

Abstract
H. haemolyticus is often misidentified as NTHi due to their close phylogenetic relationship. Differentiating between the two is important for correct identification and appropriate treatment of infective organism and to ensure any role of H. haemolyticus in disease is not being overlooked. Speciation however is not completely reliable by culture and PCR methods due to the loss of haemolysis by H. haemolyticus and the heterogeneity of NTHi. Haemophilus isolates from COPD as part of the AERIS study (ClinicalTrials - NCT01360398) were speciated by analysing sequence data for the presence of molecular markers. Further investigation into the genomic relationship was carried out using average nucleotide identity and phylogeny of allelic and genome alignments. Only 6.3% were identified as H. haemolyticus. Multiple in silico methods were able to distinguish H. haemolyticus from NTHi. However, no single gene target was found to be 100% accurate. A group of omp2 negative NTHi were observed to be phylogenetically divergent from H. haemolyticus and remaining NTHi. The presence of an atypical group from a geographically and disease limited set of isolates supports the theory that the heterogeneity of NTHi may provide a genetic continuum between NTHi and H. haemolyticus.
Introduction
Chronic obstructive pulmonary disease (COPD) is an irreversible, multifaceted disease resulting in degradation of lung function and is the third largest cause of global mortality1. Characterised by increased inflammation and mucus production in the airways alongside collapsing alveoli, the disease progresses in periods of worsening symptoms called exacerbations thought to be predominantly brought on by viral or bacterial infection2–6. Haemophilus influenzae is reportedly the most prevalent bacterial cause of exacerbations although M. catarrhalis has also been associated4,5.
H. influenzae is a Gram negative, opportunistic pathogen residing in the respiratory tract and occurs in encapsulated serotype (a-f) forms and non-encapsulated forms referred to as non-typeable H. influenzae (NTHi). Serotype b (Hib) was the largest cause of Haemophilus invasive disease before the introduction of the Hib vaccination, currently NTHi is the predominant cause and is also, furthermore, associated with the onset of COPD exacerbations7–9. Additionally, NTHi is responsible for a considerable amount of otitis media in children which can lead to chronic disease and complications resulting in hearing loss, sequelae and in rare cases, death10.
Haemophilus haemolyticus is a Gram negative commensal of the respiratory tract isolated mostly from children and COPD patients2,11. It is the closest relative of NTHi but is thought to be non-pathogenic. However, a small number have been reportedly isolated from invasive infection12,13. The two species differ in their interactions with epithelial cells, with H. haemolyticus resulting in cytotoxicity and NTHi, invasiveness14,15. Despite these differences in behaviour, the two are morphologically identical and can only be differentiated by culture due to the ability of H. haemolyticus to display beta haemolysis on certain agar2,16–19. However, this trait is not present in 100% of isolates and this has led to the misidentification of non-haemolytic H. haemolyticus as NTHi. A study isolating Haemophilus from COPD patients reported 39.5% of NTHi retrospectively re-classified as H. haemolyticus2. However, other studies have reported much lower rates of misidentification. From cystic fibrosis respiratory samples in Denmark only 0.5% were identified as H. haemolyticus whereas when clinical samples were retrospectively investigated 5.9% were reported as H. haemolyticus in Germany and 1.5% in Australia20–22.
The failure to distinguish between the two species by culture methods has driven the development of molecular methods. This has proven challenging due to the high levels of heterogeneity displayed within NTHi. Single, duplex or multiplex PCR assays all fail to provide 100% sensitivity or specificity in differentiating between NTHi and H. haemolyticus17,23–29. It has been suggested that the two species are therefore a genetic continuum ranging from NTHi to H. haemolyticus26. Encapsulated H. influenzae display lower levels of genetic diversity and are phylogenetically separable from NTHi30–32.
Alternatively, MALDI-TOF mass spectrometry has been recommended for differentiation between H. influenzae and H. haemolyticus; although there are conflicting reports of the ability to correctly identify the two species33. High levels of accuracy have been achieved due to the addition of H. haemolyticus and regional H. influenzae profiles to the MALDI-TOF database33,34. However, a more recent study using the MALDI-TOF bio-typer IVD 2.3 database, which includes 21 H. haemolyticus and 27 H. influenzae profiles, reported that 13.1% of H. influenzae and 2% of H. haemolyticus could not be correctly identified to species level35. This improved to 100% with the use of alternative newly developed software35. However, it should be noted that, MALDI-TOF does not identify the capsule status of a H. influenzae nor is there evidence of being able to distinguish the different groups.
Distinguishing H. haemolyticus from NTHi is important to understand the roles of both in the COPD lung, to ensure invasive or disease-causing strains of H. haemolyticus are not being underestimated, and to enable correct identification of the causative organism in prescribing treatment for infection. Furthermore, the question of correct taxonomical allocation of H.influenzae and H. haemolyticus and investigating how genetically related these species are is also important to further understand NTHi. The purpose of this study was to ascertain the genetic relationship between NTHi and H. haemolyticus isolated from COPD and answer the question surrounding current taxonomy. Here we use culture and in silico markers to characterise NTHi and H. haemolyticus from the COPD lung. Furthermore, we utilise whole genome sequencing to ascertain the phylogeny between the two species.
Materials and Methods
Bacterial Isolates
The Acute Exacerbation and Respiratory InfectionS in COPD (AERIS) study was a longitudinal cohort investigation of patients with moderate, severe or very severe COPD. The full study protocol has been previously detailed5. Sampling occurred monthly or on event of acute exacerbation spanning a two-year period and microbiological investigation was undertaken using traditional culture methods. The study was registered with ClinicalTrials (NCT01360398) and carried out in accordance with ethical approval granted by the Southampton and South West Hampshire Research Ethics Committee and in accordance with the Declaration of Helsinki and Good Clinical Practice. All participants provided written informed consent.
From the AERIS study, 1460 H. influenzae identified by culture, isolated from 24 patients over 134 visits were investigated5. Strains 4849, 7279 and 8467 of the National Collection of Type Cultures (NCTC) were used as reference isolates for H. influenzae. NCTC strains 10659 and 10839 were used as reference strains for H. haemolyticus.
