

Chris Boshoff, Senior Vice President of Immuno-Oncology, Translational and Early Development at Pfizer, and colleagues Samra Turajlic and Charles Swanton from the Francis Crick Institute and University College London give us their personal point of view on new insights and future therapeutic approaches for renal cancer.
Abstract
Chris Boshoff, Senior Vice President of Immuno-Oncology, Translational and Early Development at Pfizer, and colleagues Samra Turajlic and Charles Swanton from the Francis Crick Institute and University College London give us their personal point of view on new insights and future therapeutic approaches for renal cancer.
Malignant kidney tumors account for 2% of the global cancer burden, and its incidence is on the rise. In 2018, 63,000 new cases and 15,000 deaths due to kidney cancer will occur in the US and ~350,000 new cases will occur worldwide. There are multiple histological subtypes of renal cancer, each characterized by a unique molecular landscape. The most common subtype, clear cell renal cell carcinoma (ccRCC), accounts for 75% of all cases and arises from the proximal tubule cells of the kidney nephron. The prognosis of patients with metastatic ccRCC is poor, and <10% of patients are alive 5 years after diagnosis.
Development and evolution
ccRCC is defined by the loss of chromosome 3p, which is a near-ubiquitous early event resulting in the loss of heterozygosity in four tumor suppressor genes: VHL, PBRM1, SETD2, and BAP1. Mutation or methylation of the VHL gene is the most common mutational event, followed by mutations in PBRM1, SETD2, and BAP1. On the background of 3p loss, these mutations lead to bi-allelic inactivation of these genes. The most common cause of 3p loss is a chromothripsis event, which generates a concurrent 5q gain resulting in a translocation t(3,5) and can occur as early as adolescence (Mitchell et al., 2018). Distinct evolutionary trajectories have been described in established ccRCC: tumors evolving in a linear fashion with a limited number of driver events (VHL and 3p loss alone), those that evolve in a branched fashion resulting in many distinct subpopulations of cells, and finally tumors that are characterized by a punctuated burst of multiple mutational and copy number drivers leading to a clonal sweep and limited intratumor heterogeneity detectable by bulk multiregion sequencing (Turajlic et al., 2018a).
Sequencing of primary and metastatic ccRCC revealed that tumors with low genetic diversity (intra-tumor heterogeneity [ITH]) and a low fraction of the tumor genome affected by somatic copy-number alterations (SCNAs) have an overall low metastatic potential and a better overall prognosis (Turajlic et al., 2018b). Primary tumors with high ITH are associated with a tempered pattern of progression often involving single sites of metastatic disease over a protracted period. In contrast, primary tumors with low ITH but elevated SCNAs appear to be associated with rapid progression, often to multiple distant sites. Early loss of chromosome 9p, encoding CDKN2A/B among other genes, also appears to predict a more aggressive phenotype with early-onset metastases and a short overall survival (Turajlic et al., 2018a). Future clinical studies should test whether adjuvant or early combinatorial intervention is of benefit for primary tumors harboring 9p loss and/or low ITH with high SCNA.

Multiregion sequencing analyses revealed distinct evolutionary subtypes that are associated with distinct metastatic patterns. Chromosome 3p loss and VHL deficiency are nearly universal early events. Primary tumors with limited ITH and a low fraction of their genome affected by SCNAs have an overall low metastatic potential and follow a linear pattern of evolution. Primary tumors with high ITH and high genomic instability follow an attenuated progression, characterized by highly branched trees and solitary metastases. These tumors usually harbor early PBRM1 mutation followed by subclonal SCNAs, mutational activation of the PI3K–AKT–mTOR pathway, or SETD2 mutation. Both chromosome 9p loss and 14q loss are late events in tumor evolution. In primary tumors characterized by low ITH and high genomic instability, metastatic competence is acquired at the early stage of tumor evolution, which then drives rapid tumor dissemination, leading to a poor outcome and early death from disease. In this aggressive subtype, chromosome 9p and 14q loss occurs early during tumor development. VHL, von Hippel-Lindau; BAP1, BRCA1-associated protein 1; PBRM1, protein polybromo-1; PI3K, phosphatidylinositol-4,5-bisphosphate 3-kinase; SETD2, SET domain containing 2; wGII, weighted genome integrity index score, measuring the extent of genome instability.
