

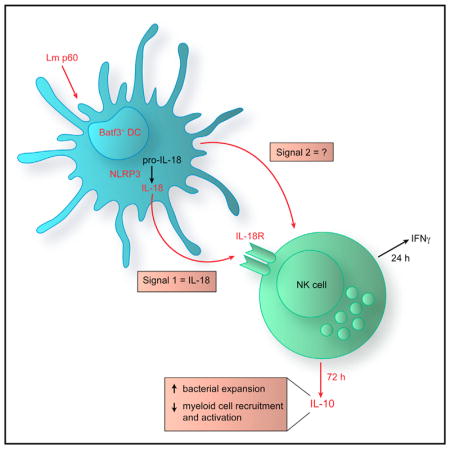
SUMMARY
The bacterial pathogen Listeria monocytogenes (Lm) capitalizes on natural killer (NK) cell production of regulatory interleukin (IL)-10 to establish severe systemic infections. Here, we identify regulators of this IL-10 secretion. We show that IL-18 signals to NK cells license their ability to produce IL-10. IL-18 acts independent of IL-12 and STAT4, which co-stimulate IFNγ secretion. Dendritic cell (DC) expression of Nlrp3 is required for IL-18 release in response to the Lm p60 virulence protein. Therefore, mice lacking Nlrp3, Il18, or Il18R fail to accumulate serum IL-10 and are highly resistant to systemic Lm infection. We further show that cells expressing or dependent on Batf3 are required for IL-18-inducing IL-10 production observed in infected mice. These findings explain how Il18 and Batf3 promote susceptibility to bacterial infection and demonstrate the ability of Lm to exploit NLRP3 for the promotion of regulatory NK cell activity.
In Brief
Clark et al. show that a bacterial pathogen suppresses protective immunity through an inflammasome-dependent pathway. Stimulation of NLRP3-dependent interleukin (IL)-18 release licenses NK cells to produce the anti-inflammatory molecule IL-10. Batf3-dependent cells are vital for IL-18 release, which drives this regulatory natural killer cell activity.
INTRODUCTION
Natural killer (NK) cells are a subset of type I innate lymphoid cells (ILCs) that respond to infection early after pathogen encounter and make important contributions to shaping the developing immune response (Vivier et al., 2011). NK cell activity is influenced by a combination of signals, including cell surface ligands, the cytokine milieu, and interactions with dendritic cells (DCs) (Cella, 2014; Lanier, 2008). Activated NK cells directly kill infected or cancerous cells and secrete diverse immune-regulatory factors, including the signature pro- and anti-inflammatory cytokines interferon γ (IFNγ) and interleukin-10 (IL-10). NK cell cytolytic activity and IFNγ production promote protective immunity during viral infections and in tumors; hence, strategies that boost these NK cell responses have direct clinical relevance (Knorr et al., 2014; Vivier et al., 2012). However, NK cell activation has deleterious effects on immune resistance in certain bacterial infection models (Kerr et al., 2005; Takada et al., 1994; Teixeira and Kaufmann, 1994). Recent work using a Listeria monocytogenes (Lm) infection model showed that the detrimental effects in this setting are dependent on NK cell production of IL-10, which suppresses accumulation and antimicrobial effector functions of inflammatory myeloid cell populations (Clark et al., 2016). IL-10 production is exploited by diverse microbial pathogens (Cyktor and Turner, 2011). However, the signals required to induce NK cell IL-10 production during bacterial infection remain undefined. One prior study identified DC secretion of IL-12 as critical for NK cell IL-10 in a murine model of Toxoplasma gondii infection (Perona-Wright et al., 2009). It has not been determined whether IL-12 contributes to NK cell IL-10 production during bacterial infections.
Lm is a bacterial pathogen responsible for foodborne human infections ranging from acute gastroenteritis to bacteremia, meningitis, and miscarriages (Hof, 2003). Systemic Lm infections are most commonly reported in elderly, immune-compromised, and pregnant individuals (Swaminathan and Gerner-Smidt, 2007). The basis for the increased susceptibility in these populations remains unclear. However, in murine models, the production of IL-10 by NK cells profoundly increases host susceptibility (Clark et al., 2016). NK cells are activated early after systemic Lm infection and are a major source of initial IFNγ (Humann et al., 2007; Kang et al., 2008). The signaling requirements for NK cell IFNγ secretion in response to Lm are well defined and include direct contact with DCs and local secretion of IL-12 and IL-18 (Humann and Lenz, 2010; Lochner et al., 2008). IL-18 was originally identified as an IFNγ-inducing factor that co-stimulates Th1-type inflammatory responses (Okamura et al., 1995). IL-18 is synthesized as an inactive pro-cytokine whose secretion and biological activity require proteolytic cleavage by one of several multi-molecular complexes termed “inflammasomes.” Inflammasomes contain the protease caspase-1, the ASC adaptor protein, and one of several different sensor molecules (Broz and Dixit, 2016). In cultured macrophages, Lm elicits IL-18 release through activation of inflammasome sensors, including NLRP3 (Hagar and Miao, 2014; Kim et al., 2010; Wu et al., 2010). Here we examine the effect of NLRP3 expression on cytokine secretion and susceptibility during in vivo Lm infection.
Lm expression of the secreted p60 protein has been shown to promote NK cell IFNγ production during systemic infection (Clark et al., 2016; Humann et al., 2007). When modeled in vitro, Lm expression of p60 increases secretion of IFNγ from NK cells co-cultured with infected bone marrow-derived DCs (BMDCs) (Schmidt et al., 2011). Treatment of BMDCs or human monocyte-derived DCs with recombinant p60 protein was also shown to promote activation of co-cultured NK cells (Clark et al., 2016; Schmidt et al., 2011). The recombinant p60 protein binds mouse BMDCs and was observed to stimulate NLRP3-dependent release of IL-1β and IL-18 (Schmidt and Lenz, 2012). These stimulatory effects of p60 map to the “L1S” region of the protein, and a recombinant protein containing just this region retains these activities (Clark et al., 2016; Schmidt et al., 2011; Schmidt and Lenz, 2012). BMDCs infected with Lm or treated with L1S require direct interaction with NK cells in addition to secretion of IL-12 and IL-18 to elicit NK cell IFNγ secretion (Schmidt et al., 2011). The mechanisms by which Lm infection stimulates NK cell IL-10 production have not been previously investigated. Thus, it remains unknown whether IFNγ and IL-10 production might be induced by different signals to NK cells.
In the current study, we identify IL-18 as a crucial factor that acts directly on NK cells to license their production of IL-10 during systemic Lm infection. We show that this response is not dependent on IL-12 or other activators of STAT4. We further reveal that NLRP3 and the basic leucine zipper ATF-like transcription factor 3 (Batf3) are crucial regulators of DC IL-18 secretion in response to Lm/p60. Thus, NK cell IL-10 production fails to occur during systemic infection of mice lacking Il18, Il18R, Nlrp3, or Batf3, and each of these strains is highly resistant to systemic Lm infection. These results demonstrate a host-detrimental role for NLRP3 and provide a mechanism to explain host-detrimental effects of Batf3, CD8α+ DCs, and IL-18 in the Lm infection model. Our results further show that Lm p60 selectively targets Batf3-dependent cells to ensure that sufficient IL-18 is produced to license NK cell IL-10 production that dampens host inflammatory and anti-microbial host responses.
