

Abstract
The hippo pathway effector, Yes-associated protein (YAP) is a transcriptional co-activator implicated in cholangiocarcinoma (CCA) pathogenesis. YAP is known to be regulated by a serine/threonine kinase relay module (MST1/2 - LATS1/2) culminating in phosphorylation of YAP at Serine 127 (S127) and cytoplasmic sequestration. However, YAP also undergoes tyrosine phosphorylation, and the role of tyrosine phosphorylation in YAP regulation remains unclear. Herein, YAP regulation by tyrosine phosphorylation was examined in human and mouse CCA cells, as well as patient-derived xenograft (PDX) models. YAP was phosphorylated on tyrosine 357 (Y357) in CCA cell lines and PDX models. SRC family kinase (SFK) inhibition with dasatinib resulted in loss of YAPY357 phosphorylation, promoted its translocation from the nucleus to the cytoplasm, and reduced YAP target gene expression; including cell lines expressing a LATS1/2-resistant YAP mutant in which all serine residues were mutated to alanine. Consistent with these observations, precluding YAPY357 phosphorylation by site-directed mutagenesis (YAPY357F) excluded YAP from the nucleus. Targeted siRNA experiments identified LCK as the SFK that most potently mediated YAPY357 phosphorylation. Likewise, inducible CRISPR/Cas9-targeted LCK deletion decreased YAPY357 phosphorylation and its nuclear localization. The importance of LCK in CCA biology was demonstrated by clinical observations suggesting LCK expression levels were associated with early tumor recurrence following resection of CCA. Finally, dasatinib displayed therapeutic efficacy in PDX models.
Keywords: dasatinib, Hippo pathway, xenograft
INTRODUCTION
Cholangiocarcinoma (CCA) is a tumor with features of biliary epithelial differentiation and extremely limited treatment options. The underlying molecular mechanisms of both tumorigenesis and progression remain incompletely understood; however, accumulating data implicates the Hippo pathway and its effector Yes-associated protein (YAP) in CCA pathogenesis.(1–3)
The Hippo pathway is a developmental pathway important for organ size control.(4) Alterations in this pathway have been implicated in the pathogenesis of multiple types of cancer, including CCA, although mutations in the pathway components themselves are rare.(5–9) The paucity of specific Hippo pathway mutations and the lack of a dedicated cell-surface receptor have increased interest in understanding the role of signaling cross-talk between the Hippo and other signal transduction pathways. Indeed, cross-regulatory networks have been demonstrated for a wide variety of cell surface receptors including G-protein linked receptors and more recently receptor tyrosine kinases such as fibroblast growth factor (FGF), endothelial growth factor (EGF), and platelet-derived growth factor (PDGF) in Hippo regulation.(2, 8, 10) The Hippo pathway itself consists of an upstream serine kinase relay module (comprised of MST1/2 and LATS1/2) which regulates the co-transcriptional activators YAP and TAZ.(4) Although YAP and TAZ are both regulated by the Hippo pathway, YAP has been more strongly implicated in CCA biology.(1, 2, 8) Hippo pathway activation acts as a “brake” to transcription. This serine phosphorylation of YAP restrains the protein in the cytosol through interactions with protein 14-3-3, limiting its function as a transcriptional co-activator. The role of serine phosphorylation regulating YAP in the context of developmental processes is well described, however emerging data suggests that tyrosine phosphorylation of YAP may also play a role in regulating YAP localization and transcriptional activity, especially in cancer.(2, 11)
We recently described cross-talk between the PDGF and Hippo signaling pathways in CCA.(2) We demonstrated regulation of YAP localization by tyrosine phosphorylation in a manner independent of the core Hippo serine/threonine kinase relay module. Inhibition of PDGF receptors induced a redistribution of YAP from the nucleus (transcriptionally active) to the cytosol (transcriptionally inactive) even when YAP was mutated at the canonical LATS 1/2 phosphorylation site, serine 127 (S127). The baseline nuclear localization of YAP appeared to be mediated by SRC family kinases (SFK), acting by phosphorylation of tyrosine 357 (Y357) on YAP. However, the specific SFK regulating YAPY357 phosphorylation was not examined in these studies. Neither was the potential therapeutic role of inhibiting SFK in CCA explored. Given the lack of therapeutic options for CCA and the role of YAP in its pathogenesis, these questions merit further consideration.