Therapeutic approaches
Deficiency in pVHL and its E3 ligase activity results in the normoxic stabilization of the transcription factors hypoxia-inducible factor (HIF) 1α and 2α, with subsequent activation of the vascular endothelial growth factor (VEGF) family, angiogenesis, tumor cell survival, and proliferation (Kaelin et al., 2015). Therefore, over the last decade, small molecules targeting VEGF receptors (VEGFR inhibitors, e.g., sunitinib, cabozantinib, sorafenib, and pazopanib) or the anti-VEGF antibody (bevacizumab used in combination with low-dose INFα) have been indispensable for treating advanced or metastatic ccRCC (Choueiri and Motzer, 2017). VEGFR inhibitors (e.g., axitinib, cabozantinib, sorafenib, or lenvatinib used in combination with the mammalian target of rapamycin [mTOR] inhibitor everolimus) are also approved for use following one prior anti-angiogenic treatment. Most of these oral VEGFR inhibitors are multi-targeted drugs, not only inhibiting VEGFR-1, -2, and -3, but also targeting related tyrosine kinases like platelet-derived growth factor receptor (sunitinib, pazopanib, sorafenib, and axitinib), fibroblast growth factor receptor (lenvatinib), or MET and AXL (cabozantinib). These target specificities are reflected in their non–cross resistance attributes (Table 1) and in their differentiated adverse event profiles. For example, increased expression of MET and AXL is implicated in the development of resistance to VEGFR inhibitors in ccRCC preclinical models, and cabozantinib demonstrates significant activity after first-line anti-angiogenic treatment.
Table 1. Current and investigative treatment options for advanced or metastatic renal cancer.
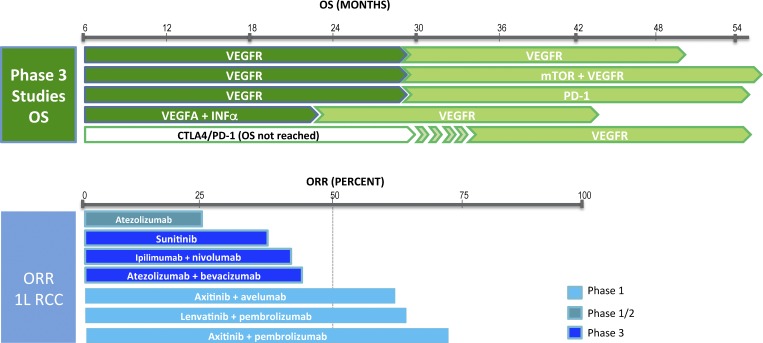 |
Top: Overall survival (OS) achieved with current approved treatment options. VEGFR inhibitors used up-front for advanced or metastatic ccRCC result in a median OS of <30 mo. The median OS for the combination of nivolumab and the CTLA-4 blocker ipilimumab has not been reached yet (FDA approved for intermediate or poor-risk advanced ccRCC). At time of disease progression, an ICB or a different VEGFR inhibitor is indicated. The combination of a VEGFR inhibitor with an mTOR inhibitor (lenvatinib and everolimus) is also an approved treatment option following one prior anti-angiogenic therapy. Bottom: ORR achieved with experimental or approved treatment options for untreated advanced or metastatic renal cancer. The ORRs achieved with the combination of a VEGFR inhibitor with an ICB are consistently >50%, although these are reported from relatively small phase 1 studies, and phase 3 randomized studies testing such combinations versus sunitinib are ongoing. CTLA-4, cytotoxic T lymphocyte–associated protein 4.