Haemolysis
All isolates of Haemophilus spp. were inoculated onto CBA agar supplemented with 5% horse blood (Oxoid, Basingstoke, UK) and incubated at 37 °C for 72 hours. This assay was to identify the capability of H. haemolyticus isolates to display beta haemolysis.
Whole Genome Sequencing
DNA extractions were prepared from isolates cultured on chocolate agar (Oxoid, UK) using the QiaAmp minikit according to manufacturer’s instructions and diluted to 0.2 ng μl−1. Library preparation was carried out using the Nextera XT DNA Prep Kit (Illumina, Saffron Walden, UK). Sequencing was done using 2 × 250 paired-end V2 chemistry on an Illumina MiSeq (Illumina, Saffron Walden, UK).
Genome Assembly and Analysis
Paired-end fastq files were trimmed of Nextera adapter sequences using trimmomatic and assembled using MaSuRCA36,37. Multi-locus Sequence Typing (MLST) and identification of genes for speciation was performed using SRST232,38. Here paired-end fastqs were mapped to reference sequences provided in Supplementary Table 1. Resulting consensus sequences were aligned in MUSCLE39 prior to maximum-likelihood phylogeny analysis using RAxML40. Phylogenies for omp6, hpd and smpB were prepared using Microreact and are stored, along with associated metadata, and viewable at URLs provided in Supplementary Table 241.
Further speciation was done using MetaPhlAn42 using the supplied 260 H. influenzae reference genes. Results were visualised using lattice in R (version 3.3.3)43. Average Nucleotide Identity (ANI) was calculated using Pyani to generate percentage genetic identity between each isolate (https://github.com/widdowquinn/pyani)44,45.
Core genome alignments generated in package ROARY were used to construct maximum likelihood phylogenies in RAxML40,46. Trees were visualised using microreact and can be accessed via http://microreact.org/project/Southampton_NTHivsHh41.
All MUSCLE alignments and RAxML phylogenies were completed on the CIPRES science gateway47. Genome assembly, MLST, gene mapping and pyani analyses were completed using the IRIDIS High Performance Computing Facility and associated IT support services at the University of Southampton.
Results
Initial characterisation
From the 1460 isolates of Haemophilus spp investigated, 12 isolates displayed haemolysis on blood agar. Further investigation demonstrated these twelve isolates could not be typed by H.influenzae MLST due to unrecognisable sequences at six loci and absence of the fucK gene. An additional 80 non-haemolytic isolates were fucK negative and did not have recognisable sequences at the six remaining loci. These 92 isolates were therefore classified as suspected H. haemolyticus. A further 54 isolates were negative for the fucK gene but did contain known alleles at the six remaining loci. These were categorised as fucK negative NTHi (f-NTHi) but were unable to be sequence typed as the MLST schema requires all seven alleles in order to allocate an ST. From the remaining 1314 isolates, 28 previously described STs were identified (n = 1076) and eight STs that were novel to this study and subsequently curated to the MLST database (STs 154, 156, 353, 356, 1314, 1441, 1442 and 1664) (n = 238). Only five STs were shared between more than one patient with ST 57, isolated from five patients, being the most prevalent.
Molecular Markers
Raw fastq sequence data for each isolate was mapped to reference sequences (Supplementary Table 1). For genes omp2, lgtC, fucP, fucK and iga, presence is expected in NTHi but not in H. haemolyticus whereas the converse is true for sodC. Due to the variation displayed in NTHi of iga a selected sequence previously identified as the beta core and reportedly highly conserved was used for gene mapping19. For a further three genes, hpd, smpB and omp6, allelic variation distinguishes between the two species. There were unexpected results for all gene markers except omp6 (Table 1).
Table 1.
Presence and absence of gene markers identified by gene mapping of 1460 Haemophilus spp against GenBank reference sequences.
| NTHi | smpB | lgtC | iga | fucP | omp2 | omp6 | sodC | hpd | fucK | Isolates with genotype (%) | No of pts (n = x/24) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| G1 | + | + | + | + | + | + | − | + | + | 1142(83) | 20 |
| G2 | + | + | + | + | −* | + | − | + | + | 84(6.1) | 7 |
| G3 | + | −* | + | + | + | + | − | + | + | 61 (4.5) | 16 |
| G4 | + | −* | + | + | −* | + | − | + | + | 5 (0.4) | 5 |
| G5 | + | + | + | + | + | + | − | −* | + | 8(0.6) | 2 |
| G6 | + | + | + | + | −* | + | − | −* | + | 12(0.9) | 1 |
| G7 | + | −* | + | + | −* | + | − | −* | + | 1(0.1) | 1 |
| G8 | + | + | + | −* | + | + | − | + | −* | 53(3.9) | 1 |
| G9 | + | −* | + | −* | + | + | − | + | −* | 1(0.1) | 1 |
| G10 | + | + | + | + | −* | + | +* | + | + | 1(0.1) | 1 |
| G11 | + | + | + | + | + | + | +* | + | + | 1(0.1) | 1 |
| Total NTHi | 1368 | 1301 | 1368 | 1314 | 1266 | 1368 | 2 | 1347 | 1314 | ||
| Hh | smpB | lgtC | iga | fucP | omp2 | omp6 | sodC | hpd | fucK | Isolates with genotype (%) | No of pts (n = x/24) |
| Hh1 | + | − | − | − | − | + | + | + | − | 78(84.8) | 11 |
| Hh2 | −* | − | − | − | − | + | + | + | − | 2(2.2) | 5 |
| Hh3 | + | − | +* | − | − | + | + | + | − | 12(13.0) | 1 |
| Total Hh | 90 | 0 | 12 | 0 | 0 | 92 | 92 | 92 | 0 |
An atypical result for the expected genotype of either NTHi or H. haemolyticus is denoted by *. Eleven different variations or ‘genotypes’ were identified in NTHi and three in H. haemolyticus. The majority 1142 (83%) of NTHi displayed the expected genotype, G1, for the molecular markers and this genotype was isolated in 20 out of the 24 patients. The majority 78 (84.8%) of H. haemolyticus also displayed the expected genotype Hh G1. The only molecular marker that did not result in an atypical result was that of omp6.
