

SUMMARY
T cell acute lymphoblastic leukemia (T-ALL) is commonly associated with activating mutations in the NOTCH1 pathway. Recent reports have shown a link between NOTCH1 signaling and intracellular Ca2+ homeostasis in T-ALL. Here, we investigate the role of store-operated Ca2+ entry (SOCE) mediated by the Ca2+ channel ORAI1 and its activators STIM1 and STIM2 in T-ALL. Deletion of STIM1 and STIM2 in leukemic cells abolishes SOCE and significantly prolongs the survival of mice in a NOTCH1-dependent model of T-ALL. The survival advantage is unrelated to the leukemic cell burden but is associated with the SOCE-dependent ability of malignant T lymphoblasts to cause inflammation in leukemia-infiltrated organs. Mice with STIM1/STIM2-deficient T-ALL show a markedly reduced necroinflammatory response in leukemia-infiltrated organs and downregulation of signaling pathways previously linked to cancer-induced inflammation. Our study shows that leukemic T lymphoblasts cause inflammation of leukemia-infiltrated organs that is dependent on SOCE.
In Brief
T cell acute lymphoblastic leukemia (T-ALL) is an aggressive cancer of T cell progenitors affecting children and adults. Saint Fleur-Lominy et al. show that calcium influx mediated by STIM1 and STIM2 promotes the proinflammatory function of leukemic cells and premature death from leukemia.
Graphical Abstract
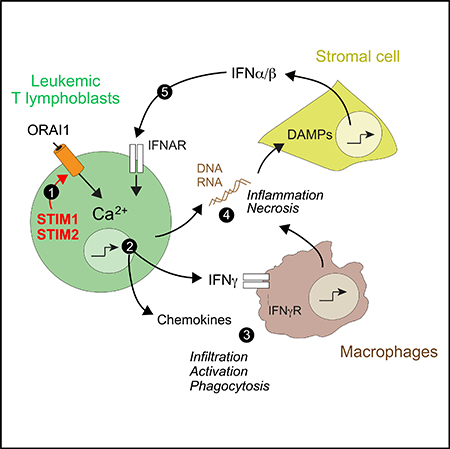
INTRODUCTION
T cell acute lymphoblastic leukemia (T-ALL) is an aggressive neoplasm of T cell progenitors that affects children and adults (Inaba et al., 2013). T-ALL is caused by activating mutations in the NOTCH1 pathway in over 50% of patients (Ferrando, 2009; Inaba et al., 2013). NOTCH1, a master regulator of T cell development, is activated by its ligands Jagged-1 and the delta-like ligand (DLL) family (Radtke et al., 2013), which initiate the proteolytic cleavage of the NOTCH1 intracellular domain (ICN1), its nuclear translocation (De Strooper et al., 1999), and transcription of NOTCH1 target genes. NOTCH1 mutations in T-ALL patients frequently occur in the proteolytic cleavage sites of NOTCH1 and/or its PEST sequence generating NOTCH1 oncogenes with autonomous signaling and/or an extended half-life (Weng et al., 2004). Despite significantly improved cure rates of pediatric T-ALL, novel therapies fail to rescue patients with relapsed or primary refractory disease (Dores et al., 2012). Clinical application of NOTCH1 inhibition has been unsuccessful because of unexpected side effects (Ryeom, 2011). It is therefore important to investigate alternative pathways as potential targets of T-ALL therapy. Multiple studies have demonstrated the importance of the leukemia microenvironment for disease development and outcome (Chiarini et al., 2016; Passaro et al., 2015; Pitt et al., 2015). A complex interaction of the leukemic cells with cells of specific niches within various organs results in tissue remodeling and modulation of leukemia biology (Hawkins et al., 2016; Pitt et al., 2015), but many key components of that interaction are not fully understood.
Calcium (Ca2+) is a versatile secondary messenger in many cell types that regulates many cell functions. In resting cells, the intracellular Ca2+ concentration ([Ca2+]i) is low (~50 nM). Stimulation of cells increases the [Ca2+]i with wide-ranging effects on cell function. Several reports have documented aberrant Ca2+ signaling in cancers in patients and animal models, and mutations in molecules that control Ca2+ homeostasis have been associated with increased tumor incidences (Bergmeier et al., 2013; Monteith et al., 2007; Roderick and Cook, 2008). In T-ALL, inhibition of calcineurin, a Ca2+-dependent serine phosphatase, with cyclosporin A slowed leukemia progression and prolonged survival in a murine model of T-ALL (Gachet et al., 2013; Medyouf et al., 2007). A small interfering RNA (siRNA) screen identified sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA) that transports Ca2+ from the cytoplasm into the ER as important regulators of oncogenic NOTCH1 signaling and survival of leukemic T cells (Roti et al., 2013). Furthermore, conditional deletion of all three inositol 1,4,5-trisphosphate receptors (IP3R), which release Ca2+ from the ER into the cytoplasm, in thymocytes resulted in spontaneous T-ALL development that was associated with increased NOTCH1 expression (Ouyang et al., 2014). These studies indicate that ER Ca2+ signaling is an important regulator of NOTCH1 expression and T-ALL development. By contrast, the role of Ca2+ influx across the plasma membrane in T-ALL pathology is unknown.
Store-operated Ca2+ entry (SOCE) is a ubiquitous Ca2+ influx pathway (Prakriya and Lewis, 2015), which is triggered by binding of receptors that activate phospholipase C and production of IP3 resulting in the release of Ca2+ from the ER via IP3Rs. The resultant reduction in the ER Ca2+ concentration activates two ER membrane proteins, stromal interaction molecule 1 (STIM1) and STIM2 (Liou et al., 2005; Roos et al., 2005). In their activated state, they bind to the Ca2+ release-activated Ca2+ (CRAC) channel protein ORAI1 in the plasma membrane, which is the main conduit of SOCE (Feske et al., 2006; Vig et al., 2006; Zhang et al., 2006). SOCE is essential for physiological T cell function and patients with null mutations in STIM1 or ORAI1 genes are severely immunodeficient (Lacruz and Feske, 2015). Several studies have implicated SOCE in various solid tumor types (Bergmeier et al., 2013; Xie et al., 2016), where it was shown to regulate the cell cycle and proliferation, angiogenesis, migration, invasiveness, and metastasis (Chen et al., 2011; Umemura et al., 2014; Wang et al., 2015). The role of SOCE in leukemia, by contrast, is unknown.
Here, we investigated SOCE in leukemia using a NOTCH1driven mouse model of T-ALL. We found that deletion of Stim1 and Stim2 genes in NOTCH1-expressing bone marrow (BM) progenitor cells abolished SOCE in leukemic cells and significantly prolonged the survival of mice with T-ALL. This survival advantage was unrelated to the proliferation and apoptosis of leukemic cells or the leukemia burden of mice but was associated with strongly attenuated inflammation and damage of organs infiltrated by leukemic cells. We identified cytokines, chemokines, and other pro-inflammatory pathways previously linked to cancer-induced inflammation to be controlled by SOCE in leukemic T lymphoblasts. Our findings indicate that SOCE is a critical signaling pathway that enables leukemic cells to cause detrimental tissue inflammation, which has important implications for T-ALL treatment and outcome.
RESULTS
Deletion of STIM1 and STIM2 Prolongs Survival in a NOTCH1-Driven Model of T-ALL
Given the importance of SOCE for T cell function (Feske et al., 2012; Lacruz and Feske, 2015) and ER Ca2+ homeostasis for T-ALL (Ouyang et al., 2014; Roti et al., 2013), we investigated the role of SOCE in T-ALL. We first compared the expression of all three ORAI and two STIM genes in T-ALL to that in other human cancer types (Barretina et al., 2012). mRNA levels of STIM1 and ORAI1 were highest in human T-ALL compared to 40 other cancers. STIM2 and ORAI2 were both more highly expressed in T-ALL than most other cancers (ranked 11 of 40) whereas ORAI3 mRNA levels were low in T-ALL (ranked 34 of 40) (Figures 1A, 1B, and S1; Tables S1 and S2). These data suggested that the SOCE pathway, an in particular ORAI1 and STIM1, is important in T-ALL. To functionally test this interpretation, we used a well-established BMtransfer model of T-ALL in which overexpression of the constitutively active intracellular domain ofNOTCH1 (ICN1) in lineage negative (lin–, i.e., CD3ε, CD4, CD5, CD8, CD11b, Gr-1, B220, and Ter-119 negative) c-kit+ BMprogenitor cells induces a lethal T-ALL-like disease in mice (Figure 1C; Li et al., 2008). Mice injected with ICN1-transduced BM progenitor cells, like human T-ALL patients, accumulate leukemic cells in their BM, spleen and other organs. We generated STIM1/STIM2-deficient BM progenitor cells by treating Stim1fl/flStim2fl/fl Mx1-Cre mice with poly(I:C) to induce Cre expression and deletion of Stim1 and Stim2 in all hematopoietic cell lineages (Figure 1C). Alternatively, we used Stim1fl/flStim2fl/fl Vav-Cre mice in which Stim1 and Stim2 are constitutively deleted in hematopoietic cells (Figure S2). These mice are collectively referred to as Stim1/2−/−mice. Deletion of Stim1/Stim2 did not affect the development of myeloid cells in the BM (Figures S2A and S2B) despite completely abolished SOCE (Figures S2C–S2E) consistent with previous findings (Vaeth et al., 2015). We tested SOCE in BM progenitor cells of Stim1fl/flStim2fl/fl Mx1-Cre mice. Stimulation of wild-type (WT) BM progenitor cells with the SERCA inhibitor thapsigargin, which induces depletion of Ca2+ from ER stores and SOCE, resulted in robust Ca2+ signals whereas BM progenitor cells from Stim1fl/flStim2fl/fl Mx1-Cre mice completely lacked SOCE (Figure 1D).
Figure 1. Deletion of Stim1 and Stim2 Prolongs the Survival of Mice with T-ALL.

(A and B) STIM1 (A) and STIM2 (B) mRNA expression in T-ALL and other human tumors. Data extracted from the cancer cell line encyclopedia (CCLE) were ranked by expression level. Numbers refer to cancer cell types (see Tables S1 and S2); hematological malignancies are in red.
(C) Experimental model of T-ALL used in (D)–(I). BM cells isolated either from poly(I:C)-treated wild-type (WT) or Stim1/2fl/fl Mx1-Cre mice and untreated WT or Stim1/2fl/fl Vav-Cre mice (Stim1/2−/−) were transduced with ICN1-IRES-GFP and cultured for 3 days in vitro. 5 × 104 ICN1-transduced (GFP+) lineage negative (lin–) BM progenitor cells were injected i.v. (together with 5 × 105 bone marrow cells from WT mice) into lethally irradiated CD45.1+ WT host mice.
(D) SOCE in c-kit+ progenitor cells isolated from WT and Stim1/2−/− mice.
(E and F) Representative flow cytometry plots of surface (s) and cytoplasmic (cy) CD3 expression (E) or CD4/CD8 expression (F) on cells isolated from the BMand spleen of healthy WT control mice (Ctrl) and mice with WT or Stim1/2−/− leukemia (gated on GFP+).
(G) SOCE in WT and Stim1/2−/− leukemic cells (GFP+) isolated from the spleen at 21 days of disease.
