1992, Semenova and Markou, 2003).
2.4. Intravenous self-administration and food responding, chambers, surgery and procedure
Methodological details of intravenous self-administration (IVSA) apparatus, catheter construction, surgery and acquisition of nicotine- and food-maintained responding have been described elsewhere (Paterson and Markou, 2004). Training and testing occurred in 24 sound-attenuated (San Diego Instruments, San Diego, CA) Plexiglas chambers (Med Associates, St. Albans, VT). Briefly, rats were food-restricted to 20 g of chow per day and trained to respond for food pellets, progressing from a fixed-ratio 1 time-out 1 s (FR1 TO1) to a FR5 TO 20 s schedule of reinforcement, with sessions lasting approximately 30 min. After the completion of food training, rats were prepared with intravenous catheters inserted into the right jugular vein under isoflurane/oxygen vapor mixture (1–2% isoflurane) anesthesia and were allowed to self-administer nicotine (0.03 mg/kg/infusion, base). Responding on the active lever (previously paired with delivery of a food pellet) resulted in the delivery of the nicotine solution in a volume of 0.1 ml over a 1 s period, and the presentation of a cue light above the active lever that remained lit for 20 s, during which time responses on the active lever had no consequences (i.e., time-out period). Responding on the inactive lever (introduced during the first self-administration session) had no consequences. Rats received 20 g rat chow per day, at least 1 h after termination of testing. Animals were allowed to self-administer nicotine for 12 h/day, 7 days/week during the dark cycle. Control rats were trained to respond for food, and then allowed to self-administer saline 12 h/day, 7 days/week. Rats were allowed to self-administer saline or nicotine for a total of 21–22 sessions.
2.5. Rating of somatic signs of nicotine withdrawal
Somatic signs of nicotine withdrawal were counted under white light conditions in cylindrical Plexiglas chambers (diameter 15 cm) with sawdust bedding on the floor. Each subject was observed for 10 min by an observer blind to the subjects’ treatments. The standard checklist used was adapted from an opiate withdrawal signs checklist (Epping-Jordan et al., 1998, Malin et al., 1992). The following signs were recorded: blinks, body and head shakes, chews, cheek tremors, teeth chattering, escape attempts, foot licks, genital licks, gasps, writhes, scratches, ptosis, and piloerection. Multiple successive counts of any sign required a distinct pause between episodes. Ptosis was counted as 1 for appearance or 0 for non-appearance during the 10 min period of observation.
2.6. Modified minipump assembly
The modified osmotic minipumps were made in our laboratory (Figure 1) as an improved modification of previously described gating device (Azar et al., 2004). The gating device consisted of a stainless steel bilateral guide cannula (C235G-2.0-SPCL, Plastics One, Roanoke, VA, USA) connected with a standard minipump via silastic tubing (Baxter Scientific, McGraw Park, IL, USA). The bilateral cannula consisted of the input cannula defined as the cannula connected to the osmotic minipump, and the output cannula defined as the cannula cut flush with the mesh that delivered drug subcutaneously. Silastic tubing was attached to the input cannula bent at a right angle and encased in dental cement anchored with a 2.2 cm2 durable plastic mesh (CMP-0500-C, Small Parts, Miami Lakes, FL, USA). The gating device was connected to the osmotic minipump by securing one side of a piece of tygon tubing to the end of the input cannula, and the other end to the minipump flow lead. Once the modified minipump was implanted, gating the device to the “ON” (drug delivery) or “OFF” (no drug delivery) position was achieved by securing a single piece of tygon tubing from the top of the input cannula to the top of output cannula. To discontinue subcutaneous drug delivery, the tygon tubing was removed from the top of the input and output cannula. A small metal cap was positioned on the top of cannula assembly to protect the tygon tubing when gating was positioned ON.
Figure 1.
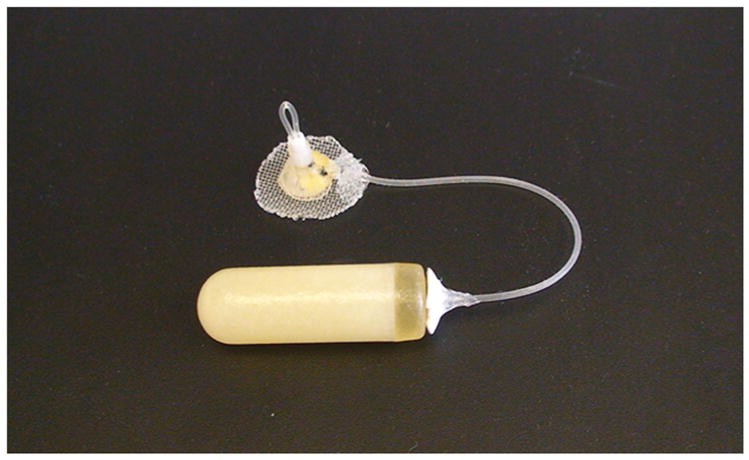
Photograph of a modified osmotic minipump showing an assembled gating device connected to a standard osmotic minipump (model 2ML4, Alzet Osmotic Pumps, Cupertino, CA).
2.7. Osmotic minipump implantation and removal
Fourteen days after ICSS electrode implantation, osmotic minipump surgery was conducted. Rats were anesthetized with an isoflurane/oxygen vapor mixture (1–2%). Standard minipumps (model 2ML4, Alzet Osmotic Pumps, Cupertino, CA) were inserted subcutaneously (back of the animal parallel to the spine) with the flow moderator directed posteriorly. For the modified minipump implantation, an incision 4 cm in length was made perpendicular to the median plane of the body. Then, subcutaneous space was made anterior to the incision to allow space for implantation of the gating device with the corresponding minipump. Once the minipump and the gating device were inserted, a small incision was made above the top of the gating device to allow for the protrusion of the gating device through the skin. After modified or standard minipump implantation, the wound was closed with 9 mm stainless steel wound clips (Becton Dickinson Primary Care Diagnostics, Sparks, MD), and triple-antibiotic ointment was applied to the incision area. On day 21, the minipumps were surgically removed using the aforementioned procedure. The concentration of nicotine was adjusted to compensate for differences in the rats’ body weights at the time of minipump implantation.
2.8. Nicotinic acetylcholine receptor binding analyses
Brains were rapidly removed 12 h after the last nicotine exposure and dissected into coronal sections using a rat brain matrix. The brain regions were identified according to the rat brain atlas (Paxinos and Watson, 1998) and dissected on an ice-cooled metal plate using a round tissue punch (1.5 mm in diameter). The bilateral brain sites dissected were the prelimbic cortex (PLC) as part of the prefrontal cortex, the ventral tegmental area (VTA), the nucleus accumbens (Acb), the amygdala (AMY), the hypothalamus (HYP), and the hippocampus (HIPP), the habenula (HB) and the interpendicular nucleus (IPN). Brain punches were promptly transferred to tubes that were immediately frozen in cooled isopentane and subsequently placed on dry ice before being stored at −80°C.
[125I]-epibatidine binding was measured as described previously (Whiteaker et al., 2000). The tissue was homogenized and in hypotonic buffer and centrifuged at 20,000 x g to obtain the particulate fraction. The particulate fraction was washed by resuspension in fresh buffer followed by recentrifugation a total of three times. The washed particulate fraction was frozen. On the day of assay the washed pellets were resuspended in the overlying buffer and centrifuged at 20,000 g for 10 min. The supernatant was discarded and the pellet was resuspended in ice-cold solution. Resuspension volume varied among brain regions and was adjusted such that less than 10% of the [125I]-epibatidine was bound to the protein at the highest ligand concentration. Samples were incubated in 96-well polystyrene plates for 2 h at room temperature in a final incubation volume of 30 μl with either buffer (total) or buffer containing cytisine (50 nM) or cytisine (100 μM), along with 200 pM [125I]-epibatidine. Buffer composition was (NaCl 144 mM; KCl, 1.5 mM; CaCl2, 2.0 mM; MgSO4, 1.0 mM, HEPES, 25 mM; pH = 7.5). The 100 μM cytisine defines non-specific binding whilst the total and 50 nM cytisine enables determination of cytisine-sensitive and cytisine-resistant binding sites. After incubation, samples were diluted with 200 μl of ice-cold wash buffer and filtered under vacuum (0.2 atm) onto glass fiber filters treated with 0.5% polyethelenimine (top filter, MFS Type B; bottom filter, TypeA/E, Pall Bioscience). An Inotech Cell Harvester (Inotech Biosystems International, Rockville, MD) collected the samples, which were subsequently washed six times with ice-cold buffer. Filters were transferred to a 96-well scintillation plate and counted using a Wallac TriLux 1450 MicroBeta scintillation counter (PerkinElmer Life and Analytical Sciences, Waltham, MA) at 30% efficiency after the addition of 150 μl of Optiphase Supramix scintillation mixture (PerkinElmer Life and Analytical Sciences, Waltham, MA). Protein was measured using the Lowry method (Lowry et al., 1951) with bovine serum albumin standard.
Cytisine-sensitive [125I]-epibatidine binding cites correspond to α4β2* nAChRs (Whiteaker, Jimenez, 2000), and were calculated after the subtraction of cytisine-resistant from total [125I]-epibatidine binding. α-bungarotoxin binding, which would assess α7 nAChR binding, was not examined because α-bungarotoxin binding returns to baseline almost immediately after chronic treatment (Marks et al., 1985) and because prior work has shown that α7 nAChRs are not involved in nicotine withdrawal/reward (Fowler et al., 2008).