All suspected H. haemolyticus were negative for omp2, lgtC, fucP and fucK. However, a truncated version of the beta core sequence of the iga gene was unexpectedly detected in twelve H. haemolyticus isolates of which only two were haemolytic (Table 1). These sequences however were very short (from 100–620 bp, on average 237 bp) in comparison to the reference sequence used (864 bp) and displayed on average 93% identity to the section of the sequence. Untruncated sequences for the iga beta core were found ubiquitously throughout the NTHi as expected. However, omp2 and lgtC were absent in 102 and 67 isolates respectively (Table 1). The 54 isolates identified as f-NTHi by MLST were also found to be fucP negative indicating that the entire fuc operon may be absent in these isolates.
For H. haemolyticus, as anticipated, sodC was found in 100% of the suspected H. haemolyticus but was unexpectedly observed in two NTHi (Table 1). These were isolated from two different patients and were not the same ST. Sequences for smpB were found in all isolates except two H. haemolyticus (Table 1). hpd was absent in 21 NTHi but present in all H. haemolyticus. All hpd negative isolates were from ST925 (n = 7) or ST819 (n = 14), furthermore, there were no hpd positive isolates for either ST. Similarly, the 102 isolates negative for omp2 were made up of ST353, ST356, ST1314 and ST819 where there were no occurrences of omp2 positive isolates from these STs. Some STs were found to include isolates that were both positive and negative for omp2. These were observed in ST11 (2 isolates out of 46), ST311(3 out of 119), ST196 (1 out of 78), ST704 (2 out of 10), ST503 (3 out of 42) and ST513 (1 out of 19). All isolates that were negative for lgtC were accompanied by positive lgtC examples within their ST. This was observed in 24 different STs and an lgtC absence was noted in a single f-NTHi isolate.
Maximum-likelihood phylogenies for hpd, smpB and omp6 were constructed from alignments. The 92 suspected H. haemolyticus identified by MLST clustered into a separate lineage from all other NTHi (Fig. 1). f-NTHi were observed to cluster with the majority of NTHi in all three phylogenies. A group of NTHi were distinct from the others in both hpd and smpB sequence phylogenies, consisting of 76 isolates from novel STs 353, 356 and 1314, and were isolated from three different patients (Fig. 1). All were negative for omp2. This group was subsequently labelled as Group III and noted as a group of interest. In the phylogeny for hpd an adjacent clade to the Group III isolates consisted of all study isolates identified as ST513 and ST409. Also, in the phylogeny for omp6 Group III are clustered together, adjacent on the same clade are isolates from ST513, ST311, ST704, ST 925 and ST154.
Figure 1.

Allelic variation of (A) smpB, (B) omp6 and (C) hpd from H. influenzae and H. haemolyticus. Unrooted maximum – likelihood phylogenetic trees constructed in RaxML and visualised in microreact. MUSCLE alignments of sequences for smpB, omp6 and hpd from 1460 Haemophilus spp. where present using the GTRGAMMA model for nucleotide substitution. Scale bar indicates number of nucleotide substitutions per site. Colours indicate study classification group as per legend. Reference NCTC 10839 and NCTC 10659 are shown in green for H. haemolyticus and cluster within the H. haemolyticus clade. NTHi reference isolate NCTC 4842 clusters within an NTHi clade in hpd as marked. URL to access phylogeny and metadata available from Supplementary Table 2.
Genetic similarity
Investigation into the genetic similarity between NTHi, f-NTHi, Group III and H. haemolyticus was carried out by comparing the sequence data of the isolates to the 260 H. influenzae reference genes held for H. influenzae on the MetaPhlAn database. The genetic similarity spanned from 82.93–100%. The f-NTHi were 93.89–100% genetically similar to the MetaPhlAn NTHi reference genes (Fig. 2). Group III were markedly less similar to the f-NTHi and NTHi groups with 82.94–89.97% identity (Fig. 2). All H. haemolyticus were found to have a much lower genetic similarity to the MetaPhlAn reference genes than the other three groups ranging from 70.94–82.09% with an average of 75.36% (Fig. 2). NTHi and f-NTHi were observed to cluster together, Group III sitting distinctly between the NTHi and H. haemolyticus (Fig. 2).
Figure 2.
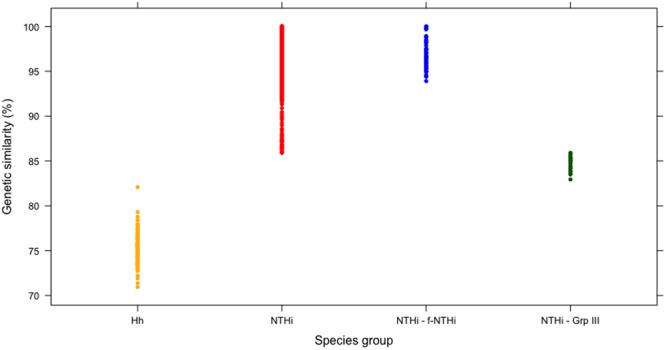
MetaPhlAn results for 1460 Haemophilus spp isolated from 24 COPD patients from 134 visits. Genetic similarity is based on percentage similarity of the Haemophilus isolates to 260 reference genes for H. influenzae. NTHi and f- NTHi can be seen to share a high degree of identity compared to Group III and H. haemolyticus.