(H) Relative weight of mice with WT and Stim1/2−/− leukemia. Values shown are mean ± SEM. Statistical analysis was performed using Student’s t test. *p < 0.05.
(I) Survival of mice with WT and Stim1/2−/− leukemia. Statistical significance was calculated with the non-parametric Kaplan-Meier estimator.
Data are from 18–20 mice per cohort in (H and I). Flow cytometry and SOCE measurements in (D)–(G) are representative of at least 3 to 10 mice per group.
To induce T-ALL, BM progenitor cells from WT and Stim1/2−/− mice were transduced with ICN1-IRES-GFP and injected into irradiated WT host mice. Mice that received either WT or Stim1/2−/−ICN1+ BM progenitors developed T-ALL as indicated by the presence of GFP+ cells in their BM and spleen 14 days after transfer. GFP+ cells expressed cytoplasmic CD3 (cyCD3) (Figure 1E), together with cell-surface expression of both CD4 and CD8 coreceptors (Figure 1F), indicating that they are leukemic T lymphoblasts. The frequencies of cyCD3+ and CD4+/CD8+ leukemic T lymphoblasts were similar in mice with WT and Stim1/2−/−leukemia (Figures 1E and 1F). GFP+ leukemic cells originated from the transferred progenitor cells as leukemic cells from the WT cohort showed robust SOCE, whereas those from the Stim1/2−/− cohort lacked SOCE (Figure 1G). These findings indicated that SOCE is not required for ICN1-induced T-ALL development. Nevertheless, mice with Stim1/2−/− leukemia lost significantly less weight compared with mice in the WT cohort, suggesting that SOCE in leukemic cells promotes cancer-associated cachexia (Figure 1H). Importantly, mice in the Stim1/2−/− cohort survived significantly longer than did those in the WT group (31 vs. 58.5 days) (Figure 1I). It is noteworthy that prolonged survival of the Stim1/2−/− cohort was independent of whether Stim1/2−/− progenitor cells were derived from poly(I:C)-treated Stim1fl/flStim2fl/fl Mx1-Cre or Stim1fl/flStim2fl/fl Vav-Cre mice (Figures S2F and S2G). We conclude that SOCE exacerbates the severity of T-ALL but is dispensable for T-ALL development.
Survival Benefit in STIM1/STIM2-Deficient Leukemia Is Unrelated to Effects of SOCE on Leukemic Cell Numbers, Proliferation, and Viability
To understand how SOCE affects T-ALL severity and progression, we analyzed tumor cell numbers and viability in the BM and spleen of mice with WT and Stim1/2−/− leukemia. Neither the frequencies (Figures 2A and 2B) nor the absolute numbers (Figures 2C and 2D) of GFP+ leukemic cells in the BM and spleen significantly differed between the two cohorts during the first 3 weeks of disease. However, at a later stage of leukemia, immediately before the death of WT mice (on day 24), we observed a sudden decline in leukemic cell numbers in the BM and spleen of the WT, but not the Stim1/2−/− group (Figures 2A–2D). In fact, the total numbers of Stim1/2−/− leukemic cells in the BM remained constant from day 24 to 46 of disease and increased by more than 4-fold in the spleen (Figures 2C and 2D), resulting in a doubling of the spleen weight (Figure 2E). By day 46, all mice with WT leukemia had already succumbed to disease. To understand the cause of the sudden loss of WT but not Stim1/2−/− leukemic cells around day 24, we investigated the role of SOCE in the proliferation and apoptosis of leukemic T lymphoblasts. Similar percentages of proliferating Ki-67+ cells were observed in Stim1/2−/− compared to WT leukemic cells at day 10 of disease, whereas a moderate increase in proliferation of Stim1/2−/− leukemic cells was observed at days 14 and 21 (Figure 2F). We further analyzed the numbers of Annexin-V+ apoptotic leukemic cells in the BM and spleen of mice, but did not observe a significant difference in the frequencies of apoptotic cells in mice with WT and Stim1/2−/− leukemia on days 14 and 21 (Figure 2G). Collectively, these data demonstrate that SOCE does not have a strong effect on the proliferation or apoptosis of leukemic cells that could explain the sudden loss of WT leukemic cells and the survival benefit of mice with Stim1/2−/− leukemia.
Figure 2. Survival Benefit of Mice with STIM1/STIM2-Deficient Leukemia Is Unrelated to Altered Leukemic Cell Numbers, Proliferation, and Survival.

(A–D) Percentages (A and B) and absolute numbers (C and D) of GFP+ leukemic cells at 14 and 24 days of disease in the BM and spleen of healthy WT control mice (ctrl) and mice with WT or Stim1/2−/− T-ALL. Flow cytometry plots (A) and cell numbers (B–D) are representative of 3–15 mice.
(E) Spleen weights of healthy WT control mice (ctrl) and mice with WT and Stim1/2−/− leukemia. Data are representative of three mice per group and time point. (F and G) Percentage of proliferating Ki-67+ (F) and Annexin-V+ apoptotic WT and Stim1/2−/− leukemic (G) cells measured by flow cytometry. Representative flow cytometry plots at day 21 after T-ALL induction (left) and quantification of proliferation and apoptosis (right). Data are representative of three to five mice per group and time point. All values represented by bar graphs are mean ± SEM. Statistical analysis was performed by Student’s t test. *p < 0.05.
STIM1/STIM2 Deficiency Protects Mice from Leukemia-Induced Tissue Damage
To investigate why Stim1/2−/− leukemia is less aggressive and results in prolonged survival, we analyzed the organs of mice infiltrated by leukemic cells. H&E stained sections of organs from mice with WT leukemia on day 24 of disease revealed extensive necrosis in the BM and granulomatous-like lesions with lympho-histiocytosis in the spleen (Figure 3A). Residual foci of leukemic cells were found within the areas of tissue damage. By contrast, no signs of necrosis or granulomatous-like lesions were apparent in the BM or spleen of mice with Stim1/2−/− leukemia (Figure 3A). Their organs were filled with monotonous lymphoid cells consistent with leukemic cells and the dominance of GFP+ cells observed by flow cytometry (Figures 2A and 2B). Although the massive infiltration of leukemic cells into the BM and spleen of mice with Stim1/2−/− leukemia effaced the overall normal tissue architecture, residual hematopoiesis was markedly reduced but detectable as evident from the presence of megakaryocytes (Figure 3A). Consistent with the collapse of leukemic cell counts in the organs of animals with WT leukemia on day 24, cells isolated from their BM and spleen showed reduced viability compared to cells isolated from mice with Stim1/2−/− eukemia (Figures 3B and 3C). To investigate death of leukemic cells in intact BM tissues, we stained femur sections with an antibody against CD3 to detect leukemic cells and used terminal deoxynucleotidyl transferase (TdT) dUTP nick-end labeling (TUNEL) to mark dead cells. Mice with WT leukemia showed a gradual increase in the frequency of TUNEL+ cells from day 14 to 24 of disease (Figure 3D). By contrast, only few TUNEL+ cells were observed in the BM of mice with Stim1/2−/− leukemia even on day 46 of disease (Figure 3D). Similar observations were made in the spleen (Figure S3). We conclude that suppression of SOCE in T-ALL cells preserves the viability of leukemia-infiltrated organs such as BM and spleen.
Figure 3. STIM1/STIM2-Deficient Leukemic Cells Fail to Cause Cell Death and Necrosis of Leukemia-Infiltrated Organs.

(A) H&E staining of BM (femur) and spleen from healthy WT control mice (Ctrl) and mice with WT and Stim1/2−/− leukemia 24 days after T-ALL induction Magnification 4003; images in bottom row represent boxed areas in top row. Images are representative of 3–5 mice per group.
(B and C) Viability of cells isolated from the BM (B) and spleen (C) of mice with WT and Stim1/2−/− leukemia measured by flow cytometry. Data are representative of 3–5 mice per cohort.
(D) Fluorescent confocal microscopy images of TUNEL staining and CD3 expression in the BM of mice with WT and Stim1/2−/− leukemia. Scale bar, 50 μm. Data are representative of three mice per group and time point.
No Anemia in Mice with Stim1/2−/− Leukemia
A common complication in patients with ALL is anemia, which has been attributed to a decline in erythropoiesis in the leukemic BM (Steele and Narendran, 2012). Because of the extensive BM necrosis in mice with WT but not Stim1/2−/− leukemia, we investigated the impact of T-ALL on erythroid precursor cells and erythropoiesis. The frequencies and especially the absolute numbers of large (FSChi) Ter119+ erythroblasts were significantly reduced in the BM and spleen of mice with WT leukemia at an advanced stage of leukemia (days 21–24) compared to mice with Stim1/2−/− leukemia or WT mice without leukemia (Figures 4A and 4B). Severely compromised erythropoiesis in the WT cohort resulted in anemia characterized by a progressive reduction of hemoglobin levels, which was less pronounced in mice with Stim1/2−/− leukemia (Figure 4C). It is noteworthy that although erythropoiesis in the BM of mice with Stim1/2−/− leukemia eventually also declined by day 46 (Figure 4A), their spleen continued to be a site of active erythropoiesis (Figure 4B). Therefore, in contrast to the WT cohort, mice with Stim1/2−/− leukemia maintained their hemoglobin levels until day 46 of disease (except for an initial decrease at day 14 that likely results from the irradiation of host mice) (Figure 4C). Taken together, deletion of STIM1/ STIM2 in leukemic cells protects mice from leukemia-associated suppression of erythropoiesis and anemia, which likely contribute to the death of mice with WT T-ALL.
Figure 4. Lack of STIM1 and STIM2 in Leukemic T Lymphoblasts Protects Mice from Anemia.

(A and B) Representative flow cytometry plots of erythropoietic precursor cells in the BM (A) and spleen (B) of mice with WT and Stim1/2−/− leukemia at day 21 of disease. Bar graphs show percentages and absolute numbers of Ter119+ forward scatter (FSC) high erythropoietic precursor cells in mice with WT and Stim1/2−/− leukemia as well as healthy WT control mice (ctrl). Data are representative of five to eight mice per group. n/a, not applicable (no cells available for analysis).
(C) Hemoglobin levels in the blood of healthy WT control mice (ctrl) and mice with WT and Stim1/2−/− leukemia. RV, range of reference values. Each dot represents a single mouse.
Cell numbers (A and B) and Hb levels (C) at days 21 and 24 of disease (D21/ D24) were combined. All values represented by bar graphs (A and B) and dot plots (C) are mean ± SEM. Statistical analysis was performed using Student’s t test. *p < 0.05.