RESULTS
Soluble Factors from BMDCs Suffice to License NK Cell IL-10 Production
To investigate the requirements for Lm-induced NK cell IL-10 production, we used an established cell co-culture model (Figure 1A). BMDCs from B6.Il10−/− mice were infected with live wild-type (WT) or p60-deficient (Δp60) Lm strains. As reported previously, WT and Δp60 Lm infect and replicate similarly in mammalian cells (Lenz et al., 2003; Schmidt et al., 2011). Alternatively, BMDCs were treated with a priming agent (lipopolysaccharide [LPS]) plus recombinant L1S protein, which is derived from the N terminus of the Lm p60 virulence protein. Purified splenic NK cells were either added to the BMDCs (“co-culture”) or cultured with filtered supernatants harvested 1 hr after BMDC treatment (“supernatant transfer”). NK cell culture supernatants were analyzed 24–72 hr later for IFNγ and IL-10. As previously shown for NK cells responding during systemic Lm infection (Clark et al., 2016), IFNγ was detected in co-cultures by 24 hr, whereas IL-10 accumulated at later time points (Figure 1B). Although IFNγ persisted in the supernatants of the co-cultures, few NK cells produced this cytokine after 24 hr (Figure 1C). Both IFNγ and IL-10 production from NK cells was greatly reduced when BMDCs were infected with p60-deficient (Δp60) Lm, confirming the important role of this Lm factor for the induction of NK cell activity. IFNγ production by NK cells during Lm infection is known to require both soluble factors released from infected accessory cells (e.g., DCs) and direct contact with these cells (Humann and Lenz, 2010; Kang et al., 2008; Schmidt et al., 2011). The requirement for contact is thought to reflect the need to form a synapse between mature dendritic cells and NK cells to direct cytokines and other signals necessary to trigger NK cell IFNγ secretion (Borg et al., 2004). Consistent with this interpretation, very little IFNγ was produced when NK cells were separated from the BMDCs using a transwell membrane or cultured with filtered supernatants from L1S+LPS-stimulated BMDCs (Figure 1D). In contrast, significant IL-10 production was still observed in cultures where NK cells and BMDCs were separated (Figure 1E). B6.Il10−/− BMDCs were used in these experiments; thus, NK cells were the only possible source of IL-10. IL-10 was produced by NK cells cultured with BMDC supernatants that were collected and filtered as early as 1 hr after stimulation with L1S+LPS (Figure 1E). BMDC-derived factors were required for this response because NK cells cultured in medium or treated directly with L1S+LPS failed to produce IL-10 or IFNγ (Figure S1). Also, supernatants from BMDCs stimulated with LPS alone were insufficient to induce NK cell IFNγ or IL-10 (Figures S1A and S1B). Together, these data indicate that L1S triggers the release of soluble factors from BMDCs that suffice to license IL-10 (but not IFNγ) secretion from NK cells in the absence of synapse formation.
Figure 1. Soluble Factors from BMDCs Suffice to License NK Cell IL-10 Production.

(A) Schematic of the in vitro co-culture and supernatant transfer systems.
(B) Supernatant IFNγ and IL-10 detected 24, 48, and 72 hr after NK cell co-culture with L1S+LPS-stimulated or Lm-infected B6.Il10−/− BMDCs (n = 3 independent experiments pooled).
(C) Intracellular IFNγ produced by NK1.1+CD3− cells 24 hr and 72 hr after co-culture (representative of n = 3 experiments).
(D and E) Supernatant cytokines detected 24 hr (IFNγ, D) or 72 hr (IL-10, E) following NK cell exposure to L1S+LPS-stimulated B6.Il10−/− BMDCs in co-culture with or without separation with a 0.4 μM transwell insert or exposed to filtered supernatants collected 1 hr post-stimulation from BMDCs, as indicated (n = 3 independent experiments pooled).
Data are displayed as mean ± SEM; *p < 0.05 and ***p < 0.001 as measured by t test.
NLRP3 Regulates NK Cell IL-10 Production and Increases Susceptibility to Lm
L1S+LPS treatment of BMDCs was shown previously to activate NLRP3-dependent release of IL-1β and IL-18 (Schmidt and Lenz, 2012). We therefore asked whether NLRP3 inflammasome activity was required for release of IL-10-inducing factors from stimulated BMDCs. NK cells purified from spleens of B6 mice were exposed to supernatants harvested 1 hr after stimulation of WT B6 or B6.Nlrp3−/− BMDCs with or without L1S+LPS. Supernatants from L1S+LPS (but not mock) stimulated B6 BMDCs induced significant IL-10 production in 72-hr NK cell cultures (Figure 2A). IL-10 production in NK cell cultures exposed to supernatants from B6 BMDCs was similar to that from B6.Il10−/− BMDCs (compare with Figure 1E). Thus, the capacity of BMDCs to produce IL-10 did not alter their ability to stimulate NK cell IL-10. The 1-hr stimulation was not sufficient to induce release of IL-10 into BMDC supernatants (Figure S2A), indicating that NK cells were the source of IL-10 in these experiments. In contrast to B6 BMDC supernatants, those collected from L1S+LPS-stimulated B6.Nlrp3−/− BMDCs failed to elicit NK cell IL-10 production (Figure 2A). Supernatants from Lm-infected B6.Nlrp3−/− BMDCs also lacked the ability to induce NK cell IL-10 production (Figure 2B). In addition, B6.Nlrp3−/− BMDCs treated with L1S+LPS or infected with Lm were very poor at inducing IFNγ production by co-cultured NK cells (Figures S2B and S2C).
Figure 2. NLRP3 Regulates NK Cell IL-10 Production and Increases Susceptibility to Lm.

(A and B) Supernatant IL-10 detected from NK cell cultures 72 hr after exposure to filtered supernatants from 1 hr L1S+LPS stimulation (A) or WT or Δp60 Lm infection (B) of B6 or B6.Nlrp3−/− BMDCs (n = 3 independent experiments pooled).(C and D) Serum IFNγ (C) and Lm burdens shown as colony-forming units (CFUs) per organ (D) from B6 or B6.Nlrp3−/− mice sacrificed 24 hpi with 104 Lm i.v.
(E and F) Serum IL-10 (E) and Lm burdens per organ (F) from B6 or B6.Nlrp3−/− mice sacrificed 72 hpi (n = 3 independent experiments pooled with 3–5 mice per group for in vivo experiments).
Data are displayed as mean ± SEM; *p < 0.05 and ***p < 0.001 as measured by t test.
To further evaluate the requirements for NLRP3 in the regulation of in vivo NK cell activation, we quantified serum IFNγ and IL-10 production 24 and 72 hr after systemic (intravenous [i.v.]) infection with 104 Lm of B6 and B6.Nlrp3−/− mice. In this model, serum IFNγ at 24 hr post-infection (hpi) is derived from both NK and T cells (Schmidt et al., 2011), whereas IL-10 at 72 hr is entirely dependent on NK cells (Clark et al., 2016). Compared with infected WT mice, serum IFNγ was significantly reduced, but still detectable, in infected B6.Nlrp3−/− mice (Figure 2C). Consistent with these results, the proportion of intracellular IFNγ+ NK cells in the spleens of infected B6.Nlrp3−/− was significantly reduced, whereas IFNγ+ T cells were unaffected (Figure S2D). Reduced NK cell IFNγ was not attributable to differences in bacterial burdens because these were equivalent in WT and B6.Nlrp3−/− mice at 24 hpi (Figure 2D). At 72 hpi, we observed an almost complete absence of serum IL-10 in the infected B6.Nlrp3−/− mice (Figure 2E). At this time point, we further observed significantly reduced Lm burdens in the spleens and livers of B6.Nlrp3−/− mice (Figure 2F). NK cells are responsible for this IL-10 production in WT B6 mice, permitting robust Lm replication during systemic infection (Clark et al., 2016). These data implicate NLRP3 as a key factor regulating the activation of NK cells during systemic Lm infection and show that the net effect of Nlrp3 expression in this model is the promotion of NK cell IL-10 production, resulting in increased host susceptibility.
NLRP3 Regulates IL-18 Release Required for NK Cell IL-10 Production in Response to Lm or L1S+LPS
The known ability of IL-18 and IL-12 to regulate NK cell IFNγ production (Fehniger et al., 1999; Tomura et al., 1998) suggested that NLRP3 might affect NK cell activity by regulating the release of IL-18. To address whether IL-18 could promote NK cell IL-10 production, we first confirmed that treatment with L1S+LPS stimulated release of IL-18 from B6 BMDCs (Figure 3A). These experiments further showed that B6.nlrp3−/− BMDCs fail to release IL-18 in response to LPS+L1S. BMDC expression of NLRP3 and Lm expression of p60 were furthermore required for secretion of IL-18 in response to Lm infection of BMDCs (Figure 3B). We next evaluated the profile of IL-18 production in B6.Nlrp3−/− mice during systemic Lm infection. In infected B6 mice, we observed that serum IL-18 concentrations increased in parallel with those of serum IL-10 (Figure 3C). Compared with B6 mice infected in parallel, B6.Nlrp3−/− mice had significantly reduced serum IL-18 at 72 hpi (Figure 3D). These data suggest that NLRP3 expression regulates the release and accumulation of mature IL-18 in the sera of Lm-infected mice.
Figure 3. NLRP3 Regulates IL-18 Release Required for NK Cell IL-10 Production in Response to Lm or L1S+LPS.

(A and B) Supernatant IL-18 detected at the indicated times post-stimulation with L1S+LPS (A) or infection with WT or Δp60 Lm (B) of B6 or B6.Nlrp3−/− BMDCs (n = 3 independent experiments pooled).