Herein, we examine tyrosine phospho-regulation of YAP cellular compartmentation and target gene expression in CCA cells and tissue. The results demonstrate enhanced YAP tyrosine phosphorylation in CCA as compared to normal human cholangiocytes. More importantly, the SFK inhibitor dasatinib blocks YAP tyrosine phosphorylation, induces YAP redistribution form the nucleus to the cytosol, and downregulates expression of YAP target genes. siRNA targeted knockdown studies suggest a pivotal role for the SFK member LCK in mediating YAPY357 phosphorylation, a previously unrecognized function for LCK. Most significantly this tyrosine phospho-regulation of YAP subcellular localization was independent of serine phospho-regulatory mechanisms; suggesting separate cytosolic and nuclear retention signals for YAP, a novel concept in YAP regulation. The efficacy of YAP downregulation by SFK inhibition translated therapeutically to increases in CCA cell death both in vitro and in vivo, providing further evidence supporting the evaluation of SFK inhibition as a therapeutic strategy in CCA.
MATERIALS AND METHODS
Cell culture
The human cholangiocarcinoma cell line HuCCT-1, mouse cholangiocarcinoma cell line SB1, and normal human cholangiocyte (NHC) cell line (12), were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 0.2% primocin under standard conditions. The murine SB1 cell line was isolated from our murine model of YAP associated CCA and has been described in detail.(13) This cell line expresses flag-tagged-YAPS127A and myr-AKT. Due to cell density and serum regulation of the Hippo pathway, cell culture experiments were performed at near confluence (~80%) in serum starved cells. Serum starved cells were cultured overnight in media without serum. The media was replaced again three hours prior to initiation of experimental conditions. For authentication of the HuCCT-1 cell line, short tandem repeat (STR) analysis was performed by the Genome Analysis Core of the Medical Genome Facility (Mayo Clinic, Rochester, MN). All cell lines underwent Mycoplasma contamination testing periodically using PlasmoTestTM -Mycoplasma Detection kit (InvivoGen). Cell lines were used within 40 passages of reanimation.
Antibodies and reagents
Dasatinib (Selleckchem), KW-2449 (Selleckchem), sodium orthovanadate (Sigma), and/or latrunculin A (Abcam) was added to cells at final concentrations from 0.1–10 µM per experimental design. The following primary antisera were used for immunoblot analysis: actin (C-11 Santa Cruz, Dallas, TX), phospho YAPY357 (ab62751 abcam, Cambridge, MA), phospho YAPS127 (4911 Cell Signaling), total YAP (63.7 Santa Cruz, 4912 CST), lamin B1 (D8P3U Cell Signaling), GAPDH (MAB374 Millipore, Temecula, CA), phospho SRCY416 (2101 Cell Signaling), SRC (L4A1 Cell Signaling), YES (3201 Cell Signaling), LCK (2657 Cell Signaling), FYN (4023 Cell Signaling), LYN (44 Santa Cruz), LATS1 (C66B Cell Signaling), phospho LATS1 (S909 Cell Signaling) and MCL-1 (S-19 Santa Cruz). The following primary monoclonal antibody were used for immunofluorescence and/or immunohistochemistry: total YAP (63.7 Santa Cruz), phospho YAPY357 (ab62751 abcam, Cambridge, MA), SOX9 (AB5535 Millipore) and anti-FLAG M2 (F1804 Sigma Adrich). ProLong Antifade with 4’,6-diamidino-2-phenylindole (DAPI, Life Technologies) was used for nuclear staining.
Immunoprecipitation and Immunoblot analyses
Whole-cell lysates were collected by adding ice cold lysis buffer (Cell Signaling Technologies) containing protease inhibitors (Sigma-Aldrich), phosphatase inhibitors (Roche Diagnostics) and 1 mM PMSF. Cells were then scraped to collect cells, vortexed and lysed on ice for approximately 20 minutes. The lysed cells were then centrifuged at 12,000g for 15 minutes to remove cellular debris. Supernatant was transferred to a clean tube and protein concentration determined by the Bradford (Sigma-Aldrich) protein assay. For immunoprecipitation, whole-cell lysates of SB1 cells that contain the FLAG-YAPS127A were incubated with equilibrated anti-FLAG M2 magnetic beads (Sigma-Aldrich) overnight at 4°C. The FLAG-YAPS127A was then eluted from the beads with FLAG peptide following manufacturer’s instructions and evaluated by mass spectroscopy (Supplemental Methods). For immunoblot, proteins were then resolved by SDS-PAGE electrophoresis and transferred to nitrocellulose membranes. Membranes were blotted with primary antibodies at 4°C overnight in 5% BSA-TBS Tween. The primary antibody dilution was 1:1000 unless otherwise indicated. After overnight incubation membranes were washed for 30 minutes in TBS Tween and then horseradish peroxidase-conjugated secondary antibodies against mouse, rabbit and goat (Santa Cruz) were added to membrane at a concentration of 1:5000 and incubated for 1 hour at room temperature. Immunoblots were visualized with enhanced chemiluminescence (ECL) or ECL prime (GE Healthcare Life Sciences). For experiments in which multiple proteins were evaluated, membranes were stripped and reblotted.