Deletion of STIM1 and STIM2 Prevents Leukemia-Associated Inflammation
Given the extensive BM necrosis in mice with WT but not Stim1/2−/− T-ALL despite similar leukemic cell burdens in both cohorts, we hypothesized that SOCE enables WT leukemia cells to cause inflammation and tissue damage. In non-malignant T cells, SOCE is required for the production of cytokines (Feske, 2007), and their ability to cause inflammation in animal models of multiple sclerosis (Kaufmann et al., 2016; Ma et al., 2010; Schuh-mann et al., 2010), colitis (McCarl et al., 2010), and graft-versus-host disease (Vaeth et al., 2017). We observed an increased presence of neutrophils in blood smears of mice with WT leukemia on day 24, which was absent in mice with Stim1/2−/− leukemia even at later stages (day 46) of disease (Figure 5A). H&E-stained BM sections from late stage (day 24) WT leukemia showed macrophages engulfing red blood cells (RBCs) (Figure 5B), which were absent in the BM of the Stim1/2−/− cohort. Phagocytosis of RBCs is a pathologic sign of immune hyperactivation reminiscent of hemophagocytic lymphohistiocytosis (HLH) (Filipovich, 2009). We found increased frequencies and total numbers of CD11b+ F4/80+ macrophages in the BM and spleen of mice with WT compared to Stim1/2−/− leukemia (Figures 5C and 5D). Tumor-associated macrophages (TAMs) have been described in many types of cancers, where they promote cancer growth through their ability to produce inflammatory and immunomodulatory cytokines (Solinas et al., 2009). We therefore measured cytokine levels in the serum of leukemic mice and observed progressively increasing inflammatory cytokine concentrations in the WT cohort over the course of disease (Figure 5E). This cytokine response was strongly reduced or delayed until day 46 of disease in mice with Stim1/2−/− leukemia. In particular, the proinflammatory cytokines interferon γg (IFNγ), tumor necrosis factor α (TNF-α) and interleukin-17 (IL-17) were significantly decreased in the serum of mice with Stim1/2−/− compared to WT leukemia. Similar reductions were observed for IL-4 and IL-10. Collectively, these data suggest that murine T-ALL cells require SOCE to induce inflammation in leukemia-infiltrated organs.
Figure 5. STIM1/STIM2 Deficiency Prevents Leukemia-Associated Inflammation.

(A) Wright-Giemsa-stained blood smears from healthy WT control mice (untreated) and mice with WT and Stim1/2−/− leukemia. Arrows indicate neutrophils. Magnification 4003. Data are representative of three mice per group.
(B) H&E-stainedBMsections of mice with WT and Stim1/2−/− leukemia at day 24 of disease. Magnification 400×. Inset for WTBMshows a macrophage engulfing red blood cells. Images are representative of 7–10 mice per cohort.
(C and D) Percentages and absolute numbers of F4/80+CD11b+ macrophages in the BM (C) and spleen (D) of healthy WT control mice (untreated, UT) and mice withWT and Stim1/2−/− leukemia. Representative flow cytometry plots at days 14 and 24 of disease (left) and quantification of macrophage numbers (right). Data are representative of 3–5 mice per cohort and time point.
(E) Serum levels of cytokines in healthy WT control mice (UT) and mice with WT and Stim1/2−/− leukemia measured by multiplex assay. Data are representative of 3–5 mice per cohort.
All values represented by bar graphs (C–E) are mean ± SEM. Statistical analysis in all experiments was performed using Student’s t test. *p < 0.05. nd, not detected; ns, not significant; nt, not tested (no cells or mice available).
SOCE Intrinsic to Leukemic T Lymphoblasts Promotes Inflammation and Tumor Progression
Our findings indicated that SOCE plays a major role in T-ALL associated tissue damage, inflammation, and anemia. To induce T-ALL, however, we had cotransferred ICN1-transduced WT or Stim1/2−/− BM progenitor cells together with WT BM cells to reconstitute the immune system of the irradiated host mice (Figure 1A). In this model, non-transduced BM progenitors of WT or Stim1/2−/− origin also contribute to immune system reconstitution and we therefore could not exclude that the delayed mortality of mice with Stim1/2−/− T-ALL was due to deletion of STIM1 and STIM2 in the reconstituted immune cells rather than the leukemic T cells. To test the specific effect of SOCE in T-ALL cells themselves on the leukemia phenotype and survival, we induced T-ALL using two additional approaches. First, we transduced c-kit+ lin– BM progenitor cells from WT and Stim1/2−/− (Stim1fl/flStim2fl/fl Vav-Cre) mice with ICN1 as before (Figure 1) and injected them intravenously (i.v.) into either irradiated WT or Stim1/2−/− (Stim1fl/flStim2fl/fl Vav-Cre) mice (Figure 6A). Regardless of whether host mice were WT or Stim1/2−/− in their immune compartment, ICN1+ WT leukemic cells induced severe weight loss and fast progressing disease with average survival times of 33.5 and 31.5 days, respectively (Figures 6B and 6C). By contrast, transfer of ICN1+ Stim1/2−/− leukemic cells induced leukemia with significantly reduced weight loss and prolonged survival regardless of whether the hosts had WT or Stim1/2−/− immune cells. These findings suggest an important cell-intrinsic role of STIM1 and STIM2 in leukemic cells rather than the host immune cells in determining the outcome of T-ALL.
Figure 6. STIM1 and STIM2 in Leukemic T Lymphoblasts Promote Inflammation, Anemia, and Leukemia-Associated Mortality.

(A) BM cells from WT or Stim1fl/flStim2fl/flVav-Cre (Stim1/2−/−) mice were transduced with ICN1-IRES-GFP and cultured for 3 days in vitro. 5 × 104 ICN1transduced (GFP+) lineage negative (lin–) BM progenitor cells were injected i.v. together with 5 × 105 BM cells from WT or Stim1/2−/− mice into lethally irradiated WT or Stim1/2−/− host mice.
(B) Relative weight of mice during disease progression. Values are mean ± SEM. Statistical analysis was performed using one-way ANOVA. ***p < 0.0001.
(C) Survival of mice with T-ALL. Statistical significance was calculated using the non-parametric Kaplan-Meier estimator. Data in (B) and (C) are from6–7 mice per cohort.
(D–K) Secondary transfer of leukemic T lymphoblasts. T-ALL in WT mice was induced using BMprogenitor cells from WT or Stim1fl/flStim2fl/flVav-Cre (Stim1/2−/−) mice as described in (A). On day 8 following injection of ICN1+ progenitor cells, cells from the BM and spleen of mice were enriched for GFP+ leukemic T lymphoblasts. 1 × 105 GFP+ cells were injected i.v. into sublethally irradiated WT host mice (D). Relative weight of mice with WT and Stim1/2−/− leukemia. Values are mean ± SEM. Statistical analysis was performed using Student’s t test. ***p < 0.0001 (E). Survival of mice with WT and Stim1/2−/− leukemia. Statistical significance was calculated with the non-parametric Kaplan-Meier estimator (F). Data in (E) and (F) are from seven mice per cohort. Representative flowcytometry plots (top) and means ± SEM (bottom) of total cell numbers as well as percentages and absolute numbers of GFP+ leukemic cells in the spleen and BM of mice at 21 days of T-ALL. Data from three mice per cohort (G). H&E stain of spleen and BM (femur) from mice with WT and Stim1/2−/− leukemia at day 21 of T-ALL. Magnification 4003; images in bottom row represent boxed areas in top row. Images are representative of three mice per cohort (H).
(I and J) Representative flow cytometry plots (I) and means ± SEM (J) of Ter119+ FSChigh erythroid progenitor cells in the spleen and BM at 21 days of disease. Percentages refer to total cells isolated from the BM and spleen. Data are representative of three mice per group.
(K and L) Hemoglobin levels (K) and percentages of monocytes (L) in the blood of healthy WT control mice (Ctrl) and mice with WT and Stim1/2−/− leukemia at 21 days of disease. Each dot represents one mouse. RV, reference values.
In the second approach, we first induced T-ALL as described above by transducing c-kit+ lin– BM progenitor cells from WT and Stim1/Stim2fl/fl Vav-cre (Stim1/2−/−) mice with ICN1 and injecting them into irradiated WT host mice (Figure 6D). Next, we purified GFP+ leukemic cells from the spleen and BM by cell sorting (Figures S4A and S4B) and injected them into sublethally irradiated WT mice to generate secondary leukemia. Host mice injected with WT leukemic cells lost weight and died on average after 20 days (Figures 6E and 6F). By contrast, host mice injected with Stim1/2−/− leukemic cells did not lose weight and died on average after 49 days. This 2.5-fold prolonged survival occurred despite an increased leukemic burden in host mice injected with Stim1/2−/− compared toWT leukemic cells at day 21 (Figure 6G). Histological analysis of host mice at day 21 after leukemic cell transfer showed pronounced necrosis and serous atrophy in the BM and prominent granulomatous-like infiltrates in the spleens of mice that had received WT leukemic cells, whereas recipients of Stim1/2−/− leukemic cells did not show these changes (Figure 6H). Recipients of WT, but not of Stim1/2−/−, leukemic cells also had reduced numbers of Ter119+ FSChi erythroid progenitor cells in BM and spleen (Figures 6I and 6J) suggesting that SOCE in leukemic cells impairs erythropoiesis. Accordingly, we observed more profound anemia in the WT than the Stim1/2−/− leukemia cohort, which was apparent by reduced hemoglobin levels (Figure 6K), red blood cell counts and hematocrit (Figure S4C). In addition, mice with WT leukemia also had a higher percentage of monocytes in their peripheral blood compared to the Stim1/2−/− leukemia cohort (Figure 6L). Collectively, these findings demonstrate that the protective effects of STIM1 and STIM2 deletion in T-ALL are intrinsic to the leukemic cells and not caused by the lack of SOCE in the non-malignant host immune cells.
SOCE Regulates the Transcription of Proinflammatory Genes in Leukemic Cells
To gain insight into the molecular regulation of SOCE-dependent inflammation in T-ALL, we investigated gene expression in WT and Stim1/2−/− GFP+ leukemic cells isolated from the BM on day 21 of disease. The expression of hundreds of genes was dysregulated in Stim1/2−/− compared to WT leukemic cells (p < 0.05, >2-fold change in expression) (Figure 7A). Because intracellular Ca2+ signals modulate the expression of NOTCH1-regulated genes (Ouyang et al., 2014; Roti et al., 2013), we first analyzed how lack of SOCE affects the expression of NOTCH1 target genes in ICN1-expressing leukemic cells. The mRNA levels of Dtx1, Hes5 and Hey family members were significantly reduced in Stim1/2−/− compared to WT leukemic cells, whereas the expression of Gata3, Nrap and Ifi204 was comparable (Figure 7B; data not shown). By contrast, expression of Myc, an important NOTCH1 target gene in T-ALL, was increased 2-fold in Stim1/2−/− compared to WT leukemic cells (Figure 7B), which may explain the moderately increased proliferation of Stim1/2−/− compared to WT leukemic cells at days 14 and 21 of disease (Figure 3A). These findings suggest that SOCE promotes the expression of many NOTCH1 target genes (with the exception of Myc), at least under conditions of constitutive NOTCH1 signaling by ectopically expressed ICN1. A further unbiased pathway analysis of differentially expressed genes (DEGs) in leukemic cells showed an upregulation of pathways associated with cell cycle, chromosome segregation, DNA repair and microtubules in Stim1/2−/− compared to WT leukemic cells, whereas pathways associated with the regulation of antiviral and innate immune responses were strongly downregulated (Figure 7C). Consistent with the later finding, gene set enrichment analysis (GSEA) revealed downregulation of genes associated with prion-flammatory cytokines and type I interferon responses in Stim1/2−/− leukemic cells (Figures 7D–7J). We observed a profound reduction in the expression of genes associated with IFNγ signaling in Stim1/2−/− T-ALL cells, including IFNγ itself (Figure 7D), which is consistent with reduced IFNγ levels in the serum of mice with Stim1/2−/− leukemia (Figure 5E). In addition, many interferon regulatory factors (IRFs), such as Irf1–2 and Irf7–9, and IFNγ-induced guanylate binding proteins (GBPs), such as Gbp1, Gbp2, and Gbp4–7, were downregulated in Stim1/2−/− leukemic cells (Figure 7E), demonstrating the absence of an IFNγ-driven inflammatory immune response in mice with Stim1/2−/− leukemia. IFNγ, which is normally produced by CD4+ T helper 1 (Th1), CD8+, and natural killer (NK) cells, is critical for the activation of macrophages and their ability to present antigen, kill phagocytosed pathogens and produce proinflammatory cytokines, such as IL-12 or IL-18. The decreased IFNγ response by Stim1/2−/− leukemic cells is consistent with the reduced numbers of F4/80+ Cd11b+ macrophages (Figures 5C and 5D) and absent signs of hemophagocytosis (Figure 5B) in mice with Stim1/2−/− compared to WT leukemia.