A subset of isolates (n = 388) was selected to represent two examples of each ST from each patient per visit alongside the suspected H. haemolyticus. This was done to reduce the computational complexity of the dataset for average nucleotide identity (ANI) calculation. The average genetic similarity to five reference strains was calculated for three groups; NTHi including f-NTHi, Group III and H. haemolyticus. The resulting heat map from ANI displays the three NTHi groups within the same species (within the red area), H. haemolyticus is depicted in blue indicating differentiation from the NTHi (Fig. 3). When using ANI, the criteria for inclusion as the same species dictates a genetic similarity threshold of 94–96% and above44,48. NTHi was observed to be within the species threshold when compared to H. influenzae NCTC reference strains 7279, 4842 and 8467 with genetic similarity averaging 96.97% ± 0.05, 96.88% ± 0.05, 96.96% ± 0.05 respectively. When compared to the H. haemolyticus NCTC reference strains 10659, 10839, the NTHi averaged 91.94% ± 0.008 and 91.94% ± 0.009 and therefore were differentiated from H. haemolyticus as this was below the species threshold (Fig. 3). Previously Group III were defined by omp2 negative status and the distance displayed in the phylogeny in smpB and hpd from the remaining study NTHi. The ANI results further demonstrated the Group III isolates to be more divergent from the NTHi reference strains than the remaining study NTHi. The genetic similarity for Group III compared to the reference strains was on average 94.74–94.84%, noticeably less than the remaining NTHi. Using the 94% threshold for species differentiation therefore, although Group III displayed more diversity to the remaining NTHi, it is still appropriate to classify them taxonomically as NTHi and distinct from H. haemolyticus with the average genetic similarity at <92% between the two44,45. Group III therefore sits distinctly between the remaining NTHi groups and the H. haemolyticus (Figs 2, 3). Interestingly, in Fig. 3 the shading gradient in between the Group III and the remaining NTHi is representative of ST311, ST513 and ST704, isolates from ST513 and 311 were seen to cluster adjacent to Group III in the hpd phylogeny and isolates from ST513, 311 and 704 in the omp6 phylogeny. However, these isolates displayed on average nucleotide identity of 95.2–95.5% genetic similarity to the NTHi reference strains and were omp2 positive in the majority of cases, differentiating them from Group III.
Figure 3.

Heatmap summarising genetic similarity ANI calculations between 388 representative samples of Haemophilus spp isolated from 24 COPD patients over two years. Blue indicates a similarity of less than 94% and therefore a distinct species. Here this represents the H. haemolyticus isolates as compared to NTHi. Dark red areas depict isolates of high genetic similarity and the dendrograms show a clustering based on ST. The lighter area represents the Group III NTHi.
Core genome alignment
Phylogeny of the core genomes from the subset of isolates confirmed the population structure as identified by ANI. The H. haemolyticus, categorised as a separate species from all NTHi by taxonomical definition, are visibly distinct. Group III are also distinct from the remaining NTHi. All instances of f-NTHi clustered with the NTHi confirming that the absence of the fucK gene does not necessarily indicate a large level of diversity in the NTHi genome (Fig. 4).
Figure 4.
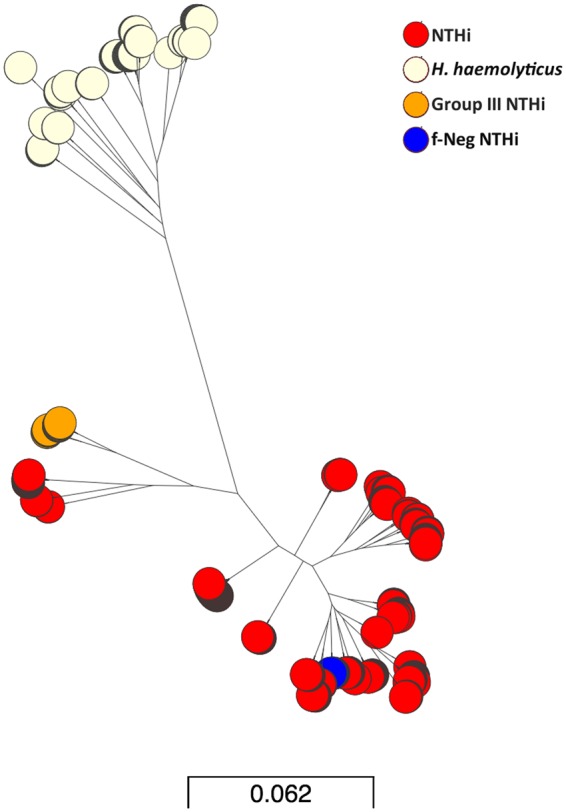
Maximum-likelihood phylogeny from core genome alignments of representative subset of 388 Haemophilus spp isolated from 24 COPD patients during the AERIS Study. Group III are distant from the majority of the NTHi however in this instance the Group III clade also contains NTHi STs 311, 704 and 513. The fucK negative isolates are observed to cluster with the majority of the NTHi and H. haemolyticus cluster together but distinctly from NTHi. Bar represents number of nucleotide substitutions per site. URL to access phylogeny and metadata available from Supplementary Table 2.
Discussion
Routine diagnostics to determine the correct causative organism in cases of infection, and in turn the appropriate prescription of antimicrobial treatment, are affected by the inability of culture methods to differentiate successfully between NTHi and H. haemolyticus. The ability for MALDI-TOF mass spectrometry to differentiate between the two has had success but there are cautionary tales of difficulties in correctly identifying regional strains and atypical fucK negative strains33–35. The heterogeneity of NTHi and its close genetic relationship with H. haemolyticus has meant the development of a successful PCR assay for differentiation has been challenging17,26,27,29,49 and has furthermore raised uncertainties about the species delineation between the two.
From the initial 1460 culture identified NTHi, 6.3% were confirmed to be H. haemolyticus and isolated from 11 patients. This ratio of H. haemolyticus to NTHi supports previous studies into misidentification in both clinical and study-based analyses. This does not fully support the results of a previous longitudinal COPD study that reported a 39.5% misidentification rate2,11,20–22. The analysis of all potential NTHi isolated from the entire AERIS study of 105 patients reported a misidentification rate of 10.6% based on lgtC/P6 duplex PCR5. Here we have confirmed the identity of H. haemolyticus by showing clear delineation between species. Furthermore, for the first time we provide evidence of a group of atypical strains that we have named Group III that show some genotypic characteristics more associated with H. haemolyticus and a reduced genetic similarity to typical NTHi (Figs 2, 3).