Immunofluorescence
Cells were grown on glass coverslips, once the cells reached desired confluency they were treated per experimental conditions. Cells were then fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100. The fixed cells were then incubated at room temperature with blocking buffer containing 5.0% bovine serum albumin (BSA) and 0.1% glycine. After blocking, the cells were incubated with primary antibody in blocking buffer overnight at 4°C. After washing, the cells were incubated with the corresponding secondary antibodies diluted in blocking buffer for 1 hour at room temperature in the dark. After secondary incubation, the coverslips were again washed and mounted onto slides using ProLong Antifade (Invitrogen-Molecular Probes) containing DAPI. The slides were analyzed by fluorescent confocal microscopy (LSM 780, Zeiss).
Immunohistochemistry
Liver sections were fixed in 10% buffered formaldehyde and embedded in paraffin. Blocks were sectioned into 3.5 µM slices. Sections were deparaffinized, hydrated, and incubated with primary antibody overnight at 4° C. Sections were stained with antibody for SOX9 (1:1000), YAP (1:50), and phospho YAPY357 (1:50). Bound antibody was detected with biotin-conjugated secondary antibody and diaminobenzidine as a substrate.
Quantitative and qualitative PCR
mRNA was isolated from cells using TRIzol Reagent (Ambion). Reverse transcription was performed using Moloney murine leukemia virus reverse transcriptase and random primers (Life Technologies). After reverse transcription, cDNA was analysed using real-time PCR (Light Cycler 480 II, Roche Diagnostics) for the quantitation of the target genes; SYBR Green (Roche Diagnostics) was used as the fluorophore. Expression was normalized to either 18 S or GAPDH and relative quantification performed according to the 2−ΔCT or 2−ΔΔCT method as previously described.(14) Data are reported as fold expression compared to calibrator as geometric mean and geometric standard deviation of expression relative to calibrator. Technical replicates were completed for each run and a minimum of three biologic replicates completed for each condition/cell line. The primers used are listed in Supplemental Table 1.
In vitro kinase assay
The LCK kinase enzyme system and ADP-Glo kinase assay (Promega) were used to asses LCKs ability to phosphorylate YAP in vitro. The protocol for the LCK kinase assay was followed as described by manufacturer. Briefly for luminescent assay, 50 µM DTT, 2 mM MnCl2, 10 mM ultra pure ATP and 0.1 µg of LCK enzyme were added to wells containing 1× reaction buffer. Recombinant YAP (abcam) 1µg or Poly (Glu4,Tyr1) peptide 1 µg were added and enzyme reaction was incubated at RT for 60 minutes. 25 µl of ADP-Glo reagent was added to well and incubated at RT for 40 minutes, 50 µl of kinase detection reagent was then added and incubated at RT for 60 minutes. Luminescence was then measured on Synergy H1 (Biotek) plate reader. For immunoblot detection 50 µM DTT, 2 mM MnCl2, 10 mM ultra pure ATP and 90 ng or recombinant YAP protein were incubated for 1 hour with and without the presence of 0.1 µg of LCK enzyme. After enzyme reaction incubation 4× laemmli sample buffer was added, samples were boiled for 10 minutes and then resolved on 10% gel for transfer and immunoblotting.
RNA Interference & transfection
SRC, YES, LCK, FYN, and LYN were transiently knocked down in the HuCCT-1 cell line with validated siRNAs (Dharmacon). Cells were grown in 6 well plates and transfected with Lipofectamine RNAiMAX (Invitrogen) following manufacturer’s instructions. After 48 hours of incubation they were used for indicated studies. The efficiency and specificity of knockdown was assayed by immunoblot and RT-PCR.