Figure 7. Deregulated Expression of Proinflammatory Genes in STIM1/STIM2-Deficient Leukemic T Lymphoblasts.

RNA sequencing (RNA-seq) analysis of GFP+ leukemic T lymphoblasts from the BM of mice with WT and Stim1/2−/− T-ALL at day 21. All data in (A)–(J) are from three mice per cohort. MA plot showing the relationship between log2-fold change of RNA reads in Stim1/2−/− versus WT leukemic cells plotted against mean expression values. Red dots represent genes with an adjusted p value < 0.05 (A). Heatmap of NOTCH1 target gene expression in Stim1/2−/− and WT leukemic cells. Shown are row minimum (blue) and maximum (red) values; percentages refer to the relative change in gene expression in Stim1/2−/− compared to WT leukemic cells (B). Gene ontology (GO) analysis of RNA-seq data using DAVID with significant GO terms (false discovery rate [FDR] < 0.0001) plotted with its corresponding gene expression data using the GO plot package (Walter et al., 2015) (C). Gene set enrichment analysis (GSEA) (D, F, and H) and heatmaps (E, G, I, and J) of differential gene expression in Stim1/2−/− versus WT leukemic cells. IFNγ signaling (D and E), cytokine signaling (F and G), type I interferon responses (H and I), and tissue remodeling pathways (J) are significantly downregulated in Stim1/2−/− leukemic cells. Heatmaps in (E), (G), (I), and (J) of selected genes show relative mRNA expression levels (maximal, red; minimal, blue) per gene (row). Adjacent tables show the percentage change in expression in Stim1/2−/− leukemic cells relative to WT with its corresponding adjusted p value calculated using DESeq2’s differential expression test.
Stim1/2−/− leukemic cells also lacked expression of many genes associated with cytokine signaling (Figure 7F). Of the cytokines whose levels were decreased in the serum of mice with Stim1/2−/− leukemia, only TNF-a and IL-10 were expressed by leukemic cells and lower in the Stim1/2−/− than the WT cohort (Figure 7G). Furthermore, levels of IL-16, IL-22, IL-34, and colony-stimulating factor 1 (CSF-1) were decreased in Stim1/2−/− compared to WT leukemic cells (Figure 7G). Of these cytokines, IL-16 was expressed most highly in WT leukemic cells, consistent with its role as a lymphocyte-derived chemoattractant that recruits and activates monocytes, eosinophils, and dendritic cells and the increased numbers of macrophages in the BM of mice with WT leukemia (Figures 5C and 5D). Besides IL-16, other chemokines mediate the recruitment of TAMs to tumors. We detected the expression of six chemokines in WT leukemic cells, of which four (CCL6, CCL9, CXCL9, and CXCL10) were significantly reduced in Stim1/2−/− leukemic cells (Figure 7G). The chemokine most highly expressed in WT leukemic cells that showed the largest defect in Stim1/2−/− T-ALL cells was CCL6, a monocyte attractant chemokine that induces inflammation (LaFleur et al., 2004). Many cytokines signal via JAK/ STAT pathways that are critical for lymphocyte activation, differentiation, and proliferation (Rawlings et al., 2004). Several hematopoietic malignancies are associated with constitutive activation of JAK/STAT signaling (Vainchenker and Constantinescu, 2013) and persistent activation of STAT3 mediates tumor-promoting inflammation (Yu et al., 2009). We found that expression of all three JAK kinases and of STAT3 was significantly impaired in Stim1/2−/− compared to WT leukemic cells (Figure 7G). These data suggest that blunted cytokine and chemokine signaling in Stim1/2−/− T-ALL cells contributes to reduced leukemia-induced inflammation in these mice. In addition to cytokines, GSEA showed a broad and strong reduction in IFNα and IFNβ response signatures in Stim1/2−/− compared to WT leukemic cells (Figure 7H). This included several IRFs (Irf1, Irf2, Irf7, and Irf9), GBPs and other interferon-induced genes such as Ifitm1, Ifi1 and Ifi44 (Figure 7I). By contrast, the expression of type I IFN receptors (IFNAR1, IFNAR2) that mediate IFNα/β signaling was comparable on Stim1/2−/− and WT leukemic cells. The strongly impaired type I IFN signature in Stim1/2−/− leukemic cells correlates with the lack of inflammation in leukemia infiltrated organs of Stim1/2−/− leukemic mice. Activation of tissue remodeling pathways has been reported in association with tumor-induced inflammation in various cancers (Johnsen et al., 1998). The analysis of DEG in leukemic cells revealed a significantly decreased expression of matrix metalloproteases (MMP) and a disintegrin and metallopeptidase domain (ADAM) family members, which are involved in the degradation of extracellular matrix (Page-McCaw et al., 2007), in Stim1/2−/− compared to WT leukemic cells (Figure 7J). Furthermore, expression of several S100 family members, which promote tumor growth and metastasis by inducing inflammation and tissue remodeling (Bresnick et al., 2015), was significantly decreased in Stim1/2−/− leukemic cells. These findings indicated that SOCE regulates genes involved in tissue remodeling and inflammation in leukemic T lymphoblasts. Taken together, our data demonstrate that SOCE regulates several pathways associated with cancer-induced inflammation including production of cytokines, chemokines, and tissue remodeling factors that promote the recruitment and activation of myeloid cells, inflammation, and cell death, which in turn induce a type I interferon response in tumor infiltrated tissues. Deletion of SOCE in leukemic T lymphoblasts suppresses these pro-inflammatory pathways and prevents inflammation-induced tissue damage and death of mice from NOTCH1-driven T-ALL.
DISCUSSION
Here, we show that abolishing SOCE in leukemic T cells by deletion of Stim1 and Stim2 results in prolonged survival of mice with T-ALL. Although leukemic burdens were not affected in the absence of SOCE, there was a striking difference between organs infiltrated with SOCE-competent or SOCE-deficient leukemic cells. Whereas mice with WT leukemia showed severe inflammation of leukemia-infiltrated organs, inflammation was absent in mice with Stim1/2−/− leukemia. The necroinflammatory process in WT leukemic mice was associated with impaired erythropoiesis, anemia, expression of proinflammatory cytokines and chemokines, and a type I interferon response. Collectively, our study demonstrates that T-ALL is associated with inflammation in leukemia-infiltrated organs, tissue damage, and suppression of erythropoiesis, which is dependent on SOCE in leukemic T lymphoblasts.
The proinflammatory function of SOCE in non-malignant T cells is well established through work by us and others (Kaufmann et al., 2016; Kim et al., 2014; Ma et al., 2010; McCarl et al., 2010; Schuhmann et al., 2010). A similar role of SOCE in malignant T lymphoblasts, however, has not been reported. The protective effects of deleting STIM proteins and SOCE from tumor-associated inflammation, T-ALL progression and death were intrinsic to the leukemic T lymphoblasts themselves and not due to impaired SOCE in non-malignant immune cells. Secondary transfer of Stim1/2−/− T-ALL cells into WT mice resulted in significantly prolonged survival of mice compared to transfer of WT leukemic cells. Similarly, transfer of ICN1-transduced WT progenitor cells into either WT or Stim1/2−/− mice resulted in the same fast leukemia progression and death of hosts, whereas transfer of Stim1/2−/− ICN+ progenitor cells resulted in slower disease progression.
Inflammation is a common feature of many solid and hematological malignancies and is widely recognized as an important factor in tumor development, progression, metastasis, and resistance to treatment, for instance through its ability to suppress antitumor immune responses (Grivennikov et al., 2010; Shalapour and Karin, 2015). By contrast, inflammation and necrosis of leukemia-infiltrated tissues are not widely recognized as a feature of T-ALL or NOTCH1-driven mouse models of T-ALL. However, multiple studies have reported BM necrosis associated with leukemia. For instance, BM necrosis was observed in 3.2% of ALL patients and was associated with a poor treatment response and prognosis (Badar et al., 2015). ALL patients with BM necrosis had complete remission rates of 70% compared to 92% in ALL patients without BM necrosis. The fact that deletion of STIM1 and STIM2 completely prevents BM necrosis in ICN1-induced T-ALL suggests that inhibition of SOCE is a possible therapeutic approach in patients with ALL and BM necrosis that otherwise have a poor prognosis. Our findings are consistent with recent studies that emphasize the complex interaction between leukemic cells and specific niches in various organs that have profound effects on leukemia biology including disease development and outcome (Chiarini et al., 2016; Hawkins et al., 2016; Passaro et al., 2015; Pitt et al., 2015).
Inflammation in mice with T-ALL was characterized by several SOCE-dependent hallmarks:
1. T-ALL in the WT, but not the Stim1/2−/−, cohort was associated with increased numbers of neutrophils and monocytes in the blood and TAMs in leukemia-infiltrated tissues. TAMs establish a local inflammatory environment that supports cancer growth, angiogenesis, and metastasis, and their presence is strongly correlated with a poor cancer prognosis (Solinas et al., 2009). Consistent with increased TAM infiltration, we detected hemophagocytosis in mice with WT leukemia, which likely contributes to their anemia. Hemophagocytosis is usually seen in hyperactive immune reactions such as systemic HLH. Whereas primary HLH is a rare genetic disorder caused by defects in the degranulation of T or NK cells resulting in ineffective clearance of infection and immune hyperactivation (Otrock and Eby, 2015), secondary HLH is associated with certain infections or malignancies including T-ALL (Falini et al., 1990; O’Brien et al., 2008; Otrock and Eby, 2015; Su et al., 1993; Trebo et al., 2005). Importantly, we did not observe hemophagocytosis in the BM of mice with Stim1/2−/− leukemia, suggesting that SOCE in leukemic cells is required for TAM recruitment and/or activation.