Using MLST we were able to successfully distinguish NTHi from H. haemolyticus as well as identify fucK negative isolates of NTHi. The latter represented 4% of all isolates and were isolated from the same patient over different time points. fucP was also observed to be absent in the f-NTHi isolates, implying the deletion of the entire operon as previously reported which raises an important question to the suitability of this loci in the MLST schema (Table 1)50,51. These isolates were distinguishable from H. haemolyticus by the presence of other identifiable alleles at the six remaining MLST loci. From the MLST analysis it was observed that only one patient carried the same ST throughout the different time points and only five STs were found in more than one patient. In total 36 different STs were identified from 24 patients. This included eight that were novel to this study and subsequently submitted for curation to the MSLT database. Three of the novel STs were characterised as the Group III (ST 353, 356, 1314). The minimal sharing of ST between patients demonstrates a large level of diversity of NTHi in this study group. Therefore, the presence of Group III isolated from three different patients and made up of three different sequence types yet displaying the same atypical genomic content is very interesting. The isolates were not only omp2 negative but were also found to be genetically divergent to the NTHi and f-NTHi as well as closely clustering together in both cases of phylogeny of molecular markers and whole genome content. The absence of the fucK and fucP genes from f-NTHi did not predict genetic diversity from the NTHi.
Molecular markers used in isolation would have resulted in incorrect speciation which concurs with previous reports26,50,52. Unexpected results for species type were observed for all molecular markers that were based on presence/absence (Table 1). Recently, duplex PCR assays such as using purT and hpd or hpd and fucP have been developed. However, these have also not been found to be 100% accurate in differentiating between the two species24,25. The absence of more than one marker gene in some isolates was noted in this study (Table 1). This supports previously reported absence of hpd and fucose genes, and emphasizes the issues around using two molecular markers for accurate speciation50–52. Within this study, all markers were required to give a robust speciation. The allelic variation in smpB exhibited the separation of NTHi and H. haemolyticus; however, Group III displayed a significant diversity from the NTHi therefore interpreting the allelic information from smpB required supporting information. Allelic variation between species in omp6 and hpd was also not deemed suitable to be used in isolation for speciation purposes. The hpd gene was found to be absent in 21 isolates and the diversity demonstrated by the omp6 gene not easily interpretable between species when viewed on its own (Fig. 1). It should be noted that the lgtC results were questionable due to the large inconsistency of lgtC negative isolates with ST type in time points. This may highlight potentially false negatives due to heterogeneity within the gene, over sensitivity of the mapping method or level of gene coverage in the sequencing data which would require further phenotypic investigation or traditional PCR to ascertain gene presence. However, there were no lgtC occurrences identified in H. haemolyticus. Additionally, it is very doubtful that the iga beta core sequences identified in twelve H. haemolyticus would be instrumental in Iga protein expression, the identified sequences were on average around 236 bp long compared to the 864 bp long sequence for the beta core which in itself is a selected sequence, reportedly conserved, from a much larger gene19. This again could be a result of over sensitivity or previously highlighted limitations of the mapping method.
Surface exposed proteins resulting from some of the genes discussed in this study, Protein D, Omp2 and Omp6, have also been investigated as potential vaccine candidates52–54. The importance for vaccination for NTHi does not come from its ability to cause invasive disease but is driven by the larger morbidity caused by NTHi in cases of otitis media and the role NTHi plays in triggering exacerbations in COPD. Figures differ globally but it is thought that 80% of children will suffer from acute OM before the age of five10. In the UK in 2016, the prevalence of OM among children under the age of five was reported as 2469.5 per 100,0001. COPD in the UK is reported to have attributed to 6.4% of all deaths in 20161. A vaccination for NTHi, therefore, could result in a reduction of disease and associated economic burden. However, designing a vaccine for a subspecies as heterogeneic as NTHi has not proven straight forward. Changes in protein expression mediated by sequence variation, genotype absence and phase variation all hinder the discovery of a successful candidate. The absence of omp2 and, to a lesser extend hpd, from NTHi, both in this study and in previous reports, raises the question of the validity of these targets for vaccine candidature26,52. The sequence variation throughout omp6 demonstrated within this study supports other previous reports and may result in variability of exposed regions and thus, potentially, a risk to vaccine efficacy (Fig. 1)55.
It has been suggested that H. haemolyticus and NTHi are two ends of a genetic continuum of one species26. Using whole genome analysis, the isolates in this study fell into three groups (NTHi including f-NTHi, Group III and H. haemolyticus) rather than a continuation of genome diversity but this does consider the limitations of this study derived from bias drawn from limited isolation site and disease, and limited geographical area of patient recruitment. H. haemolyticus was distinguishable from NTHi in phylogeny for hpd, smpB and omp6 sequences and identified as a different species from NTHi using ANI, which has been deemed comparable for taxonomy purposes to the gold standard: DNA-DNA hybridization (Fig. 2)45. Group III were calculated as approximately 92% genetically similar to H. haemolyticus, a similar divergence from H. haemolyticus to the remaining NTHi (Figs 3, 4). However, ANI is only one taxonomy tool and does not consider orthologous content of the genomes, it also relies on alignment for comparison which may disregard some of the genome; however, it gives a good indication of the phylogenetic relationship between strains.
Conclusion
In this study, H. haemolyticus and NTHi are definable as two species, but the presence of Group III demonstrates the diversity of NTHi in a limited study set. These isolates are genetically divergent from NTHi, sit phylogenetically between the remaining NTHi and H. haemolyticus and display an unexpected genotype for NTHi. Group III represent a lineage of atypical strains composed of three novel STs (353, 356, 1314) that were identified from three separate patients. With the addition of genomic data from NTHi isolated from different geographical locations, disease states and carriage samples, other examples of differing atypical groups may occur and further muddy the boundaries between the two species. Interestingly though, genetically the similarity to H. haemolyticus was no greater than NTHi or f-NTHi. Hypothetically, Group III may represent a lineage that requires further investigation to ascertain whether survival techniques have developed in these isolates specific to the COPD niche. Further research is also required to understand their potential in colonisation and virulence.
