

SUMMARY
To address the significance of enhancing myelination for functional recovery after white matter injury (WMI) in preterm infants, we characterized hypomyelination associated with chronic hypoxia and identified structural and functional deficits of excitatory cortical synapses with a prolonged motor deficit. We demonstrate that genetically delaying myelination phenocopies the synaptic and functional deficits observed in mice after hypoxia, suggesting that myelination may possibly facilitate excitatory presynaptic innervation. As a gain of function experiment, we specifically ablated the muscarinic receptor 1 (M1R), a negative regulator of oligodendrocyte differentiation in oligodendrocyte precursor cells. Genetically enhancing oligodendrocyte differentiation and myelination rescued the synaptic loss after chronic hypoxia and promoted functional recovery. As a proof of concept, drug-based myelination-therapies also resulted in accelerated differentiation and myelination with functional recovery after chronic hypoxia. Together, our data indicates that myelination-enhancing strategies in preterm infants may represent a promising therapeutic approach for structural/functional recovery after hypoxic WMI.
Keywords: White matter injury, M1R, Clemastine, U-50488, Hypomyelination, Beam walking test, Olig2
Graphical Abstract
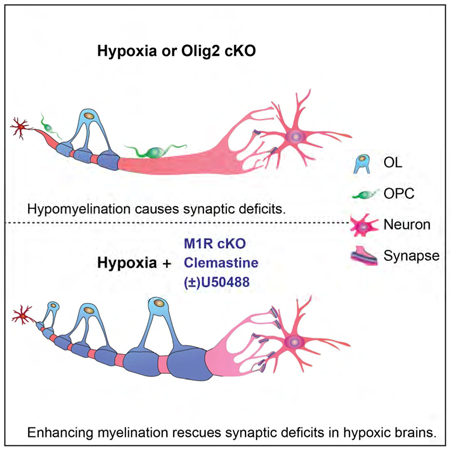
IN BRIEF:
Wang et al., demonstrate that hypomyelination specifically contributes to the significant loss of excitatory synapses and prolonged functional deficits after chronic hypoxia and that myelination-enhancing strategies represent a promising approach for functional recovery following hypoxic WMI.
INTRODUCTION
Preterm birth is estimated to affect approximately 5–18% of newborns worldwide with an increasing incidence rate each year (Blencowe et al., 2012). About 5–10% of preterm infants, including 50% of low-birth-weight (<1kg) preterm survivors, are diagnosed with white matter injury (WMI), a major form of brain injury (Curtis et al., 2002; Ferriero, 2004; Maureen et al., 2002; Woodward et al., 2006). Accumulating evidence has shown that WMI can lead to long-term functional impairments, including cognitive delay, motor deficits, cerebral palsy and neurosensory impairments (Curtis et al., 2002; Ferriero, 2004; Maureen et al., 2002; Woodward et al., 2006). Unfortunately, relevant therapies to promote functional recovery are still unavailable.
The major component of white matter is the myelin sheath, generated by mature oligodendrocytes (OLs) that differentiate from oligodendrocyte precursor cells (OPCs) in the CNS (Chang et al., 2016; Taveggia et al., 2010). Myelin ensures the precise and rapid conduction of action potentials while OLs provide metabolic support to meet the energy demand of axons (Morrison et al., 2013; Nave, 2010). Increasing evidence has demonstrated that hypomyelination and insufficient numbers of mature OLs are prominent after WMI, presumably owing to a wave of acute cell death of immature OLs and inhibitory environmental cues preventing OPC differentiation (Fancy et al. 2014., Back et al., 2002; Billiards et al., 2008; Buser et al., 2012; Riddle et al., 2011; Segovia et al., 2008). It is noteworthy that OPC density is increased in WMI regions but that these OPCs are arrested from differentiating into OLs (Back, 2017; Back, 2014; Buser et al., 2012). This suggests that a failure in differentiation rather than survival or proliferation of OPCs contributes to hypomyelination in WMI brains. We hypothesize that enhancing endogenous oligodendroglial differentiation can rescue hypomyelination defects and consequently improve neuronal functional recovery following WMI.
It is generally accepted that perinatal hypoxia is one leading cause of WMI (Back, 2017, 2015; Ortega et al., 2016; Rumajogee et al., 2016; Salmaso et al., 2014). A rodent animal model of chronic hypoxia has been well-established and widely used to mimic hypoxia-induced diffuse brain injury in preterm children and infants (Cree et al., 2018; Scafidi et al., 2013; Yuen et al., 2014; Zonouzi et al., 2015). Hypomyelination typically coincides with other pathological changes. For instance, a number of studies have demonstrated that a delay in synaptogenesis accompanies hypomyelination in hypoxic brains (Curristin et al., 2002; Liu et al., 2016; Otellin et al., 2014; Piorkowska et al., 2014). Therefore, determining the specific functional significance of myelination requires oligodendroglial-specific genetic manipulations that alter oligodendroglial differentiation in the CNS. Previously, we and others have reported that the transcription factor Olig2 acts as a positive regulator of oligodendroglial differentiation and that the muscarinic receptor 1 (M1R) acts as a negative regulator (Mei et al., 2013; Mei et al., 2016a; Yue et al., 2006; Zhou et al., 2001). Genetic manipulation of these genes in oligodendroglia provides us with a unique opportunity to uncouple the functional importance of myelination in hypoxia brains.
In this study, we examine the functional significance of myelination on WMI by OPC-specific deletion of Olig2 (loss of function) or M1R (gain of function). Our results demonstrate that hypomyelination specifically contributes to the significant loss of excitatory synapses and prolonged functional deficits after chronic hypoxia and that myelination-enhancing strategies represent a promising approach for functional recovery following WMI.
RESULTS
1. Chronic hypoxia impairs myelination and synaptogenesis in developing brains.
To determine whether chronic hypoxia is a reasonable model to recapitulate hypoxia induced diffuse brain injury in prenatal infants, we set out to examine the effect of chronic hypoxia (10% oxygen) on newborn mice from postnatal day 3 (P3) to P10, corresponding to gestation week 32 to 40 in humans (Ortega et al., 2016; Rumajogee et al., 2016) (Figure 1A). To exclude variations from different litters, we exposed half of each litter to hypoxia with their birth mother, and the other half to normoxia (Room air, 21% oxygen) with a foster mother. At P10, we examined myelination and quantified the number of mature oligodendrocytes (OLs) in the CNS by immunostaining for myelin basic protein (MBP) and, CC1 respectively (Figures1B–E). As expected, MBP expression was significantly diminished and the numbers of CC1 positive OLs were greatly reduced in hypoxic brains as compared to normoxia littermates (Figures 1B–E). Consistent with the changes, the number of nodes of Ranvier was greatly decreased in the hypoxic spinal cords, indicated by immunostaining for Caspr, a paranodal marker and sodium channel 1.6 (Nav1.6), a nodal marker (Figure 1F) (Chang et al., 2016). Similar to the immunostaining results, transmission electronic microscopy (TEM) showed much fewer myelinated axons in the corpus callosum of hypoxic brains as compared to the normoxic control littermates (Figure 1G). It is important to note that the NF200 positive axon densities were not altered in the brains after hypoxia (Figures 1B’, B”, C’, C”, D) suggesting that chronic hypoxia may not cause significant axonal degeneration. Further, we examined axonal degeneration by labeling for SMI-32 and apoptosis by TUNEL assay respectively (Figures S1A, B), and neither axonal degeneration nor neuronal apoptosis was apparent in the hypoxic brains (Figures S1A, B). In response to hypoxia, NG2-positive OPC numbers were overtly increased in the hypoxic brains (Figure S1D). These results suggest that chronic hypoxia inhibits oligodendroglial differentiation and myelination in the developing CNS.
Figure 1. Chronic hypoxia results in hypomyelination in neonatal mice.

(A) Schematic diagram displaying the time course of hypoxic exposure and assays; (B, C) Representative immunofluorescent images displaying MBP expression in the normoxic (B) and hypoxic (C) brains; (B’, C’, B”, C”) Magnified images showing MBP and NF200 expression in the corpus callosum (B’, C’) and striatum (B”, C”). Scale bar, 0.5mm (B, C) and 50μm (B’-C”); (D) Quantification of MBP and NF200 density in the cortex and corpus callosum; (E) Immunostaining for CC1 displaying mature oligodendrocytes in the hypoxic and normoxic brains. (F) Representative images showing Nav1.6 positive nodal (green) and Caspr positive (red) paranodal regions on the longitudinal sections of spinal cords at P10. Numbers of CC1 positive cells (E) and Nav1.6/Caspr double positive nodes (F) were quantified in the brains and spinal cords respectively; (G) Representative EM images of corpus callosum at P15. Myelinated axon numbers were quantified. Scale bar, 100μm (E), 10μm (F) and 2μm (G). Error bars represent mean ± s.e.m. *p<0.05 or **p<0.01, significance based on Student’s t-test comparing hypoxia with normoxia conditions. n = 3 normoxic mice, n = 5 hypoxic mice for all the experiments. See also Figure S1.