CRSPR/Cas9 knockdown and doxycycline induction
The sequences of single guide RNA (sgRNA) for LCK was designed using online CRISPR design tool (http://crispr.mit.edu/) and was 5′-TCCCGACCCACTGGTTACCTACGA -3′. DNA oligonucleotides for the sgRNA were synthesized and cloned into the FgH1tUTG plasmid, a kind gift of Dr. Marco Herold.(15) To produce lentiviruses, FgH1tLCK or FUCas9Cherry plasmids were transiently co-transfected into HEK293T cells with the packaging plasmids pRSV-Rev, pMDLg/pRRE and pCMV-VSV-G (all from Addgene, Cambridge, MA, USA) using FuGENE® HD Transfection Reagent (Promega Corporation, Madison, WI, USA). After 48 and 72 hours, supernatant containing lentiviruses was harvested and passed through a 0.45-µM filter. Target HuCCT-1 cells were transduced with the lentiviruses in the presence of 8 µg/mL polybrene for 24 hours. After the additional 3 days culturing, m-cherry (Cas9) and GFP (inducible sgRNA) dual-positive cells were sorted on a single cell-basis into a 96 well plate by flow cytometry. To induce sgRNA expression, doxycycline hyclate (SIGMA) was added to culture medium at a final concentration of 7.5 µg/mL. The efficacy of CRISPR/Cas9-mediated gene deletion was determined by immunoblot analysis.
YAP serine mutant generation
pCMV-Flag-YAP-5SA/S94A was a gift from Kunliang Guan (Addgene plasmid # 33103). HuCCT-1 cells were transfected using FugeneHD (Promega) following the manufacturer’s instructions. Following 48 hour transfection the cells were used for subsequent experiments.
YAP tyrosine mutant generation
To construct pEGFP-YAP, pENTR-YAP (a generous gift from Dr. Xin Chen, UCSF) was digested with BglII and EcoRI and purified YAP fragment was ligated into a fragment of BglII-EcoRI pEGFP-C3 vector. For site-directed mutagenesis of tyrosine residues, primers were designed using NEBaseChanger on-line tool and PCR was performed using Q5 Site-Directed Mutagenesis Kit (New England Biolabs). To make pEGFP-YAP Y357F, PCR was performed with pENTR-YAP as a template using oligonucleotides 5’- ATGAGCAGCTTCAGTGTCCCTC -3’ and 5’-GCTTAGTCCACTGTCTGTAC-3’ as primers (the complementary sequences underlined). Triple mutant pEGFP-YAP Y341,357,394F was prepared in a similar way by consecutive mutations of pENTR-YAP Y357F using primers 5’-AGTGGCACCTTTCACTCTCGAG-3’ and 5’-GTTAAGGAAAGGATCTGAGC-3’, followed by TTCCCAGACTTCCTTGAAGCCATTC and 5’-ACGGTTCTGCTGTGAGGG-3’. Finally, pENTR plasmids with mutated YAP were digested with BglII and EcoRI and YAP fragments were inserted into a BglII-EcoRI pEGFP-C3 vector. All constructs of YAP contain a FLAG epitope at the amino-terminus.
RNA sequencing and bioinformatics
Following IRB approval, patient derived xenografts of resected intrahepatic cholangiocarcinoma tumors were created as described previously(2). RNA sequencing was undertaken in 7 resected ICC PDX. Clinicopathologic features and recurrence free and overall survival were abstracted for the patients. Differentially expressed genes were identified between early recurrence (<10 months) and late recurrence with a log2 threshold of 1.5 with a p-value of <0.01. For this cohort of patients spliced aligned was carried out using TopHat v2.1.0 (TopHat2) with two conditions, one corresponding to early recurrence (L10 group) and the second condition corresponding to late recurrence (G10 group) with 4 and 3 replicates, respectively.(16) The reference genome and the annotation transcriptome utilized in this work for all the alignments were based on the iGenome package provided by Illumina. TopHat2 package utilized Bowtie v2.2.3.0 as the underlying read-alignment software.(17) For downstream analysis we utilized cufflinks v2.2.1 to assemble transcripts for all the samples.(18–21) These programs rely on the accepted_hitms.bam generated after running TopHat2. Finally, Cuffdiff v2.2.1 was used to generate differential gene expression and the results were analyzed via the cummeRbund package v2.10.0.(22) Differential gene expression was measured using fragments per kilobase of transcript per million fragments mapped (FPKM). We calculated the fragment per kilobase of transcript per million (FPKM) of each transcript and gene using statistical significance in Cuffdiff, which reports statistical significance based on whether the p-value is greater than the false discovery rate (FDR) after applying the Benjamini-Hochberg correction.(22) The Biomarker Discovery RNA-seq (BMD_RNA-seq) pipeline workflow was utilized to carry out alignment and differential gene expression.(23)
Quantification of cell death/proliferation
Cell death and proliferation was quantified by counting using Celigo (Nexcelom) after staining with propidium iodide (Sigma) and Hoechst 33342 (Invitrogen), respectively.