(C) Serum IL-10 and IL-18 detected in uninfected (naive) or Lm-infected (104 i.v.) B6 mice at the indicated time points.
(D) Serum IL-18 detected in B6 or B6.Nlrp3−/− mice sacrificed 72 hpi.
(E and F) Serum IL-10 (E) and Lm burdens per organ (F) from B6 or B6.Il18−/− mice sacrificed 72 hpi (n = 3 independent experiments pooled with 3–5 mice per group for in vivo experiments).
(G and H) Supernatant IL-10 detected in NK cell cultures 72 hr after exposure to filtered supernatants from 1 hr L1S+LPS stimulation (G) or WT or Δp60 Lm infection (H) of B6 or B6.Il18−/− BMDCs with or without 50 pg/mL rIL-18 added to NK cell cultures.
(I and J) Supernatant IL-10 detected in NK cell cultures 72 hr after exposure to filtered supernatants from 1 hr L1S+LPS stimulation (I) or WT or Δp60 Lm infection (J) of B6 or B6.Nlrp3−/− BMDCs + 50 pg/mL rIL-18 added to NK cell cultures (n = 3 independent experiments pooled for in vitro experiments).
Data are displayed as mean ± SEM; *p < 0.05, **p < 0.01, and ***p < 0.001 as measured by t test.
Consistent with a critical role for IL-18 in regulation of NK cell IL-10 production, we further observed that, at 72 hpi, serum IL-10 concentrations (Figure 3E) and bacterial burdens (Figure 3F) were significantly lower in B6.Il18−/− versus control B6 mice. Lm burdens were not significantly different in B6 and B6.Il18−/− mice at 24 hpi despite reduced serum IFNγ in the latter (Figure S3A). These data suggest that impaired induction of NK cell IL-10 is responsible for the previously reported resistance of Il18−/− animals (Lochner et al., 2008). Cell culture experiments further demonstrated that 1-hr supernatants from L1S+LPS-stimulated (Figure 3G) or Lm-infected (Figure 3H) B6.Il18−/− BMDCs did not support NK cell IL-10 production. In contrast, B6 and B6.Il18−/− NK cells secreted equivalent levels of IL-10 in response to supernatants from L1S+LPS-stimuated B6 BMDCs (Figure S3B). Importantly, recombinant IL-18 restored the ability of supernatants from B6.Il18−/− BMDCs to promote NK cell IL-10 production (Figures 3G and 3H). However, the addition of recombinant IL-18 alone did not induce NK cell IL-10 production in the absence of conditioned BMDC supernatants. These data demonstrate that BMDC release of IL-18 and an additional factor(s) together elicit IL-10 production by NK cells. To more directly assess whether this additional factor(s) might also be dependent on NLRP3, B6 or B6.Nlrp3−/− BMDCs were stimulated with L1S+LPS or infected with Lm. Supernatants were harvested at 1 hr, filtered, and added to purified NK cells in the presence of recombinant IL-18. The results indicated that IL-18 fully complemented the ability of stimulated B6.Nlrp3−/− BMDC supernatants to induce NK cell IL-10 production (Figures 3I and 3J). IL-18 also rescued NK cell IFNγ production in co-cultures with L1S+LPS-stimulated or Lm-infected B6.Nlrp3−/− BMDCs (Figures S3C and S3D). Unlike L1S+LPS, activation of BMDC NLRP3 using ATP or alum with or without LPS did not support NK cell IL-10 production (Figures S3E and S3F). These data show that NLRP3 regulation of NK cell IL-10 production is due to its ability to facilitate IL-18 release, although additional NLRP3-independent factors are also required for the activation of NK cell IL-10 in response to Lm or L1S+LPS.
IL-18 Acts on NK Cells to License IL-10 Secretion Independent of IL-12/STAT4 Signaling
To address whether IL-18 must directly act on NK cells to promote their production of IL-10, we first evaluated cell surface IL-18R1 staining on NK cells from tissues of Lm-infected B6.tiger mice, which carry an IL-10 GFP reporter (Kamanaka et al., 2006). Essentially all of the GFP+ cells observed at 72 hpi were found to co-stain positively for cell surface IL-18R1 (Figure 4A). We therefore studied the effects of Il18r1 deficiency. B6 and B6.Il18r−/− mice were infected with 104 Lm i.v. and evaluated 72 hr later. As for B6.Il18−/− and B6.Nlrp3−/− mice, the absence of IL-18R1 was associated with loss of serum IL-10 and significantly reduced bacterial burdens at 72 hpi (Figure 4B). Next, B6.Il10−/− BMDCs were infected with WT Lm or treated with L1S+LPS and cultured with purified splenic B6 or B6.Il18r1−/− NK cells. In contrast to co-cultures with B6 NK cells, no IL-10 was produced in cultures containing only B6.Il18r1−/− NK cells (Figure 4C). Expression of IL-18R1 by NK cells was also required for IL-10 production in response to filtered supernatants from L1S+LPS-stimulated B6.Il10−/− BMDCs (Figure 4D). Thus, IL-18 acts on NK cells, rather than BMDCs, to license NK cell production of IL-10.
Figure 4. IL-18 Acts on NK Cells to License IL-10 Secretion Independent of IL-12/STAT4 Signaling.

(A) Percentage of NK1.1+CD3− cells from the spleen, liver, and blood positive for IL-18R1 expression from IL-10 GFP+ and IL-10 GFP− populations at the indicated time points with 104 Lm i.v. in IL-10 GFP reporter mice (representative of n = 3 experiments with 3–5 mice per group).
(B) Serum IL-10 and Lm burdens per liver from B6 or B6.Il18R−/− mice sacrificed 72 hpi (n = 3 independent experiments pooled with 3–5 mice per group).
(C) Supernatant IL-10 detected in B6 or B6.Il18R−/− NK cells 72 hr after co-culture with B6.Il10−/− BMDCs stimulated with L1S+LPS or infected with Lm.
(D) Supernatant IL-10 detected from B6 or B6.Il18R−/− NK cells 72 hr after co-culture with or exposure to filtered supernatants from B6.Il10−/− BMDCs stimulated with L1S+LPS for 1 hr.
(E) Supernatant IFNγ detected in B6 or B6.Il18R−/− NK cells 24 hr after co-culture with B6 BMDCs stimulated with L1S+LPS.
(F and G) Supernatant cytokines detected 24 hr (IFNγ, F) or 72 hr (IL-10, G) in NK cells in co-culture with B6.Il10−/− BMDCs stimulated with L1S+LPS with or without 1 μg/mL anti-IL-12p70 or 50 μg/mL anti-IL-12R added with NK cells to co-cultures.
(H) Supernatant IL-10 detected 72 hr after stimulation of NK cells with 50 pg/mL rIL-12 + 50 pg/mL rIL-2 with or without 80 μM STAT4 inhibitor lisofylline.
(I) Supernatant IL-10 detected at 72 hr from NK cells in co-culture with B6.Il10−/− BMDCs stimulated with L1S+LPS or infected with Lm with or without 80 μM STAT4 inhibitor lisofylline added with NK cells to co-cultures.
(J) Supernatant IL-10 detected in B6 or B6.Il18R−/− NK cells 72 hr post-stimulation with 50 pg/mL rIL-12 + 50 pg/mL rIL-2 (n = 3 independent experiments pooled for in vitro experiments).
Data are displayed as mean ± SEM; *p < 0.05 and ***p < 0.001 as measured by t test.