The misidentification rate of H. haemolyticus as NTHi was relatively low in this study echoing previous studies investigating this matter20–22. As previously reported, further investigation required to confidently distinguish the two species may not warrant the time consuming and expensive assays when faced with the need for timely treatment for potential Haemophilus infection20–22. Ultimately the heterogeneity in NTHi leaves single molecular markers insufficient to delineate with 100% accuracy.
Electronic supplementary material
Acknowledgements
The authors acknowledge the role of the volunteers who made this study possible by committing their time. We acknowledge the Directors and staff of the Southampton NIHR Respiratory Biomedical Research Unit and the NIHR Wellcome Trust Clinical Research Facility and express appreciation to Alice Still and Rebecca Anderson for sequencing and laboratory support. The authors acknowledge the use of the IRIDIS High Performance Computing Facility, and associated IT support services at the University of Southampton, in the completion of this work.This publication made use of the Haemophilus influenzae MLST website (https://pubmlst.org/hinfluenzae/) sited at the University of Oxford (Jolley & Maiden 2010, BMC Bioinformatics, 11:595). The development of this site has been funded by the Wellcome Trust. The authors would like to also thank Geraldine Drevon and Regis Azizieh (XPE Pharma & Science, on behalf of GSK Vaccines) for coordination and editorial support. This study was funded by GlaxoSmithKline as part of the AERIS study ClinicalTrials.gov NCT01360398.
Author Contributions
K.O. had access to the AERIS data and takes responsibility for the accuracy of further data generated and the data analysis. J.M.D., N.D., T.P., S.C.C. and T.M.A.W. conceived and designed the AERIS study. J.M.D., D.W.C., S.C.C. and T.M.A.W. collected or generated the data for the AERIS study. J.M.D., N.D., T.P., M.C.M., D.W.C., S.C.C. and T.M.A.W. analysed or interpreted the data for the AERIS study. All authors contributed to the development of the manuscript and approved the final version. All members of the AERIS Study Group were involved in the planning, conduct, and/or reporting of the work described in the article.
Data Availability
The results summary for this study (GSK study number 114378 NCT 01360398) is available on the GSK Clinical Study Register and can be accessed at www.gsk-clinicalstudyregister.com. For interventional studies that evaluate our medicines, anonymized patient-level data will be made available to independent researchers, subject to review by an independent panel, at www.clinicalstudydatarequest.com within six months of publication. To protect the privacy of patients and individuals involved in our studies, GSK does not publicly disclose patient- level data.
Competing Interests
K.O. was funded by G.S.K. to conduct this study as part of a studentship. T.M.A.W. has received reimbursement for travel and meeting attendance from Boehringer Ingelheim and AstraZeneca, outside of the submitted work. S.C. reports grants from G.S.K., during the conduct of the study; grants from Pfizer, outside the submitted work. N.D., T.P. and J.M.D. are employees of the G.S.K. group of companies, and own shares/restricted shares in the G.S.K. group of companies. D.W.C. and T.M.A.W. received an institutional grant from the G.S.K. group of companies to conduct this study. The other authors have nothing to disclose.
Footnotes
A comprehensive list of consortium members appears at the end of the paper.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
David W. Cleary and Stuart C. Clarke jointly supervised this work.
Contributor Information
Stuart C. Clarke, Email: S.C.Clarke@soton.ac.uk
AERIS Study Group:
J. Alnajar, R. Anderson, E. Aris, W. R. Ballou, A. Barton, S. Bourne, M. Caubet, C. Cohet, N. Coombs, V. Devine, E. Dineen, T. Elliott, R. Gladstone, S. Harden, V. Kim, S. Mesia Vela, P. Moris, K. Ostridge, M. Peeters, S. Schoonbroodt, K. J. Staples, A. Tuck, L. Welch, V. Weynants, A. P. Williams, N. Williams, M. Wojtas, and S. Wootton
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-32973-3.
References
- 1.Collaborators GBOD. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. 2017;390:1151–1210. doi: 10.1016/S0140-6736(17)32152-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murphy TF, et al. Haemophilus haemolyticus: a human respiratory tract commensal to be distinguished from Haemophilus influenzae. The Journal of infectious diseases. 2007;195:81–89. doi: 10.1086/509824. [DOI] [PubMed] [Google Scholar]
- 3.Bandi V, et al. Infectious exacerbations of chronic obstructive pulmonary disease associated with respiratory viruses and non-typeable Haemophilus influenzae. FEMS immunology and medical microbiology. 2003;37:69–75. doi: 10.1016/S0928-8244(03)00100-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilkinson TM, et al. Effect of interactions between lower airway bacterial and rhinoviral infection in exacerbations of COPD. Chest. 2006;129:317–324. doi: 10.1378/chest.129.2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilkinson Tom M A, Aris Emmanuel, Bourne Simon, Clarke Stuart C, Peeters Mathieu, Pascal Thierry G, Schoonbroodt Sonia, Tuck Andrew C, Kim Viktoriya, Ostridge Kristoffer, Staples Karl J, Williams Nicholas, Williams Anthony, Wootton Stephen, Devaster Jeanne-Marie. A prospective, observational cohort study of the seasonal dynamics of airway pathogens in the aetiology of exacerbations in COPD. Thorax. 2017;72(10):919–927. doi: 10.1136/thoraxjnl-2016-209023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chung KF, Adcock IM. Multifaceted mechanisms in COPD: inflammation, immunity, and tissue repair and destruction. The European respiratory journal. 2008;31:1334–1356. doi: 10.1183/09031936.00018908. [DOI] [PubMed] [Google Scholar]
- 7.Anderson EC, et al. Epidemiology of invasive Haemophilus influenzae infections in England and Wales in the pre-vaccination era (1990-2) Epidemiology and infection. 1995;115:89–100. doi: 10.1017/S0950268800058155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Collins S., Litt D. J., Flynn S., Ramsay M. E., Slack M. P. E., Ladhani S. N. Neonatal Invasive Haemophilus influenzae Disease in England and Wales: Epidemiology, Clinical Characteristics, and Outcome. Clinical Infectious Diseases. 2015;60(12):1786–1792. doi: 10.1093/cid/civ194. [DOI] [PubMed] [Google Scholar]
- 9.PHE. Laboratory reports of Haemophilus influenzae by age group and serotype (England and Wales): annual report for 2015. (Public Health England, 2015).