Since a delay in synaptogenesis may be coincident with hypomyelination in hypoxic brains (Curristin et al., 2002), we next examined whether hypoxia alters synaptogenesis in layers 3–4 of the cortex. Given that motor deficits are consistently observed in WMI patients (Misumi et al., 2016), we selected the motor cortex (M2) to immunostain for the pan-presynaptic marker, Synapsin1 (De Camilli et al., 1983) and the pan-postsynaptic marker, Homer1 (Tu et al., 1998) at P10 (Figures 2A–C). The pre- and postsynaptic markers are closely associated with the somatodendritic membranes of MAP2 positive neurons (Figures 2A–C). Strikingly, the densities of both Synapsin1 and Homer1 positive puncta were significantly decreased in the hypoxic brains without altering the number of MAP2 positive neurons (Figure 2E), suggesting that hypoxia can inhibit synaptogenesis. Next, we examined the excitatory and inhibitory presynaptic terminals by labeling vesicular glutamate transporter 1 (vGlut1) and vesicular GABA transporter (vGAT) respectively (Minelli et al., 2003; Paolicelli et al., 2017). We found that the number of vGlut1 puncta but not vGAT was greatly reduced in hypoxic brains at P10 as compared to the normoxia littermates (Figures 2D, S1C), suggesting that chronic hypoxia greatly inhibits excitatory presynaptic innervation. Since the chronic hypoxia mouse model is characterized by mild and global diffuse brain injury without prominent axonal degeneration or neuronal death (Figures S1A, B), the synaptic loss does not seem to be attributed to neuron or axonal degeneration.
Figure 2. Synaptogenesis in hypoxic mouse brains.

(A) Schematic diagram showing axonal terminals and synapses in the M2 cortex; (B-E) Representative images and quantification of synaptic puncta (red, white arrows) using a pan-presynaptic marker, synapsin-1 (B), a panpostsynaptic marker, homer1 (C) and an excitatory presynaptic marker, vGlut1 (D) in the normoxic and hypoxic M2 cortex at P10. Neurons were co-labeled with MAP2 staining and the numbers of MAP2 positive cells were quantified (E); Error bars represent mean ± s.e.m. *p<0.05 or **p<0.01, significance based on Student’s t-test as compared with the respective controls, n = 3 mice for all experiments. Scale bar, 40μm (upper panels in A-C) and 10μm (lower panels in A-C). See also Figure S1.
2. Disrupted synaptic function and prolonged motor deficits in adolescent hypoxic mice.
Since chronic hypoxia inhibits excitatory presynaptic innervation, we next assessed excitatory synaptic function by recording miniature excitatory postsynaptic currents (mEPSC) from pyramidal neurons in the M2 cortex at P21 (Figure 3A). The mEPSC amplitudes were unchanged, whereas the frequency was significantly decreased in the hypoxic brains as compared to the normoxic controls (Figures 3B–D), indicating that chronic hypoxia severely attenuates excitatory synaptic function. Considering that both synaptogenesis and myelination are actively developing throughout late pregnancy and during the postnatal period, we next asked whether hypoxic insult could result in long-term impairments on synaptogenesis and myelination in adolescent mice. The expression of MBP was greatly reduced in the hypoxic cortex at P40 as well as levels of Synapsin1 and Homer1 expression (Figures S2A–E). These results indicate that neonatal hypoxic exposure can result in long-term impairment of myelination and synaptogenesis. Next, we examined functional deficits in adolescent mice by applying a beam-walking test to assess motor coordination in the hypoxic mice at P40 and calculating the frequency of foot slippage while walking the length of the beam (Figure 3E). Consistent with the pathological observations, the frequency of foot slippage from the hypoxic mice was significantly increased as compared to the normoxic controls (Figure 3E), suggesting that chronic hypoxia results in prolonged behavioral deficits (Scafidi et al., 2013; Zonouzi et al., 2015). Together, these results demonstrate that chronic hypoxia results in hypomyelination and disruption of synaptogenesis, which may lead to functional impairment in adolescent mice.
Figure 3: Chronic hypoxia impairs synaptic transmission and prolongs motor deficits.

(A) Schematic illustration showing the whole-cell patch clamp in the M2 cortex; (B-C) Sample recordings (3 s) of miniature EPSCs (mEPSC) recorded from the cortical pyramidal neurons of normoxic and hypoxic cortex at P21 (B), and average amplitude (C) and frequency (D) of mEPSCs were calculated (n = 9 cells from 2 normoxic mice, n=12 cells from 2 hypoxic mice). Error bars represent mean ± s.e.m. **p<0.01 significance based on Student’s t-test. (E) Mean number of foot slips of hypoxic and normoxic mice by using the beam walking test at P40. Error bars represent mean ± s.e.m. *p<0.05 significance based on non-parametric Mann-Whitney test comparing hypoxic to normoxic mice (n = 5 mice in each group). See also Figure S2.
3. Hypomyelination disrupts synaptogenesis and synaptic transmission, and impairs neuronal function.
Given that hypomyelination is accompanied by disrupted synaptogenesis and functional deficits after chronic hypoxia, we decided to ask whether hypomyelination could specifically contribute to synaptic loss and behavioral impairments. To determine the direct effect of hypomyelination and examine its functional consequence, we ablated Olig2 specifically in oligodendroglia to mimic the hypomyelination phenotype observed in hypoxic brains. It has been previously shown that Olig2 is an essential transcriptional factor that is important in regulating the specification and differentiation of oligodendrocytes (Mei et al., 2013; Yue, 2006; Zhou et al., 2001). Briefly, the Olig2 floxed mouse line was crossed with the CNP-Cre line to induce Olig2 deletion in OPCs (Mei et al., 2013). As expected, conditional deletion of Olig2 in OPCs resulted in a decrease in MBP expression in the corpus callosum and cortex of the Olig2 cKO brains (CNP-Cre; Olig2 fl/fl) at P10 when compared to wildtype littermates (Olig2 fl//fl) (Figure 4A). Consistent with this evidence, the number of nodes of Ranvier, indicated by immunostaining for Caspr and Nav1.6, were significantly decreased in the Olig2 cKO spinal cords at P10 (Figure 4B) and the numbers of myelinated axons were also reduced in the Olig2 cKO corpus callosum at P15 when examined by TEM (Figure 4C). By P42, MBP expression in the corpus callosum of the Olig2 cKO brains was comparable to the wildtype littermates, but decreased MBP expression persisted in the cortex (Figures 4D, E). It is notable that NF200 positive axonal densities were not significantly altered (Figure 4E) and SMI-32 expression was undetectable (Figure S3E) in the Olig2 cKO brains, indicating that the delay in myelination does not result in axonal degeneration in the adolescent mice.
Figure 4. Deletion of Olig2 in OPCs delays myelination.

(A, B) Representative images and quantification of MBP expression (A) in the brains and Caspr/Nav1.6 positive nodes (B) in the spinal cords of the Olig2 cKO mice (CNP-Cre; Olig2 fl/fl) and wildtype (Olig2 fl/fl) littermates at P10, Scale bar, 0.2mm (A) and 10μm (B); (C) Representative EM images of corpus callosum at P15. Myelinated axon numbers were quantified. (D) Representative images showing MBP in the Olig2 cKO and littermate wildtype brains at P42. (E) Magnified images showing MBP positive myelin and NF200 positive neurofilament in the cortex at P42. MBP and NF200 density was quantified in the cortex. Scale bar, 2μm (C), 0.5mm (D) and 0.2mm (E). Error bars represent mean ± s.e.m. *p<0.05, significance based on Student’s t-test comparing Olig2 cKO to wildtype, n = 3 mice for all experiments. See also Figure S3.
Since Olig2 deletion in OPCs phenocopies the change in myelination of hypoxic brains, we next asked if hypomyelination could alter synaptogenesis in the M2 cortex. Interestingly, coincident with hypomyelination, the levels of Synapsin1 and Homer1 positive puncta were significantly decreased in the hypoxic brains without altering the number of MAP2 positive neurons (Figures 5A, B, D), suggesting that hypomyelination may directly inhibit synaptogenesis. Consistent with these findings, vGlut1 positive puncta were significantly decreased in the M2 cortex of Olig2 cKO brains as compared to wildtype mice at P10 (Figures 5C, D), suggesting that disrupted excitatory presynaptic innervation was a direct result of hypomyelination. Next, we assessed synaptic function by recording mEPSCs from the pyramidal excitatory neurons in the M2 cortex at P14 (Figure 5E). Consistent with the changes in synaptic markers, the mEPSC frequency was significantly decreased while the amplitude was unchanged in the Olig2-cKO brains (Figure 5E), indicating disrupted excitatory synaptic function. Once again, the beam walking examination displayed a higher frequency of foot slippage in the Olig2-cKO mice as compared to the wildtype littermates even at P42 (Figure 5F). Consistent with these findings, the rotarod test revealed that the Olig2 cKO mice exhibited an increased probability to fall from the spinning rod than the wildtype littermates (Figure 5G). To exclude the possibility that Olig2 in mature OLs is required for the maintenance of myelin or axonal integrity, we crossed the Olig2 floxed line to the PLP-CreERt driver line to induce Olig2 deletion in mature OLs by tamoxifen at P40 (Figures S3A, B). No signs of demyelination or axonal degeneration were observed in the Olig2 cKO (PLP-CreERt; Olig2 fl/fl) brains at P80, indicated by immunostaining for MBP and TEM (Figures S3C, D). These results demonstrate that a delay in myelination disrupts the proper timing of synaptogenesis and results in prolonged motor deficits.
Figure 5. Hypomyelination attenuates synaptogenesis and synaptic transmission, and prolongs motor deficits in Olig2 cKO mice.