2.9. Nicotine and cotinine concentration analyses
On days 21–22 of the experiment, blood samples (300 μL) were collected from the tip of the tail after exposure to nicotine for 12 h. The trunk blood samples were collected from the site where animal was decapitated at the 12 h nicotine withdrawal time point (i.e., 12 h after the last nicotine exposure). Nicotine and cotinine levels in the blood were measured (Apredica, Watertown, MA) in both nicotine- and saline-exposed rats to ensure blinding and verification of assay sensitivity. Briefly, plasma samples were analyzed with liquid chromatography/mass spectrometry technique using an Agilent 6410 mass spectrometer coupled with an Agilent 1200 HPLC and a CTC PAL chilled autosampler, all controlled by MassHunter software (Agilent). After separation on a Polar silica HILIC (Sepax) HPLC column using an acetonitrile-water gradient system, peaks were analyzed by mass spectrometry (MS) using ESI ionization in multiple reaction monitoring (MRM) mode. The lower limit of quantitation for nicotine and cotinine was 27 nM in plasma.
2.10. Experimental design
Experimental design is presented in Figure 2.
Figure 2. Experimental design.
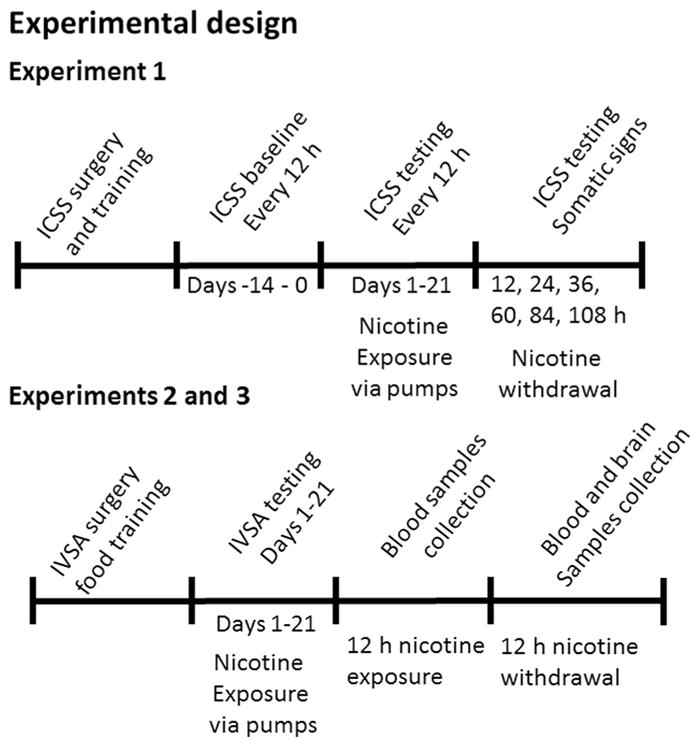
Timeline of experimental design for Experiment 1 (top panel) and Experiments 2 and 3 (bottom panel) showing the sequence of experimental procedures, nicotine exposure and blood/brain samples collection. See text for details. ICSS, intracranial self-stimulation; IVSA, intravenous self-administration; h, hour.
2.10.1. Experiment 1. The effects of temporal pattern and nicotine dose on brain reward function and somatic signs during chronic nicotine exposure and nicotine withdrawal
Rats were prepared with ICSS electrodes into the lateral hypothalamus and trained in the ICSS threshold procedure as described above once daily until stable reward thresholds were established, defined as 10% variation in thresholds for five consecutive days. Rats had reward thresholds assessed twice daily for 14 consecutive days to habituate them to multiple daily threshold assessments. Then, rats prepared with osmotic minipumps were exposed to different doses of nicotine administered intermittently for 12 h (1.2, 3.16 and 6.32 mg/kg/12 h, n=6–14/group) or continuously for 24 h (1.2, 3.16 and 6.32 mg/kg/24 h, n=6–9/group) for 21 days using modified or standard minipumps. The higher nicotine doses were selected because they were shown previously to produce reliable affective (i.e., threshold elevations) and somatic signs of nicotine withdrawal upon cessation of nicotine exposure (Epping-Jordan et al., 1998, Skjei and Markou, 2003); while the lower nicotine dose was selected to approximate the dose self-administered in a 12 h daily session (Kenny and Markou, 2006). Twenty one days of nicotine/saline exposure were selected because 20–28 days of exposure to nicotine self-administration (Kenny and Markou, 2006) induced threshold lowering upon cessation of nicotine self-administration. The use of modified minipumps allowed us to start and stop the nicotine/saline infusion every 12 h (i.e., intermittent non-contingent nicotine exposure). Rats were tested either once (continuous nicotine exposure) or twice (intermittent nicotine exposure) daily in the ICSS procedure. In rats exposed to nicotine intermittently, the first daily ICSS session assessed effects of 12 h nicotine withdrawal; while the second daily ICSS session assessed the effects of 12 h nicotine/saline self-administration. Thresholds continued to be assessed after pump removal at 12 h and every 24 h thereafter until return to baseline levels. Somatic signs were assessed 12 and 36 h post-nicotine. The experiment involving intermittent nicotine exposure was conducted in two replications, with each cohort comprised of all experimental groups. ≪ Figure 2 ≫
2.10.2. Experiments 2 and 3. The effects of administration contingency, temporal pattern and nicotine dose on nAChR binding in various brain areas (Experiment 2), and blood nicotine and cotinine levels (Experiment 3)
Rats initially trained to respond for food reinforcement were allowed to intravenously self-administer nicotine (0.03 mg/kg/infusion, n=12) or saline (n=11) for 21–22 days on a FR5 TO 20 s schedule of reinforcement for 12 h/day, 7 days/week. The other rats were exposed to nicotine (1.2 or 3.16 mg/kg/day) for 21–22 days via 28-day standard (nicotine-1.2: n=10; nicotine-3.16: n=10) or modified (nicotine-1.2: n=8; nicotine 3.16: n=18) osmotic minipumps. Control rats self-administered saline. Due to the large number of rats and the need to closely control the nicotine exposure regimen (i.e., 12 h after the last nicotine exposure), half of the rats in each experimental group were euthanized after 21 days of nicotine exposure and the other half after 22 days of nicotine exposure.
2.11. Statistical Analyses
Statistical analyses were conducted using the Statistical Package for the Social Sciences version 15.0 (SPSS Inc., Chicago, IL). All behavioral data were analyzed with the appropriate mixed-design ANOVAs, with factors Temporal pattern (intermittent vs continuous nicotine exposures) and Nicotine dose being a between-subjects factors, and factor Day of nicotine exposure (19 levels)/nicotine withdrawal (6 levels for ICSS thresholds/latencies and 2 levels for somatic signs) being a within-subject factors. Thresholds and response latencies were expressed as a percentage of the mean of the last five baseline values before the nicotine/saline exposure. Somatic sign data were expressed as the total number of somatic signs observed during the 10 min observation period. The number of nicotine/saline infusions and the number of active/inactive lever presses during the 21 days of nicotine/saline self-administration were analyzed by two-way repeated measures ANOVAs with Nicotine dose as a between-subjects factor and Day of exposure and Lever as a within-subjects factor. Plasma nicotine/cotinine levels and nAChRs binding data were analyzed using one-way ANOVAs with factors Experimental Group (7 levels), Contingency (contingent vs non-contingent), or Temporal pattern (continuous 24 h exposure vs intermittent 12 h exposure) being a between-subjects factors and a two-way ANOVA with Nicotine dose and Temporal pattern being a between-subjects factors. Post hoc comparisons among means were conducted after significant effects using Fisher's Least Significant Difference (LSD) test after statistically significant effects were observed in the ANOVAs. The level of significance was set at 0.05.
3. Results
3.1. Experiment 1. The effects of temporal pattern and nicotine dose on brain reward function and somatic signs during chronic nicotine exposure and nicotine withdrawal
Before any treatments began, no statistically significant differences were observed among mean absolute values of baseline thresholds among the various treatment groups [average group mean thresholds ± SEM: group with intermittent exposure to saline (136.30±12.25 μA), nicotine-1.2 (132.79 ±11.43 μA), nicotine-3.16 (132.29±11.11 μA), nicotine-6.32 (131.43±19.08 μA); group with continuous exposure to saline (114.22±11.92 μA), nicotine-1.2 (118.05±16.89 μA); nicotine-3.16 (112.37±10.93 μA); nicotine-6.32 (128.43±8.68 μA)]. No statistically significant differences were observed among mean absolute values of baseline response latencies for the various treatment groups [average group mean latencies ±SEM: group with intermittent exposure to saline (3.43±0.10 s), nicotine-1.2 (3.72±0.13 s), nicotine-3.16 (3.34±0.09 s), nicotine-6.32 (3.59±0.23 s); group with continuous exposure to saline (2.95±0.15 s), nicotine-1.2 (3.15±0.10 s), nicotine-3.16 (3.27±0.14 s); nicotine-6.32 (3.15±0.13 s)].
3.1.1. The effects of 20-day intermittent and continuous nicotine/saline exposure on reward thresholds and response latencies
During the 20 days of either 12 h intermittent or 24 h continuous nicotine/saline administration, there were no significant changes in ICSS thresholds or response latencies for any of the treatment groups (data not shown). The ANOVA revealed no significant effect of Temporal pattern, Nicotine dose, Day or any interactions.
During the 20 days of repeated 12 h withdrawal from intermittent nicotine/saline administration there were no significant changes in ICSS thresholds or response latencies in any of the treatment groups (data not shown). The ANOVA revealed no effect of Nicotine dose, Day, Temporal pattern or their interaction.