It is well established that IL-18 signaling synergizes with IL-12 to promote NK cell secretion of IFNγ (Chaix et al., 2008; Fehniger et al., 1999). Consequently, little IFNγ was detected in L1S+LPS-stimulated co cultures of B6.Il10−/− BMDCs with B6.Il18r1−/− NK cells (Figure 4E). IL-12 was also required for IFNγ production in L1S+LPS-stimulated co-cultures of B6.Il10−/− BMDC and B6 NK cells because antibody blockade of IL-12p70 or IL-12R prevented IFNγ production (Figure 4F). However, blockade of IL-12/ IL-12R did not prevent similarly treated NK cells from secreting IL-10 (Figure 4G). Thus, IL-12 selectively co-stimulates IFNγ, but not IL-10, production by NK cells. This result demonstrated an additional distinction in the regulation of NK cell IFNγ and IL-10 secretion but was somewhat surprising given prior results that IL-12 and STAT4 signaling induces IL-10 production by cultured NK cells and NK cells from parasite-infected mice (Grant et al., 2008; Perona-Wright et al., 2009). We thus confirmed that culture of purified B6 NK cells with recombinant IL-12 and IL-2 elicited IL-10 secretion and that this was inhibited by treatment with the STAT4 inhibitor lisofylline (Figure 4H). However, when co-cultured NK cells were treated with lisofylline, they retained the ability to produce IL-10 in response to both L1S+LPS-stimulated and Lm-infected BMDCs (Figure 4I). Thus, neither IL-12-nor other STAT4-activating factors were required for LPS+L1S-elicited IL-10 production by NK cells. Moreover, although NK cell IL-10 production in response to Lm infection or L1S+LPS treatment of BMDCs requires IL-18, as shown above, we found that IL-12 stimulation readily induced IL-10 production by cultured B6.Il18r1−/− NK cells (Figure 4J). These results show that IL-18 signaling to NK cells is critical for their production of IL-10 during Lm infection and in response to L1S+LPS stimulation of BMDCs. They further show that NK cell IFNγ and IL-10 secretion are differentially dependent on IL-12 and STAT4.
Batf3 Expression Licenses DC IL-18 Production in Response to L1S+LPS
To determine whether, like BMDCs, primary DCs respond to L1S+LPS stimulation, positive selection with magnetic beads was used to enrich CD11c+ cells from spleens of B6 mice (Figure 5A). When isolated CD11c+ cells and CD11c-depleted splenocytes were assayed for responsiveness to L1S+LPS, we observed that only splenic CD11c+ cells secreted IL-18 (Figure 5B). Approximately 20% of the CD11c+ cells isolated in this manner co-stained positively for CD8α (Figure 5C). The transcription factor Batf3 promotes development of the CD8α+ DC population (Hildner et al., 2008); thus, we asked how loss of Batf3 expression affects the responsiveness of CD11c+ splenocytes to L1S+LPS stimulation. Following a 24-hr stimulation, the quantity of IL-18 released from B6.Batf3−/− CD11c+ splenocytes was found to be significantly less than that from control B6 cells despite comparable secretion of LPS-induced IL-6 (Figure 5D). These data indicate that Batf3-deficient cells selectively fail to release IL-18 in response to L1S stimulation. Consistent with the absence of IL-18, B6 NK cells failed to secrete IFNγ or IL-10 when co-cultured or treated with supernatants from L1S+LPS-stimulated B6.Batf3−/− CD11c+ cells (Figures 5E and 5F). Some splenic CD11c+ cells purified from Batf3−/− spleens remained positive for CD8α, as reported by others (Figure S4A; Edelson et al., 2011). Furthermore, Il18 and nlpr3 transcript abundance and pro-IL-18 and NLRP3 protein amounts were similar in purified CD11c+ splenocytes from B6 and B6.Batf3−/− mice, indicating a defect in IL-18 secretion rather than expression (Figures S4B and S4C). Batf3 expression was also required for licensing of IL-12 production by CD11c+, but not CD11c−, spleen cells (Figures 5G and 5H). These data argue that Batf3 expression is not essential for responsiveness to LPS or for expression of NLRP3 or IL-18, but selectively licenses the ability of CD11c+ splenocytes to release soluble factors, including IL-18, that are essential for NK cell IFNγ and IL-10 production.
Figure 5. Batf3 Expression Licenses DC IL-18 Production in Response to L1S+LPS.

(A) Schematic of CD11c+ cell stimulation in vitro.
(B) Supernatant IL-18 detected from CD11c+/− cells 24 hr post-stimulation with L1S+LPS (n = 3 independent experiments pooled).
(C) CD8α+CD11c+ cells detected by flow cytometry from the B220−CD3−CD11c+ gate of purified CD11c+ cells (representative of n = 3 experiments).
(D) Supernatant IL-18 (left) and IL-6 (right) detected in CD11c+ cells purified from B6 or B6.Batf3−/− mice 24 hr after stimulation with L1S+LPS.
(E and F) Supernatant cytokines detected following co-culture of NK cells with B6 or B6.Batf3−/− CD11c+ cells 24 hr after stimulation with L1S+LPS (IFNγ, E) or 72 hr following NK cell exposure to filtered supernatants from B6 or B6.Batf3−/− CD11c+ cells stimulated with L1S+LPS for 1 hr (IL-10, F).
(G and H) Supernatant IL-12p70 detected 24 hr after stimulation of B6 or B6.Batf3−/− CD11c+ (G) or CD11c− (H) cells with L1S+LPS (n = 3 independent experiments pooled for in vitro experiments).
Data are displayed as mean ± SEM; *p < 0.05 and ***p < 0.001 as measured by t test.
Batf3 Expression Promotes Bacterial Expansion during Systemic Lm Infection by Licensing NK Cell IL-10 Secretion
To further evaluate the effect of Batf3 on NK cell activity in vivo, B6 and B6.Batf3−/− mice were infected i.v. with 104 Lm. Equivalent Lm burdens were recovered from tissues of the infected mice at 24 hpi (Figure 6A). However, significantly reduced Lm burdens were recovered from the spleens and livers of B6.Batf3−/− mice at 72 hpi (Figure 6B). Indeed, Lm burdens increased ~1,000-fold in B6 mice between 24 and 72 hpi, whereas those in B6.Batf3−/− mice remained unchanged. These results suggest that bacteria seed target organs normally but that, in the absence of Batf3, their survival and/or growth is subsequently impaired. Similar phenotypes are seen in Nlrp3−/−, Il18−/−, and Il18r−/− mice (above) and in mice depleted of NK cells prior to infection (Clark et al., 2016), all of which demonstrate loss of NK cell activation. Consistent with a loss of NK cell activity in infected B6.Batf3−/− mice, serum IFNγ was significantly lower at 24 hpi (Figure 6C), and little or no serum IL-10 was detected at 72 hpi (Figure 6D). These results show that Batf3 expression is critical for in vivo activation of NK cells, contributing to early bacterial expansion during systemic Lm infection.
Figure 6. Batf3 Expression Promotes Bacterial Expansion during Systemic Lm Infection by Licensing NK Cell IL-10 Secretion.

(A and B) Lm burdens per organ of B6 or B6.Batf3−/− mice harvested at 24 hpi (A) or 72 hpi (B) with 104 Lm i.v.
(C and D) Serum cytokines detected from B6 or B6.Batf3−/− mice harvested at 24 hpi (IFNγ, C) or 72 hpi (IL-10, D).
(E) Schematic of NK cell adoptive transfer.
(F) CD3−NK1.1+CD45.2+ donor cells (or [−] = no transfer) detected by flow cytometry from the spleens of B6.Il10−/− recipient mice at 96 hpi.
(G and H) Total spleen homogenate IL-10 (G) and Lm burdens per livers (H) detected in B6.Il10−/− recipients at 96 hpi. NT, no transfer.
Data were pooled from n = 3 independent experiments with 3–5 mice per group. Data are displayed as mean ± SEM; *p < 0.05 and ***p < 0.001 as measured by t test (B–D) or ANOVA (G and H). See also Figure S5.
We considered two possible explanations for the defective NK cell response in the infected B6.Nlrp3−/− mice. Either NK cells failed to develop properly in a host lacking Batf3, or, as suggested by our cell culture experiments above, Batf3 expression licensed the production of IL-18 and/or other factors critical for stimulation of NK cell activity during Lm infection. To distinguish between these possibilities, we first asked whether B6.Batf3−/− NK cells were capable of responding to Lm infection and producing IL-10 during systemic infection. Here naive splenic NK cells from CD45.2+ B6 or B6.Batf3−/− mice were purified and transferred into CD45.1+ B6.Il10−/− recipients 24 hpi (Figure 6E). In mice given CD45.2+ NK cells, a small population of the donor cells was detected upon harvest at 96 hpi (Figure 6F). Further, elevated IL-10 was detected in spleen homogenates from B6.Il10−/− mice that received either type of donor NK cell (Figure 6G). This NK cell-dependent IL-10 production was further confirmed to increase Lm burdens in the B6.Il10−/− recipients (Figure 6H). As we showed previously (Clark et al., 2016), NK cell IL-10 production and increased Lm burdens were associated with reduced accumulation of inflammatory myeloid cells in Lm-infected spleens. Similarly, we observed increased recruitment of inflammatory myeloid cells to the spleens of IL-10-deficient mice at 96 hpi with 104 Lm compared with IL-10-deficient recipients of donor WT B6 or B6.Batf3−/− NK cells (Figure S5). These data argue that NK cells develop normally in B6.Batf3−/− mice and that Batf3 expression by a non-NK cell population is required to activate NK cell production of IL-10, which suppresses inflammatory myeloid cell recruitment and increases Lm burdens.