2. The expression of proinflammatory cytokines and chemokines was elevated in WT, but not (or to a lesser degree) in Stim1/2−/−, leukemic T lymphoblasts. This included IFNγ, which is normally produced by CD4+ Th1 and CD8+ T cells and whose main function is to activate macrophages. The reduced levels of IFNγ protein in the serum and IFNγ mRNA in leukemic T lymphoblasts is consistent with the lack of hemophagocytosis in Stim1/2−/− leukemic mice. Furthermore, our global gene expression analysis in T-ALL cells revealed that many inflammation-associated cytokines and chemokines required to recruit TAMs were significantly downregulated in STIM1/2-deficient leukemic cells. Among these were IL-16, IL-34, and CSF-1. IL-16 is a lymphocyte-derived chemoattractant that recruits and activates monocytes, eosinophils, and dendritic cells and IL-34 functions as a monocyte growth stimulatory factor. CSF-1 is critical for TAM expansion in tumors; inhibition of the CSF-1 receptor in tumor-bearing mice prevents TAM infiltration and has been investigated as a therapeutic approach in patients with refractory Hodgkin lymphoma (von Tresckow et al., 2015). In addition, we found that CCL6 and CCL9 are highly expressed in WT, but not Stim1/2−/−, leukemic cells. CC chemokines are major determinants of TAM infiltration into tumors based on studies of melanoma, carcinoma, glioma, and breast cancers (Lewis and Pollard, 2006; Murdoch et al., 2004). CCL6 is a proinflammatory chemokine that attracts monocytes and promotes tumor metastasis (LaFleur et al., 2004). Collectively, the reduced expression of chemokines and of IL-16, IL-34, and CSF1 is consistent with the decreased numbers of F4/80+ Cd11b+ macrophages in theBMof mice with Stim1/2−/− leukemia and an important proinflammatory role of SOCE in leukemic cells via the recruitment and activation of TAMs into leukemia-infiltrated tissues.
3. WT, but not Stim1/2-/-, leukemic T lymphoblasts featured a type I interferon response gene expression signature. The expression of IFNα and IFNβ is regulated by IRFs in response to danger-associated molecular patterns (DAMPs), such as nucleotides released from necrotic tumor cells. IFNα and IFNβ signal through IFNAR1 and IFNAR2 and control expression of a variety of interferon response genes. Depending on the biological context, type I interferons were shown to have anti- or protumor effects. Impaired production of type I interferons results in immune evasion and enhanced progression of several types of cancer (Bidwell et al., 2012; Katlinski et al., 2017; Zitvogel et al., 2015). In chronic myeloid leukemia (CML), other types of leukemia (Cull et al., 2003; Quesada et al., 1984), and lymphoma (Armitage et al., 2006; Aviles et al., 2015; Olsen, 2003), type I interferons have been shown to possess strong anti-tumor effects and were a first line treatment before the era of kinase inhibitors (Bonifazi et al., 2001; Kantarjian et al., 2003). In Stim1/2−/− leukemic T lymphoblasts, type I interferon response genes were strongly downregulated despite normal expression of IFNAR1 and IFNAR2, suggesting that IFNα and IFNβ levels are low in Stim1/2−/− leukemic mice. Given the generally tumor-suppressing role of type I interferons and the downregulation of type I interferon responses in Stim1/2−/− leukemic T lymphoblasts, it was surprising to find prolonged survival of mice with Stim1/2−/− T-ALL. However, type I interferons can also promote tumor growth as has been demonstrated in several tumor types. A recent study showed that type I interferons promote stemness of cancer cells as well as resistance to therapy and poor outcome in adenocarcinomas and squamous cell carcinoma (Qadir et al., 2017). In human glioblastoma, increased expression of IRF7 led to a more aggressive therapy resistant tumor and shorter overall survival of patients (Li et al., 2017). Likewise, in viral infections such as influenza excessive type I IFN responses can have detrimental effects with increased inflammation and cell death that are associated with increased mortality (Iwasaki and Pillai, 2014). The role of IFNα and IFNβ in T-ALL has not yet been defined. Our finding of a strong type I interferon signature in WT, but not Stim1/2−/−, T-ALL cells suggests that IFNα and IFNβ are produced in the leukemia-infiltrated tissues, in response to SOCE-dependent inflammation and cell death, where they may modulate T-ALL progression.
Recent reports have shown an important link between Ca2+ homeostasis in the ER and T-ALL (Ouyang et al., 2014; Roti et al., 2013). Deletion of all three IP3R homologs in T cells, which prevents Ca2+ release from the ER, causes spontaneous T-ALL development that is associated with increased NOTCH1 expression (Ouyang et al., 2014). By contrast, inhibition of SERCA pumps to prevent reuptake of Ca2+ from the cytosol into the ER decreased ICN1 levels, induced a G0/G1 arrest in NOTCH1-mutated human leukemia cells and attenuated tumor growth in two human xenograft models of T-ALL (Roti et al., 2013). Together, these studies suggest that Ca2+ release from the ER suppresses leukemia likely by interfering with NOTCH1 signaling. Importantly, ER Ca2+ dynamics are coupled to SOCE because depletion of ER Ca2+ stores promotes activation of STIM1, STIM2, and SOCE, which, in turn, mediates the refilling of ER Ca2+ stores (Feske, 2007). Here, we show that abolishing SOCE in leukemic T lymphoblasts prolongs the survival of mice with Stim1/2−/− compared to WT T-ALL. The fact that interfering with SOCE, IP3R expression, or SERCA function affects the course of leukemia indicates that Ca2+ signaling is a critical factor in T-ALL pathophysiology. However, inhibition of IP3R, SERCA, and STIM1/STIM2 function affects different aspects of T-ALL pathology. Whereas modulation of ER Ca2+ homeostasis via inhibition of IP3R and SERCA function affected T-ALL development (Ouyang et al., 2014; Roti et al., 2013), interfering with SOCE by deletion of STIM1/STIM2 did not have an overt effect on T-ALL development but instead attenuated inflammation in leukemia-infiltrated organs. These data suggest that the origin of Ca2+ signals critically determines the effects on T-ALL.
Here, we propose a model in which leukemic T lymphoblasts cause a necroinflammatory response in the organs they infiltrate. This response is dependent on SOCE, which controls the expression of proinflammatory cytokines and chemokines, such as INFg, IL-16, CCL6, and others that recruit TAMs into the leukemia-infiltrated tissues. Activated TAMs amplify inflammation, cause tissue damage, and mediate hemophagocytosis, resulting in impaired erythropoiesis and anemia. Tissue necrosis and the death of leukemic cells result in the release of nucleic acids that act as danger signals and cause the production of type I interferons by cells in leukemia-infiltrated organs, which further exacerbates inflammation. In the absence of SOCE, leukemic T lymphoblasts fail to produce chemokines and cytokines that would normally recruit and activate TAMs, resulting in attenuated inflammation and a suppressed type I interferon response. Our findings shed light on the pathogenesis of T-ALL and provide a rationale for targeting SOCE as a therapeutic approach to improve the long-term outcome of T-ALL through the suppression of detrimental leukemia-induced inflammation.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT OR RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CD3 (17A2; 145–2C11) | ebioscience | Cat# 48–0032-82, Cat# 12–0031-81 |
| Anti-mouse CD4 (GK1.5) | ebioscience | Cat# 17–0041-82, Cat# 48–0041-82, Cat# 12–0041-82 |
| Anti-mouse CD5 (53–7.3) | ebioscience | Cat# 12–0051-81 |
| Anti-mouse CD8 (53–6.7) | ebioscience | Cat# 12–0081-81, Cat# 17–0081-82, Cat# 48–0081-82 |
| Anti-mouse CD11b (M1/70) | ebioscience | Cat# 12–0112-81, Cat# 48–0112-82 |
| Anti-mouse CD45.1 (A20) | ebioscience | Cat# 17–0453-82 |
| Anti-mouse CD45.2 (104) | ebioscience | Cat# 48–0454-82 |
| Anti-mouse CD71 (R17217) | ebioscience | Cat# 17–0711-80 |
| Anti-mouse B220 (RA3–6B2) | ebioscience | Cat# 12–0452-82 |
| Anti-mouse GR-1 (RB6–8C5) | ebioscience | Cat# 12–5931-82 |
| Anti-mouse Ter119 (TER-119) | ebioscience | Cat# 12–5921-82 |
| Anti-mouse F4/80 (BM8) | Biolegend | Cat# 123127 |
| Anti-mouse Ki67 (16A8) | Biolegend | Cat# 652404, Cat# 652421 |
| Annexin V | Biolegend | Cat# 640918, Cat# 640920 |
| Anti-mouse CD3 (2GV6) | Ventana Medical Systems | Cat# 790–4341 |
| Chemicals, Peptides and Recombinant Proteins | ||
| DMEM medium | Mediatech | Cat# 10–013-CV |
| Opti-MEM medium | Gilbeco | Cat# 11058–021 |
| L-glutamine | Mediatech | Cat# 25–005-CI |
| Fetal bovine serum | Mediatech | Cat# 35–011-CV |
| Penicillin/streptomycin | Mediatech | Cat# 30–002-CI |
| GenJET transfection reagent | SignaGen | Cat# SL100489 |
| Polybrene | Sigma Aldrich | Cat# 107689–10G |
| Poly-L lysine | Sigma Aldrich | Cat# P8920 |
| Poly (I:C) | Sigma-Aldrich | Cat# P0913–50MG |
| Mouse IL-6 | Peprotech | Cat# 216–16 |
| Mouse IL-7 | Peprotech | Cat# 217–17 |
| Mouse FLT3-L | Peprotech | Cat# 250–31L |
| Mouse SCF | Peprotech | Cat# 250–03 |
| Thapsigargin | EMD millipore | Cat# 586005–1MG |
| Fura-2-AM | Invitrogen | Cat# F1201 |
| Fluo-4-AM | Invitrogen | Cat# F14201 |
| Critical Commercial Assays | ||
| Annexin V Binding buffer | Biolegend | Cat# 422201 |
| Foxp3 permeabilization kit | ebioscience | Cat# 00–5521-00 |
| EasySep mouse CD117 (cKIT) positive selection kit | StemCell Technologies | Cat# 18757 |
| Ventana’s reagents and detection kits | Ventana Medical Systems | N/A |
| In Situ Cell Death TMR Detection Kit | Roche Diagnostics | Cat# 12156792910 |
| MILLIPLEX MAG mouse cytokine / chemokine magnetic bead kit | Millipore | MCYTOMAG-70K-10 |
| RNeasy Micro Kit | QIAGEN | Cat# 74004 |
| Deposited Data | ||
| RNA-seq data | This paper | GEO: GSE116776 |
| Experimental Models: Cell Lines | ||
| Platinum E cells | Cell Biolabs | Cat# RV-101 |
| Experimental Models: Organisms/Strains | ||
| WT CD45.1+ C57BL/6 mice | Jackson Laboratories | Cat# 002014 |
| WT CD45.2+ C57BL/6 mice | Jackson Laboratories | Cat# 000664 |
| Stim1fl/flStim2fl/fl Mx1-Cre | Vaeth et al., 2015 | N/A |
| Stim1fl/flStim2fl/fl Vav-Cre | Vaeth et al., 2015 | N/A |
| Recombinant DNA | ||
| MSCV-based ICN1-IRES-GFP vector | Li et al., 2008 | N/A |
| pCL-10A1 | Miller and Chen, 1996 | N/A |
| Software and Algorithms | ||
| FACSDiva software | BD Bioscience | N/A |
| Flowjo v.8.7 and v.10.0.8r1 | FlowJo, LLC | https://www.flowjo.com/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Prism 7 | GraphPad | https://www.graphpad.com/ |
| LCS software package | Leica | N/A |
| Illustrator CS5 | Adobe Systems | N/A |
| Photoshop CS5 | Adobe Systems | N/A |
| Other | ||
| Amicon Ultra-15 centrifugal filters | Merck Millipore | Cat# UFC910024 |
| LSRII flow cytometer | BD Bioscience | N/A |
| FlexStation 3 multi-mode microplate reader | Molecular Devices | N/A |
| 96-well imaging plates | Fisher Scientific | Cat# 08–772-225 |
| IX81 epifluorescence microscope | Olympus | N/A |
| BX53 light microscope | Olympus | N/A |
| LSM 800 Meta confocal microscope | Zeiss | N/A |
| Hemavet 950 instrument | Drew Scientific | N/A |
| MAGPIX system | Luminex Corporation | N/A |
| Ventana Medical Systems Discovery XT | Ventana Medical Systems | N/A |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and request for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Stefan Feske (feskes01@nyumc.org). The mouse lines described in this study are available from our laboratory and require a Material Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
MICE
Stim1fl/flStim2fl/fl Mx1-Cre and Stim1fl/flStim2fl/fl Vav-Cre mice have been described previously (Vaeth et al., 2015). All mice were on the C57BL/6 background. Mice were maintained under specific pathogen free conditions in accordance with institutional guidelines for animal welfare approved by the Institutional Animal Care and Use Committee at New York University School of Medicine
METHOD DETAILS
Cells, cell culture and production of retroviral supernatants
Platinum E (Plat-E) cells for retroviral packaging were cultured in DMEM medium (Mediatech) supplemented with 10% (v/v) fetal bovine serum (FBS), 2 mM L-glutamine, 100 U/mL penicillin, and 100 μg/ml streptomycin (all from Mediatech) at 37°C, 10% CO2. Retroviral supernatant was produced by co-transfecting Plat-E packing cells with ICN1-IRES-GFP and pCL-10A1 retroviral packing constructs using the GenJET transfection reagent (SignaGen) according to the manufacturer’s instructions. Supernatants were collected 2 and 3 days later and concentrated using Amicon Ultra-15 centrifugal filters (Merck Millipore).