pVHL loss is also linked to an altered chromatin profile that promotes ccRCC tumorigenesis and metastases; for example, VHL loss induces tumor-specific enhancer gains around angiogenic and metabolic targets through the stabilization of HIF2α/HIF1β heterodimers (Yao et al., 2017). Preclinical data support the key role for HIF2α in pVHL-defective ccRCC, and a human HIF2α genetic polymorphism is associated with an increased risk of developing ccRCC. Inhibition of HIF2α may result in the down-regulation of activated HIF2α-dependent enhancer regions within ccRCC, and HIF2α-specific inhibitors in clinical development may provide another targeted therapeutic opportunity (Chen et al., 2016; Yao et al., 2017).
The FDA approval in 2015 of nivolumab (anti–PD-1 immune checkpoint blocker [ICB]) for patients with advanced ccRCC who have received prior anti-angiogenic therapy heralded a new era in the management of ccRCC (Motzer et al., 2015). Subsequently, it was reported in a single arm experience that pembrolizumab (anti–PD-1) resulted in a 38% objective response rate (ORR) in first-line metastatic ccRCC (McDermott, D.F., et al. 2018. ASCO Annual Meeting. Abstract 4500). This is a noticeable ORR for an ICB considering that the tumor mutational burden (TMB) in ccRCC is low compared with other tumors such as melanoma and lung cancer. ccRCC may be an exception among tumors, where TMB does not correlate with ORR, suggesting that those genetic alterations associated with ccRCC yield a disproportionately higher amount of neoantigens and elicit robust adaptive immune responses. In this regard, a high number of frameshift insertion deletion mutations were reported in ccRCC, and such indels appear to act as potent neoantigens and are also associated with a high cytolytic CD8+ T cell expression signature (Turajlic et al., 2017). Other mechanisms, e.g., reactivated endogenous retroviral elements (ERVs), have also been postulated to play a role in ccRCC immunity, and a subclass of ERVs whose derepression triggers innate immune signaling has been identified in certain tumors, including ccRCC (Cañadas et al., 2018). Discovering the high-affinity neoantigens enabling T cell recognition will be critical to further advance immunotherapy for ccRCC, including autologous vaccine and cellular approaches.
Loss of function of two genes on chromosome 3p (PBRM1 and BAP1) is associated with an immune microenvironment and potentially with increased responses to ICBs (Miao et al., 2018). PBRM1 and BAP1 are mutated in ~40% and 15% of patients, respectively, and are largely non-overlapping (Voss et al., 2017). Truncating mutations in PBRM1 are associated with a distinct immune-related gene expression profile. PBRM1 acts as a tumor suppressor gene with roles in DNA repair, maintenance of genome stability, and control of cell proliferation. BAP1 mutant ccRCC tumors are also correlated with an inflammatory tumor microenvironment (Wang et al., 2018), which is noteworthy because BAP1 deficiency is also linked to an inflammatory phenotype in primary uveal melanoma. This inflamed subtype of ccRCC correlates with systemic inflammatory manifestations such as anemia and thrombocytosis, which predict a poor prognosis (Wang et al., 2018). These observations emphasize the importance of efforts to correlate genetic alterations in ccRCC with outcome and potentially develop such genetic features as biomarkers to select for the use of immunotherapy.