(A-D) Representative images of synaptic puncta (Red, white arrowheads) using synapsin-1 (A), homer1 (B), or vGlut1 (C) in the M2 cortex of Olig2 cKO and wildtype littermates at P10. Neurons were identified by MAP2 staining. Scale bar, 40μm (upper panels in A-C) and 10μm (lower panels in A-C). The numbers of MAP2 positive cells and the density of synaptic puncta were quantified (D), n = 3 mice in each group; (E) Sample recordings (3 s) of EPSCs from cortical pyramidal neurons at P14 and the average amplitude (left), and frequency (right) of EPSCs were quantified (n = 12 cells from 3 Olig2 fl/fl mice, n = 15 cells from 3 CNP-Cre; Olig2 fl/fl mice). Error bars represent mean ± s.e.m. *p<0.05, significance based on Student’s t-test with the respective controls. (F) Mean number of foot slips of Olig2 cKO mice and wildtype littermates revealed by the beam walking test at P40, n = 5 mice in each group; (G) The rotarod test revealed the latency in the time to fall from the spinning drum at P56, n = 7 mice in each group. *p<0.05 or **p<0.01, significance based on non-parametric Mann-Whitney test comparing with the respective controls.
4. Enhancing myelination rescues synaptic deficits and improves functional recovery in hypoxic mice.
Given the necessity of myelination for neuronal functional development, we sought to ask whether enhancing oligodendroglial differentiation is sufficient to rescue the hypomyelination phenotype and consequently the synaptic deficits and behavioral response in hypoxic mice. To accelerate myelination without altering other cell types, we induced the cell-specific deletion of the muscarinic receptor 1 (M1R) in OPCs. We previously identified the M1R as a negative regulator for oligodendroglial differentiation, and deletion of M1R in OPCs resulted in the enhanced intrinsic ability to differentiate and myelinate axons (Mei et al., 2016a). Briefly, the M1R floxed line was crossed with the NG2-CreERt driver line to generate inducible OPC-specific M1R knockout mice (NG2-CreERt; M1R fl/fl). Recombination was induced by administration of tamoxifen concomitantly with chronic hypoxia treatment from P3 to P10 and histology was performed at P10 (Figure 6A). M1R deletion in OPCs resulted in enhanced MBP expression and increased CC1 positive OL numbers in the corpus callosum and cortex as compared to the wildtype (M1R fl/fl) littermates (Figures 6B, C). Additionally, the formation of nodes of Ranvier, indicated by immunostaining for Caspr and Nav1.6, was enhanced in the spinal cord at P10 (Figure 6D). Under TEM, increased myelinated axons were observed in the optic nerves and corpus callosum of the M1R cKO mice compared to wildtype littermates at P15 (Figures 6E–H). To determine the percentage of myelinated axons that were generated from the differentiation of M1R null OPCs after the hypoxic challenge, we crossed the NG2-CreERt; M1R fl/fl mice with the Tau-mGFP mice (NG2-CreERt; M1R fl/fl; Tau-mGFP) (Figure S4A). As only mature oligodendrocytes express tau, newly formed OLs and myelin can be visualized by expression of the membrane-associated isoform of GFP (mGFP) after tamoxifen-induced recombination in OPCs (NG2) (Figure S4B). Immunostaining showed that mGFP expression is exclusively co-localized with MBP, Myelin Oligodendrocyte Glycoprotein (MOG), and the mature OL marker, CC1 (Figures S4C, D) in the corpus callosum of the mice. The fact that more than 90% of CC1 positive cells were mGFP positive (arrows in Figure S4D) indicates that the increased myelination is largely attributed to the enhanced differentiation of M1R null OPCs. To directly quantify the recombination efficiency of the NG2-CreERt line in OPCs, we crossed the mice to a fluorescent reporter line (mT/mG) (Figure S4E). The expression of mGFP can be visualized in OPCs, upon the expression of Cre recombinase (Figures S4F, G). Quantification of the mGFP positive cells indicated that more than 85% of NG2 positive OPCs also express mGFP seven days after tamoxifen induction (Figure S4F, G). Taken together, these results suggest that M1R deletion in OPCs enhances myelination in the hypoxic brains.
Figure 6. Conditional knockout of M1R in OPCs rescues hypomyelination caused by hypoxia.

(A) Schematic diagram showing the time course of tamoxifen induction and hypoxic exposure; (B-D) Representative images showing MBP positive myelin (B), CC1 positive mature OLs (C) in the corpus callosum and Caspr/Nav1.6 positive nodes (D) in the spinal cord of M1R cKO (NG2-CreERt; M1R fl/fl) and wildtype (M1R fl/fl) mice at P10. Scale bar, 100μm (B), 200μm (C) and 10μm (D). MBP density, CC1 positive cell numbers and Caspr/Nav1.6 double positive nodes were quantified; (E, F) Representative EM images showing corpus callosum (E) and optic nerve (F) in the hypoxic M1R cKO mice and littermates. Scale bar, 1μm (E) and 2μm (F). Myelinated axon numbers in the corpus callosum (E) and optic nerve (G) were quantified. (H) Quantification of myelin sheath thickness and the scatterplot displays g-ratios of individual axons as a function of axonal diameter. Error bars represent mean ± s.e.m. *p<0.05 or **p<0.01, significance based on Student’s t-test by comparing M1R cKO to wildtype. n = 3 mice for all experiments. See also Figure S4.
We next asked whether enhancing myelination could also rescue the synaptic deficits and promote functional recovery in the hypoxic brains. Interestingly, we found that the densities of Synapsin1 and Homer1 positive puncta were rescued in the M1R cKO hypoxic brains compared to the hypoxic wildtype littermates (Figures 7A, B, D). Strikingly, the vGlut1 positive puncta in the M1R cKO hypoxic brains were recovered to similar levels as in the normoxic controls (Figure 7C, D), suggesting that enhancing myelination can directly rescue disrupted excitatory presynaptic innervation in the cortex of hypoxic brains. In support of the synaptic changes, the mEPSC frequency was significantly increased in the hypoxic M1R cKO brains compared to the hypoxic wildtype littermates at P21, reaching a level that was similar to the normoxic controls, while the mEPSC amplitudes were unchanged (Figure 7E). Consistent with these findings, the M1R cKO mice also displayed a lower frequency of foot slippage as compared to the wildtype littermates in the beam-walking examination (Figure 7F). Collectively, our data indicate that enhancing myelination rescues synaptic deficits and improves motor behavior in mice after chronic hypoxia.
Figure 7. Enhanced myelination rescues hypoxia induced synaptic deficits and promotes functional neuronal recovery.

(A-D) Representative images and quantification of synaptic puncta (red) using synapsin-1 (A), homer1 (B) and vGlut1 (C) in the M2 cortex of normoxic wildtype, hypoxic M1R cKO and wildtype littermates at P10. Neurons were identified by MAP2 (green) staining. Scale bar, 40μm (upper panels in A-C) and 10μm (lower panels in A-C). The number of synaptic puncta were quantified (D), n = 3 mice for all experiments, *p<0.05, ** p<0.01, significance based on Student’s t-test by comparing to respective control; (F) Sample recordings (3 s) of miniature EPSCs (mEPSC) recorded from cortical pyramidal neurons (n = 9 cells from 2 animals per group) and average amplitude (left), and frequency (right) of mEPSCs at P21. Error bars represent mean ± s.e.m. *p<0.05, ** p<0.01, significance based on Student’s t-test with the respective controls; (F) Beam walking test reveal the frequency of foot slips in the normoxic wildtype, hypoxic M1R cKO mice and wildtype littermates in at P40. ** p<0.01, *p<0.05, significance based on non-parametric Mann-Whitney by comparing to respective control, n = 5 mice in each group. See also Figure S5.
It is well documented that microglial cells prune synapses and over-activated glial cells may strip synapses in developing brains (Chung and Barres, 2012; Paolicelli et al., 2017; Schafer et al., 2012; Vainchtein et al., 2018). We examined microglial cells and astrocytes by immunostaining for Iba-1 and GFAP respectively in the hypoxic brains (Figures S5A, B). The density and morphology of Iba-1 and GFAP positive cells in the cortex of hypoxic M1R cKO mice were similar to the wildtype controls (Figures S5A, B). To further examine the phagocytic ability of microglia to prune synapses, we calculated the number of PSD-95 puncta, a postsynaptic marker, engulfed by microglia (Figure S5C). The engulfed PSD-95 puncta were similar between the M1R cKO and wildtype hypoxic brains (Figure S5C), suggesting that glial cells are unlikely contributing to the change in the number of synapses. To better understand whether SVZ proliferation is altered by M1R deletion, we injected a dose of BrdU 24 hrs before sacrificing the mice and examined BrdU incorporation in the SVZ (Figure S5D). The percentage of BrdU positive cells in the SVZ was not changed between the hypoxic M1R cKO and wildtype controls.