Plasmids and Reagents
The MSCV-based ICN1-IRES-GFP vector (Li et al., 2008) and the retroviral packaging construct pCL-10A1 (Miller and Chen, 1996) have been previously described.
Flow cytometry
Fluorescently conjugated antibodies against mouse CD3 (17A2), CD4 (GK1.5), CD5 (53–7.3), CD8 (53–6.7), CD11b (M1/70), CD71 (R17217), B220 (RA3–6B2), Gr-1 (RB6–8C5) and Ter-119 (TER-119) (all from eBiosciences), F4/80 (BM8), Ki-67 (16A8) and Annexin V (all from Biolegend) were used. Surface staining was performed on freshly isolated cells in phosphate buffered saline (PBS). Apoptosis was assessed using an Annexin-V staining kit (Biolegend). Intracellular CD3 and Ki-67 staining was performed using a Foxp3 permeabilization kit (eBioscience). All stains were done using 2 μg/ml of antibody. Cell viability was assessed by flow-cytometry by measuring forward scatter (FSC) and side scatter (SSC) of isolated bone marrow and spleen cell suspensions after lysis of erythrocytes in hypotonic buffer. Briefly, FSClow/SSClow cells were defined as cell debris and excluded from the analysis. FSChigh/ SSChigh cells were defined as viable, whereas FSClow/SSChigh cells as necrotic or late apoptotic. Percent of viable cells was defined as percent of FSChigh/SSChigh in the SSChigh population. Flow cytometry was conducted using a LSRII flow cytometer and FACSDiva software (BD Biosciences). Data were further analyzed with FloJo software (Tree Star).
T-ALL model
BM progenitor cells were isolated from the bone marrow (BM) of Stim1/Stim2fl/fl Mx1-Cre mice or Stim1fl/flStim2fl/fl Vav-Cre mice. To inducibly delete the loxP-flanked Stim1 and Stim2 alleles in hematopoietic lineage cells of Stim1fl/flStim2fl/fl Mx1-Cre mice, animals were injected i.p. with 10 mg/g body weight poly(I:C) (Sigma Aldrich) every other day for one week, followed by a one month resting period. Deletion of the loxP-flanked exons in Stim1 and Stim2 was confirmed by genomic PCR (Vaeth et al., 2015) and deletion efficiency by measuring SOCE in hematopoietic lineages. BM progenitor cells were isolated from the BM using the EasySep mouse CD117 (cKIT) positive selection kit (StemCell Technologies, Inc.). BM progenitor cells were plated in a density of 1×106 cells/ml and cultured in OPTI-MEM media (GIBCO) supplemented with 10% FBS (v/v) in the presence of 10 ng/ml IL-6, 10 ng/ml IL-7, 50 ng/ml FLT3-L and 50 ng/ml SCF (all from Peprotech). On days 1 and 2 after isolation, BM progenitor cells were transduced with concentrated retroviral supernatant containing pseudotyped ICN1-IRES-GFP plasmids in the presence of 6 μg/ml polybrene (Sigma Aldrich) using a spin infection protocol (2500 rpm, 30°C, 90 minutes). On day 4 after isolation, BM progenitor cells were stained with antibodies against CD3, CD4, CD5, CD8, CD11b, B220, Ter119 and Gr-1 to determine the frequency of ICN1 transduced (GFP+) lineage negative (Lin–) BM progenitor cells (CD3- CD4- CD5- CD8- CD11b- B220- Ter119- Gr-1-). 5×104 GFP+ Lin– cells were co-transferred together with 5×105 freshly isolated BM cells from wild-type (WT) C57BL/6 donors into lethally irradiated (1100 cGy) 6–8 weeks old female WT host mice by retro-orbital injection. Alternatively, for experiments shown in Figures 6A–6C, we either cotransferred 5×104 ICN1-transduced, GFP+ Lin– WT BM progenitor cells together with 5×105 freshly isolated WT BM cells into lethally irradiated WT or Stim1fl/flStim2fl/fl Vav-Cre host mice or 5×104 ICN1-transduced, GFP+ Lin– Stim1fl/flStim2fl/fl Vav-Cre progenitor cells together with 5×105 freshly isolated Stim1fl/flStim2fl/fl Vav-Cre BM cells into lethally irradiated WT or Stim1fl/flStim2fl/fl Vav-Cre host mice. Mice were monitored daily after transfer over the course of the disease and analyzed for leukemia as described.
For the secondary transfer of T-ALL cells, WT or Stim1/2−/− ICN1-transduced (GFP+) leukemic cells were isolated from the spleen and BM of leukemic mice on day 8 after leukemia induction as shown in Figure 6D and enriched for GFP+ leukemic cells by flow cytometry. 1×105 GFP+ cells were transferred into sublethally irradiated (600 cGy) female WT host mice by retroorbital injection to induce leukemia. Mice were analyzed for weight loss, survival and organ histopathology.
Intracellular Ca2+ measurements
To measure intracellular Ca2+ concentrations ([Ca2+]i), we used three different methods. The deletion efficiency of Stim1 and Stim2 in hematopoietic lineage cells was analyzed by flow cytometry. Splenocytes and BM cells were loaded with 1 mg/ml of the fluorescent Ca2+ indicator dye Fluo-4-AM and stained with fluorescently labeled antibodies against CD3, CD4, CD8, B220, CD11b. Baseline [Ca2+]i was measured in Ca2+-free Ringer solution (containing in mM: 155 NaCl, 4.5 KCl, 3 MgCl2, 10 D-glucose, 5 Na-HEPES pH 7.4), followed by addition of 1 mM thapsigargin (EMD Millipore) and 1 mM Ca2+-containing Ringer solution to induce store-operated Ca2+ entry (SOCE). Fluo-4 signals were recorded for 400–800 s and further analyzed by FloJo software. Alternatively, the [Ca2+]i in BM progenitor cells was analyzed using a FlexStation 3 multi-mode microplate reader (Molecular Devices). BM progenitor cells were loaded with 1 μg/ml of Fura-2-AM (Life Technologies) for 30 min, washed and plated in 0.01% poly-L-lysine (w/v) (Sigma-Aldrich) 96-well imaging plates (Fisher) and. Baseline [Ca2+]i was measured in Ca2+-free Ringer solution followed by addition of 1 μM thapsigargin and 2 mM Ca2+-containing Ringer solution. [Ca2+]i was analyzed as the ratio of Fura-2 emission after excitation at 340 and 380 nm. Lastly, [Ca2+]i in GFP+ leukemic cells isolated from the spleen of mice with T-ALL was analyzed by single cell Ca2+ imaging as described (Feske et al., 2006). Briefly, cells were loaded with 1 μM Fura-2-AM, plated onto UV-sterilized coverslips coated with poly-L-lysine and analyzed by time-lapse imaging on an IX81 epifluorescence microscope (Olympus). After stimulation with 1 mM thapsigargin in Ca2+-free Ringer solution, cells were perfused with Ringer solution containing 2 mM Ca2+. F340/F380 emission ratios of Fura-2 were recorded every 5 s for 10 min. All experiments were performed at room temperature (22–25°C). Analysis of > 20 cells per experiment was conducted using Slidebook imaging software v4.2.
Histology
Femurs, and spleen were dissected and fixed overnight in 4% paraformaldehyde. Samples were washed 3 times in PBS, dehydrated by incubation in 50% ethanol for 2h followed by overnight incubation in 70% ethanol. For bone samples, decalcification was performed by immersion in decalcification solution (10% disodium EDTA; pH 7.2) for two weeks, replacing the solution every other day. Samples were embedded in paraffin blocks, cut into 5 μm sections and stained with hematoxylin and eosin (H&E) using standard procedures. H&E stained tissue samples and Wright-Giemsa stained blood smears were analyzed using an Olympus BX53 light microscope at 400X magnification or using a SCN400 F slide scanner with SlidePath digital image hub software (Leica).
TUNEL staining and fluorescence microscopy
Immunohistochemistry was performed on paraformaldehyde-fixed, paraffin-embedded, 4-μm sections of spleens and decalcified femurs using unconjugated rabbit anti-mouse CD3 (clone 2GV6; Ventana Medical Systems Inc.). Immunofluorescence was performed on a Ventana Medical Systems Discovery XT instrument with online deparaffinization using Ventana’s reagents and detection kits unless otherwise noted. Antigen retrieval was performed using pre-diluted Ventana Cell Conditioner 1 (Tris, EDTA, Borate pH 8.0) for 36 minutes. Endogenous peroxidase activity was blocked with hydrogen peroxide. Pre-diluted CD3 was applied for 30 minutes at 37°C. Primary antibody was detected with anti-rabbit horseradish peroxidase-conjugated multimer incubated for 8 minutes and then visualized with tyramide conjugated FITC. Slides were washed with instrument buffer and Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) was performed on the Discovery XT platform using the In Situ Cell Death TMR Detection Kit (Roche Diagnostics) following the protocol recommended by the manufacture with a modified reaction mixture incubation time of 2 hours at 37°C. Appropriate positive and negative controls were included with the study section. Images were captured on a Zeiss LSM 800 Meta confocal microscope using a 20X objective and analyzed using ImageJ (NIH) (Schneider et al., 2012).
Cytokine measurements
Blood was collected by submandibular vein puncture into a sterile Eppendorf tube. Serum was isolated by centrifugation and used for analysis of serum cytokines using a multiplex assay and a custom MILLIPLEX MAG mouse cytokine / chemokine magnetic bead kit for the following cytokines IFNγ, IL-1a, IL-1β, IL-2, IL-4, IL-6, IL-10, IL12p70, IL-17, TNFα (cat.no. MCYTOMAG-70K-10; Millipore) according to the manufacturer’s instructions. Cytokine levels were analyzed using a MAGPIX system (Luminex Corporation).