3.1.2. The effects of spontaneous withdrawal from intermittent and continuous nicotine exposure on reward thresholds and response latencies
The reward thresholds and response latencies during spontaneous withdrawal from chronic non-contingent nicotine exposure (intermittent and continuous) are presented in Figure 3. The ANOVA revealed statistically significant effect of Temporal pattern (thresholds: F(1,73)=11.53, p<0.001; latencies: F(1,73)=24.27, p<0.001), Day of withdrawal (thresholds: F(5,365)=17.46, p<0.05; latencies: F(5,365)=12.57, p<0.01) and Nicotine dose for latencies (F(3,73)=3.06, p<0.05), but no effect of Nicotine dose for thresholds, and no 3-way interaction effect for either thresholds or latencies.
Figure 3.

The effects of withdrawal from intermittent and continuous nicotine exposure on brain reward thresholds (A and C, respectively) and response latencies (B and D, respectively). Data are presented as a percentage of baseline thresholds/latencies (mean ± SEM) before nicotine/saline exposure. Asterisks denote statistically significant differences between corresponding groups of nicotine- and saline-exposed rats (**p < 0.01, *p < 0.05, LSD post hoc test).
To further analyse the effects of each temporal pattern on nicotine withdrawal, two-way ANOVAs were performed on thresholds and response latencies. Significant differences between rats receiving nicotine intermittently or continuously were found. Withdrawal from intermittent nicotine exposure had no effect on reward thresholds (Fig. 3A) or response latencies (Fig. 3B) for any nicotine treatment dose as demonstrated by the ANOVA (no significant effect of Nicotine Dose, or Day of withdrawal and no interaction effect). In contrast and in replication of our previous findings (Epping-Jordan et al., 1998, Semenova and Markou, 2003, Skjei and Markou, 2003), rats that received continuous nicotine infusions exhibited threshold elevations (Fig. 3C) and increases in response latencies (Fig. 3D) during nicotine withdrawal. The ANOVA revealed significant main effects of Nicotine dose (thresholds: F(3,28)=3.06, p<0.05; latencies: F(3,28)=4.77, p<0.01) and Day of withdrawal (thresholds: F(5,140)=3.06, p<0.05; latencies: F(5,140)=7.90, p<0.05). There was no significant Nicotine dose by Day of withdrawal interaction effect for thresholds and latencies. Pre-planned comparisons revealed that rats that received continuous nicotine infusions at the doses of 3.16 and 6.32 mg/kg/day exhibited threshold elevations at all withdrawal times between 12–84 h post-nicotine. Significantly increased response latencies were also observed 12 h and 36 h post-nicotine compared to thresholds and latencies of the saline control group for rats that received continuous nicotine infusions at the doses of 3.16 and 6.32 mg/kg/day.
3.1.3. The effects of spontaneous withdrawal from intermittent and continuous nicotine exposure on somatic signs
Figure 4 shows number of somatic signs during spontaneous withdrawal from chronic non-contingent nicotine exposure. The ANOVA revealed a statistically significant effect of Temporal pattern (F(1,73)=10.01, p<0.05), Day of withdrawal (F(1,73)=22.55, p<0.05) and Nicotine dose (F(3,73)=4.67, p<0.05). Furthermore, there were significant interaction effects of Temporal pattern by Day of withdrawal (F(1,73)=18.75, p<0.05), and Temporal pattern by Nicotine dose (F(3,73)=3.24, p<0.05).
Figure 4.

The effects of withdrawal from the intermittent (A) and continuous (B) nicotine exposure on somatic signs of nicotine withdrawal (mean ± SEM). Asterisks denote statistically significant differences between nicotine- and saline-exposed rats (**p < 0.01, *p < 0.05, LSD post hoc test).
To further analyse the effects of each temporal pattern on the expression somatic signs after spontaneous nicotine withdrawal, separate two-way ANOVAs were performed. No effect on the number of somatic signs were observed following withdrawal from intermittent nicotine exposure to any nicotine dose (Fig. 4A) as reflected by non-significant effect of Day of withdrawal, Nicotine dose and Day by Nicotine dose interaction. In contrast, withdrawal from continuous nicotine exposure resulted in increased number of somatic signs (Fig. 4B) as reflected in significant effect of Day of withdrawal (F(1,28)=23.46, p<0.05) and Nicotine dose (F(3,28)=4.87, p<0.05) effects. However, no significant Day by Nicotine dose interaction was observed. Post hoc comparisons revealed that continuous exposure to all nicotine doses induced increases in somatic signs 12 h post-nicotine compared to saline exposure (Fig. 4B). Furthermore, somatic signs were significantly increased 36 h post-nicotine after exposure to the highest dose (6.32 mg/kg/day; Fig. 4B).
3.2. Experiment 2. The effects of administration contingency, temporal pattern and nicotine dose on nAChR bindings in various brain areas
3.2.1. Nicotine self-administration
Figure 5 presents the time courses for both active and inactive lever presses of rats self-administering either saline (n=21) or nicotine (n=22) for 12 h per day for 21 days. Those rats self-administering nicotine received an average dose of 1.04±0.06 mg/kg per day of nicotine base ranging from 0.77 to 1.27 mg/kg during days 1–21 of self-administration. A two-way ANOVA on the number of nicotine/saline infusions revealed significant main effects of Infusions (F(1,41)=88.9, p<0.0001), Day (F(20,820)= 17.26, p<0.0001), and an Infusions by Day interaction (F(20,820)=2.49, p<0.001). Post hoc comparisons showed that the number of nicotine infusions was significantly higher than the number of saline infusions during 12 h daily sessions from day 3 to day 21 of self-administration (p<0.001). The relatively high rates of responding on the first day resulted from initial food training prior to nicotine self-administration. A two-way repeated measures ANOVA on the number of active and inactive lever presses revealed significant effects of Nicotine/saline exposure (F(1,82) =79.45, p<0.0001), Lever (F(1,82)=162.15, p<0.0001), Day (F(20,1640)=23.02, p<0.0001) as well as Nicotine/saline exposure by Lever (F(1,82)=45.54, p<0.0001) and Day by Lever (F(20,1640)=9.22, p<0.001) interactions. Both saline and nicotine maintained more responding on the active than the inactive lever, and nicotine maintained significantly more active lever pressing than saline (Fig. 5A&B).
Figure 5.
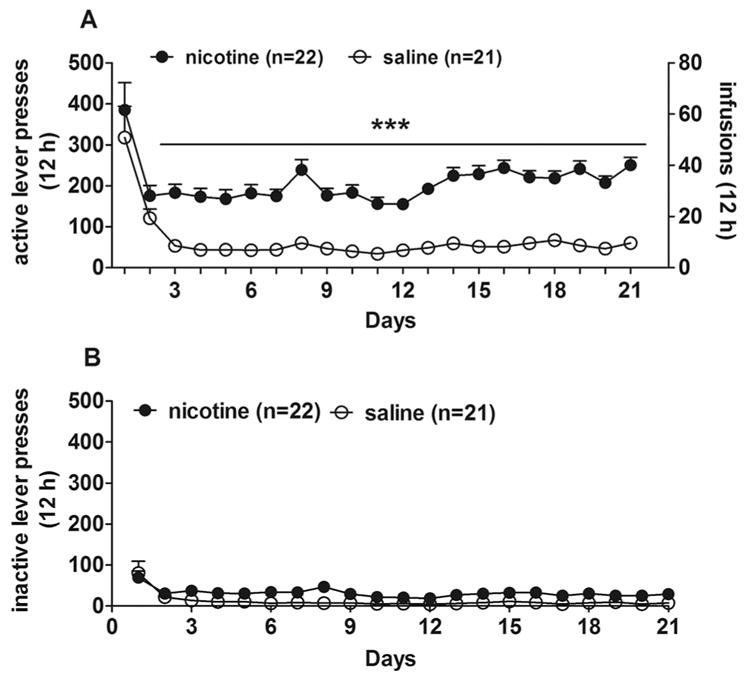
Nicotine self-administration in rats. (A) The number of infusions and active lever presses obtained daily during the 21-day self-administration period. (B) The number of inactive lever presses obtained daily during the 21-day self-administration period. Data are expressed as mean ± SEM. ***p < 0.001, compared with saline using LSD post hoc comparisons.
3.2.2. Nicotinic acetylcholine receptor binding analyses
This experiment was performed in two replications because several samples were spoiled during sample collection and processing. Cystine-sensitive and resistant nAChR binding was measured in the PLC, AMY, Acb, VTA, HIPP, HYP, HAB and IPN 12 h after the termination of nicotine exposure via pumps (continuously or intermittently) or self-administration. Rats were exposed to nicotine at doses equal to both contingently self-administered (1.2 mg/kg/12 h) and non-contingently administered via minipumps (1.2 mg/kg/12 h and 1.2 mg/kg/24 h), as well as the minimal nicotine dose that produced nicotine withdrawal signs (3.16 mg/kg/12 h and 3.16 mg/kg/24 h). nAChRs binding assays were not conducted on brain samples from rats treated with the highest nicotine dose (6.32 mg/kg/24 h or 6.32 mg/kg/12 h base) because medium nicotine dose (3.16 mg/kg/12 h and 3.16 mg/kg/24 h) produced upregulation of nAChRs in all brain areas assessed independent of contingency.
Figure 6 presents the levels of cytisine-sensitive epibatidine binding in corticolimbic brain areas. One-way ANOVAs for all seven treatment groups revealed significant effects of Treatment Group in PLC (F(6,60)=4.35, p<0.001), AMY (F(6,55)=15.86, p<0.001), Acb (F(6,51)=4.43, p<0.01), VTA (F(6,60)=4.35, p<0.001), HIPP (F(6,125)=27.01, p<0.001) and HYP (F(6,131)=9.67, p<0.001), but not in HAB (F(6,59)=0.89, p>0.05) or IPN (F(6,76)=0.79, p>0.05). There were no significant differences in nAChR binding between control groups exposed to saline via self-administration or pump.