Batf3-Dependent Cells Are a Vital Source of IL-18 that Regulates NK Cell IL-10 Responses during Lm Infection
The data above indicated that Batf3 expression by CD11c+ cells regulates their production of IL-12 and IL-18 and their ability to stimulate NK cell activity. Batf3 and IL-18 were likewise required for NK cell activation during systemic infection. However, because IL-18 can be produced by multiple cell types, it remained unclear whether Batf3+ cells were an essential source of IL-18 production during systemic Lm infection. To evaluate this, we constructed and infected a series of bone marrow chimeras (Figure 7A). Lethally irradiated B6.Ptprca (CD45.1) mice were reconstituted with bone marrow (BM) from WT or mutant B6 mice. Initially, reciprocal chimeras were constructed in which irradiated B6 or B6.Il18−/− mice were reconstituted with BM from the opposite strain. These chimeric animals were infected with 104 Lm and analyzed at 72 hpi. At the time of harvest, mice in which only hematopoietic cells could produce IL-18 supported ~100 times higher bacterial burdens than mice where only non-hematopoietic cells could produce IL-18 (Figure 7B). Infected mice whose hematopoietic cells were Il18−/− also failed to accumulate serum IL-18 (Figure 7C). These results showed that hematopoietic cells are the major source of detectable IL-18 during systemic Lm infection and that their IL-18 production is required for increasing host susceptibility.
Figure 7. Batf3-Dependent Cells Are a Vital Source of IL-18 that Regulates NK Cell IL-10 Responses during Lm Infection.

(A) Schematic of the bone marrow (BM) chimera experiment.
(B and C) Bacterial burdens per liver (B) and serum IL-18 (C) in lethally irradiated B6 versus B6.Il18−/− recipients of B6.Il18−/− or B6 BM, respectively, harvested at 72 hpi with 104 Lm i.v. (n = 2 independent experiments with 3–5 mice per group).
(D and E) Serum IL-18 (D) and IL-10 (E) detected in lethally irradiated BM recipients at 72 hpi.
(F) Lm burdens per the livers of BM recipients at 72 hpi (data pooled from n = 2 independent experiments with 3–5 mice per group, repeated a third time with similar results).
Data are displayed as mean ± SEM; *p < 0.05 and **p < 0.01 as measured by t test (B and C) or ANOVA (D–F).
Subsequently, irradiated B6.Ptprca mice reconstituted with B6.Il18−/− BM were compared directly with those reconstituted with WT B6 BM, B6.Batf3−/− BM, or a mixture of BM from each source. Following Lm infection, mice reconstituted with donor BM from WT mice produced ample IL-18 (Figure 7D). By comparison, little or no IL-18 was observed in sera from infected mice reconstituted individually with B6.Il18−/− or B6.Batf3−/− BM. These data are consistent with the results above and further indicate that hematopoietic cells require Batf3 to license their production of systemic IL-18. Strikingly, mice reconstituted with 1:1 mixtures of donor BM from B6.Il18−/− and B6.Batf3−/− mice (in which batf3-dependent cells develop but are the only cell type that cannot produce IL-18) also failed to produce substantial IL-18 in response to the 72-hr Lm infection (Figure 7D). By comparison, when WT hematopoietic cells were present, serum IL-18 was present at WT concentrations regardless of the presence of B6.Il18−/− or B6.Batf3−/− cells. This result indicated that cells lacking Batf3 or IL-18 did not suppress IL-18 production by neighboring cells. Further, it showed that an ~50% reduction in the population of IL-18-producing hematopoietic cells did not significantly reduce serum IL-18 during the infection. Consistent with our data above implicating IL-18 as a key regulator of NK cell IL-10 production during Lm infection, we further found that serum IL-10 concentrations paralleled those of IL-18 in the various chimeric animals (Figure 7E). The amounts of IL-18 and IL-10 detected in the sera of chimeric animals were also found to correlate with differences in Lm burdens (Figure 7D). Hence, we conclude that hematopoietic cells are a critical source the IL-18 required to license NK cell IL-10 secretion, increasing bacterial burdens during systemic Lm infection, and that cell-intrinsic expression of Batf3 is vital for the development and/or activity of IL-18-secreting cells that respond to Lm.
DISCUSSION
IL-10 production potently affects inflammation, infection, cancer, and autoimmune diseases. Lm is one of several pathogens known to benefit from early host IL-10 production, and we recently showed that NK cells are a key source of this IL-10 (Clark et al., 2016). We now identify a critical host pathway required for this bacterially induced NK cell IL-10 secretion. Our data show that Lm infection or stimulation with a recombinant fragment of the Lm p60 protein (L1S) induces release of IL-18 from DCs. IL-18 acts directly on NK cell IL-18R in conjunction with a currently unknown second factor to promote IL-10 secretion. We further identify Batf3 and NLRP3 as essential regulators of this NK cell-stimulatory IL-18. These findings suggest that Lm targets a Batf3-dependent cell population to induce NLRP3-dependent IL-18 secretion, which promotes an immune-suppressive response mediated by NK cell IL-10 production.
Our studies reveal that host NLRP3 plays a key role in the regulation of the IL-18 production critical for NK cell IL-10 secretion and increased susceptibility during Lm infection. NLRP3 is a sensor component of a multi-protein complex termed the inflammasome. Inflammasomes regulate the proteolytic cleavage of pro-IL-18 and release of the mature biologically active protein, although inflammasome-independent mechanisms can also contribute to IL-18 processing (Broz and Dixit, 2016; Dinarello et al., 2013). Although crucial for accumulation of serum IL-18 and IL-10 at 72 hpi, NLRP3 was only partly required for serum IFNγ at 24 hpi. However, IL-18 was required for both responses. This discrepancy suggests that there is NLRP3-independent IL-18 production early after Lm infection. Indeed, the Lm proteins LLO and flagellin and bacterial DNA have previously been implicated in activation of NLRP3, NLRC4, and AIM2 inflammasomes in cultured macrophages (Meixenberger et al., 2010; Warren et al., 2008; Wu et al., 2010). The increased resistance of Nlrp3−/− mice was somewhat surprising given that they are more susceptible to infections by Citrobacter rodentium and group B Streptococcus (Costa et al., 2012; Liu et al., 2012). Evasion of NLRP3 or other inflammasome activation is also thought to be a pathogenic strategy for Streptococcus pneumoniae, Yersinia, Francisella tularensis, and Mycobacterium tuberculosis (Brodsky et al., 2010; Huang et al., 2010; Master et al., 2008; Witzenrath et al., 2011). These data and the fact that inflammasome activity promotes clearance of certain non-pathogenic environmental bacteria such as Chromobacterium violaceum has led to the proposition that non-pathogens activate the inflammasome, whereas the success of true pathogens relies on evasion or suppression of inflammasome activity (Maltez et al., 2015). However, this model fails to incorporate the fact that certain pathogens have evolved strategies to exploit inflammasome activation. For example, inflammasome activity was previously shown to impair bacterial clearance and host survival in a Pseudomonas aeruginosa infection model (Cohen and Prince, 2013). Further, Mycobacterium marinum and influenza virus encode proteins that promote NLRP3 activation, increasing replication of both pathogens in animal infection models (Carlsson et al., 2010; Tate et al., 2016). We show that Lm also exploits NLRP3 inflammasome activation and indicate that the mechanism by which this likely benefits the pathogen is through release of IL-18 to promote production of anti-inflammatory IL-10.
IL-18 had not been previously implicated in the regulation of NK cell IL-10 production. This cytokine was originally identified as an “IFNγ-inducing factor” that promotes IFNγ production during sepsis (Okamura et al., 1995). However, IL-18 alone induces little or no IFNγ. IL-18 instead acts synergistically with IL-12 to promote IFNγ secretion by NK and T lymphocytes in the context of Lm and other infections (Berg et al., 2002; Humann and Lenz, 2010; Kupz et al., 2014; Soudja et al., 2012). We found that NK cell IL-10 production elicited in response to L1S or Lm-stimulated DCs was dependent on DC production of IL-18 and NK cell expression of IL-18R1. However, IL-18 alone failed to elicit this IL-10 production in the absence of other DC factors. These findings argue that IL-18 also acts in synergy with a second Lm-induced DC factor to promote NK cell IL-10 secretion. Prior studies have shown that IL-12-induced STAT4 signaling alone can promote IL-10 secretion from human and mouse NK cells (Grant et al., 2008; Mehrotra et al., 1998). However, our data showed that IL-18 was not required for this IL-12 induced IL-10 secretion and that blockade of IL-12 or STAT4 did not inhibit NK cell IL-10 production induced by Lm/L1S-stimulated DC. Hence, there exist at least two distinct pathways that can drive NK cell production of IL-10. Further characterization of the unknown second factor that synergizes with IL-18 will be an important goal of future studies.