PDX generation and treatment
Mayo Clinic, Rochester, has an ongoing IRB approved protocol for collection and xenotransplantation of resected biliary tract cancers. In accordance with this protocol and an established IACUC protocol, human CCA tumors were implanted in NOD/SCID mice as previously described by us.(8) Once the tumors had reached palpable size (~125 mm3), size was recorded and treatment with dasatinib (15 mg/kg) or vehicle begun daily for 3 days or 2 weeks via gavage as previously described.(24) At the end of the treatment period, all mice were sacrificed and tumor tissue obtained for further studies.
TUNEL staining
The fluorescent TUNEL assay (in situ cell death detection kit, Roche) was utilized on tissue sections. Sections were paraformaldehyde-fixed and hydrated. The TUNEL assay was then performed using the manufacturer’s protocol. Slides were mounted with ProLong Antifade (Invitrogen-Molecular Probes) containing DAPI. The slides were analyzed by fluorescent confocal microscopy (LSM 780, Zeiss). Dead cells were quantified by counting TUNEL-positive nuclei in 20 random microscopic fields (20×).
Methods regarding mass spectrometry, protein identification, and statistical methods are located in the supplemental methods.
RESULTS
Tyrosine 357 phosphorylation is increased in cholangiocarcinoma cells and tissue
Initially, we examined the phosphorylation status of YAP tyrosine 357 (YAPY357) at baseline in multiple models of cholangiocarcinoma as compared to normal human cholangiocytes. YAPY357 phosphorylation was readily identified in HuCCT-1 and SB1 cells as compared to normal human cholangiocytes (Figure 1A). The YAPY357 phosphorylation observed in these cell lines was consistent with the YAPY357 phosphorylation observed in both CCA patient derived xenografts (PDX) (Figure 1B) and parental tumors from which the SB1 cell line was derived (Figure 1C). Similar to our previous observations that phosphorylated YAPY357 was enriched in the nucleus of HuCCT-1 cells, immunohistochemistry also demonstrated significant enrichment of the phosphorylated YAPY357 in the nuclei of tumors from the PDX and our YAP-driven murine model of CCA (Figures 1B,C). Consistent with the minimal phosphorylated YAPY357 noted in normal human cholangiocytes by immunoblot analysis; immunohistochemistry of normal adjacent liver from SB1 tumor bearing mice also revealed minimal nuclear phosphorylated YAPY357 in cholangiocytes despite the presence of unphosphorylated YAP in this cell type (Fig. 1C). These data indicate that although nuclear YAP is present in normal cholangiocytes, the YAP is not Y357 phosphorylated. In contrast, in CCA a significant proportion of YAP localized to the nucleus is phosphorylated at Y357.
Figure 1. YAPY357 is phosphorylated in cholangiocarcinoma cell lines and tumors.

(A) Cell lysates from the NHC, HuCCT-1, and SB1 were subjected to immunoblot for phosphorylated YAP tyrosine 357 (pYAPY357). Total YAP and actin were used as loading controls. (B,C) Representative images of immunohistochemistry used to detect SOX9, YAP, and phosphorylated YAP (pYAPY357) expression in CCA patient derived xenografts (PDX)(B), parental tumor from which the SB1 cell line was derived (C), and from normal adjacent liver (C). Secondary only controls are included. N=5 tumors/livers were stained. Original magnification 40×. Abbreviations: H&E, hematoxylin and eosin. Scale bar = 50 microns.