Figure 6.

Cytisine-sensitive nicotinic acetylcholine receptor binding data in different brain regions. Data are expressed as mean ± SEM. Asterisks denote a significant difference between exposures to nicotine (nic) and saline (sal) in corresponding treatments groups (*, p<0.05, **, p<0.01, ***, p<0.001, LSD post hoc test). (#) denotes a significant difference between exposures to low nicotine dose via self-administration (SA) and pumps with intermittent (12 h) or continuous (24 h) nicotine delivery (#, p<0.05, ##, p<0.01, ###, p<0.001, LSD post hoc test). (^) denotes a significant difference between intermittent exposure to 1.2 mg/kg/12 h and 3.16 mg/kg/12 h nicotine doses via pumps (^, p<0.05, LSD post hoc test).
Analysis of the effect of nicotine treatment for rats self-administering nicotine was assessed using t-tests (Figure 6). These analyses revealed a significant effect of nicotine treatment on cytisine-sensitive [125I]-epibatidine binding in AMY (t14 = 5.54, p<0.001), HIPP (t37 = 4.35, p<0.001), HYP (t39 = 3.35, p<0.001), Acb (t14 = 2.48, p=0.026), PLC (t39 = 4.32, p<0.001), but not in HAB (t21 = 0.34, p>0.05), IPN (t21 = 0.08, p>0.05) or VTA (t21 = 0.02, p>0.05).
Additional statistical analyses compared nAChR binding after exposure to the low 1.2 mg/kg nicotine dose via self-administration and minipumps (intermittently). Consistent with the results of the overall one-way ANOVA, significant effects of nicotine exposure on cytisine-sensitive [125I]-epibatidine binding were revealed by two-way ANOVA (with nicotine dose and route of administration as the independent variables): AMY (F(1,28)=45.11, p<0.001), HIPP (F(1,60)=46.39, p<0.001), HYP (F(1,63)=45.27, p<0.001), Acb (F(1,24)=6.99, p=0.014), PLC (F(1,62)=69.08, p<0.001). However, the main effect of nicotine dose was not significant in VTA (F(1,32)=3.67, p=0.064), HAB (F(1,28)=0.88, p>0.05) and IPN (F(1,34)=0.27, p>0.05). No significant difference between the treatment groups on cytisine-sensitive [125I]-epibatidine binding (self-administration vs. minipump) was detected in any brain region. However, a significant treatment group by nicotine dose interaction was noted for HIPP (F(1,60)=7.83, p=0.007), HYP (F(1,63)=8.01, p=0.006) and PLC (F(1,62)=8.46, p≤0.005) reflecting difference in the magnitude of increase between for the self-administration and minipump groups elicited by nicotine.
Further analyses compared nAChR binding after intermittent and continuous exposure to nicotine via minipumps. A two-way ANOVA revealed a significant difference between rats treated for 12 h or 24 h in three brain regions with higher binding noted for the 24 h treatment group: HIPP (F(1,75)=11.51, p<0.001), Acb (F(1,28)=8.65, p=0.006), and PLC (F(1,62)=9.32, p=0.006). The two-way ANOVA also indicated a significant effect of nicotine dose in HIPP (F(1,75)=7.88, p=0.006) where higher binding was noted following treatment with 3.6 mg/kg/day: HIPP (F(1,75)=7.88, p=0.006).
The levels of cytisine-resistant binding were relatively small compared to cytisine-sensitive epibatidine binding in all brain structures except the AMY, VTA, HAB and IPN (Table 1). All analyses revealed no effect of Experimental group for any of these four brain structures.
Table 1.
Cytisine-resistant nicotinic acetylcholine receptor binding (fmol/mg protein) in different brain structures at 12 h post-nicotine/saline exposure.
| Group | PLC | AMY | Acb | VTA | HYP | HIPP |
|---|---|---|---|---|---|---|
| SA-sal | 81.2±14.2 | 1.89±0.3 | −0.38±0.9 | 37.5±14.2 | 1.78±0.8 | 2.10±0.1 |
| SA-nic | 57.7±10.1 | 2.36±0.4 | 0.00±0.0 | 32.04±10.1 | 1.52±0.9 | 2.51±0.1 |
| Pump-sal | 64.3±9.7 | 1.89±0.3 | 1.3±1.7 | 25.78±9.7 | 3.40±0.4 | 1.3±0.2# |
| 1.2-nic-12h | 77.0±7.4 | 2.79±0.4 | 0.00±0.0 | 21.04±7.4 | 0.98±1.0 | 2.81±0.2* |
| 1.2-nic-24h | 74.8±10.1 | 2.65±0.3 | 0.00±0.0 | 26.8±10.1 | 0.35±1.5 | 3.23±0.2* |
| 3.16-nic-12h | 76.1±7.3 | 2.58±0.3 | 0.00±0.0 | 20.76±7.3 | 1.11±1.05 | 3.00±0.2* |
| 3.16-nic-24h | 71.6±18.5 | 2.90±0.3 | 0.00±0.0 | 48.92±18.5 | 1.16±0.8 | 3.88±0.3*^ |
Data are expressed as mean ± SEM.3
p<0.001 nicotine exposure via pumps compared to saline exposure via pumps;
p<0.05 saline exposure via self-administration compare to saline exposure via pumps;
p<0.01 intermittent (12 h) compared to continuous (24 h) exposure to 3.16 mg/kg/day nicotine dose via pumps.
Abbreviations: PLC, prelimbic cortex, AMY-amygdala, Acb, nucleus accumbens, VTA, ventral tegmental area, HYP, hypothalamus, HIPP, hippocampus, nic, nicotine, sal, saline, SA, self-administration.
3.3. Experiment 3. The effects of administration contingency, temporal pattern and nicotine dose on plasma nicotine and cotinine levels
After continuous or intermittent exposure to nicotine for 21 days, levels of both nicotine and cotinine were detected in plasma (Table 2) immediately after nicotine exposure for 12 h. One-way ANOVA revealed a significant effect of Treatment Group on level of nicotine (F(4,48)=3.46, p<0.05) and cotinine (F(4,48)=3.66, p<0.01). During exposure to the similar nicotine dose 1.2 mg/kg/day intermittently via self-administration and pump or continuously via pump, ANOVA revealed a significant effect of Contingency for nicotine (F(2,29)=5.58, p<0.01), but not for cotinine. Post hoc comparisons revealed that nicotine levels were significantly higher after nicotine self-administration compared to intermittent exposures to a similar nicotine dose via pumps; while cotinine levels did not differ between these experimental groups (Table 2). During intermittent and continuous exposure to low and high nicotine doses via pumps, an ANOVA revealed no effect of Temporal Pattern or Temporal Pattern by Nicotine dose interaction, although there was a significant effect of Nicotine dose for nicotine (F(1,36)=11.45, p<0.01) and cotinine (F(1,36)=8.78, p<0.01) levels in plasma. Group comparisons showed that intermittent or continuous exposure to low nicotine dose via pumps resulted in significantly lower levels of both nicotine and cotinine compared to intermittent or continuous exposure to high nicotine dose via pumps (Table 2).
Table 2.
Plasma nicotine and cotinine levels following exposure to nicotine or nicotine withdrawal in rats exposed to nicotine/saline for 21 days.
| Temporal Pattern | Nicotine dose | N | Cotinine (ng/ml) | Nicotine (ng/ml) |
|---|---|---|---|---|
| Exposure to nicotine | ||||
| Intermittent-SA | 1.2 mg/kg/12h | 12 | 666.4±78.2a | 35.4±7.1 |
| Intermittent-pump | 1.2 mg/kg/12h | 9 | 510.25±158.4a | 14.45±5.2ab |
| Continuous-pump | 1.2 mg/kg/24h | 9 | 450.73±30.9a | 13.53±1.44ab |
| Intermittent-pump | 3.16 mg/kg/12h | 9 | 825.04±358.5a | 29.69±11.6 |
| Continuous-pump | 3.16 mg/kg/24h | 10 | 1211.12±40.6 | 38.69±2.1 |
| Exposure to 12h nicotine withdrawal | ||||
| Intermittent-SA | 1.2 mg/kg/12h | 12 | 141.6±17.1 | |
| Intermittent-pump | 1.2 mg/kg/12h | 9 | 162.6±32.4ac | |
| Continuous-pump | 1.2 mg/kg/24h | 9 | 104.9±9.2ac | |
| Intermittent-pump | 3.16 mg/kg/12h | 9 | 540.8±87.7a | |
| Continuous-pump | 3.16 mg/kg/24h | 10 | 284.5±14.4 | |
Data are expressed as mean ± SEM.
denotes a significant group differences from continuous exposure to 3.16 mg/kg/24h nicotine dose (a, p<0.05, LSD test);
denotes a significant group differences from nicotine self-administration (b, p<0.05, LSD test);
denoted a significant group differences from intermittent exposure to 3.16 mg/kg/12h nicotine dose (c, p<0.05, LSD test).
During nicotine withdrawal, no nicotine was detected in plasma. No nicotine or cotinine were detected in plasma in control rats exposed to saline either via minipumps or self-administration. SA, self-administration, N, number of samples per group.