5. Drug-based myelination strategies promote functional recovery in hypoxic mice.
Given the beneficial effects of enhancing myelination on the motor behavior in hypoxic mice, drug-based strategies may be the most feasible and realistic way to translate the findings into clinical therapies for WMI patients. Previous efforts on screening for remyelination compounds have yielded a number of potent candidates that can enhance oligodendroglial differentiation. Among them, clemastine and (±)U50488 are two powerful representatives. Clemastine is a muscarinic receptor antagonist that has proven effective for remyelination in MS patients (Deshmukh et al., 2013; Green et al., 2017) and (±)U50488 is a kappa opioid receptor agonist (Du et al., 2016; Mei et al., 2016b). Despite the fact that myelination-enhancing drugs are not approved for children less than 12 years of age, as a proof of concept for the utility of myelin-enhancing strategy in hypoxia-related WMI, we treated the mice with either clemastine or (±)U50488 at 10mg/kg per day during exposure of hypoxia (Figure 8A). As expected, MBP staining for myelin and CC1 positive OL numbers were greatly enhanced at P10 as compared to the vehicle controls (Figures 8B,C). Consistently, the number of nodes (Caspr and Nav1.6 double positive) in the hypoxic spinal cords and myelinated axons in the corpus callosum were significantly increased under TEM after treatment with clemastine or (±)U50488 (Figures 8D, E), indicating that these compounds promote oligodendroglial differentiation and myelination in the hypoxic brains. As expected, the drug treatment rescued the number of synapses revealed by labeling for Synapsin1 and Homer1 (Figures S6A, B) without changing the proliferation of cells in the SVZ (Figures S6C-E). The cell density of GFAP positive astrocytes and Iba1 positive microglial cells was unchanged in the hypoxic cortex treated with drugs as compared to vehicle controls (Figures S7A, B). Next, we subjected the mice to the beam walking examination at P40 and found that the drug-treated hypoxic mice have a lower frequency of foot slippage than the vehicle control mice (Figure 8F) and that motor coordination was unaltered in the normoxic mice treated with the drugs (Figure S7C). Since cognitive deficits are also prevalent in children with WMI (Maureen et al., 2002; Woodward et al., 2006), we also assessed whether cognition was also changed after drug treatment in the hypoxic mice by novel object recognition (Figure 8G). Our results indicate that hypoxic mice treated with clemastine or (±)U50488 displayed an improved performance in this task as compared to the vehicle controls, indicating that myelination-enhancing drugs can promote myelination and functional recovery against chronic hypoxia.
Figure 8. Myelin-enhancing drug treatment during hypoxia rescues hypoxia-induced hypomyelination and improves functional recovery.

(A) Experimental paradigm showing drug treatment and hypoxic exposure; (B, C) Representative images and quantification of MBP expression (red, B), CC1 positive OLs (green, C) in the brains and Caspr (red)/Nav1.6 (green) positive nodes (D) in the spinal cords of clemastine, (±)U50488 or vehicle treated mice. Error bars represent mean ± s.e.m. *p<0.05 or **p<0.01, significance based on Student’s t-test with the respective controls, n = 3 mice for each group. (E) Representative images and quantification of myelinated axons in the corpus callosum at P15. Error bars represent mean ± s.e.m. **p<0.01, significance based on Student’s t-test by comparing clemastine or (±)U50488 to vehicle, n = 3 mice for all experiments. Scale bar, 200μm (B), 100μm (C), 10μm (D) and 2μm (E). (F) The beam-walking test showing the frequency of foot slips of the clemastine, (±)U50488 or vehicle treated mice at P40, n = 7 mice for each group. *p<0.05, significance based on non-parametric Mann-Whitney test by comparing clemastine or (±)U50488 to vehicle; (G) The hypoxic mice treated with clemastine or (±)U50488 displayed significantly enhanced novel object recognition as compared to the vehicle controls, assessed 2 hours later after introduced to the objects, n = 7 mice for each group. *p<0.05, significance based on non-parametric Mann-Whitney test by comparing clemastine or (±)U50488 to vehicle. See also Figure S6–8.
Considering that brain injuries may occur in infants and children prior to being detectable by imaging, we next investigated whether enhancing myelination after hypoxic injury could promote recovery of myelination and functional deficits. We treated mice from P11 to P18 with either clemastine or (±)U50488 at 10mg/kg per day after hypoxic exposure (Figure S8A). Interestingly, MBP positive myelin density and CC1 positive OL numbers were greatly increased in the cortex and corpus callosum respectively at P18 as compared to the vehicle controls (Figures S8B–E), indicating that these compounds are still effective in promoting oligodendroglial differentiation and myelination post hypoxia. Once again, we subjected these mice to the beam walking examination at P40 and found that the mice that received the drug treatment displayed a lower frequency of foot slippage than the vehicle control mice (Figure S8F). These results indicate that enhancing myelination after hypoxic exposure can also promote functional recovery against chronic hypoxia. All together, our data provide novel insights into the importance of myelination in regulating synaptogenesis, and that myelination-enhancing approaches may represent a promising strategy to rescue synaptogenesis and behavioral deficits after hypoxic insult (Figure S8G).
DISCUSSION
Advances in the care of premature infants have resulted in an increasing survival rate of preterm infants, especially for the very low birth weight infants (<1.5 kg), leading to a decrease in severe brain injuries, such as intraventricular hemorrhage (IVH) and intraparenchymal hemorrhage (Back and Rosenberg, 2014; Fowlie and Davis, 2003; Volpe, 2001). However, the incidence of WMI in preterm infants and children remains unchanged (Ballabh, 2010). Immature white matter in preterm newborns is highly vulnerable to hypoxia, ischemia and inflammation (Back, 2017; Hagberg et al., 2015). Despite that oxygenation disturbance is generally considered a major cause of WMI, multiple other etiologies also emerge as critical risk factors for WMI as well. For instance, inflammation induced by chorioamnionitis and fetal sepsis has been shown to be an important risk factor for WMI (Kuypers et al., 2012). In this study, we focus our efforts on chronic neonatal hypoxia-induced brain injury which attempts to mimic diffuse WMI in prenatal infants (Salmaso et al., 2014). Admittedly, while there are no perfect animal models to duplicate the human condition, our study begins to provide definitive clues into the role of myelination using the chronic hypoxia model in neonatal mice.
Currently, there are no effective clinical therapies for WMI. The hypoxia-induced WMI and hypoxic brains are characterized by a cohort of pathological changes, including hypomyelination, synaptic loss, inflammation and cellular injury (Back, 2017; Rumajogee et al., 2016). Development of therapies against WMI necessities a thorough understanding of the functional consequences of the pathological changes. Disrupted neuronal circuit formation leads to long-term neuronal dysfunction, and may be due to inhibited synaptogenesis (Kiss et al., 2014; Luciana, 2003). However, there are no direct interventions available to strengthen neuronal circuit wiring. Hypomyelination and arrested OPC differentiation are prominent in the WMI regions and hypoxic brains (Buser et al., 2012; Segovia et al., 2008). Determining the functional importance of myelin requires OPC-specific manipulation that solely alters myelination. Using the Olig2 conditional knockout mice, we clearly demonstrate that hypomyelination can directly impair neuronal functional development. By using the OPC-specific M1R conditional knockout line as a gain of function experiment, our results indicate that M1R deletion in OPCs improves myelination and subsequently promotes functional recovery after chronic hypoxia. Thus, we conclude that enhancing myelination represents a promising approach to promote neuronal functional recovery after hypoxia-induced WMI.
Given that postnatal myelination occurs in a spatiotemporal manner in rodent brains, how myelination alters neuronal architecture and function are still yet unclear. The function of myelin ensures the rapid and efficient transmission of action potentials along axons (Chang et al., 2016; Pan and Chan, 2017). Deposition of myelin alters the axonal cytoskeleton and induces a rearrangement of ion channels on the axolemma (Pan and Chan, 2017). Recent evidence indicates that oligodendroglia dictate to different neuronal demands and deposit variable amounts of myelin sheaths on the axons of cortical neurons and optic nerves (Etxeberria et al., 2016; Tomassy et al., 2014). Thus, it is plausible that the delay of myelination may disrupt or slow signal transmission along axons and ultimately impart negative effects on neuronal development. Given that rapid progression of myelination overlaps with a burst of synaptogenesis in the first three weeks (Sarnat et al., 2015), is it possible that myelination participates in regulating synaptogenesis? A number of studies demonstrate that a delay in synapse formation is accompanied by hypomyelination in the brains of hypoxic mice (Curristin et al., 2002; Liu et al., 2016; Otellin et al., 2014; Piorkowska et al., 2014) and other rodent models with developmental neuronal functional deficits (Hanics et al., 2012; Sarnat and Flores-Sarnat, 2016; Tohda et al., 2007). It is well documented that microglia and astrocytes are responsible for pruning synapses, and over-activated microglia could lead to excessive loss of synapses in the developing CNS (Chung and Barres, 2012; Schafer et al., 2012; Vainchtein et al., 2018). Our results provide direct evidence that myelination can facilitate synaptogenesis, especially excitatory presynaptic innervation. In addition, enhancing myelination is sufficient to rescue the structural and functional deficits of synaptic loss in hypoxic brains. Considering that the timing of neuronal activity plays a crucial role in synapse plasticity, so-called spike timing-dependent plasticity (STDP), it is plausible that myelination may participate in the synaptic integration and neuronal network remodeling by facilitating the efficiency and velocity of action potential propagation in the developing CNS (Caporale and Dan, 2008; Dan and Poo, 2004). Therefore, enhanced myelination improves neuronal functional recovery at least partially through facilitating synaptogenesis in the M1R cKO brains after chronic hypoxia.
To translate these findings into clinical therapies, drug-based strategies may be the most feasible and realistic way to enhance myelination for WMI patients. Recently, epidermal growth factor (EGF) and a GABA transporter inhibitor were shown to increase OPC proliferation, migration and specification, and promote myelination after chronic hypoxia (Aguirre et al., 2007; Scafidi et al., 2013; Zonouzi et al., 2015). Recent efforts in high-throughput screening have identified a number of compounds potent in promoting remyelination (Deshmukh et al., 2013; Du et al., 2016; Mei et al., 2016b, 2014). Clemastine and (±)U50488 are two compounds that seem to display the most potential for remyelination. Here we have demonstrated as a proof of concept the utility of these drugs for WMI as a result of hypoxia, although no myelin-enhancing drugs are available for children less than 12 years of age. We demonstrate that treatment of these two drugs during or after hypoxia promotes myelination and functional recovery. As a proof of concept, a recent phase-two clinical trial has revealed that clemastine significantly promotes remyelination in chronic multiple sclerosis patients (Green et al., 2017). Therefore, if myelinating-inducing compounds become available for children, drug-based myelin-enhancing strategies may become a promising therapeutic approach to promote functional recovery for pediatric WMI patients.