Multiple SFK are expressed in cholangiocarcinoma
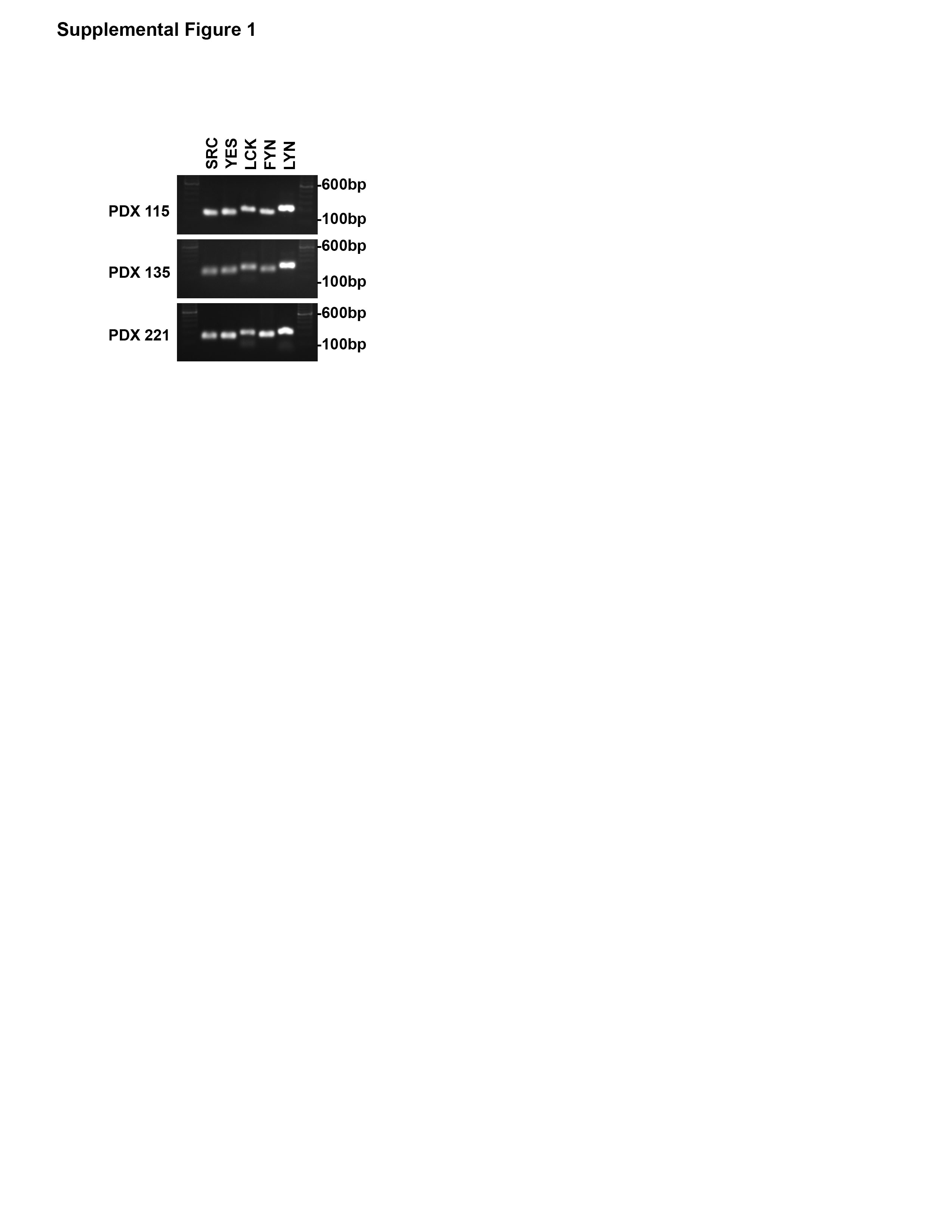
We next evaluated the expression pattern of SFK in the SB1 cell line and multiple CCA patient-derived xenografts. We have previously demonstrated that five SFK are expressed in the HuCCT-1 cell line similar to the normal human cholangiocyte cell line. These five include the ubiquitously expressed SRC, YES, and FYN, as well LCK, and LYN. Evaluation of the SB1 cell line demonstrated expression of these five SFK as well (Figure 2A). We then profiled CCA patient derived xenografts from multiple unique patients. These intrahepatic CCA xenografts all expressed these five SFK similar to the CCA cell lines (Figure 2A and Supplemental 1). We next evaluated the effect of SFK activity on YAP target gene expression. Incubation of both the HuCCT-1 and SB1 cell lines with the SFK inhibitor dasatinib (clinically utilized as a BCR-ABL inhibitor but with nanomolar affinity for SRC family kinases) led to downregulation of the YAP target genes CTGF, CYR61, and MCL-1 (Figure 2B). Similar to our previous observations in HuCCT-1 cells, incubation of the SB1 cell line with dasatinib also blocked the activating phosphorylation of SFK at tyrosine 419 (SFKY419) (Figure 2C). The positive autoregulatory catalytic domain is highly conserved across the SFK members precluding definitive identification of individual members by immunoblot; however, inhibition of SFK tyrosine phosphorylation was accompanied by a parallel decrease in YAPY357 phosphorylation (Figure 2D). Given that c-Abl kinase inhibition could also account for the observations with dasatinib treatment, we also examined YAPY357 phosphorylation and YAP subcellular localization following treatment with KW-2449, a structurally dissimilar c-Abl kinase inhibitor. This more specific c-Abl kinase inhibitor displayed minimal effect on YAPY357 phosphorylation, and no effect on YAP subcellular localization (Supplemental Figure 2). We next evaluated the effects of SFK inhibition by dasatinib in vivo, in a CCA PDX model following a short course of treatment (3 days). Similar on-target effects were noted in the PDX including reduced SFKY419 phosphorylation, YAPY357 phosphorylation, and YAP target gene expression (Figures 2B–D). Importantly, a significant decrease in YAPY357 phosphorylation was also observed by immunohistochemistry (Figure 2E). These data strongly suggest that in CCA, SFK activity is linked to YAPY357 phosphorylation, and pharmacologic inhibition can decrease YAPY357 phosphorylation and YAP target gene expression both in vitro and in vivo.