ANOVA on cotinine levels at the 12 h nicotine withdrawal time-point, revealed a significant effect of Treatment Group (F(4,48)=20.82, p<0.001). During exposure to a similar nicotine dose (1.2 mg/kg/day) administered either intermittently via self-administration or pump, an ANOVA revealed no effect of Contingency for cotinine levels. During intermittent and continuous exposure to low and high nicotine doses via pumps, the ANOVA revealed a significant effect of Temporal Pattern (F(1,36)=12.91, p<0.001), Nicotine dose (F(1,36)=40.71, p<0.001) and their interaction (F(1,36)=5.16, p<0.05). Group comparisons showed that intermittent or continuous exposure to a low nicotine dose via pumps resulted in significantly lower levels of cotinine compared to intermittent or continuous exposure to the high nicotine dose administered via pumps during withdrawal (Table 2).
4. Discussion
4.1. Main findings
The results of the present studies showed that the termination of intermittent non-contingent nicotine exposure (1.2, 3.16 and 6.32 mg/kg/12 h) had no effect on reward thresholds, response latencies or somatic signs indicating no nicotine withdrawal after this intermittent pattern of nicotine exposure. In contrast and in replication of our previous findings, withdrawal from continuous non-contingent nicotine exposure at higher doses (3.16 and 6.32 mg/kg/24 h) induced threshold elevations and increased number of somatic signs. Termination from continuous exposure to a low nicotine dose (1.2 mg/kg/24 h) did not induce threshold elevations but increased number of somatic signs. Chronic nicotine exposure induced upregulation of α4β2* nAChR in contingency-dependent manner that varied among brain regions. Contingent intermittent nicotine exposure resulted in a higher plasma nicotine levels compared to levels seen after nicotine exposure at the same dose non-contingently independent of temporal pattern. Plasma nicotine levels after non-contingent exposure depended on nicotine dose but not on temporal pattern. The same nicotine dose resulted in higher cotinine levels after intermittent exposure compared to continuous exposure via minipumps.
4.2. Expression of nicotine withdrawal
We showed previously that dose and duration of nicotine exposure significantly influenced the duration and/or overall severity of the subsequent nicotine withdrawal (Epping-Jordan et al., 1998, Skjei and Markou, 2003). In replication of these findings, withdrawal from continuous nicotine exposure administered at the doses of 3.16 or 6.32 mg/kg/24 h elevated reward thresholds, increased response latencies and the number of somatic signs of withdrawal. Importantly, these same nicotine doses (3.16 or 6.32 mg/kg/12 h) administered intermittently did not induce threshold elevations or increases in somatic signs. Further, withdrawal from the lowest nicotine dose administered continuously (1.2 mg/kg/24 h) or intermittently (1.2 mg/kg/12 h) via minipumps had no effect on reward threshold, response latencies or somatic signs of withdrawal. In contrast to our findings, one published study (Brynildsen et al., 2016), using a different intermittent pattern of exposure (1 h ON, 1 h OFF) via a modified minipump, reported lasting effects of intermittent nicotine (1.2, 2,4 and 4.8 mg/kg/day for 14 days) on mecamylamine-precipitated somatic withdrawal signs. Similarly, moderate increases in somatic signs were observed during spontaneous or mecamylamine-precipitated nicotine withdrawal in rats exposed to daily nicotine self-administration for 1 h or 6 h (Paterson and Markou, 2004), or 23 h (Harris et al., 2011, O'Dell et al., 2007). These results demonstrate the importance of temporal pattern of nicotine exposure in inducing the negative affective and somatic signs of spontaneous, but not mecamylamine-precipitated nicotine withdrawal. The reported mecamylamine-precipitated withdrawal after exposure to low nicotine doses may be attributed to acute blockade of nAChRs; while during spontaneous nicotine withdrawal other neuroplasticity mechanisms are involved.
The findings that intermittent non-contingent nicotine administration does not affect measures of nicotine withdrawal are in contrast to the effects observed following contingent 12 h nicotine self-administration of the same nicotine dose reported previously (Kenny and Markou, 2006) that produced long-lasting enhancement of brain reward function. However, withdrawal from unlimited 23 h access to nicotine self-administration resulted in transient small threshold elevations (below 10 % of baseline levels) during the first three days of withdrawal followed by modest short-lasting threshold lowering on days 5–6 of withdrawal/extinction (Harris et al., 2011), a result that contrasts with the current study and the previous report (Kenny and Markou, 2006). Thus, it appears that intermittent access to nicotine self-administration produced differential effects on brain reward function compared to unlimited access.
The expression of nicotine withdrawal in rodents is complex (for review, Jackson et al., 2015). Studies with genetically modified mice have established that somatic signs of withdrawal are mediated by β4 (Salas et al., 2004; Stoker et al., 2012), α2 (Salas et al., 2009) and α5 nAChR subunits (Jackson et al., 2008; Salas et al., 2009), and also involve receptors in the medial habenula-interpeduncular nucleus system (Salas et al., 2009). While the β2 nAChR subunit is not implicated in mediating somatic signs of withdrawal, this subunit in involved in affective signs of withdrawal including reward deficits or anhedonia (Stoker et al., 2015), fear conditioning (Portugal et al., 2008) and anxiety-like behavior (Jackson et al., 2008). Nicotine withdrawal is also influenced by age, sex, the environment indicating that, in addition to genetic factors, complex neuropsychological mechanisms mediate the withdrawal syndrome (for review, Jackson et al., 2015). Our findings indicate that both contingency and a temporal pattern of exposure are important determinants in modeling nicotine dependence in Wistar male rats. These results may differ somewhat from results in human smokers who experience well-characterized withdrawal syndrome following smoking cessation (Hughes, 2007). The differential effects of temporal pattern of non-contingent nicotine exposure on signs of nicotine withdrawal may be attributed to different pharmacokinetic mechanisms of exposure. However, limited assessment of nicotine levels at one time point was not sufficient to draw meaningful conclusions. Further, considering that chronic nicotine exposure produces a wide range of adaptations in neuroplasticity, the absence of spontaneous withdrawal signs from intermittent non-contingent nicotine exposure can be attributed to multiple factors. The role of these factors in the expression of nicotine withdrawal induced by non-contingent intermittent nicotine exposure was not investigated in the present studies. Future work in this area is warranted.
4.3. Upregulation of α4β2* nAChR
Cytisine-sensitive [125I]-epibatidine binding results demonstrated that α4β2* nAChR upregulation was brain region-specific and depended on both contingency and temporal pattern of nicotine exposure. Consistent with previous reports, the cytisine-sensitive [125I]-epibatidine binding in HAB and IPN were unaffected by any of the nicotine treatments performed in the present study (Marks et al., 2011, Nguyen et al., 2003). In contrast, nicotine treatment elicited up-regulation of cytisine-sensitive [125I]-epibatidine binding in the other brain regions tested, although some differences in the response to nicotine exposure among these regions were noted. Self-administration of nicotine elicited significant up-regulation of cytisine-sensitive [125I]-epibatidine binding in PLC, AMY, Acb, HIPP and HYP but not in VTA, HAB or IPN. When the extent of up-regulation of cytisine-sensitive [125I]-epibatidine binding elicited by contingent nicotine administration was compared to that elicited by the most comparable non-contingent nicotine treatment (1.2 mg/kg/12 h), significant effects of nicotine in PLC, AMY, Acb, HIPP and HYP were noted, but there were no main effects of treatment pattern were found. However, the significant treatment by nicotine interactions observed for HIPP, HYP and PLC indicated a different pattern of response in these brain regions. Of particular interest was the difference in response to nicotine treatment between VTA and Acb, brain structures directly involved in nicotine reward and withdrawal (D'Souza and Markou, 2011, Koob and Volkow, 2010). Nicotine self-administration produced α4β2* nAChR upregulation in the Acb, but not in the VTA. Consistent with our results, upregulation of α4β2* nAChR was observed in the Acb after 1 h (Moretti et al., 2010) and 23 h (Parker et al., 2004) of nicotine self-administration in rats. Interestingly, however, exposure to 23 h of nicotine self-administration also up-regulated α4β2* nAChR in the VTA (Parker et al., 2004), indicating that longer duration of contingent nicotine exposure was needed to produce α4β2* nAChR up-regulation in the VTA. The opposite pattern of results was observed in studies with mice, where increased levels of α4β2* nAChR binding were observed in the VTA, but not in the Acb, after 1 h self-administration (Metaxas et al., 2010). These discrepancies between mouse and rat studies may be attributed to species or procedural differences (e.g., the lower nicotine dose self-administered by mice compared to those self-administered by rats; shorter duration of nicotine exposure).
In contrast to contingent nicotine self-administration, intermittent non-contingent exposure to the same nicotine dose (1.2 mg/kg/ 12 h) upregulated α4β2* nAChR in the VTA and PLC, but not in the Acb. Similar to our findings, non-contingent repeated nicotine injections (4 x 0.4 mg/kg base for 7 days) upregulated α4β2* nAChRs in the VTA, but not in the Acb or prefrontal cortex (Baker et al., 2013). Thus, it appears that contingent nicotine self-administration is associated with specific upregulation of α4β2* nAChR in the Acb; while non-contingent intermittent nicotine exposure upregulates α4β2* nAChR in the VTA and the cortex. Non-contingent exposure to the higher nicotine doses tested, administered continuously or intermittently up-regulated α4β2* nAChR in all six brain areas. These findings are consistent with earlier work showing increased ligand binding after non-contingent intermittent or continuous nicotine exposure in various brain regions including the prefrontal cortex, the VTA and the Acb (for review see (Vezina et al., 2007)).