Anti-angiogenic therapies have been associated with inducing an antitumor microenvironment. For example, blocking VEGF augments intra-tumoral T cell infiltration, partly through vascular normalization and endothelial cell activation. Anti-VEGF antibodies may also enhance the antitumor activity of ICB by promoting T cell infiltration, up-regulating major histocompatibility complex class I expression, and reversing myeloid immunosuppression. Therefore, combining an ICB with an anti-angiogenic targeted therapy is a rational combination to test in the clinic. The ORR (Table 1) and duration of responses reported for such combinations infer that they could become a future paradigm for managing advanced ccRCC. The combination of the anti–PD-L1 ICB atezolizumab with bevacizumab improves the clinical outcome compared with sunitinib in tumors enriched for T effector cells (correlating with PD-L1 positivity), whereas sunitinib appears to be more favorable in highly angiogenic tumors. ICB monotherapy is effective in tumors with pre-existing immunity and a relatively lower expression of myeloid inflammation–associated genes but is less effective in T cell–enriched tumors with concurrently high myeloid inflammation (McDermott et al., 2018). Integrating such observations based on cellular profiles with the genetic architecture of ccRCC will be essential to advance a precision medicine approach in which either an angiogenic inhibitor or an ICB, or a combination of these, is selected for advanced ccRCC. For example, VHL and PBRM1 mutant tumors are associated with both an immune profile and an angiogenic signature, suggesting that tumors harboring both defects could benefit most from a combinatorial approach. In contrast, loss of BAP1 associates with decreased angiogenic signaling and an adverse outcome to angiogenic inhibitors, and such tumors may benefit mostly from an ICB (Voss et al., 2017). Effective combination versus sequencing approaches will also be critical to advance the overall outcome for patients with ccRCC.
Due to obesity and an ageing population, the incidence of ccRCC is predicted to increase significantly during the next decade. During the next couple of years, we will see a rapidly evolving treatment paradigm, and treatment decision-making will become more multifaceted as new therapies are launched. As the treatment armamentarium expands, including the combination of ICBs with angiogenic inhibitors, the need for predictive biomarkers to guide the use of such combinations versus single-agent approaches will become essential for treatment decision-making. The detection of primary tumors with a linear evolutionary trajectory and less genomic instability may also infer a more wait-and-see approach. As the genetic events initiating and driving ccRCC evolution also shapes the tumor microenvironment (TME), integrating information from the genetic architecture and evolutionary potential of ccRCC with the cellular composite of the TME and treatment outcomes will be crucial to advance rational treatment decisions.
Acknowledgments
Charles Swanton consults for Boehringer Ingelheim, Novartis, Eli Lilly, Roche Ventana, GlaxoSmithKline, Pfizer, Genentech, and Celgene and is a co-founder of Achilles Therapeutics. Samra Turajlic has received funding from Roche Ventana. Chris Boshoff is a full-time employee of Pfizer, Inc.
References
- Cañadas I., et al. 2018. Nat. Med. 24:1143–1150. 10.1038/s41591-018-0116-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W., et al. 2016. Nature. 539:112–117. 10.1038/nature19796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choueiri T.K., and Motzer R.J.. 2017. N. Engl. J. Med. 376:354–366. 10.1056/NEJMra1601333 [DOI] [PubMed] [Google Scholar]
- Kaelin W., et al. 2015. Kidney Cancer: Principles and Practice. Second edition. 31–57. [Google Scholar]
- McDermott D.F., et al. 2018. Nat. Med. 24:749–757. 10.1038/s41591-018-0053-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao D., et al. 2018. Science. 359:801–806. 10.1126/science.aan5951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell T.J., et al. 2018. Cell. 173:611–623.e17. 10.1016/j.cell.2018.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motzer R.J., et al. 2015. N. Engl. J. Med. 373:1803–1813. 10.1056/NEJMoa1510665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turajlic S., et al. 2017. Lancet Oncol. 18:1009–1021. 10.1016/S1470-2045(17)30516-8 [DOI] [PubMed] [Google Scholar]
- Turajlic S., et al. 2018a Cell. 173:581–594.e12. 10.1016/j.cell.2018.03.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turajlic S., et al. 2018b Cell. 173:595–610.e11. 10.1016/j.cell.2018.03.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss M.H., et al. 2017. J. Clin. Oncol. 35:484 10.1200/JCO.2017.35.6_suppl.484 [DOI] [PubMed] [Google Scholar]
- Wang T., et al. Cancer Discov. 2018 doi: 10.1158/2159-8290.CD-17-1246. [DOI] [Google Scholar]
- Yao X., et al. 2017. Cancer Discov. 7:1284–1305. 10.1158/2159-8290.CD-17-0375 [DOI] [PubMed] [Google Scholar]