Blood smears
Blood was collected by submandibular vein puncture into an EDTA-containing tube for 2 types of analyses. First, blood smears were prepared by spreading 20 ul of blood on a glass slide. Slides were air-dried and stained twice with Wright-Giemsa stains. Second, a complete blood count (CBC) was acquired using an automated Hemavet 950 instrument (Drew Scientific) according to the manufacturer’s instructions.
RNA sequencing and data processing
BM cells were isolated from leukemic mice (3 per cohort) 21 days after disease induction and GFP+ cells were isolated using a cell sorter Moflo XDP. Total RNA was extracted using the RNeasy Micro Kit (QIAGEN). RNA quality and quantity were analyzed using a 2100 Bioanalyzer (Agilent) and PICO chip. Samples with an RNA integrity number (RIN) of > 9 were used for library preparation. RNaseq libraries were prepared using the TruSeq RNA sample prep v2 kit (Illumina), starting from 100 ng of DNase I (QIAGEN) treated total RNA with 15 PCR cycles and following the manufacturer’s protocol. The amplified libraries were purified using AMPure beads (Beckman Coulter), quantified by Qubit 2.0 fluorometer (Life Technologies), and visualized in an Agilent Tapestation 2200. The libraries were pooled equimolarly, and loaded on the HiSeq 2500 Sequencing System (Illumina) and run as single 50 nucleotide reads generating about 30 million reads per sample. For gene expression analysis, reads were aligned to the NCBIM37 (iGenome) mouse genome using Bowtie software (Version 1.0.0) with 2 mismatches allowed. Uniquely-mapped reads were further processed by removing PCR duplicates with Picard MarkDuplicates (http://broadinstitute.github.io/picard/). Transcripts were counted using HTSeq. Analysis of differential gene expression between WT and Stim1/2−/− leukemic cells was performed separately for each sample using DESeq2 Bioconductor package in the R statistical programming environment. Differences in gene expression were considered significant if p < 0.05. Data are deposited in Gene Expression Omnibus (GEO) database.
QUANTIFICATION AND STATISTICAL ANALYSIS
All numerical results are reported as mean ± standard error of the mean (SEM). We used the Kaplan-Meier estimator for evaluating the statistical significance of the difference between the survival curves of mice. For all other experiments, the statistical difference between experimental groups was determined by two-tailed unpaired Student’s t test assuming equal variance. Differences were considered significant for p < 0.05 (noted in the figures by an *). The exact sample size (n) is indicated in each figure and corresponds to individual animals unless otherwise stated.
DATA AND SOFTWARE AVAILABILITY
The GEO accession number for RNaseq data is GSE116776.
Supplementary Material
Highlights.
STIM1 and STIM2 mediate calcium influx in Notch1-induced leukemic T lymphoblasts
Deletion of STIM1 and STIM2 prolongs the survival of mice with T-ALL
Similar leukemic burden but no tissue necrosis in mice with STIM1/STIM2-negative T-ALL
STIM1 and STIM2 control multiple proinflammatory pathways in leukemic T lymphoblasts
ACKNOWLEDGMENTS
We dedicate this article to our late colleague Dr. Lawrence B. Gardner with gratitude for his support. We thank Miriam Eckstein for help with bone decalcification and the following core facilities at NYUSOM: Immune Monitoring (multiplex cytokine analysis), Histopathology (H&E and TUNEL stains), the Genome Technology Center (GTC) (RNA sequencing), and the Applied Bioinformatics Laboratory (ABL) (help with analysis of RNA-seq data). The GTC and ABL are shared resources at NYUSOM, which are partially supported by a Cancer Center Support grant (P30CA016087). We thank Drs. William Carroll, George Miller, and Meike Dittmann for critically reading the manuscript. This work was funded by a Feinberg Lymphoma pilot grant (Laura and Isaac Perlmutter Cancer Center at NYUSOM) and the NIH/NIAID (R01AI097302) (to S.F.); the NIH/NCI (R01CA202025 and R01CA133379), a Leukemia and Lymphoma Society grant (6480–16), and a Chemotherapy Foundation grant (to I.A.); the Luciano Research Fund, the Conquer Cancer Foundation of ASCO-YIA, and a Hematology Training grant from the NIH/NHLBI (T32HL007151–35) (to S.S.F.-L.); and a Young Investigator Award by the Alex’s Lemonade Stand Foundation (to M.M.). NYUSOMcore facilities are supported, in part, by grants from the NIH/ORIP (S10OD01058 and S10OD018338) and the National Center for Advancing Translational Sciences (NIH/NCATS) (UL1 TR00038).
Footnotes
DECLARATION OF INTERESTS
S.F. is a scientific cofounder of Calcimedia. All other authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes four figures and two tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.08.030.
REFERENCES
- Armitage AE, Armitage JD, and Armitage JO (2006). Alpha-interferon for relapsed non-Hodgkin’s lymphoma. Bone Marrow Transplant. 38, 701–702. [DOI] [PubMed] [Google Scholar]
- Aviles A, Neri N, Fernandez-Diez J, Silva L, and Nambo MJ (2015). Interferon and low doses of methotrexate versus interferon and retinoids in the treatment of refractory/relapsed cutaneous T-cell lymphoma. Hematology 20, 538–542. [DOI] [PubMed] [Google Scholar]
- Badar T, Shetty A, Bueso-Ramos C, Cortes J, Konopleva M, Borthakur G, Pierce S, Huang X, Chen HC, Kadia T, et al. (2015). Bone marrow necrosis in acute leukemia: clinical characteristic and outcome. Am. J. Hematol. 90, 769–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmeier W, Weidinger C, Zee I, and Feske S (2013). Emerging roles of store-operated Ca2+ entry through STIM and ORAI proteins in immunity, hemostasis and cancer. Channels (Austin) 7, 379–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidwell BN, Slaney CY, Withana NP, Forster S, Cao Y, Loi S, Andrews D, Mikeska T, Mangan NE, Samarajiwa SA, et al. (2012). Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat. Med 18, 1224–1231. [DOI] [PubMed] [Google Scholar]
- Bonifazi F, de Vivo A, Rosti G, Guilhot F, Guilhot J, Trabacchi E, Hehlmann R, Hochhaus A, Shepherd PC, Steegmann JL, et al. ; Europena Study Group on Interferon in Chronic Myeloid Leukemia; Italian Cooperative Study Group on CML; France Intergroup of CML; German CML Study Group; UK Medical Research Council Working Party on CML; Spanish CML Study Group; Australian CML Study Group; Swedish CML Study Group (2001). Chronic myeloid leukemia and interferon-alpha: a study of complete cytogenetic responders. Blood 98, 3074–3081. [DOI] [PubMed] [Google Scholar]
- Bresnick AR, Weber DJ, and Zimmer DB (2015). S100 proteins in cancer. Nat. Rev. Cancer 15, 96–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YF, Chiu WT, Chen YT, Lin PY, Huang HJ, Chou CY, Chang HC, Tang MJ, and Shen MR (2011). Calcium store sensor stromal-interaction molecule 1-dependent signaling plays an important role in cervical cancer growth, migration, and angiogenesis. Proc. Natl. Acad. Sci. USA 108, 15225–15230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarini F, Lonetti A, Evangelisti C, Buontempo F, Orsini E, Evangelisti C, Cappellini A, Neri LM, McCubrey JA, and Martelli AM (2016). Advances in understanding the acute lymphoblastic leukemia bone marrow microenvironment: From biology to therapeutic targeting. Biochim. Biophys. Acta 1863, 449–463. [DOI] [PubMed] [Google Scholar]
- Cull VS, Tilbrook PA, Bartlett EJ, Brekalo NL, and James CM (2003). Type I interferon differential therapy for erythroleukemia: specificity of STAT activation. Blood 101, 2727–2735. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, et al. (1999). A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature 398, 518–522. [DOI] [PubMed] [Google Scholar]
- Dores GM, Devesa SS, Curtis RE, Linet MS, and Morton LM (2012). Acute leukemia incidence and patient survival among children and adults in the United States, 2001–2007. Blood 119, 34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falini B, Pileri S, De Solas I, Martelli MF, Mason DY, Delsol G, Gatter KC, and Fagioli M (1990). Peripheral T-cell lymphoma associated with hemophagocytic syndrome. Blood 75, 434–444. [PubMed] [Google Scholar]
- Ferrando AA (2009). The role of NOTCH1 signaling in T-ALL. Hematology Am. Soc. Hematol. Educ. Program 2009, 353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feske S (2007). Calcium signalling in lymphocyte activation and disease. Nat. Rev. Immunol 7, 690–702. [DOI] [PubMed] [Google Scholar]
- Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, and Rao A (2006). A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441, 179–185. [DOI] [PubMed] [Google Scholar]
- Feske S, Skolnik EY, and Prakriya M (2012). Ion channels and transporters in lymphocyte function and immunity. Nat. Rev. Immunol 12, 532–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipovich AH (2009). Hemophagocytic lymphohistiocytosis (HLH) and related disorders. Hematology Am. Soc. Hematol. Educ. Program 2009, 127–131. [DOI] [PubMed] [Google Scholar]
- Gachet S, Genescá E, Passaro D, Irigoyen M, Alcalde H, Clémenson C, Poglio S, Pflumio F, Janin A, Lasgi C, et al. (2013). Leukemia-initiating cell activity requires calcineurin in T-cell acute lymphoblastic leukemia. Leukemia 27, 2289–2300. [DOI] [PubMed] [Google Scholar]
- Grivennikov SI, Greten FR, and Karin M (2010). Immunity, inflammation, and cancer. Cell 140, 883–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins ED, Duarte D, Akinduro O, Khorshed RA, Passaro D, Nowicka M, Straszkowski L, Scott MK, Rothery S, Ruivo N, et al. (2016). T-cell acute leukaemia exhibits dynamic interactions with bone marrow microenvironments. Nature 538, 518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba H, Greaves M, and Mullighan CG (2013). Acute lymphoblastic leukaemia. Lancet 381, 1943–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A, and Pillai PS (2014). Innate immunity to influenza virus infection. Nat. Rev. Immunol 14, 315–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnsen M, Lund LR, Rømer J, Almholt K, and Danø K (1998). Cancer invasion and tissue remodeling: common themes in proteolytic matrix degradation. Curr. Opin. Cell Biol 10, 667–671. [DOI] [PubMed] [Google Scholar]
- Kantarjian HM, O’Brien S, Cortes JE, Shan J, Giles FJ, Rios MB, Faderl SH, Wierda WG, Ferrajoli A, Verstovsek S, et al. (2003). Complete cytogenetic and molecular responses to interferon-alpha-based therapy for chronic myelogenous leukemia are associated with excellent long-term prognosis. Cancer 97, 1033–1041. [DOI] [PubMed] [Google Scholar]