Associations between nAChRs upregulation and expression of nicotine withdrawal signs are not well-understood. Our finding demonstrated that α4β2* nAChRs upregulation can be observed in the absence of reward deficit and somatic withdrawal after exposure to non-contingent intermittent nicotine exposure. Consistent with our findings, β2* nAChR availability in abstinent smokers during early abstinence did not correlate with the severity of nicotine withdrawal assessed with the Minnesota Withdrawal Questionnaire (Staley et al., 2006). Nevertheless, these data doesn’t preclude a more direct relationship with individual features of nicotine withdrawal such as depression, anxiety or other symptoms because the severity of nicotine withdrawal is likely determined by the complex interplay between α4β2* nAChRs and multiple neurochemical systems as well as the large number of signaling pathways (for review, Jackson et al., 2015).
Cytisine-resistant [125I]-epibatidine binding sites are related to other nAChRs subtypes that may contain α2, α3, α4, α6, β2 and β4 subunits (Baddick and Marks, 2011, Marks et al., 2006). Independent of contingency or temporal pattern, exposure to nicotine had no effect in the four brain areas expressing reliably measurable cytisine-resistant [125I]-epibatidine binding. Consistent with our findings, no differences in cytisine-resistant nAChRs binding between self-administering mice and yoked control mice in corticolimbic brain areas were reported (Metaxas et al., 2010). Altogether, consistent with previous work (Nguyen et al., 2003), our findings on cytisine-sensitive (primarily α4β2*-nAChR sites) and cytisine-resistant nAChRs (non- α4β2*-nAChR sites, primarily α3β4*-nAChR sites in HAB and IPN) binding suggest that chronic nicotine exposure upregulates mainly α4β2* nAChRs, but not nAChRs subtypes containing α2, α3, α4, α6, β2 and β4 subunits in the corticolimbic brain areas, but the effects on α4β2*-nAChR sites depend on contingency and temporal pattern of nicotine exposure. Our observation that chronic nicotine treatment has differential effects on α4β2*-nAChR sites in various brain regions is also consistent with previous reports (Marks et al., 2011, Nguyen et al., 2003). The reasons underlying these differential responses to chronic nicotine remain unclear. However, differences in receptor composition, such as the resistance of α4β2α5-nAChR subtype to up-regulation following nicotine treatment (Mao et al., 2008) and potential differences in responses of receptors differing in α4/β2 stoichiometry (Moroni et al., 2006) may contribute to the differential responses among brain regions.
4.4. Plasma nicotine and cotinine levels
Our results extend previous findings showing differential effects of contingency of nicotine exposure on nicotine/cotinine levels in plasma. In our studies, nicotine plasma levels after the 12 h self-administration session (39.8±2.8 infusions/12 h, 35.4±7.1 ng/ml) were lower compared to previously reported plasma nicotine levels measured after fewer infusions obtained during 1 h (7 infusions, ~95 ng/ml nicotine arterial levels (Donny et al., 2000)) or 2 h (10 infusions, 65.4 ng/ml nicotine venous levels (Shoaib and Stolerman, 1999)) self-administration cessions. These results are not surprising because the highest nicotine intake usually occurs during the first hour of the self-administration session, and then a steady low intake is maintained during 22 h unlimited access to nicotine (O'Dell et al., 2007). When nicotine was delivered continuously via minipumps, plasma nicotine levels depended on nicotine dose but not on temporal pattern. The highest nicotine plasma levels (38.7±2.1 ng/ml) were achieved during continuous nicotine exposure to the highest nicotine dose (3.16 mg/kg/24 h) via minipumps. Similarly, dose-dependent increases in plasma nicotine levels can also be gathered from the literature reporting nicotine levels during continuous exposure to nicotine at different doses via minipumps (Ghosheh et al., 2001, O'Dell et al., 2006, Winders et al., 1998). Interestingly, in the present work, the plasma nicotine levels after 12 h of self-administration were higher compared to levels seen after nicotine exposure at the same dose via minipumps (intermittently or continuously). To reach plasma nicotine levels comparable to those observed during nicotine self-administration, nicotine dose delivered via minipumps had to be 3-fold higher than the dose administered non-contingently independent of temporal pattern of administration (intermittent or continuous). Consistent with our findings, nicotine plasma levels in smokers usually peak at 15–40 ng/ml following cigarette smoking and the levels decline thereafter to around 10 ng/ml (Feyerabend and Russell, 1990, Russell et al., 1986). However, some smokers can achieve nicotine plasma levels as high as 100 ng/ml depending on how quickly the cigarette is smoked (Benowitz et al., 1983), as well as the time of blood sampling (Rose et al., 1999).
Further, in smokers, the plasma cotinine levels are in average approximately 300 ng/ml (Benowitz et al., 1997), and increase during the day proportionally to the number of cigarettes smoked reaching up to 600 ng/ml in some smokers (Benowitz et al., 1983). In the present study, the highest cotinine levels were detected during continuous exposure to nicotine at the highest dose via minipumps (1211.1±40.6 ng/ml); while all other nicotine exposures yielded plasma cotinine levels ranging from 450 to 666 ng/ml. Similar to nicotine levels, dose-dependent increases in plasma cotinine levels were reported in the literature during continuous exposure to various nicotine doses via minipumps (Ghosheh et al., 2001, O'Dell et al., 2006, Winders et al., 1998). During nicotine withdrawal, plasma cotinine levels were decreased in all experimental groups. However, the decreases in cotinine levels during nicotine withdrawal were dose-dependent within each temporal pattern with no effect of contingency. Interestingly, however, the same nicotine dose resulted in higher cotinine levels after intermittent exposure compared to continuous exposure via minipumps.
5. Conclusions
Our findings demonstrated that contingency and temporal pattern of nicotine exposure induced differential behavioral adaptations and neuroplasticity related to α4β2* nAChRs expression. Although intermittent pattern of nicotine exposure closely mimics smoking patterns in humans, cessation of non-contingent intermittent nicotine exposure via minipumps did not result in reward deficit or somatic withdrawal compared to non-contingent continuous nicotine exposure in rats. Thus, temporal pattern of nicotine exposure should be taken into consideration when modeling reward deficit and somatic aspects of nicotine withdrawal in rats. Upregulation of α4β2* nAChRs was brain site-specific and depended on both temporal pattern and contingency of nicotine exposure suggesting that different brain circuits may mediate behavioral adaptations in response to different patterns of nicotine exposure. Future research is warranted to understand potential mechanisms underlying differential effects of temporal pattern of nicotine exposure on reward deficit and somatic aspects of nicotine withdrawal in rats.
Highlights.
Non-contingent continuous nicotine produced withdrawal and upregulated α4β2 nAChRs.
Non-contingent intermittent nicotine exposure did not produce nicotine withdrawal.
Non-contingent intermittent nicotine upregulated α4β2 nAChRs in the VTA and PLC.
Contingent intermittent nicotine exposure upregulated α4β2* nAChRs in the Acb.
Acknowledgments
This work was supported by the National Institutes on Drug Abuse grants R56DA011946 (AM) and R01 DA003194 to MJM. The authors would like to thank Mrs. Jessica Benedict for technical assistance. The authors thank Dr. Neil E. Paterson for conducting preliminary studies investigating the effects of intermittent and continuous nicotine exposure on brain reward function and somatic signs during spontaneous nicotine withdrawal that facilitated the design of the reported studies.
Footnotes
Authors contributions
SS, MJM and AM were responsible for the study concept and design. SS, XJ contributed to the acquisition of animal data. MJM, TDMCB, MPT contributed to the acquisition of molecular data. SS, XJ, MJM completed data analysis and interpretation of findings. SS drafted the manuscript. SS, AM and MJM provided critical revision of the manuscript for important intellectual content. All authors critically reviewed the content and approved the final version for publication.