Together, our findings provide novel insights into our understanding of the functional significance of myelination in hypoxic brains, and indicate that myelination-enhancing strategies represent a promising approach to promote functional recovery after WMI.
STAR★METHODS
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit anti-NG2 | Millipore | Cat# AB5320, RRID:AB_11213678 |
| rabbit anti-NF200 | Sigma-Aldrich | Cat# N4142, RRID:AB_477272 |
| rabbit anti-MOG | Abcam | Cat# ab32760, RRID:AB_2145529 |
| rat anti-MBP | Millipore | Cat# MAB395–1ML, RRID:AB_240845 |
| mouse anti-CC1 | Millipore | Cat# OP80, RRID:AB_2057371 |
| Guinea anti-vGlut1 | Synaptic Systems | Cat# 135 304, RRID:AB_887878 |
| rabbit anti-NeuN | Abcam | Cat# ab177487, RRID:AB_2532109 |
| rabbit anti-Nav1.6 | alomone labs | Cat# ASC-009, RRID:AB_2040202 |
| mouse anti-Caspr | UC Davis/NIH NeuroMab Facility | Cat# 75–001, RRID:AB_2083496 |
| mouse anti-vGAT | Synaptic Systems | Cat# 131 011, RRID:AB_887872 |
| rabbit anti-Homer1 | Synaptic Systems | Cat# 160 003, RRID:AB_887730 |
| mouse anti-PSD95 | Millipore | Cat# MAB1596, RRID:AB_2092365 |
| rabbit anti-Synapsin1 | CST | Cat: 5297 |
| goat anti-GFAP | Santa Cruz Biotechnology | Cat# sc-6170, RRID:AB_641021 |
| rabbit anti-lba1 | Wako | Cat# 019–19741, RRID:AB_839504 |
| chicken anti-MAP2 | Millipore | Cat# AB5543, RRID:AB_571049 |
| Alexa Fluor 488 donkey anti-rabbit IgG | Thermo Fisher Scientific | Cat# A-21206, RRID:AB_2535792 |
| Alexa Fluor 488 donkey anti-mouse IgG | Thermo Fisher Scientific | Cat# A-21202, RRID:AB_141607 |
| Alexa Fluor 488 goat anti-rat IgG | Thermo Fisher Scientific | Cat# A-11006, RRID:AB_2534074 |
| Alexa Fluor 568 donkey anti-mouse IgG | Thermo Fisher Scientific | Cat# A10037, RRID:AB_2534013 |
| Alexa Fluor 568 donkey anti-rabbit IgG | Thermo Fisher Scientific | Cat# A10042, RRID:AB_2534017 |
| Alexa Fluor 568 donkey anti-goat IgG | Thermo Fisher Scientific | Cat# A-11057, RRID:AB_2534104 |
| Alexa Fluor 568 goat anti-guinea pig IgG | Thermo Fisher Scientific | Cat# A-11075, RRID:AB_2534119 |
| Alexa Fluor 594 donkey anti-rat IgG | Thermo Fisher Scientific | Cat# A-21209, RRID:AB_2535795 |
| Alexa Fluor 647 donkey anti-mouse IgG | Thermo Fisher Scientific | Cat# A-31571, RRID:AB_162542 |
| Alexa Fluor 647 goat anti-chicken IgG | Thermo Fisher Scientific | Cat# A-21449, RRID:AB_2535866 |
| Chemicals | ||
| Clemastine Fumarate | SelleckChem | Cat: S1847 |
| (±)U-50488 | Tocris | Cat: 0495 |
| Tamoxifen | Sigma-aldrich | Cat: T5648 |
| BrdU | Sigma-aldrich | Cat: B5002 |
| Critical commercial assays | ||
| In situ cell death detection kit | Roche | Cat: 11684795910 |
| Experimental Models: organisms/strains | ||
| Mouse: M1R fl/fl | Kamsler etal., 2010 | N/A |
| Mouse: NG2-CreERT2 | Jackson Laboratory | Catalog: 008538 |
| Mouse: CNP-Cre | Lappe-Siefke et al., 2003 | N/A |
| Mouse: mT/mG | Jackson Laboratory | Catalog: 007676 |
| Mouse: Tau-mGFP | Jackson Laboratory | Catalog: 021162 |
| Mouse: PLP-CreERT2 | Jackson Laboratory | Catalog: 005975 |
| Mouse: Olig2 fl/fl | Yue etal., 2006 | N/A |
| Software and Algorithms | ||
| Image-Pro Plus software 5.0 | Media Cybernetics | http://www.mediacy.com/imageproplus;RRID:SCR_007369 |
| ImageJ | NIH | https://imagej.nih.gov/ij/;RRID:SCR_003070 |
| FV10-ASW 4.2 Viewer | Olympus | N/A |
| ZEN | Zeiss | N/A |
| Imaris 7.6.3 | Bitplane | http://www.bitplane.com/imaris/imaris;RRID:SCR_007370 |
| pClamp 10.0 | Axon Instruments | N/A |
| Mini-analysis 6.0 | Synaptosoft | N/A |
| GraphPad prism 6 | GraphPad Software | https://www.graphpad.com/scientificsoftware/prism/;RRID:SCR_015807 |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents and resources should be directed to and will be fulfilled by the Lead Contact, Feng Mei (meif@tmmu.edu.cn).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
For all studies, both male and female mice from postnatal day 10–80 on a C57BL/J background were used. All the mice were housed in temperature- and humidity-controlled environments with free access to standard chow and water and on a 12h/12h light/dark cycle according to the guidelines of Third Military Medical University. All mouse strains were maintained in the Third Military Medical University specific pathogen free animal facility. The C57BL/6 wildtype mice were purchased from the animal breeding center of our university. The M1R fl/fl mice have been described previously (Mei et al., 2016b). Briefly, the M1R floxed line was crossed with the NG2-CreERT2 (The Jackson Laboratory, Catalog # 008538) to generate NG2-CreERT2; M1R fl/fl mice and littermate controls (M1R fl/fl) (Zhu et al., 2011; Kamsler et al., 2010). The mT/mG (Catalog # 007676) mice, PLP-CreERT2 (Catalog # 005975) and Tau-mGFP mouse line (Catalog # 021162) were obtained from The Jackson Laboratory. The CNP-Cre and Olig2 floxed mice have been described previously (Mei et al., 2013). Genotypes of all mice were determined by using a PCR analysis of tail genomic DNA with appropriate primers. All the transgenic mice were backcrossed >10 generations on a C57BL/6 background. Experiments were performed using double transgenic mice and respective littermate controls. All the animal experiments were handled in accordance with an approved protocol from the Laboratory Animal Welfare and Ethics Committee of the Third Military Medical University.
Hypoxia model
Neonatal mice with their parents were subjected to chronic sublethal hypoxia by receiving 10% fraction of inspired oxygen from postnatal day 3 (P3) to postnatal day 10 (P10). The oxygen concentration was monitored and maintained by an oxygen modulator (Biospherix, ProOx110).
METHOD DETAILS
Experimental design
The sample size per experiment was determined according to previous publications with similar methodologies. All experiments were performed for 3 times. The findings were successfully replicated in individual experiment. All the data were included for analysis.
Administration of tamoxifen
To induce Cre recombination in newborn pups, Tamoxifen (Sigma-Aldrich, Cat: T5648) was administered to the mother at 100mg/kg per day (dissolved in sunflower oil at 30 mg/ml) for two consecutive days by oral gavage from P3, as tamoxifen can be delivered to pups through lactation (Figure S5C). In addition, the pups were administered with Tamoxifen for two consecutive days by oral gavage at 10mg/kg daily from P5. For the PLP-CreERT; Olig2 fl/fl mice, Tamoxifen was administered at the dosage of 100mg/kg per day for five consecutive days from P40.
Drug treatment
The hypoxic pups were treated with either clemastine (SelleckChem, Cat: S1847) or (±)U-50488 (Tocris, Cat: 0495) daily at 10mg/kg or the equivalent volume of vehicle by oral gavage from P3 to P10. Each pup was orally administered with the drug or vehicle solution starting from 10 microliters at P3 and the daily volume was determined by body weight. In the post-hypoxia treatment experiment, mice pups were also treated with clemastine, (±)U-50488 at 10mg/kg/day or the equivalent volume of vehicle for 8 consecutive days from P11 to P18. Both male and female mice were pooled for analysis in this study, since we did not observe sexual dimorphism of myelination defects in the hypoxia mice.
Tissue processing
Mice were deeply anesthetized with 1% pentobarbital and transcardially perfused with 4% paraformaldehyde in 0.1M PB after an initial flush with 0.01M PBS. Brains and spinal cords were collected and postfixed with 4% paraformaldehyde in 0.1M PB overnight, then these tissues were dehydrated in 30% sucrose in 0.01M PBS. Brains were embedded in optimal cutting temperature compound (O.C.T. Compound, SAKURA, 4583, USA) and then sliced coronally (20 m) on a cryostat microtome (MS 1850, Leica, Wetzlar, Germany). Spinal cords were sliced longitudinally (20 m).