One previous report suggested that Il18−/− mice resist systemic Lm infection, consistent with our finding that IL-18 increases host susceptibility (Lochner et al., 2008). Murine infections with Pseudomonas aeruginosa and Ehrlichia also revealed detrimental roles for IL-18 (Ghose et al., 2011; Schultz et al., 2003), and mice lacking IL-18 have increased susceptibility to Mycobacterium tuberculosis and Helicobacter infections (Hitzler et al., 2012; Schneider et al., 2010). Given our findings here, we speculate that, in some or all of these cases, IL-18 may promote susceptibility through the licensing of regulatory NK cell activity. Conversely, IL-18 has been suggested in at least one prior study to play a protective role during Lm infection. In that study, mice were treated with recombinant IL-18 at 24, 48, and 72 hpi. This was found to reduce Lm bacterial burdens upon harvest at 4 days post-infection (dpi) (Maltez et al., 2015). We propose that, in this context, inflammasome activation and IL-18 production might have exerted positive effects on host resistance given its earlier timing and distinct conditions. Specifically, systemic IL-18 early during Lm infection (24 hpi) occurs in the presence of few inflammatory cells and low bacterial burdens. This IL-18 normally synergizes with IL-12 and other factors to stimulate a wave of NK and memory T cell activation and IFNγ production. Supplementation of this early response with recombinant IL-18 might thus be expected to further boost resistance. The higher concentrations of endogenous IL-18 seen at later times after infection (72 hpi) are instead normally associated with a high number of recruited inflammatory cells and higher bacterial burdens. In this context, endogenous IL-18 production primarily synergizes with a non-IL-12 factor to trigger IL-10 production and suppress inflammation.
Several prior reports have suggested a vital role for DCs in promoting Lm infection. Depletion of CD11c+ cells in CD11c-diptheria toxin receptor (DTR) mice reduced splenic Lm burdens during systemic infection (Neuenhahn et al., 2006). CD8α+ DCs were identified as a cell population that is infected with Lm at early time points post-infection (Neuenhahn et al., 2006). Batf3-deficient mice were later shown to have reduced numbers of CD8α+ and CD103+ DCs and to support reduced Lm burdens (Edelson et al., 2011). However, it has remained unclear precisely how Batf3 expression might increase host susceptibility to Lm. Our studies here indicate that Batf3 plays a vital role in regulating the production of IL-18 in response to Lm/L1S. Despite there being many potential sources of IL-18 production in vivo, our mixed chimera experiments showed that high concentrations of serum IL-18 at 72h after Lm infection are only observed when Batf3 and IL-18 are co-expressed by the same cell. The presence of Batf3+ cells in vivo failed to increase host susceptibility when these cells could not produce IL-18 (even when all other cell types could produce IL-18). Similarly, in response to L1S treatment, we observed little IL-18 secretion and NK cell activation by CD11c+ cells purified from spleens of Batf3−/− mice. These data together argue that Batf3 does not increase bacterial burdens during Lm infection solely by providing a cellular niche for bacterial replication. Rather, Batf3+ cells appear to be a vital source of IL-18 production during in vivo Lm infection. We do not find a requirement for Batf3 in transcription of Nlrp3 and Il18 or expression of NLRP3 and IL-18 proteins. Indeed, other splenic DC populations similarly expressed both of these factors regardless of Batf3 expression. Instead, we propose that Batf3 expression and/or an abundance of CD8α+ DCs may increase host sensitivity to the Lm-derived p60/L1S protein. Our in vitro data using purified splenic CD11c+ populations are consistent with the model that batf3 directly regulates the responsiveness of DCs to L1S. However, we cannot, at this point, exclude the alternative possibility that, during Lm infection, the presence of IL-18-producing Batf3+ DCs might initiate IL-18 production that also requires other cell types. In this light, a previous study showed that, in cultured Ly6C+ monocytes activate Lm memory CD8+ T cells in an IL-18R-dependent manner (Soudja et al., 2012). Nevertheless, our work shows a crucial importance for IL-18 production by Batf3-dependent cell population(s) in the induction of NK cell IL-10 secretion and the suppressive effects of this IL-10 on host resistance. As such, our findings further provide a revised mechanistic paradigm to account for the previously reported deleterious role of Batf3 and CD8α+ DCs in the early immune response to Lm.
In summary, our work has identified several critical components of a pathway exploited by Lm to elicit an immune-suppressive NK cell response. Although there are currently immunotherapy approaches focused on the improvement of NK cell responsiveness to tumors (Lowry and Zehring, 2017), strategies to dampen NK cell anti-inflammatory responses remain unexplored. Our findings reveal several possible targets for interventions to limit NK cell IL-10 production during Lm and potentially other bacterial infections. Because NK cell regulatory activity has broad implications for host immunity, further study of the mechanisms contributing to this process may reveal new targets for therapy of infectious and inflammatory diseases.
EXPERIMENTAL PROCEDURES
Animals
Adult male and female mice were used at 8–12 weeks of age. C57BL/6J (B6), B6.tiger (IL-10 GFP reporter), B6.Il10−/−, B6.Ptprca (CD45.1), B6.Il18r−/−, B6.Il18−/−, B6.Nlrp3−/−, and B6.Batf3−/− mice were purchased from The Jackson Laboratory. Mice were maintained in the University of Colorado Office of Laboratory Animal Resources.
Infections
WT Lm, strain 10403s) or Δp60 Lm (Lenz et al., 2003) were thawed from frozen stocks and diluted in tryptic soy broth (MP Biomedicals) with streptomycin (50 μg/mL) for growth to log phase. Log phase bacteria were diluted in PBS and injected i.v. in the lateral tail vein. Mice received a single sublethal dose of 104 colony-forming units (CFUs). For CFU determinations, organs were harvested in 0.02% Nonidet P-40 and homogenized for 1 min with a tissue homogenizer (IKA Works). Serial dilutions were plated on trypticase soy broth (TSB) agar plates with streptomycin (50 μg/mL) and grown overnight at 37°C.
Generation of Chimeric Mice
Mice received a dose of 500 rads for irradiation. For BM cell transfers, each mouse received 106 BM cells delivered i.v. in 200 μL. Mice were allowed to reconstitute for 5–8 weeks before infection with 104 Lm i.v. Reconstitution of the hematopoietic system by donor-derived cells was in all cases >80%, as determined by staining for CD45.1/2 and flow cytometric analysis.
Cell Isolation and Stimulations
NK cells were purified by negative selection from spleens of naive mice using the EasySep NK Cell Enrichment Kit (19855, STEMCELL Technologies). NK cell negative sort isolations were >80% NK1.1+CD3− cells. CD11c+ cells were sorted by positive selection using the EasySep Mouse CD11c Positive Selection Kit (18758, STEMCELL Technologies). CD11c+ sort isolations were >90% CD11c+ cells. CD11c+ cells were plated 18 hr prior to stimulation.
For co-culture experiments, BMDCs were cultured and infected with Lm or stimulated with 10 ng/mL LPS (L8274, Sigma-Aldrich, St. Louis, MO) and 30 μg/mL recombinant L1S protein as previously described (Humann and Lenz, 2010; Schmidt et al., 2011). For transwell experiments, NK cells were separated from BMDCs by 0.4 μM membranes. For supernatant transfer experiments, BMDCs were cultured or CD11c+ cells were purified, and 3 × 105 cells were plated overnight in 24-well plates. One hr post-infection, cells were washed, and gentamycin was added at 10 μg/mL. One hr after this, supernatants were harvested and filtered (0.22 μM). Filtered supernatants were added to a new 24-well plate. NK cells purified as above were then added to the new plates with filtered supernatants. For L1S+LPS stimulations, cells were stimulated as above for 1 hr. Subsequently, supernatants were harvested, filtered, and added to a new 24-well plate, and then purified NK cells were added.
Study Approval
The Animal Care and Use Committee of the University of Colorado School of Medicine (protocols 105614(05)1E and #105617(04)1E) approved these studies.