Financial disclosures/conflict of interest
There are no actual or potential financial conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Azar MR, Ahmed SH, Lintz R, Gutierrez T, Stinus L, Koob GF. A non-invasive gating device for continuous drug delivery that allows control over the timing and duration of spontaneous opiate withdrawal. Journal of neuroscience methods. 2004;135:129–35. doi: 10.1016/j.jneumeth.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Baddick CG, Marks MJ. An autoradiographic survey of mouse brain nicotinic acetylcholine receptors defined by null mutants. Biochem Pharmacol. 2011;82:828–41. doi: 10.1016/j.bcp.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker LK, Mao D, Chi H, Govind AP, Vallejo YF, Iacoviello M, et al. Intermittent nicotine exposure upregulates nAChRs in VTA dopamine neurons and sensitises locomotor responding to the drug. Eur J Neurosci. 2013;37:1004–11. doi: 10.1111/ejn.12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benowitz NL, Hall SM, Herning RI, Jacob P, 3rd, Jones RT, Osman AL. Smokers of low-yield cigarettes do not consume less nicotine. N Engl J Med. 1983;309:139–42. doi: 10.1056/NEJM198307213090303. [DOI] [PubMed] [Google Scholar]
- Benowitz NL, Zevin S, Jacob P., 3rd Sources of variability in nicotine and cotinine levels with use of nicotine nasal spray, transdermal nicotine, and cigarette smoking. British journal of clinical pharmacology. 1997;43:259–67. doi: 10.1111/j.1365-2125.1997.00566.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benwell ME, Balfour DJ, Anderson JM. Evidence that tobacco smoking increases the density of (−)-[3H]nicotine binding sites in human brain. J Neurochem. 1988;50:1243–7. doi: 10.1111/j.1471-4159.1988.tb10600.x. [DOI] [PubMed] [Google Scholar]
- Breese CR, Marks MJ, Logel J, Adams CE, Sullivan B, Collins AC, et al. Effect of smoking history on [3H]nicotine binding in human postmortem brain. J Pharmacol Exp Ther. 1997;282:7–13. [PubMed] [Google Scholar]
- Brynildsen JK, Najar J, Hsu LM, Vaupel DB, Lu H, Ross TJ, et al. A novel method to induce nicotine dependence by intermittent drug delivery using osmotic minipumps. Pharmacology, biochemistry, and behavior. 2016;142:79–84. doi: 10.1016/j.pbb.2015.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Changeux JP. Nicotine addiction and nicotinic receptors: lessons from genetically modified mice. Nat Rev Neurosci. 2010;11:389–401. doi: 10.1038/nrn2849. [DOI] [PubMed] [Google Scholar]
- Collins AC, Romm E, Wehner JM. Dissociation of the apparent relationship between nicotine tolerance and up-regulation of nicotinic receptors. Brain Res Bull. 1990;25:373–9. doi: 10.1016/0361-9230(90)90222-l. [DOI] [PubMed] [Google Scholar]
- Collins AC, Salminen O, Marks MJ, Whiteaker P, Grady SR. The road to discovery of neuronal nicotinic cholinergic receptor subtypes. Handb Exp Pharmacol. 2009:85–112. doi: 10.1007/978-3-540-69248-5_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosgrove KP, Batis J, Bois F, Maciejewski PK, Esterlis I, Kloczynski T, et al. beta2-Nicotinic acetylcholine receptor availability during acute and prolonged abstinence from tobacco smoking. Arch Gen Psychiatry. 2009;66:666–76. doi: 10.1001/archgenpsychiatry.2009.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryan JF, Bruijnzeel AW, Skjei KL, Markou A. Bupropion enhances brain reward function and reverses the affective and somatic aspects of nicotine withdrawal in the rat. Psychopharmacology (Berl) 2003;168:347–58. doi: 10.1007/s00213-003-1445-7. [DOI] [PubMed] [Google Scholar]
- D'Souza MS, Markou A. Neuronal mechanisms underlying development of nicotine dependence: implications for novel smoking-cessation treatments. Addict Sci Clin Pract. 2011;6:4–16. [PMC free article] [PubMed] [Google Scholar]
- Dani JA, Heinemann S. Molecular and cellular aspects of nicotine abuse. Neuron. 1996;16:905–8. doi: 10.1016/s0896-6273(00)80112-9. [DOI] [PubMed] [Google Scholar]
- Donny EC, Caggiula AR, Rowell PP, Gharib MA, Maldovan V, Booth S, et al. Nicotine self-administration in rats: estrous cycle effects, sex differences and nicotinic receptor binding. Psychopharmacology (Berl) 2000;151:392–405. doi: 10.1007/s002130000497. [DOI] [PubMed] [Google Scholar]
- Epping-Jordan MP, Watkins SS, Koob GF, Markou A. Dramatic decreases in brain reward function during nicotine withdrawal. Nature. 1998;393:76–9. doi: 10.1038/30001. [DOI] [PubMed] [Google Scholar]
- Fasoli F, Moretti M, Zoli M, Pistillo F, Crespi A, Clementi F, et al. In vivo chronic nicotine exposure differentially and reversibly affects upregulation and stoichiometry of alpha4beta2 nicotinic receptors in cortex and thalamus. Neuropharmacology. 2016;108:324–31. doi: 10.1016/j.neuropharm.2016.04.048. [DOI] [PubMed] [Google Scholar]
- Feyerabend C, Russell MA. A rapid gas-liquid chromatographic method for the determination of cotinine and nicotine in biological fluids. The Journal of pharmacy and pharmacology. 1990;42:450–2. doi: 10.1111/j.2042-7158.1990.tb06592.x. [DOI] [PubMed] [Google Scholar]
- Fowler CD, Arends MA, Kenny PJ. Subtypes of nicotinic acetylcholine receptors in nicotine reward, dependence, and withdrawal: evidence from genetically modified mice. Behav Pharmacol. 2008;19:461–84. doi: 10.1097/FBP.0b013e32830c360e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry CL, Lukas RJ. Regulation of nicotinic acetylcholine receptor numbers and function by chronic nicotine exposure. Curr Drug Targets CNS Neurol Disord. 2002;1:359–85. doi: 10.2174/1568007023339184. [DOI] [PubMed] [Google Scholar]
- Ghosheh OA, Dwoskin LP, Miller DK, Crooks PA. Accumulation of nicotine and its metabolites in rat brain after intermittent or continuous peripheral administration of [2'-(14)C]nicotine. Drug Metab Dispos. 2001;29:645–51. [PubMed] [Google Scholar]
- Jackson KJ, Martin BR, Changeux JP, Damaj MI. Differential role of nicotinic acetylcholine receptor subunits in physical and affective nicotine withdrawal signs. J Pharmacol Exp Ther. 2008;325:302–312. doi: 10.1124/jpet.107.132977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson KJ, Muldoon PP, De Biasi M, Damaj MI. New mechanisms and perspectives in nicotine withdrawal. Neuropharmacology. 2015;96:223–234. doi: 10.1016/j.neuropharm.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris AC, Pentel PR, Burroughs D, Staley MD, Lesage MG. A lack of association between severity of nicotine withdrawal and individual differences in compensatory nicotine self-administration in rats. Psychopharmacology (Berl) 2011;217:153–66. doi: 10.1007/s00213-011-2273-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison AA, Gasparini F, Markou A. Nicotine potentiation of brain stimulation reward reversed by DH beta E and SCH 23390, but not by eticlopride, LY 314582 or MPEP in rats. Psychopharmacology (Berl) 2002;160:56–66. doi: 10.1007/s00213-001-0953-6. [DOI] [PubMed] [Google Scholar]
- Harrison AA, Liem YT, Markou A. Fluoxetine combined with a serotonin-1A receptor antagonist reversed reward deficits observed during nicotine and amphetamine withdrawal in rats. Neuropsychopharmacology. 2001;25:55–71. doi: 10.1016/S0893-133X(00)00237-2. [DOI] [PubMed] [Google Scholar]
- Hughes JR. Effects of abstinence from tobacco: valid symptoms and time course. Nicotine Tob Res. 2007;9:315–327. doi: 10.1080/14622200701188919. [DOI] [PubMed] [Google Scholar]
- Kenny PJ, Markou A. Nicotine self-administration acutely activates brain reward systems and induces a long-lasting increase in reward sensitivity. Neuropsychopharmacology. 2006;31:1203–11. doi: 10.1038/sj.npp.1300905. [DOI] [PubMed] [Google Scholar]
- Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35:217–38. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornetsky C, Esposito RU, McLean S, Jacobson JO. Intracranial self-stimulation thresholds: a model for the hedonic effects of drugs of abuse. Arch Gen Psychiatry. 1979;36:289–92. doi: 10.1001/archpsyc.1979.01780030055004. [DOI] [PubMed] [Google Scholar]
- Lindblom N, de Villiers SH, Semenova S, Kalayanov G, Gordon S, Schilstrom B, et al. Active immunisation against nicotine blocks the reward facilitating effects of nicotine and partially prevents nicotine withdrawal in the rat as measured by dopamine output in the nucleus accumbens, brain reward thresholds and somatic signs. Naunyn Schmiedebergs Arch Pharmacol. 2005;372:182–94. doi: 10.1007/s00210-005-0019-0. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–75. [PubMed] [Google Scholar]
- Malin DH, Lake JR, Newlin-Maultsby P, Roberts LK, Lanier JG, Carter VA, et al. Rodent model of nicotine abstinence syndrome. Pharmacol Biochem Behav. 1992;43:779–84. doi: 10.1016/0091-3057(92)90408-8. [DOI] [PubMed] [Google Scholar]
- Mao D, Perry DC, Yasuda RP, Wolfe BB, Kellar KJ. The alpha4beta2alpha5 nicotinic cholinergic receptor in rat brain is resistant to up-regulation by nicotine in vivo. J Neurochem. 2008;104:446–56. doi: 10.1111/j.1471-4159.2007.05011.x. [DOI] [PubMed] [Google Scholar]
- Markou A. Review. Neurobiology of nicotine dependence. Philos Trans R Soc Lond B Biol Sci. 2008;363:3159–68. doi: 10.1098/rstb.2008.0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markou A, Koob GF. Construct validity of a self-stimulation threshold paradigm: effects of reward and performance manipulations. Physiol Behav. 1992;51:111–9. doi: 10.1016/0031-9384(92)90211-j. [DOI] [PubMed] [Google Scholar]