Immunofluorescence staining and image acquisition
For immunofluorescence staining, floating sections were blocked with 5% bovine serum albumin (BSA) and 0.2% Triton-X 100 for 2h at room temperature and then sequentially incubated with primary antibodies overnight at 4°C and the fluores cent secondary antibodies for 2h at room temperature. Primary antibodies include: rabbit anti-NG2 (1:500, Millipore, Cat: MAB5320), rabbit anti-NF200 (1:1000, Sigma-Aldrich, Cat: N4142), rabbit anti-MOG (1:1000; abcam; Cat: ab32760); rat anti-MBP (1:500, Millipore, Cat: MAB395), mouse anti-CC1 (1:500, Calbiochem, Cat: OP80), Guinea anti-vGlut1 (1:2000, Synaptic Systems, Cat: 135304); rabbit anti-NeuN (1:1000, abcam, Cat: ab177487); rabbit anti-Nav1.6 (1:2000, alomone labs, Cat:ASC-009); mouse anti-Caspr (1:2000, NeuoMab; Cat:75–001); mouse anti-vGAT (1:1000, Synaptic Systems, Cat: 131011); rabbit anti-Homer1 (1:1000, Synaptic Systems, Cat: 160003); mouse anti-PSD95 (1:1000, Millipore, Cat: MAB1596); rabbit anti-Synapsin-1 (1:1000, CST, Cat: 5297); goat anti-GFAP (1:50, Santa Cruz, Cat: sc-6170); rabbit anti-Iba1 (1:200, Wako, Cat: 019–19741) and chicken anti-MAP2 (1:2000, EMD Millipore, Cat: AB5543). Secondary antibodies included the following: AlexaFluor-488-, AlexaFluor-568-, or AlexaFluor-647-conjugated secondary antibodies against goat, rabbit, mouse, guinea pig, chicken or rat (1:500; Invitrogen) antibodies. Nuclei were counterstained with DAPI. Fluorescent images were captured using an Axio Imager M2 fluorescence microscope (Zeiss, Oberkochen, Germany) and a confocal laser-scanning microscope (Olympus, FV1000, Shinjuku, Tokyo) with excitation wavelengths appropriate for Alexa Fluor 488 (488 nm), 596 (568 nm), 647 (628 nm) or DAPI (380 nm).
BrdU Labeling
The mice were given a dose of BrdU (Sigma, Cat: B5002) intraperitoneal at 100mg/kg 24 hours before sacrifice. After rinsing in 0.01M PBS, the specimens were treated with 2 N HCl for 30 min at 37°C for partial denaturalization of double-stra nded DNA. To reveal BrdU, the specimens were incubated with a rat anti-BrdU (1:200, BIO-RAD, Cat: OBT0030) overnight, then the sections were incubated with Alexa 488-conjugated secondary antibody.
TUNEL assays
To detect apoptosis, floating sections were washed twice with PBS for 30 min, and then treated with 0.1% Triton X-100 for 20 min on ice (2–8°C). T he sections were incubated with the TUNEL reaction buffer, containing 45 L of the labeling solution and 5 L of the enzyme solution, at 37°C for 1 h in a humidified and dark atmosphere. T he sections were then washed three times for 15 min each with PBS to remove unincorporated fluorescein-dUTP. The specimens were observed with a fluorescence microscope with an excitation wavelength in the range of 450–500 nm.
Microglial in vivo engulfment assay
The entire microglia were scanned and imaged on Z-stacks at an interval 0.56μm by using a confocal laser-scanning microscope (Olympus, FV1000) with a 63x objective (oil, NA1.3). All the parameters were consistent throughout all experiments. 3D surface rendering of microglia Images and PSD-95 positive puncta were created by using Imaris software (Bitplane). The PSD-95 positive puncta within the Iba1 positive cells were calculated.
Electron microscopy
For electron microscopy, animals were perfused with 1.25% glutaraldehyde, 2% paraformaldehyde in 0.1 M PB after an initial flush with 0.01 M PBS. Brains and optic nerves were rapidly removed and postfixed at least a week at 4°C. Tissues were rinsed, postfixed with 1% OsO4 in PB for 2 h, counterstained with uranyl acetate, dehydrated with a graded acetone series, infiltrated with propylene oxide, and embedded in Epon. Ultrathin sections (~60 nm) were cut by using an ultramicrotome (LKB-V, LKB Produkter AB, Bromma) and observed under a transmission electron microscope (HT7700, TITACHI, Japan). G-ratios of myelinated fibers were calculated as the ratio of the diameter of the axon to the diameter of the axon with the myelin sheath by using Image-Pro Plus software. Measurements were made on electron micrographs from three pairs of mice in all cases.
In vitro Electrophysiology
The whole-cell patch clamp recordings were conducted as previously described (Paz et al., 2010). Coronal slices (300–400 m) were cut with a VT1200 vibrotome (Leica, Germany) and removed to a chamber which was superfused with oxygenated artificial cerebrospinal fluid. Whole-cell patch clamp recordings were performed in the layer 5/6 neurons of the M2 region verified with BX51WI microscope (Olympus, Japan) with infrared differential interference contrast (IR-DIC) illumination. The electrophysiological signals were fed into a computer through a Digidata-1440A interface (Axon Instruments, USA) for data acquisition (pClamp 10.0, Axon Instruments, USA). Neurons were held at a membrane potential of −70 mV and characterized by injection of rectangular voltage pulse (5 mV, 50 ms) to monitor the whole-cell membrane capacitance, series resistance and membrane resistance. Neurons were excluded from the study if the series resistance changed by more than 15%. Miniature excitatory postsynaptic current (mEPSCs) were recorded with 1 M TTX and 100 M picrotoxin pre-added to the normal ACSF for at least 15 minutes. Data analyses were conducted using Mini-analysis software (version 6.0, Synaptosoft, USA). Experimenter was blinded to genotype.
Behavioral tests
All mice were fed in a controlled environment (25 °C) with free accesses to food and water and housed on a 12h/12h day/night cycle. All tests were performed from 12:00 to 18:00. After each trial, the apparatuses were wiped with 30% ethanol to avoid smell cues. For all behavioral experiments, investigators were blinded for genotype and mice were handled gently to avoid stress.
Beam walking test: In the modified beam walking test, a beam (0.5cm wide) was placed 50 cm above the floor in a dark room, with one end being lightened by a lamp and the other end in a box (non-transparent, 20 × 10 × 20 cm3). Padding material from cages was placed in the box to attract the mice to walk through in a sequence of 30-, 50- and 70-cm long distances on the beam during the training days. Mice were placed at the end with light and trained three times per day for 3 days before testing. Videos were recorded from both sides of the beam on the testing day. The frequency of hind limb slippage were calculated as the index of beam walking performance when the mice walk through an 80-cm long distance (Brooks and Dunnett, 2009).
Rotarod test: The mice were placed on the rod, and the speed of rotating rod was gradually increased from 4 to 40 rpm within 5 minute. Five trials were completed with an inter-trial interval of 5 minutes, the duration that the mice can stay on the rod was calculated as the index of the rotatod test (Brooks and Dunnett, 2009).
Novel object recognition test: The experiment was conducted as described previously (Grayson et al., 2015). Briefly, the experimental apparatus consisted of a white polyvinyl plastic square open field (25 cm × 25 cm × 40 cm). Two rectangular plastic objects were placed 8 cm away from the walls in the apparatus and were introduced to a mouse for 5 minutes. Two hours later, one of the rectangular objects was replaced by a cylinder (a new toy) before they were introduced to the same mouse again. The discrimination index was determined as a ratio of the time spent exploring the novel object to the total time spent exploring the two objects.
QUANTIFICATION AND STATISTICAL ANALYSIS
Stereology and quantification
Brain sections were collected consecutively from Bregma 1.1mm to Bregma −1.34mm of the adult mouse brains or of the P10 and P18 mouse brains correspondingly. Cervical spinal cords from C4-C7 were sliced longitudinally and all the sections were collected in 24-well plates. To sample in a systematic random manner, a set of 5–8 sections were sampled from the consecutive sections of each brain or spinal cord. In practice, every twentieth section was systematically sampled, the first one was randomly sampled from the first 20 sections. All the quantification was carried out by using Image-Pro Plus software 5.0 (Media Cybernetics, Silver Spring, MD, USA). Experimenter was blinded to genotype.
Cell and node quantification
For the cell number quantification, at least three representative fields were randomly acquired from layer 2–5 of M2 cortex, SVZ or corpus callosum of brain sections by using the Zeiss M2 fluorescence microscope (20x). The nodes of Ranvier were imaged from the spinal cord white matter (40x), at least five representative fields were acquired. The Nav1.6 positive nodal segments that were paired with two neighboring Caspr positive paranodal segments were counted as nodes of Ranvier.
Puncta quantification
To quantify synaptic puncta density, at least three representative fields (oil, 63x) were randomly acquired from the layer 2–5 of M2 cortex by using a confocal laser-scanning microscope (Olympus, FV1000). All the images were acquired from the same Z positive using standardized imaging parameters throughout. To separate puncta from background, threshold was set at least two times the background intensity. Puncta were defined by distinct intensity peaks above threshold.
Fluorescent density and area quantification
To quantify fluorescent positive area and density, the entire corpus callosum and cortex were selected and then were automatically imaged (20x) and tiled by using the Zeiss M2 fluorescence microscope. The areas of interest (AOI) were separated by setting threshold at least two times the background.
Statistical analysis
All graph data are presented as the mean ± s.e.m. n represents the number of mice used per experiment unless indicated otherwise. No methods were used to determine whether the data met the assumptions of the statistical approach. The unpaired t-test was used to determine the significance between two experimental groups. The non-parametric Mann-Whitney test was used when the raw data displayed an abnormal distribution. Significance was reported as *p <0.05 or **p<0.01.