Statistical Analysis
Prism (GraphPad) was used for graphing and statistical analysis. Statistical tests included t tests and ANOVA. p < 0.05 was considered significant.
Supplementary Material
Highlights.
NLRP3 inflammasome-dependent IL-18 limits host protection against Listeria
Batf3-dependent cells are a critical source of immune-suppressive IL-18
IL-18 acts directly on NK cells to license their secretion of IL-10
IL-18 promotion of NK cell IL-10 is independent of IL-12 and cell contact
Acknowledgments
We thank other current and former members of the lab for critical discussion of these data. This work was funded by National Institute of Allergy and Infectious Diseases grants AI114075 (to S.E.C.), AI065638, and AI131662 (to L.L.L.).
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and five figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.04.106.
AUTHOR CONTRIBUTIONS
S.E.C., R.L.S., and L.L.L. conceived and designed the experiments. S.E.C., R.L.S., D.S.M., and L.L.L. performed the experiments. S.E.C. and L.L.L. wrote the paper and edited the manuscript.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Berg RE, Cordes CJ, Forman J. Contribution of CD8+ T cells to innate immunity: IFN-gamma secretion induced by IL-12 and IL-18. Eur J Immunol. 2002;32:2807–2816. doi: 10.1002/1521-4141(2002010)32:10<2807::AID-IMMU2807>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Borg C, Jalil A, Laderach D, Maruyama K, Wakasugi H, Charrier S, Ryffel B, Cambi A, Figdor C, Vainchenker W, et al. NK cell activation by dendritic cells (DCs) requires the formation of a synapse leading to IL-12 polarization in DCs. Blood. 2004;104:3267–3275. doi: 10.1182/blood-2004-01-0380. [DOI] [PubMed] [Google Scholar]
- Brodsky IE, Palm NW, Sadanand S, Ryndak MB, Sutterwala FS, Flavell RA, Bliska JB, Medzhitov R. A Yersinia effector protein promotes virulence by preventing inflammasome recognition of the type III secretion system. Cell Host Microbe. 2010;7:376–387. doi: 10.1016/j.chom.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16:407–420. doi: 10.1038/nri.2016.58. [DOI] [PubMed] [Google Scholar]
- Carlsson F, Kim J, Dumitru C, Barck KH, Carano RAD, Sun M, Diehl L, Brown EJ. Host-detrimental role of Esx-1-mediated inflammasome activation in mycobacterial infection. PLoS Pathog. 2010;6:e1000895. doi: 10.1371/journal.ppat.1000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cella M, Miller H, Song C. Beyond NK cells: the expanding universe of innate lymphoid cells. Front Immunol. 2014;5:282. doi: 10.3389/fimmu.2014.00282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaix J, Tessmer MS, Hoebe K, Fuséri N, Ryffel B, Dalod M, Alexopoulou L, Beutler B, Brossay L, Vivier E, Walzer T. Cutting edge: Priming of NK cells by IL-18. J Immunol. 2008;181:1627–1631. doi: 10.4049/jimmunol.181.3.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark SE, Filak HC, Guthrie BS, Schmidt RL, Jamieson A, Merkel P, Knight V, Cole CM, Raulet DH, Lenz LL. Bacterial Manipulation of NK Cell Regulatory Activity Increases Susceptibility to Listeria monocytogenes Infection. PLoS Pathog. 2016;12:e1005708–e1005721. doi: 10.1371/journal.ppat.1005708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen TS, Prince AS. Activation of inflammasome signaling mediates pathology of acute P. aeruginosa pneumonia. J Clin Invest. 2013;123:1630–1637. doi: 10.1172/JCI66142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa A, Gupta R, Signorino G, Malara A, Cardile F, Biondo C, Midiri A, Galbo R, Trieu-Cuot P, Papasergi S, et al. Activation of the NLRP3 inflammasome by group B streptococci. J Immunol. 2012;188:1953–1960. doi: 10.4049/jimmunol.1102543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyktor JC, Turner J. Interleukin-10 and immunity against prokaryotic and eukaryotic intracellular pathogens. Infect Immun. 2011;79:2964–2973. doi: 10.1128/IAI.00047-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA, Novick D, Kim S, Kaplanski G. Interleukin-18 and IL-18 binding protein. Front Immunol. 2013;4:289. doi: 10.3389/fimmu.2013.00289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelson BT, Bradstreet TR, Hildner K, Carrero JA, Frederick KE, Kc W, Belizaire R, Aoshi T, Schreiber RD, Miller MJ, et al. CD8α(+) dendritic cells are an obligate cellular entry point for productive infection by Listeria monocytogenes. Immunity. 2011;35:236–248. doi: 10.1016/j.immuni.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehniger TA, Shah MH, Turner MJ, VanDeusen JB, Whitman SP, Cooper MA, Suzuki K, Wechser M, Goodsaid F, Caligiuri MA. Differential cytokine and chemokine gene expression by human NK cells following activation with IL-18 or IL-15 in combination with IL-12: implications for the innate immune response. J Immunol. 1999;162:4511–4520. [PubMed] [Google Scholar]
- Ghose P, Ali AQ, Fang R, Forbes D, Ballard B, Ismail N. The interaction between IL-18 and IL-18 receptor limits the magnitude of protective immunity and enhances pathogenic responses following infection with intracellular bacteria. J Immunol. 2011;187:1333–1346. doi: 10.4049/jimmunol.1100092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant LR, Yao ZJ, Hedrich CM, Wang F, Moorthy A, Wilson K, Ranatunga D, Bream JH. Stat4-dependent, T-bet-independent regulation of IL-10 in NK cells. Genes Immun. 2008;9:316–327. doi: 10.1038/gene.2008.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagar JA, Miao EA. Detection of cytosolic bacteria by inflammatory caspases. Curr Opin Microbiol. 2014;17:61–66. doi: 10.1016/j.mib.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322:1097–1100. doi: 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitzler I, Sayi A, Kohler E, Engler DB, Koch KN, Hardt WD, Müller A. Caspase-1 has both proinflammatory and regulatory properties in Helicobacter infections, which are differentially mediated by its substrates IL-1β and IL-18. J Immunol. 2012;188:3594–3602. doi: 10.4049/jimmunol.1103212. [DOI] [PubMed] [Google Scholar]
- Hof H. History and epidemiology of listeriosis. FEMS Immunol Med Microbiol. 2003;35:199–202. doi: 10.1016/S0928-8244(02)00471-6. [DOI] [PubMed] [Google Scholar]
- Huang MTH, Mortensen BL, Taxman DJ, Craven RR, Taft-Benz S, Kijek TM, Fuller JR, Davis BK, Allen IC, Brickey WJ, et al. Deletion of ripA alleviates suppression of the inflammasome and MAPK by Francisella tularensis. J Immunol. 2010;185:5476–5485. doi: 10.4049/jimmunol.1002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humann J, Lenz LL. Activation of naive NK cells in response to Listeria monocytogenes requires IL-18 and contact with infected dendritic cells. J Immunol. 2010;184:5172–5178. doi: 10.4049/jimmunol.0903759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humann J, Bjordahl R, Andreasen K, Lenz LL. Expression of the p60 autolysin enhances NK cell activation and is required for listeria monocytogenes expansion in IFN-gamma-responsive mice. J Immunol. 2007;178:2407–2414. doi: 10.4049/jimmunol.178.4.2407. [DOI] [PubMed] [Google Scholar]
- Kamanaka M, Kim ST, Wan YY, Sutterwala FS, Lara-Tejero M, Galán JE, Harhaj E, Flavell RA. Expression of interleukin-10 in intestinal lymphocytes detected by an interleukin-10 reporter knockin tiger mouse. Immunity. 2006;25:941–952. doi: 10.1016/j.immuni.2006.09.013. [DOI] [PubMed] [Google Scholar]