- 10.Monasta Lorenzo, Ronfani Luca, Marchetti Federico, Montico Marcella, Vecchi Brumatti Liza, Bavcar Alessandro, Grasso Domenico, Barbiero Chiara, Tamburlini Giorgio. Burden of Disease Caused by Otitis Media: Systematic Review and Global Estimates. PLoS ONE. 2012;7(4):e36226. doi: 10.1371/journal.pone.0036226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kirkham LA, et al. Nasopharyngeal carriage of Haemophilus haemolyticus in otitis-prone and healthy children. Journal of clinical microbiology. 2010;48:2557–2559. doi: 10.1128/JCM.00069-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morton DJ, Hempel RJ, Whitby PW, Seale TW, Stull TL. An invasive Haemophilus haemolyticus isolate. Journal of clinical microbiology. 2012;50:1502–1503. doi: 10.1128/JCM.06688-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anderson R, et al. Haemophilus haemolyticus isolates causing clinical disease. Journal of clinical microbiology. 2012;50:2462–2465. doi: 10.1128/JCM.06575-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pickering JL, et al. Haemophilus haemolyticus Interaction with Host Cells Is Different to Nontypeable Haemophilus influenzae and Prevents NTHi Association with EpithelialCells. Frontiers in cellular and infection microbiology. 2016;6:50. doi: 10.3389/fcimb.2016.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singh NK, Kunde D, Tristram S. Inability of H. haemolyticus to invade respiratory epithelial cells in vitro. Journal of medical microbiology. 2016 doi: 10.1099/jmm.0.000349. [DOI] [PubMed] [Google Scholar]
- 16.Kilian M. The haemolytic activity of Haemophilus species. Acta pathologica et microbiologica Scandinavica. Section B. Microbiology. 1976;84B:339–341. doi: 10.1111/j.1699-0463.1976.tb01950.x. [DOI] [PubMed] [Google Scholar]
- 17.McCrea KW, et al. Relationships of nontypeable Haemophilus influenzae strains to hemolytic and nonhemolytic Haemophilus haemolyticus strains. Journal of clinical microbiology. 2008;46:406–416. doi: 10.1128/JCM.01832-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.PHE. in UK Standards for Microbiology Investigations. Vol. ID 12 (2015).
- 19.Sandstedt SA, et al. Comparison of laboratory-based and phylogenetic methods to distinguish between Haemophilus influenzae and H. haemolyticus. Journal of microbiological methods. 2008;75:369–371. doi: 10.1016/j.mimet.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fenger MG, Ridderberg W, Olesen HV, Norskov-Lauritsen N. Low occurrence of ‘non-haemolytic Haemophilus haemolyticus’ misidentified as Haemophilus influenzae in cystic fibrosis respiratory specimens, and frequent recurrence of persistent H. influenzae clones despite antimicrobial treatment. International journal of medical microbiology: IJMM. 2012;302:315–319. doi: 10.1016/j.ijmm.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 21.Zhang B, Kunde D, Tristram S. Haemophilus haemolyticus is infrequently misidentified as Haemophilus influenzae in diagnostic specimens in Australia. Diagnostic microbiology and infectious disease. 2014;80:272–273. doi: 10.1016/j.diagmicrobio.2014.08.016. [DOI] [PubMed] [Google Scholar]
- 22.Frickmann H, Podbielski A, Essig A, Schwarz NG, Zautner AE. Difficulties in species identification within the genus Haemophilus - A pilot study addressing a significant problem for routine diagnostics. European journal of microbiology & immunology. 2014;4:99–105. doi: 10.1556/EuJMI.4.2014.2.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hiltke TJ, Sethi S, Murphy TF. Sequence stability of the gene encoding outer membrane protein P2 of nontypeable Haemophilus influenzae in the human respiratory tract. The Journal of infectious diseases. 2002;185:627–631. doi: 10.1086/339362. [DOI] [PubMed] [Google Scholar]
- 24.Hu Fang, Rishishwar Lavanya, Sivadas Ambily, Mitchell Gabriel J., Jordan I. King, Murphy Timothy F., Gilsdorf Janet R., Mayer Leonard W., Wang Xin. Comparative Genomic Analysis of Haemophilus haemolyticus and Nontypeable Haemophilus influenzae and a New Testing Scheme for Their Discrimination. Journal of Clinical Microbiology. 2016;54(12):3010–3017. doi: 10.1128/JCM.01511-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Gier Camilla, Pickering Janessa L., Richmond Peter C., Thornton Ruth B., Kirkham Lea-Ann S. Duplex Quantitative PCR Assay for Detection of Haemophilus influenzae That Distinguishes Fucose- and Protein D-Negative Strains. Journal of Clinical Microbiology. 2016;54(9):2380–2383. doi: 10.1128/JCM.00982-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Binks MJ, et al. Molecular surveillance of true nontypeable Haemophilus influenzae: an evaluation of PCR screening assays. PloS one. 2012;7:e34083. doi: 10.1371/journal.pone.0034083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Price EP, et al. Haemophilus influenzae: using comparative genomics to accurately identify a highly recombinogenic human pathogen. BMC genomics. 2015;16:641. doi: 10.1186/s12864-015-1857-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fung WW, et al. Presence of copper- and zinc-containing superoxide dismutase in commensal Haemophilus haemolyticus isolates can be used as a marker to discriminate them from nontypeable H. influenzae isolates. Journal of clinical microbiology. 2006;44:4222–4226. doi: 10.1128/JCM.01376-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Latham R, Zhang B, Tristram S. Identifying Haemophilus haemolyticus and Haemophilus influenzae by SYBR Green real-time PCR. Journal of microbiological methods. 2015;112:67–69. doi: 10.1016/j.mimet.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 30.Connor TR, Corander J, Hanage WP. Population subdivision and the detection of recombination in non-typable Haemophilus influenzae. Microbiology. 2012;158:2958–2964. doi: 10.1099/mic.0.063073-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Power PM, Bentley SD, Parkhill J, Moxon ER, Hood DW. Investigations into genome diversity of Haemophilus influenzae using whole genome sequencing of clinical isolates and laboratory transformants. Bmc Microbiol. 2012;12:273. doi: 10.1186/1471-2180-12-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meats E, et al. Characterization of encapsulated and noncapsulated Haemophilus influenzae and determination of phylogenetic relationships by multilocus sequence typing. Journal of clinical microbiology. 2003;41:1623–1636. doi: 10.1128/JCM.41.4.1623-1636.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu B, et al. MALDI-TOF MS distinctly differentiates nontypable Haemophilus influenzae from Haemophilus haemolyticus. PloS one. 2013;8:e56139. doi: 10.1371/journal.pone.0056139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bruin JP, et al. Identification of Haemophilus influenzae and Haemophilus haemolyticus by matrix-assisted laser desorption ionization-time of flight mass spectrometry. European journal of clinical microbiology & infectious diseases: official publication of the European Society of Clinical Microbiology. 2014;33:279–284. doi: 10.1007/s10096-013-1958-x. [DOI] [PubMed] [Google Scholar]