- Marks MJ, Burch JB, Collins AC. Effects of chronic nicotine infusion on tolerance development and nicotinic receptors. J Pharmacol Exp Ther. 1983;226:817–25. [PubMed] [Google Scholar]
- Marks MJ, McClure-Begley TD, Whiteaker P, Salminen O, Brown RW, Cooper J, et al. Increased nicotinic acetylcholine receptor protein underlies chronic nicotine-induced up-regulation of nicotinic agonist binding sites in mouse brain. J Pharmacol Exp Ther. 2011;337:187–200. doi: 10.1124/jpet.110.178236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks MJ, Stitzel JA, Collins AC. Time course study of the effects of chronic nicotine infusion on drug response and brain receptors. J Pharmacol Exp Ther. 1985;235:619–28. [PubMed] [Google Scholar]
- Marks MJ, Whiteaker P, Collins AC. Deletion of the alpha7, beta2, or beta4 nicotinic receptor subunit genes identifies highly expressed subtypes with relatively low affinity for [3H]epibatidine. Mol Pharmacol. 2006;70:947–59. doi: 10.1124/mol.106.025338. [DOI] [PubMed] [Google Scholar]
- Metaxas A, Bailey A, Barbano MF, Galeote L, Maldonado R, Kitchen I. Differential region-specific regulation of alpha4beta2* nAChRs by self-administered and non-contingent nicotine in C57BL/6J mice. Addict Biol. 2010;15:464–79. doi: 10.1111/j.1369-1600.2010.00246.x. [DOI] [PubMed] [Google Scholar]
- Moretti M, Mugnaini M, Tessari M, Zoli M, Gaimarri A, Manfredi I, et al. A comparative study of the effects of the intravenous self-administration or subcutaneous minipump infusion of nicotine on the expression of brain neuronal nicotinic receptor subtypes. Mol Pharmacol. 2010;78:287–96. doi: 10.1124/mol.110.064071. [DOI] [PubMed] [Google Scholar]
- Moroni M, Zwart R, Sher E, Cassels BK, Bermudez I. alpha4beta2 nicotinic receptors with high and low acetylcholine sensitivity: pharmacology, stoichiometry, and sensitivity to long-term exposure to nicotine. Mol Pharmacol. 2006;70:755–68. doi: 10.1124/mol.106.023044. [DOI] [PubMed] [Google Scholar]
- Nguyen HN, Rasmussen BA, Perry DC. Subtype-selective up-regulation by chronic nicotine of high-affinity nicotinic receptors in rat brain demonstrated by receptor autoradiography. J Pharmacol Exp Ther. 2003;307:1090–7. doi: 10.1124/jpet.103.056408. [DOI] [PubMed] [Google Scholar]
- O'Dell LE, Bruijnzeel AW, Smith RT, Parsons LH, Merves ML, Goldberger BA, et al. Diminished nicotine withdrawal in adolescent rats: implications for vulnerability to addiction. Psychopharmacology (Berl) 2006;186:612–9. doi: 10.1007/s00213-006-0383-6. [DOI] [PubMed] [Google Scholar]
- O'Dell LE, Chen SA, Smith RT, Specio SE, Balster RL, Paterson NE, et al. Extended access to nicotine self-administration leads to dependence: Circadian measures, withdrawal measures, and extinction behavior in rats. J Pharmacol Exp Ther. 2007;320:180–93. doi: 10.1124/jpet.106.105270. [DOI] [PubMed] [Google Scholar]
- Parker SL, Fu Y, McAllen K, Luo J, McIntosh JM, Lindstrom JM, et al. Up-regulation of brain nicotinic acetylcholine receptors in the rat during long-term self-administration of nicotine: disproportionate increase of the alpha6 subunit. Mol Pharmacol. 2004;65:611–22. doi: 10.1124/mol.65.3.611. [DOI] [PubMed] [Google Scholar]
- Paterson NE, Balfour DJ, Markou A. Chronic bupropion attenuated the anhedonic component of nicotine withdrawal in rats via inhibition of dopamine reuptake in the nucleus accumbens shell. Eur J Neurosci. 2007;25:3099–108. doi: 10.1111/j.1460-9568.2007.05546.x. [DOI] [PubMed] [Google Scholar]
- Paterson NE, Balfour DJ, Markou A. Chronic bupropion differentially alters the reinforcing, reward-enhancing and conditioned motivational properties of nicotine in rats. Nicotine Tob Res. 2008;10:995–1008. doi: 10.1080/14622200802097571. [DOI] [PubMed] [Google Scholar]
- Paterson NE, Markou A. Prolonged nicotine dependence associated with extended access to nicotine self-administration in rats. Psychopharmacology (Berl) 2004;173:64–72. doi: 10.1007/s00213-003-1692-7. [DOI] [PubMed] [Google Scholar]
- Pellegrino L, Pellegrino A, Cushman A. A Stereotaxic Atlas of the Rat Brain. 2. New York: Plenum Press; 1986. [Google Scholar]
- Perry DC, Davila-Garcia MI, Stockmeier CA, Kellar KJ. Increased nicotinic receptors in brains from smokers: membrane binding and autoradiography studies. J Pharmacol Exp Ther. 1999;289:1545–52. [PubMed] [Google Scholar]
- Picciotto MR, Kenny PJ. Molecular mechanisms underlying behaviors related to nicotine addiction. Cold Spring Harbor perspectives in medicine. 2013;3:a012112. doi: 10.1101/cshperspect.a012112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portugal GS, Kenney JW, Gould TJ. Beta2 subunit containing acetylcholine receptors mediate nicotine withdrawal deficits in the acquisition of contextual fear conditioning. Neurobiol Learn Mem. 2008;89:106–113. doi: 10.1016/j.nlm.2007.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose JE, Behm FM, Westman EC, Coleman RE. Arterial nicotine kinetics during cigarette smoking and intravenous nicotine administration: implications for addiction. Drug Alcohol Depend. 1999;56:99–107. doi: 10.1016/s0376-8716(99)00025-3. [DOI] [PubMed] [Google Scholar]
- Rowell PP, Li M. Dose-response relationship for nicotine-induced up-regulation of rat brain nicotinic receptors. J Neurochem. 1997;68:1982–9. doi: 10.1046/j.1471-4159.1997.68051982.x. [DOI] [PubMed] [Google Scholar]
- Russell MA, Jarvis MJ, Feyerabend C, Saloojee Y. Reduction of tar, nicotine and carbon monoxide intake in low tar smokers. Journal of epidemiology and community health. 1986;40:80–5. doi: 10.1136/jech.40.1.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas R, Pieri F, De Biasi M. Decreased signs of nicotine withdrawal in mice null for the beta4 nicotinic acetylcholine receptor subunit. J Neurosci. 2004;24:10035–10039. doi: 10.1523/JNEUROSCI.1939-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas R, Sturm R, Boulter J, De Biasi M. Nicotinic receptors in the habenulo-interpeduncular system are necessary for nicotine withdrawal in mice. J Neurosci. 2009;29:3014–3018. doi: 10.1523/JNEUROSCI.4934-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson EM, Drasdo AL, McCrea K, Wonnacott S. Upregulation of nicotinic receptors following continuous infusion of nicotine is brain-region-specific. Brain Res. 1993;617:349–52. doi: 10.1016/0006-8993(93)91104-z. [DOI] [PubMed] [Google Scholar]
- Schwartz RD, Kellar KJ. Nicotinic cholinergic receptor binding sites in the brain: regulation in vivo. Science. 1983;220:214–6. doi: 10.1126/science.6828889. [DOI] [PubMed] [Google Scholar]
- Semenova S, Markou A. Clozapine treatment attenuated somatic and affective signs of nicotine and amphetamine withdrawal in subsets of rats exhibiting hyposensitivity to the initial effects of clozapine. Biol Psychiatry. 2003;54:1249–64. doi: 10.1016/s0006-3223(03)00240-3. [DOI] [PubMed] [Google Scholar]
- Shoaib M, Stolerman IP. Plasma nicotine and cotinine levels following intravenous nicotine self-administration in rats. Psychopharmacology (Berl) 1999;143:318–21. doi: 10.1007/s002130050954. [DOI] [PubMed] [Google Scholar]
- Skjei KL, Markou A. Effects of repeated withdrawal episodes, nicotine dose, and duration of nicotine exposure on the severity and duration of nicotine withdrawal in rats. Psychopharmacology (Berl) 2003;168:280–92. doi: 10.1007/s00213-003-1414-1. [DOI] [PubMed] [Google Scholar]
- Staley JK, Krishnan-Sarin S, Cosgrove KP, Krantzler E, Frohlich E, Perry E, et al. Human tobacco smokers in early abstinence have higher levels of beta2* nicotinic acetylcholine receptors than nonsmokers. J Neurosci. 2006;26:8707–14. doi: 10.1523/JNEUROSCI.0546-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoker AK, Marks MJ, Markou A. Null mutation of the beta2 nicotinic acetylcholine receptor subunit attenuates nicotine withdrawal-induced anhedonia in mice. Eur J Pharmacol. 2015;753:146–150. doi: 10.1016/j.ejphar.2014.05.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoker AK, Olivier B, Markou A. Role of alpha7- and beta4-containing nicotinic acetylcholine receptors in the affective and somatic aspects of nicotine withdrawal: studies in knockout mice. Behav Genet. 2012;42:423–436. doi: 10.1007/s10519-011-9511-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich YM, Hargreaves KM, Flores CM. A comparison of multiple injections versus continuous infusion of nicotine for producing up-regulation of neuronal [3H]-epibatidine binding sites. Neuropharmacology. 1997;36:1119–25. doi: 10.1016/s0028-3908(97)00107-x. [DOI] [PubMed] [Google Scholar]
- Vezina P, McGehee DS, Green WN. Exposure to nicotine and sensitization of nicotine-induced behaviors. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:1625–38. doi: 10.1016/j.pnpbp.2007.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteaker P, Jimenez M, McIntosh JM, Collins AC, Marks MJ. Identification of a novel nicotinic binding site in mouse brain using [(125)I]-epibatidine. Br J Pharmacol. 2000;131:729–39. doi: 10.1038/sj.bjp.0703616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winders SE, Grunberg NE, Benowitz NL, Alvares AP. Effects of stress on circulating nicotine and cotinine levels and in vitro nicotine metabolism in the rat. Psychopharmacology (Berl) 1998;137:383–90. doi: 10.1007/s002130050634. [DOI] [PubMed] [Google Scholar]
- Wonnacott S. The paradox of nicotinic acetylcholine receptor upregulation by nicotine. Trends Pharmacol Sci. 1990;11:216–9. doi: 10.1016/0165-6147(90)90242-z. [DOI] [PubMed] [Google Scholar]