- Kang SJ, Liang HE, Reizis B, Locksley RM. Regulation of hierarchical clustering and activation of innate immune cells by dendritic cells. Immunity. 2008;29:819–833. doi: 10.1016/j.immuni.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr AR, Kirkham LAS, Kadioglu A, Andrew PW, Garside P, Thompson H, Mitchell TJ. Identification of a detrimental role for NK cells in pneumococcal pneumonia and sepsis in immunocompromised hosts. Microbes Infect. 2005;7:845–852. doi: 10.1016/j.micinf.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Kim S, Bauernfeind F, Ablasser A, Hartmann G, Fitzgerald KA, Latz E, Hornung V. Listeria monocytogenes is sensed by the NLRP3 and AIM2 inflammasome. Eur J Immunol. 2010;40:1545–1551. doi: 10.1002/eji.201040425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knorr DA, Bachanova V, Verneris MR, Miller JS. Clinical utility of natural killer cells in cancer therapy and transplantation. Semin Immunol. 2014;26:161–172. doi: 10.1016/j.smim.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupz A, Curtiss R, 3rd, Bedoui S, Strugnell RA. In vivo IFN-γ secretion by NK cells in response to Salmonella typhimurium requires NLRC4 inflammasomes. PLoS ONE. 2014;9:e97418. doi: 10.1371/journal.pone.0097418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol. 2008;9:495–502. doi: 10.1038/ni1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz LL, Mohammadi S, Geissler A, Portnoy DA. SecA2-dependent secretion of autolytic enzymes promotes Listeria monocytogenes pathogenesis. Proc Natl Acad Sci USA. 2003;100:12432–12437. doi: 10.1073/pnas.2133653100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Zaki MH, Vogel P, Gurung P, Finlay BB, Deng W, Lamkanfi M, Kanneganti TD. Role of inflammasomes in host defense against Citrobacter rodentium infection. J Biol Chem. 2012;287:16955–16964. doi: 10.1074/jbc.M112.358705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochner M, Kastenmüller K, Neuenhahn M, Weighardt H, Busch DH, Reindl W, Förster I. Decreased susceptibility of mice to infection with Listeria monocytogenes in the absence of interleukin-18. Infect Immun. 2008;76:3881–3890. doi: 10.1128/IAI.01651-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry LE, Zehring WA. Potentiation of Natural Killer Cells for Cancer Immunotherapy: A Review of Literature. Front Immunol. 2017;8:1061. doi: 10.3389/fimmu.2017.01061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltez VI, Tubbs AL, Cook KD, Aachoui Y, Falcone EL, Holland SM, Whitmire JK, Miao EA. Inflammasomes Coordinate Pyroptosis and Natural Killer Cell Cytotoxicity to Clear Infection by a Ubiquitous Environmental Bacterium. Immunity. 2015;43:987–997. doi: 10.1016/j.immuni.2015.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Master SS, Rampini SK, Davis AS, Keller C, Ehlers S, Springer B, Timmins GS, Sander P, Deretic V. Mycobacterium tuberculosis prevents inflammasome activation. Cell Host Microbe. 2008;3:224–232. doi: 10.1016/j.chom.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrotra PT, Donnelly RP, Wong S, Kanegane H, Geremew A, Mostowski HS, Furuke K, Siegel JP, Bloom ET. Production of IL-10 by human natural killer cells stimulated with IL-2 and/or IL-12. J Immunol. 1998;160:2637–2644. [PubMed] [Google Scholar]
- Meixenberger K, Pache F, Eitel J, Schmeck B, Hippenstiel S, Slevogt H, N’Guessan P, Witzenrath M, Netea MG, Chakraborty T, et al. Listeria monocytogenes-infected human peripheral blood mononuclear cells produce IL-1beta, depending on listeriolysin O and NLRP3. J Immunol. 2010;184:922–930. doi: 10.4049/jimmunol.0901346. [DOI] [PubMed] [Google Scholar]
- Neuenhahn M, Kerksiek KM, Nauerth M, Suhre MH, Schiemann M, Gebhardt FE, Stemberger C, Panthel K, Schröder S, Chakraborty T, et al. CD8α+ dendritic cells are required for efficient entry of Listeria monocytogenes into the spleen. Immunity. 2006;25:619–630. doi: 10.1016/j.immuni.2006.07.017. [DOI] [PubMed] [Google Scholar]
- Okamura H, Tsutsi H, Komatsu T, Yutsudo M, Hakura A, Tanimoto T, Torigoe K, Okura T, Nukada Y, Hattori K, et al. Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature. 1995;378:88–91. doi: 10.1038/378088a0. [DOI] [PubMed] [Google Scholar]
- Perona-Wright G, Mohrs K, Szaba FM, Kummer LW, Madan R, Karp CL, Johnson LL, Smiley ST, Mohrs M. Systemic but not local infections elicit immunosuppressive IL-10 production by natural killer cells. Cell Host Microbe. 2009;6:503–512. doi: 10.1016/j.chom.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt RL, Lenz LL. Distinct licensing of IL-18 and IL-1β secretion in response to NLRP3 inflammasome activation. PLoS ONE. 2012;7:e45186–e45189. doi: 10.1371/journal.pone.0045186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt RL, Filak HC, Lemon JD, Potter TA, Lenz LL. A LysM and SH3-domain containing region of the Listeria monocytogenes p60 protein stimulates accessory cells to promote activation of host NK cells. PLoS Pathog. 2011;7:e1002368. doi: 10.1371/journal.ppat.1002368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider BE, Korbel D, Hagens K, Koch M, Raupach B, Enders J, Kaufmann SHE, Mittrücker HW, Schaible UE. A role for IL-18 in protective immunity against Mycobacterium tuberculosis. Eur J Immunol. 2010;40:396–405. doi: 10.1002/eji.200939583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz MJ, Knapp S, Florquin S, Pater J, Takeda K, Akira S, van der Poll T. Interleukin-18 impairs the pulmonary host response to Pseudomonas aeruginosa. Infect Immun. 2003;71:1630–1634. doi: 10.1128/IAI.71.4.1630-1634.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soudja SM, Ruiz AL, Marie JC, Lauvau G. Inflammatory monocytes activate memory CD8(+) T and innate NK lymphocytes independent of cognate antigen during microbial pathogen invasion. Immunity. 2012;37:549–562. doi: 10.1016/j.immuni.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan B, Gerner-Smidt P. The epidemiology of human listeriosis. Microbes Infect. 2007;9:1236–1243. doi: 10.1016/j.micinf.2007.05.011. [DOI] [PubMed] [Google Scholar]
- Takada H, Matsuzaki G, Hiromatsu K, Nomoto K. Analysis of the role of natural killer cells in Listeria monocytogenes infection: relation between natural killer cells and T-cell receptor gamma delta T cells in the host defence mechanism at the early stage of infection. Immunology. 1994;82:106–112. [PMC free article] [PubMed] [Google Scholar]
- Tate MD, Ong JD, Dowling JK, McAuley JL, Robertson AB, Latz E, Drummond GR, Cooper MA, Hertzog PJ, Mansell A. Reassessing the role of the NLRP3 inflammasome during pathogenic influenza A virus infection via temporal inhibition. Sci Rep. 2016;6:27912. doi: 10.1038/srep27912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira HC, Kaufmann SHE. Role of NK1.1+ cells in experimental listeriosis. NK1+ cells are early IFN-gamma producers but impair resistance to Listeria monocytogenes infection. J Immunol. 1994;152:1873–1882. [PubMed] [Google Scholar]
- Tomura M, Zhou XY, Maruo S, Ahn HJ, Hamaoka T, Okamura H, Nakanishi K, Tanimoto T, Kurimoto M, Fujiwara H. A critical role for IL-18 in the proliferation and activation of NK1.1+ CD3- cells. J Immunol. 1998;160:4738–4746. [PubMed] [Google Scholar]
- Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, Yokoyama WM, Ugolini S. Innate or adaptive immunity? The example of natural killer cells. Science. 2011;331:44–49. doi: 10.1126/science.1198687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L. Targeting natural killer cells and natural killer T cells in cancer. Nat Rev Immunol. 2012;12:239–252. doi: 10.1038/nri3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren SE, Mao DP, Rodriguez AE, Miao EA, Aderem A. Multiple Nod-like receptors activate caspase 1 during Listeria monocytogenes infection. J Immunol. 2008;180:7558–7564. doi: 10.4049/jimmunol.180.11.7558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witzenrath M, Pache F, Lorenz D, Koppe U, Gutbier B, Tabeling C, Reppe K, Meixenberger K, Dorhoi A, Ma J, et al. The NLRP3 inflammasome is differentially activated by pneumolysin variants and contributes to host defense in pneumococcal pneumonia. J Immunol. 2011;187:434–440. doi: 10.4049/jimmunol.1003143. [DOI] [PubMed] [Google Scholar]
- Wu J, Fernandes-Alnemri T, Alnemri ES. Involvement of the AIM2, NLRC4, and NLRP3 inflammasomes in caspase-1 activation by Listeria monocytogenes. J Clin Immunol. 2010;30:693–702. doi: 10.1007/s10875-010-9425-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.