Supplementary Material
HIGHLIGHTS:
Chronic hypoxia causes hypomyelination, synaptic loss and functional deficits.
Hypomyelination results in synaptic and functional deficits.
Enhancing myelination rescues hypoxia-induced synaptic and functional deficits.
Myelination-enhancing drugs improve functional recovery against hypoxia.
ACKNOWLEDGEMENTS:
We would like to thank J. Josh Lawrence (Texas Tech University) and Susumu Tonegawa (MIT) for the M1R floxed mice, Dr. Richard Lu (University of Cincinnati) for the Olig2 floxed mice and Dr. Klaus Nave for the CNP-Cre mice. Funding: This work was supported by the National Natural Science Foundation of China (31771120, 81471346), the Chongqing Scientific and Technical Innovation Foundation of China (CSTCKJCXLJRC07) to L.X., Cheung Kong Scholars Program, the March of Dimes Foundation (6-FY17–551) to S.P.J.F., the NIH/NINDS (R01NS062796, R01NS097428, R01NS095889), the Adelson Medical Research Foundation (A130141) and the Rachleff Family endowment to J.R.C.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental Information includes one table and nine figures can be found with this article.
DECLARATION OF INTERESTS: The authors declare no competing financial interests.
REFERENCES
- Aguirre A, Dupree JL, Mangin JM, Gallo V, 2007. A functional role for EGFR signaling in myelination and remyelination. Nat. Neurosci 10, 990–1002. [DOI] [PubMed] [Google Scholar]
- Back S. a, Han BH, Luo NL, Chricton C. a, Xanthoudakis S, Tam J, Arvin KL, Holtzman DM, 2002. Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J. Neurosci 22, 455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, 2017. White matter injury in the preterm infant : pathology and mechanisms. Acta Neuropathol. 134, 331–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, 2015. Pediatric Neurology Brain Injury in the Preterm Infant : New Horizons for Pathogenesis and Prevention. Pediatr. Neurol 53, 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Rosenberg PA, 2014. Pathophysiology of glia in perinatal white matter injury. Glia 62, 1790–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballabh P, 2010. Intraventricular hemorrhage in premature infants: mechanism of disease. Pediatr. Res 67, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billiards SS, Haynes RL, Folkerth RD, Borenstein NS, Trachtenberg FL, Rowitch DH, Ligon KL, Volpe JJ, Kinney HC, 2008. Myelin abnormalities without oligodendrocyte loss in periventricular leukomalacia. Brain Pathol. 18, 153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blencowe H, Cousens S, Oestergaard MZ, Chou D, Moller AB, Narwal R, Adler A, Vera Garcia C, Rohde S, Say L, Lawn JE, 2012. National, regional, and worldwide estimates of preterm birth rates in the year 2010 with time trends since 1990 for selected countries: A systematic analysis and implications. Lancet 379, 2162–2172. [DOI] [PubMed] [Google Scholar]
- Brooks SP, Dunnett SB, 2009. Tests to assess motor phenotype in mice: A user’s guide. Nat. Rev. Neurosci 10, 519–529. [DOI] [PubMed] [Google Scholar]
- Buser JR, Maire J, Riddle A, Gong X, Nguyen T, Nelson K, Luo NL, Ren J, Struve J, Sherman LS, Miller SP, Chau V, Hendson G, Ballabh P, Grafe MR, Back SA, 2012. Arrested preoligodendrocyte maturation contributes to myelination failure in premature infants. Ann. Neurol 71, 93–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporale N, Dan Y, 2008. Spike Timing–Dependent Plasticity: A Hebbian Learning Rule. Annu. Rev. Neurosci 31, 25–46. [DOI] [PubMed] [Google Scholar]
- Chang K, Redmond SA, Chan JR, 2016. Remodeling myelination: implications for mechanisms of neural plasticity. Nat. Neurosci 19, 190–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WS, Barres BA, 2012. The role of glial cells in synapse elimination. Curr. Opin. Neurobiol 22, 438–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cree BAC, Niu J, Hoi KK, Zhao C, Caganap SD, Henry RG, Dao DQ, Zollinger DR, Mei F, Shen Y-AA, Franklin RJM, Ullian EM, Xiao L, Chan JR, Fancy SPJ, 2018. Clemastine rescues myelination defects and promotes functional recovery in hypoxic brain injury. Brain 141, 85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curristin SM, Cao A, Stewart WB, Zhang H, Madri JA, Morrow JS, Ment LR, 2002. Disrupted synaptic development in the hypoxic newborn brain. Proc. Natl. Acad. Sci. U. S. A 99, 15729–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis WJ, Lindeke LL, Georgieff MK, Nelson CA, 2002. Neurobehavioural functioning in neonatal intensive care unit graduates in late childhood and early adolescence. Brain 125, 1646–59. [DOI] [PubMed] [Google Scholar]
- Dan Y, Poo MM, 2004. Spike timing-dependent plasticity of neural circuits. Neuron 44, 23–30. [DOI] [PubMed] [Google Scholar]
- De Camilli P, Harris SM, Huttner WB, Greengard P, 1983. Synapsin I (protein I), a nerve terminal-specific phosphoprotein. II. Its specific association with synaptic vesicles demonstrated by immunocytochemistry in agarose-embedded synaptosomes. J. Cell Biol. 96, 1355–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh V. a, Tardif V, Lyssiotis C. a, Green CC, Kerman B, Kim HJ, Padmanabhan K, Swoboda JG, Ahmad I, Kondo T, Gage FH, Theofilopoulos AN, Lawson BR, Schultz PG, Lairson LL, 2013. A regenerative approach to the treatment of multiple sclerosis. Nature 502, 327–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du C, Duan Y, Wei W, Cai Y, Chai H, Lv J, Du X, Zhu J, Xie X, 2016. Kappa opioid receptor activation alleviates experimental autoimmune encephalomyelitis and promotes oligodendrocyte-mediated remyelination. Nat. Commun 7, 11120. doi: 10.1038/ncomms11120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etxeberria A, Hokanson KC, Dao DQ, Mayoral SR, Mei F, Redmond SA, Ullian EM, Chan JR, 2016. Dynamic Modulation of Myelination in Response to Visual Stimuli Alters Optic Nerve Conduction Velocity. J. Neurosci 36, 6937–6948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancy SPJ, Harrington EP, Yuen TJ, Silbereis JC, Zhao C, Baranzini SE, Bruce CC, Otero JJ, Huang EJ, Nusse R, Franklin RJM, Rowitch DH, 2011. Axin2 as regulatory and therapeutic target in newborn brain injury and remyelination. Nat. Neurosci 14, 1009–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancy SPJ, Harrington EP, Baranzini SE, Silbereis JC, Shiow LR, Yuen TJ, Huang EJ, Lomvardas S, Rowitch DH, 2014. Parallel states of pathological Wnt signaling in neonatal brain injury and colon cancer. Nat. Neurosci 17, 506–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferriero DM, 2004. Neonatal brain injury. N. Engl. J. Med 351, 1985–95. [DOI] [PubMed] [Google Scholar]
- Fowlie PW, Davis PG, 2003. Prophylactic indomethacin for preterm infants: a systematic review and meta-analysis. Arch. Dis. Child. Fetal Neonatal Ed 88, F464–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayson B, Leger M, Piercy C, Adamson L, Harte M, Neill JC, 2015. Assessment of disease-related cognitive impairments using the novel object recognition (NOR) task in rodents. Behav. Brain Res. 285, 176–193. [DOI] [PubMed] [Google Scholar]
- Green AJ, Gelfand JM, Cree BA, Bevan C, Boscardin WJ, Mei F, Inman J, Arnow S, Devereux M, Abounasr A, Nobuta H, Zhu A, Friessen M, Gerona R, von Büdingen HC, Henry RG, Hauser SL, Chan JR, 2017. Clemastine fumarate as a remyelinating therapy for multiple sclerosis (ReBUILD): a randomised, controlled, double-blind, crossover trial. Lancet 390, 2481–2489. [DOI] [PubMed] [Google Scholar]
- Hagberg H, Mallard C, Ferriero DM, Vannucci SJ, Levison SW, Vexler ZS, Gressens P, 2015. The role of inflammation in perinatal brain injury. Nat. Rev. Neurol 11, 192–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanics J, Barna J, Xiao J, Millán JL, Fonta C, Négyessy L, 2012. Ablation of TNAP function compromises myelination and synaptogenesis in the mouse brain. Cell Tissue Res. 349, 459–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamsler A, McHugh TJ, Gerber D, Huang SY, Tonegawa S, 2010. Presynaptic m1 muscarinic receptors are necessary for mGluR long-term depression in the hippocampus. PNAS 107:1618–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss JZ, Vasung L, Petrenko V, 2014. Process of cortical network formation and impact of early brain damage. Curr. Opin. Neurol 27, 133–141. [DOI] [PubMed] [Google Scholar]
- Kuypers E, Ophelders D, Jellema RK, Kunzmann S, Gavilanes AW, Kramer BW, 2012. White matter injury following fetal inflammatory response syndrome induced by chorioamnionitis and fetal sepsis: Lessons from experimental ovine models. Early Hum. Dev 88, 931–936. [DOI] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA, 2003. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nature Genetics 33:366–374. [DOI] [PubMed] [Google Scholar]