Figure 2. SFK inhibition downregulates YAP target gene expression, YAPY357 phosphorylation, and YAP nuclear localization in cholangiocarcinoma cell lines and tumors.

(A) RT-PCR identified five SFK expressed in the HuCCT-1, NHC, and SB1 cell lines, as well as PDX42. (B) mRNA expression in HuCCT-1 and SB1 CCA cell lines, and PDX42 +/− SFK inhibitor dasatinib (1 µM, 2 hours) for cell lines and dasatinib (15mg/kg) daily × 3 days for PDX. Fold expression treated/control is depicted as geometric mean +/−geometric standard deviation (n=3). *p<0.05 (C) Cell lysates from the HuCCT-1 and SB1 cell lines, and PDX42 tumors treated with dasatinib at the same concentration as (B) were subjected to immunoblot for phosphorylated SRC (Y416). Total SRC and Actin were used as loading controls. (D) Cell lysates from the HuCCT-1 and SB1 cell lines, and PDX42 tumors treated with dasatinib at the same concentration as (B) were subjected to immunoblot for phosphorylated YAP (pYAPY357). Total YAP and Actin were used as loading controls. (E) (Upper panel)Representative images of immunohistochemistry used to detect pYAPY357 localization in intrahepatic CCA xenografts +/− dasatinib (15mg/kg, 3days). Secondary only controls are included. Original magnification 40×. Scale bar = 10 microns. N=5 tumors stained per treatment. (Lower panel) Quantification of nuclei with nuclear pYAPY357. *p<0.002
The SFK member LCK selectively regulates YAP tyrosine phosphorylation and target gene expression, and LCK expression levels are associated with tumor recurrence
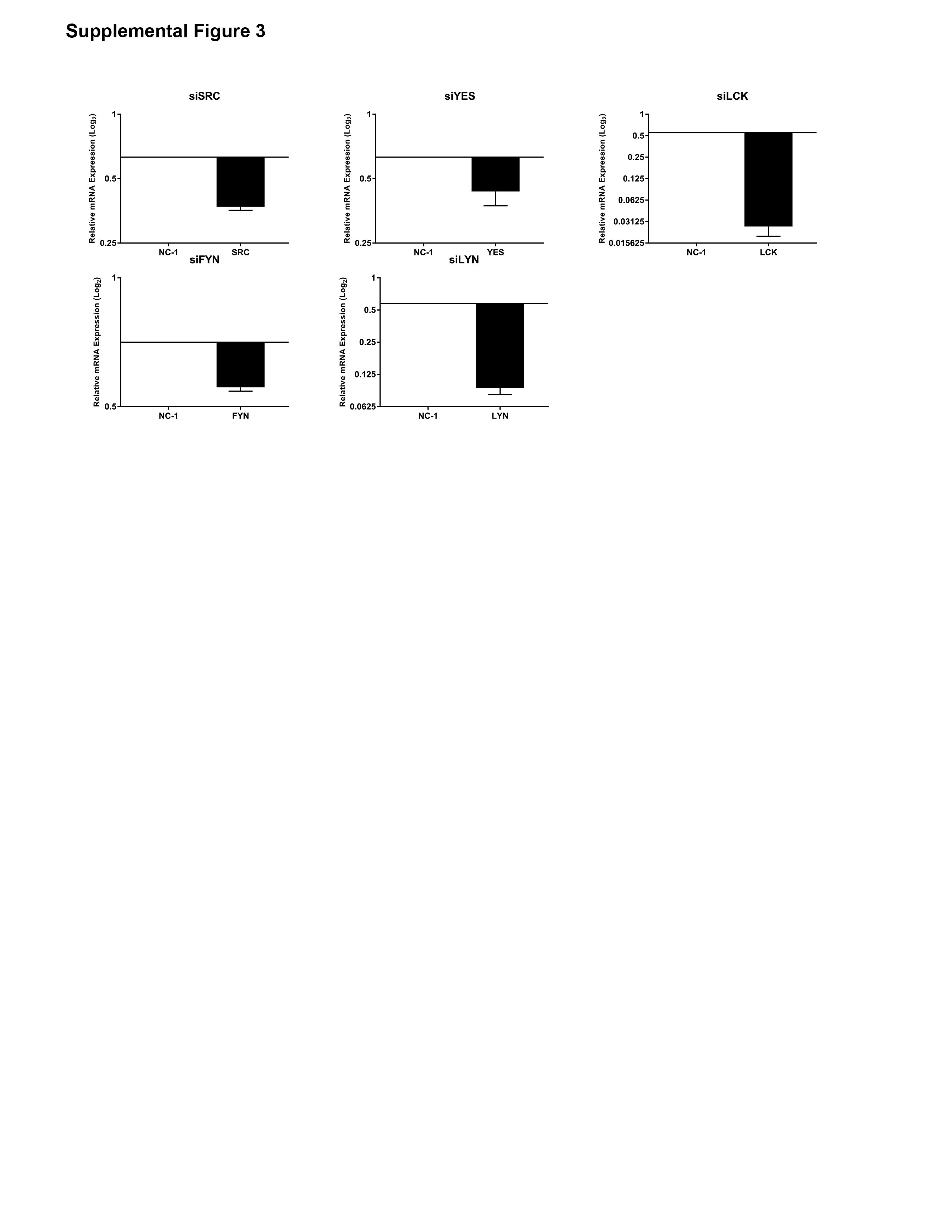
Given the multiple members of the SFK expressed in CCA, we next sought to determine which member was responsible for YAPY357 phosphorylation. Each previously identified family member was targeted by siRNA in HuCCT-1 cells. Targeted knockdown was verified for each member by immunoblot analysis and RT-PCR (Figure 3A and Supplemental 3). Only knockdown of LCK was associated with a significant decrease in YAPY357 phosphorylation (Figure 3B). Utilizing the YAP target genes CTGF and CYR61 as markers of YAP co-transcriptional activity we demonstrated downregulation of co-transcriptional activity with knockdown of multiple SFKs; however the most dramatic downregulation of YAP co-transcriptional activity was noted in the siLCK-HuCCT1 cells as compared to a non-targeted control (Figure 3C). Cell proliferation was evaluated by cell counting following transfection with siSFK or a non-targeting siRNA control (NC-1) in HuCCT-1 cells. Only siLCK-HuCCT-1 cells demonstrated a significant downregulation