- Katlinski KV, Gui J, Katlinskaya YV, Ortiz A, Chakraborty R, Bhattacharya S, Carbone CJ, Beiting DP, Girondo MA, Peck AR, et al. (2017). Inactivation of interferon receptor promotes the establishment of immune privileged tumor microenvironment. Cancer Cell 31, 194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann U, Shaw PJ, Kozhaya L, Subramanian R, Gaida K, Unutmaz D, McBride HJ, and Feske S (2016). Selective ORAI1 inhibition ameliorates autoimmune central nervous system inflammation by suppressing effector but not regulatory T cell function. J. Immunol 196, 573–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KD, Srikanth S, Tan YV, Yee MK, Jew M, Damoiseaux R, Jung ME, Shimizu S, An DS, Ribalet B, et al. (2014). Calcium signaling via Orai1 is essential for induction of the nuclear orphan receptor pathway to drive Th17 differentiation. J. Immunol 192, 110–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacruz RS, and Feske S (2015). Diseases caused by mutations in ORAI1 and STIM1. Ann. N Y Acad. Sci 1356, 45–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFleur AM, Lukacs NW, Kunkel SL, and Matsukawa A (2004). Role of CC chemokine CCL6/C10 as a monocyte chemoattractant in a murine acute peritonitis. Mediators Inflamm. 13, 349–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis CE, and Pollard JW (2006). Distinct role of macrophages in different tumor microenvironments. Cancer Res. 66, 605–612. [DOI] [PubMed] [Google Scholar]
- Li X, Gounari F, Protopopov A, Khazaie K, and von Boehmer H (2008). Oncogenesis of T-ALL and nonmalignant consequences of overexpressing intracellular NOTCH1. J. Exp. Med 205, 2851–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Huang Q, Chen H, Lin Z, Zhao M, and Jiang Z (2017). Interferon regulatory factor 7 promoted glioblastoma progression and stemness by modulating IL-6 expression in microglia. J. Cancer 8, 207–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE Jr., and Meyer T (2005). STIM is a Ca2+ sensor essential for Ca2+-store-depletiontriggered Ca2+ influx. Curr. Biol 15, 1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, McCarl CA, Khalil S, Lüthy K, and Feske S (2010). T-cell-specific deletion of STIM1 and STIM2 protects mice from EAE by impairing the effector functions of Th1 and Th17 cells. Eur. J. Immunol 40, 3028–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarl CA, Khalil S, Ma J, Oh-hora M, Yamashita M, Roether J, Kawasaki T, Jairaman A, Sasaki Y, Prakriya M, and Feske S (2010). Store-operated Ca2+ entry through ORAI1 is critical for T cell-mediated autoimmunity and allograft rejection. J. Immunol 185, 5845–5858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medyouf H, Alcalde H, Berthier C, Guillemin MC, dos Santos NR, Janin A, Decaudin D, de Thé H, and Ghysdael J (2007). Targeting calcineurin activation as a therapeutic strategy for T-cell acute lymphoblastic leukemia. Nat. Med. 13, 736–741. [DOI] [PubMed] [Google Scholar]
- Miller AD, and Chen F (1996). Retrovirus packaging cells based on 10A1 murine leukemia virus for production of vectors that use multiple receptors for cell entry. J. Virol 70, 5564–5571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteith GR, McAndrew D, Faddy HM, and Roberts-Thomson SJ (2007). Calcium and cancer: targeting Ca2+ transport. Nat. Rev. Cancer 7, 519–530. [DOI] [PubMed] [Google Scholar]
- Murdoch C, Giannoudis A, and Lewis CE (2004). Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood 104, 2224–2234. [DOI] [PubMed] [Google Scholar]
- O’Brien MM, Lee-Kim Y, George TI, McClain KL, Twist CJ, and Jeng M (2008). Precursor B-cell acute lymphoblastic leukemia presenting with hemophagocytic lymphohistiocytosis. Pediatr. Blood Cancer 50, 381–383. [DOI] [PubMed] [Google Scholar]
- Olsen EA (2003). Interferon in the treatment of cutaneous T-cell lymphoma. Dermatol. Ther 16, 311–321. [DOI] [PubMed] [Google Scholar]
- Otrock ZK, and Eby CS (2015). Clinical characteristics, prognostic factors, and outcomes of adult patients with hemophagocytic lymphohistiocytosis. Am. J. Hematol 90, 220–224. [DOI] [PubMed] [Google Scholar]
- Ouyang K, Leandro Gomez-Amaro R, Stachura DL, Tang H, Peng X, Fang X, Traver D, Evans SM, and Chen J (2014). Loss of IP3R-dependent Ca2+ signalling in thymocytes leads to aberrant development and acute lymphoblastic leukemia. Nat. Commun 5, 4814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page-McCaw A, Ewald AJ, and Werb Z (2007). Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol 8, 221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passaro D, Irigoyen M, Catherinet C, Gachet S, Da Costa De Jesus C, Lasgi C, Tran Quang C, and Ghysdael J (2015). CXCR4 is required for leukemia-initiating cell activity in T cell acute lymphoblastic leukemia. Cancer Cell 27, 769–779. [DOI] [PubMed] [Google Scholar]
- Pitt LA, Tikhonova AN, Hu H, Trimarchi T, King B, Gong Y, Sanchez-Martin M, Tsirigos A, Littman DR, Ferrando AA, et al. (2015). CXCL12-producing vascular endothelial niches control acute t cell leukemia maintenance. Cancer Cell 27, 755–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakriya M, and Lewis RS (2015). Store-operated calcium channels. Physiol. Rev 95, 1383–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qadir AS, Ceppi P, Brockway S, Law C, Mu L, Khodarev NN, Kim J, Zhao JC, Putzbach W, Murmann AE, et al. (2017). CD95/Fas increases stemness in cancer cells by inducing a STAT1-dependent type I interferon response. Cell Rep. 18, 2373–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesada JR, Reuben J, Manning JT, Hersh EM, and Gutterman JU (1984). Alpha interferon for induction of remission in hairy-cell leukemia. N. Engl. J. Med 310, 15–18. [DOI] [PubMed] [Google Scholar]
- Radtke F, MacDonald HR, and Tacchini-Cottier F (2013). Regulation of innate and adaptive immunity by Notch. Nat. Rev. Immunol 13, 427–437. [DOI] [PubMed] [Google Scholar]
- Rawlings JS, Rosler KM, and Harrison DA (2004). The JAK/STAT signaling pathway. J. Cell Sci 117, 1281–1283. [DOI] [PubMed] [Google Scholar]
- Roderick HL, and Cook SJ (2008). Ca2+ signalling checkpoints in cancer: remodelling Ca2+ for cancer cell proliferation and survival. Nat. Rev. Cancer 8, 361–375. [DOI] [PubMed] [Google Scholar]
- Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, et al. (2005). STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol 169, 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roti G, Carlton A, Ross KN, Markstein M, Pajcini K, Su AH, Perrimon N, Pear WS, Kung AL, Blacklow SC, et al. (2013). Complementary genomic screens identify SERCA as a therapeutic target in NOTCH1 mutated cancer. Cancer Cell 23, 390–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryeom SW (2011). The cautionary tale of side effects of chronic Notch1 inhibition. J. Clin. Invest 121, 508–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuhmann MK, Stegner D, Berna-Erro A, Bittner S, Braun A, Kleinschnitz C, Stoll G, Wiendl H, Meuth SG, and Nieswandt B (2010). Stromal interaction molecules 1 and 2 are key regulators of autoreactive T cell activation in murine autoimmune central nervous system inflammation. J. Immunol 184, 1536–1542. [DOI] [PubMed] [Google Scholar]
- Shalapour S, and Karin M (2015). Immunity, inflammation, and cancer: an eternal fight between good and evil. J. Clin. Invest 125, 3347–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solinas G, Germano G, Mantovani A, and Allavena P (2009). Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J. Leukoc. Biol 86, 1065–1073. [DOI] [PubMed] [Google Scholar]
- Steele M, and Narendran A (2012). Mechanisms of defective erythropoiesis and anemia in pediatric acute lymphoblastic leukemia (ALL). Ann. Hematol 91, 1513–1518. [DOI] [PubMed] [Google Scholar]
- Su IJ, Hsu YH, Lin MT, Cheng AL, Wang CH, and Weiss LM (1993). Epstein-Barr virus-containing T-cell lymphoma presents with hemophagocytic syndrome mimicking malignant histiocytosis. Cancer 72, 2019–2027. [DOI] [PubMed] [Google Scholar]
- Trebo MM, Attarbaschi A, Mann G, Minkov M, Kornmüller R, and Gadner H (2005). Histiocytosis following T-acute lymphoblastic leukemia: a BFM study. Leuk. Lymphoma 46, 1735–1741. [DOI] [PubMed] [Google Scholar]
- Umemura M, Baljinnyam E, Feske S, De Lorenzo MS, Xie LH, Feng X, Oda K, Makino A, Fujita T, Yokoyama U, et al. (2014). Store-operated Ca2+ entry (SOCE) regulates melanoma proliferation and cell migration. PLoS ONE 9, e89292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaeth M, Zee I, Concepcion AR, Maus M, Shaw P, Portal-Celhay C, Zahra A, Kozhaya L, Weidinger C, Philips J, et al. (2015). Ca2+ signaling but not store-operated Ca2+ entry is required for the function of macrophages and dendritic cells. J. Immunol 195, 1202–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaeth M, Yang J, Yamashita M, Zee I, Eckstein M, Knosp C, Kaufmann U, Karoly Jani P, Lacruz RS, Flockerzi V, et al. (2017). ORAI2 modulates store-operated calcium entry and T cell-mediated immunity. Nat. Commun 8, 14714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainchenker W, and Constantinescu SN (2013). JAK/STAT signaling in hematological malignancies. Oncogene 32, 2601–2613. [DOI] [PubMed] [Google Scholar]
- Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan-Huberson M, Kraft S, Turner H, Fleig A, Penner R, and Kinet JP (2006). CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 312, 1220–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Tresckow B, Morschhauser F, Ribrag V, Topp MS, Chien C, Seetharam S, Aquino R, Kotoulek S, de Boer CJ, and Engert A (2015). An open-label, multicenter, phase I/II study of JNJ-40346527, a CSF-1R inhibitor, in patients with relapsed or refractory Hodgkin lymphoma. Clin. Cancer Res 21, 1843–1850. [DOI] [PubMed] [Google Scholar]
- Walter W, Sánchez-Cabo F, and Ricote M (2015). GOplot: an R package for visually combining expression data with functional analysis. Bioinformatics 31, 2912–2914. [DOI] [PubMed] [Google Scholar]
- Wang JY, Sun J, Huang MY, Wang YS, Hou MF, Sun Y, He H, Krishna N, Chiu SJ, Lin S, et al. (2015). STIM1 overexpression promotes colorectal cancer progression, cell motility and COX-2 expression. Oncogene 34, 4358–4367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng AP, Ferrando AA, Lee W, Morris JP 4th, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, and Aster JC (2004). Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306, 269–271. [DOI] [PubMed] [Google Scholar]
- Xie J, Pan H, Yao J, Zhou Y, and Han W (2016). SOCE and cancer: Recent progress and new perspectives. Int. J. Cancer 138, 2067–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Pardoll D, and Jove R (2009). STATs in cancer inflammation and immunity: a leading role for STAT3. Nat. Rev. Cancer 9, 798–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SL, Yeromin AV, Zhang XH, Yu Y, Safrina O, Penna A, Roos J, Stauderman KA, and Cahalan MD (2006). Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release-activated Ca(2+) channel activity. Proc. Natl. Acad. Sci. USA 103, 9357–9362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, and Kroemer G (2015). Type I interferons in anticancer immunity. Nat. Rev. Immunol 15, 405–414. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.