- Liu X-B, Shen Y, Pleasure DE, Deng W, 2016. The vulnerability of thalamocortical circuitry to hypoxic-ischemic injury in a mouse model of periventricular leukomalacia. BMC Neurosci. 17, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luciana M, 2003. Cognitive development in children born preterm: implications for theories of brain plasticity following early injury, Development and Psychopathology.15, 1017–1047 [DOI] [PubMed] [Google Scholar]
- Maureen Hack, Flannery Daniel J., Schluchter Mark, Cartar Lydia, Borawski Elaine, N.K., 2002. Outcomes in young adulthood for very-low-birth-weight infants. N. Engl. J. Med 346, 149–157. [DOI] [PubMed] [Google Scholar]
- Mei F, Fancy SPJ, Shen Y.-A. a, Niu J, Zhao C, Presley B, Miao E, Lee S, Mayoral SR, Redmond S. a, Etxeberria A, Xiao L, Franklin RJM, Green A, Hauser SL, Chan JR, 2014. Micropillar arrays as a high-throughput screening platform for therapeutics in multiple sclerosis. Nat. Med 20, 954–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei F, Lehmann-Horn K, Shen Y-AA, Rankin KA, Stebbins KJ, Lorrain DS, Pekarek K, A Sagan S, Xiao L, Teuscher C, von Büdingen H-C, Wess J, Lawrence JJ, Green AJ, Fancy SP, Zamvil SS, Chan JR, 2016a. Accelerated remyelination during inflammatory demyelination prevents axonal loss and improves functional recovery. Elife 5, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei F, Mayoral SR, Nobuta H, Wang F, Desponts C, Lorrain DS, Xiao L, Green AJ, Rowitch D, Whistler J, Chan JR, 2016b. Identification of the Kappa-Opioid Receptor as a Therapeutic Target for Oligodendrocyte Remyelination. J. Neurosci 36, 7925–7935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei F, Wang H, Liu S, Niu J, Wang L, He Y, Etxeberria A, Chan JR, Xiao L, 2013. Stage-Specific Deletion of Olig2 Conveys Opposing Functions on Differentiation and Maturation of Oligodendrocytes. J. Neurosci 33, 8454–8462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minelli A, Alonso-Nanclares L, Edwards RH, Defelipe J, Conti F, 2003. Postnatal development of the vesicular GABA transporter in rat cerebral cortex. Neuroscience 117, 337–346. [DOI] [PubMed] [Google Scholar]
- Misumi S, Ueda Y, Nishigaki R, Suzuki M, Ishida A, Jung C, Hida H, 2016. Dysfunction in Motor Coordination in Neonatal White Matter Injury Model without Apparent Neuron Loss. Cell Transplant. 25, 1381–1393. [DOI] [PubMed] [Google Scholar]
- Morrison BM, Lee Y, Rothstein JD, 2013. Oligodendroglia: metabolic supporters of axons. Trends Cell Biol. 23, 644–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nave K-A, 2010. Myelination and the trophic support of long axons. Nat. Rev. Neurosci 11, 275–283. [DOI] [PubMed] [Google Scholar]
- Ortega SB, Kong X, Venkataraman R, Savedra AM, Kernie SG, Stowe AM, Raman L, 2016. Perinatal chronic hypoxia induces cortical inflammation, hypomyelination, and peripheral myelin-specific T cell autoreactivity. J. Leukoc. Biol 99, 21–29. [DOI] [PubMed] [Google Scholar]
- Otellin VA, Khozhai LI, Shishko TT, 2014. Responses of Rat Brain Interneuronal Synapses to Hypoxia in the Early Neonatal Period. Neurosci. Behav. Physiol 44, 1082–1087. [Google Scholar]
- Pan S, Chan JR, 2017. Regulation and dysregulation of axon infrastructure by myelinating glia. J. Cell Biol 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicelli RC, Jawaid A, Henstridge CM, Spires-jones T, Schulz PE, Paolicelli RC, Jawaid A, Henstridge CM, Valeri A, Merlini M, Robinson JL, Lee EB, Rose J, Appel S, Lee VM, Trojanowski JQ, Spires-jones T, Schulz PE, Rajendran L, 2017. TDP-43 Depletion in Microglia Promotes Amyloid Clearance but Also Induces Synapse Loss Article TDP-43 Depletion in Microglia Promotes Amyloid Clearance but Also Induces Synapse Loss. Neuron 95, 297–308. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz JT, Christian CA, Parada I, Prince DA, Huguenard JR, 2010. Focal Cortical Infarcts Alter Intrinsic Excitability and Synaptic Excitation in the Reticular Thalamic Nucleus. J. Neurosci 30, 5465–5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piorkowska K, Thompson J, Nygard K, Matushewski B, Hammond R, Richardson B, 2014. Synaptic development and neuronal myelination are altered with growth restriction in fetal guinea pigs. Dev. Neurosci 36, 465–476. [DOI] [PubMed] [Google Scholar]
- Riddle A, Dean J, Buser JR, Gong X, Maire J, Chen K, Ahmad T, Cai V, Nguyen T, Kroenke CD, Hohimer AR, Back SA, 2011. Histopathological correlates of magnetic resonance imaging-defined chronic perinatal white matter injury. Ann. Neurol 70, 493–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumajogee P, Bregman T, Miller SP, Yager JY, Fehlings MG, 2016. Rodent Hypoxia-Ischemia Models for Cerebral Palsy Research: A Systematic Review. Front. Neurol. 7, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmaso N, Jablonska B, Scafidi J, Vaccarino FM, Gallo V, 2014. Neurobiology of premature brain injury. Nat. Neurosci 17, 341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarnat HB, Flores-Sarnat L, 2016. Synaptogenesis and Myelination in the Nucleus/Tractus Solitarius. J. Child Neurol 31, 722–732. doi: 10.1177/0883073815615227 [DOI] [PubMed] [Google Scholar]
- Sarnat HB, Philippart M, Flores-Sarnat L, Wei XC, 2015. Timing in Neural Maturation: Arrest, Delay, Precociousness, and Temporal Determination of Malformations. Pediatr. Neurol 52, 473–486. doi: 10.1016/j.pediatrneurol.2015.01.020 [DOI] [PubMed] [Google Scholar]
- Scafidi J, Hammond TR, Scafidi S, Ritter J, Jablonska B, Roncal M, Szigeti-Buck K, Coman D, Huang Y, McCarter RJ, Hyder F, Horvath TL, Gallo V, 2013. Intranasal epidermal growth factor treatment rescues neonatal brain injury. Nature 506, 230–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B, 2012. Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-Dependent Manner. Neuron 74, 691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segovia KN, McClure M, Moravec M, Luo NL, Wan Y, Gong X, Riddle A, Craig A, Struve J, Sherman LS, Back SA, 2008. Arrested oligodendrocyte lineage maturation in chronic perinatal white matter injury. Ann. Neurol 63, 520–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taveggia C, Feltri ML, Wrabetz L, 2010. Signals to promote myelin formation and repair. Nat. Rev 6, 276–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohda C, Nakanishi R, Kadowaki M, 2007. Learning deficits and agenesis of synapses and myelinated axons in phosphoinositide-3 kinase-deficient mice. NeuroSignals 15, 293–306. [DOI] [PubMed] [Google Scholar]
- Tomassy GS, Berger DR, Chen H-H, Kasthuri N, Hayworth KJ, Vercelli A, Seung HS, Lichtman JW, Arlotta P, 2014. Distinct Profiles of Myelin Distribution Along Single Axons of Pyramidal Neurons in the Neocortex. Science (80-.). 344, 319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu JC, Xiao B, Yuan JP, Lanahan AA, Leoffert K, Li M, Linden DJ, Worley PF, 1998. Homer binds a novel proline-rich motif and links group I metabotropic glutamate receptors with IP3 receptors. Neuron 21, 717–726. [DOI] [PubMed] [Google Scholar]
- Vainchtein ID, Chin G, Cho FS, Kelley KW, Miller JG, Chien EC, Liddelow SA, Nguyen PT, Nakao-Inoue H, Dorman LC, Akil O, Joshita S, Barres BA, Paz JT, Molofsky AB, Molofsky AV, 2018. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science (80-.). 359, 1269–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe JJ, 2001. The Developing Nervous System : A Series of Review Articles Neurobiology of Periventricular Leukomalacia in the Premature Infant. Pediatr. Res 50, 553–562. [DOI] [PubMed] [Google Scholar]
- Woodward LJ, Anderson PJ, Austin NC, Howard K, Inder TE, 2006. Neonatal MRI to predict neurodevelopmental outcomes in preterm infants. N. Engl. J. Med 355, 685–94. [DOI] [PubMed] [Google Scholar]
- Yue T, Xian K, Hurlock E, Xin M, Kernie SG, Parada LF, Lu QR, 2006. A critical role for dorsal progenitors in cortical myelination. J. Neurosci 26, 1275–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen TJ, Silbereis JC, Griveau A, Chang SM, Daneman R, Fancy SPJ, Zahed H, Maltepe E, Rowitch DH, 2014. Oligodendrocyte-Encoded HIF Function Couples Postnatal Myelination and White Matter Angiogenesis. Cell 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Choi G, Anderson DJ, 2001. The bHLH transcription factor Olig2 promotes oligodendrocyte differentiation in collaboration with Nkx2.2. Neuron 31, 791–807. [DOI] [PubMed] [Google Scholar]
- Zhu X, Hill R. a, Dietrich D, Komitova M, Suzuki R, Nishiyama A, 2011. Age-dependent fate and lineage restriction of single NG2 cells. Development 138, 745–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zonouzi M, Scafidi J, Li P, McEllin B, Edwards J, Dupree JL, Harvey L, Sun D, Hübner CA, Cull-Candy SG, Farrant M, Gallo V, 2015. GABAergic regulation of cerebellar NG2 cell development is altered in perinatal white matter injury. Nat. Neurosci 18, 674–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.