- 35.Chen JHK, et al. Rapid Differentiation of Haemophilus influenzae and Haemophilus haemolyticus by Use of Matrix-Assisted Laser Desorption Ionization-Time of Flight Mass Spectrometry with ClinProTools Mass Spectrum Analysis. Journal of clinical microbiology. 2017;55:2679–2685. doi: 10.1128/JCM.00267-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zimin AV, et al. The MaSuRCA genome assembler. Bioinformatics. 2013;29:2669–2677. doi: 10.1093/bioinformatics/btt476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Inouye M, et al. SRST2: Rapid genomic surveillance for public health and hospital microbiology labs. Genome medicine. 2014;6:90. doi: 10.1186/s13073-014-0090-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic acids research. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Argimon S, et al. Microreact: visualizing and sharing data for genomic epidemiology and phylogeography. Microb Genom. 2016;2:e000093. doi: 10.1099/mgen.0.000093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Segata N, et al. Metagenomic microbial community profiling using unique clade-specific marker genes. Nature methods. 2012;9:811–814. doi: 10.1038/nmeth.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Team, R. RStudio: Integrated development for R, http://www.rstudio.com (2015).
- 44.Richter M, Rossello-Mora R. Shifting the genomic gold standard for the prokaryotic species definition. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:19126–19131. doi: 10.1073/pnas.0906412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goris J, et al. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. International journal of systematic and evolutionary microbiology. 2007;57:81–91. doi: 10.1099/ijs.0.64483-0. [DOI] [PubMed] [Google Scholar]
- 46.Page AJ, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31:3691–3693. doi: 10.1093/bioinformatics/btv421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miller MA, et al. A RESTful API for Access to Phylogenetic Tools via the CIPRES Science Gateway. Evolutionary bioinformatics online. 2015;11:43–48. doi: 10.4137/EBO.S21501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chan JZ, Halachev MR, Loman NJ, Constantinidou C, Pallen MJ. Defining bacterial species in the genomic era: insights from the genus Acinetobacter. Bmc Microbiol. 2012;12:302. doi: 10.1186/1471-2180-12-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reddington K, et al. Comparison of Established Diagnostic Methodologies and a Novel Bacterial smpB Real-Time PCR Assay for Specific Detection of Haemophilus influenzae Isolates Associated with Respiratory Tract Infections. Journal of clinical microbiology. 2015;53:2854–2860. doi: 10.1128/JCM.00777-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.de Gier C, Kirkham LA, Norskov-Lauritsen N. Complete Deletion of the Fucose Operon in Haemophilus influenzae Is Associated with a Cluster in Multilocus Sequence Analysis-Based Phylogenetic Group II Related to Haemophilus haemolyticus: Implications for Identification and Typing. Journal of clinical microbiology. 2015;53:3773–3778. doi: 10.1128/JCM.01969-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ridderberg W, Fenger MG, Norskov-Lauritsen N. Haemophilus influenzae may be untypable by the multilocus sequence typing scheme due to a complete deletion of the fucose operon. Journal of medical microbiology. 2010;59:740–742. doi: 10.1099/jmm.0.018424-0. [DOI] [PubMed] [Google Scholar]
- 52.Smith-Vaughan HC, et al. Absence of an important vaccine and diagnostic target in carriage- and disease-related nontypeable Haemophilus influenzae. Clinical and vaccine immunology: CVI. 2014;21:250–252. doi: 10.1128/CVI.00632-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Neary JM, Murphy TF. Antibodies directed at a conserved motif in loop 6 of outer membrane protein P2 of nontypeable Haemophilus influenzae recognize multiple strains in immunoassays. FEMS immunology and medical microbiology. 2006;46:251–261. doi: 10.1111/j.1574-695X.2005.00033.x. [DOI] [PubMed] [Google Scholar]
- 54.Hua CZ, et al. Serum Concentrations of Antibodies against Outer Membrane Protein P6, Protein D, and T- and B-Cell Combined Antigenic Epitopes of Nontypeable Haemophilus influenzae in Children and Adults of Different Ages. Clinical and vaccine immunology: CVI. 2016;23:155–161. doi: 10.1128/CVI.00506-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chang A, Kaur R, Michel LV, Casey JR, Pichichero M. Haemophilus influenzae vaccine candidate outer membrane protein P6 is not conserved in all strains. Human vaccines. 2011;7:102–105. doi: 10.4161/hv.7.1.13351. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The results summary for this study (GSK study number 114378 NCT 01360398) is available on the GSK Clinical Study Register and can be accessed at www.gsk-clinicalstudyregister.com. For interventional studies that evaluate our medicines, anonymized patient-level data will be made available to independent researchers, subject to review by an independent panel, at www.clinicalstudydatarequest.com within six months of publication. To protect the privacy of patients and individuals involved in our studies, GSK does not publicly disclose patient- level data.