of proliferation as compared to controls (Figure 3D). In order to overcome the possible effect of residual LCK expression following siRNA knockdown, we next evaluated YAPY357 phosphorylation and nuclear localization in HuCCT-1 cells utilizing an inducible CRISPR/Cas9 system. Incubation of the sgLCK-Cas9-HuCCT-1 cell line with doxycycline lead to a more complete loss of LCK (Figure 3E). Genetic deletion of LCK was paralleled by a near complete repression of YAPY357 phosphorylation, and subsequent redistribution of nuclear YAP to the cytosol, with undetectable levels at 72 hours (Figures 3E,F). To confirm our findings implicating LCK as a direct mediator of YAP tyrosine phosphorylation, we next examined the ability of LCK to phosphorylate YAP directly in vitro. Utilizing recombinant YAP as a phospho-substrate with LCK in a cell free assay, we were able to directly demonstrate efficient YAP Y357 phosphorylation by LCK (Figure 3G). Finally the role of SFK expression in patients with resected intrahepatic cholangiocarcinoma was examined by comparing RNA sequencing profiles in patient-derived xenografts from patients who had undergone curative-intent resection of their tumors. Differential expression was evaluated for patients with early tumor recurrence (less than 10 months) as compared to patients with late tumor recurrence (greater than 10 months) and identified 200 genes. The patient cohort with early recurrence had a markedly upregulated expression of LCK, log2 fold change 7.77, p=0.00005, representing the third highest upregulated gene in this patient group (Supplemental Table 2). No other SFK were noted to be differentially expressed. Interestingly, tumor specimens from patients with early recurrence also displayed upregulation of ARID3a, a known binding partner of the YAP-TEAD4 complex (Figure 3H). Taken together, these data suggest a role for LCK in regulating YAPY357 phosphorylation and YAP co-transcriptional activity in cholangiocarcinoma.
Figure 3. The SFK member LCK regulates YAP tyrosine phosphorylation and transcriptional activity in cholangiocarcinoma.

(A) Cell lysates from HuCCT-1 cell lines transfected with targeting and non-targeting siRNA were subjected to immunoblot for SRC, YES, LCK, FYN, LYN. GAPDH was used as a loading control. (B) Cell lysates from HuCCT-1 cell lines transfected with non-targeting siRNA and siRNA for five SFKs were subjected to immunoblot for phosphorylated YAP (pYAPY357). Total YAP and GAPDH were used as loading controls. (C) CTGF and CYR61 mRNA expression in HuCCT-1 cell line transfected with non-targeting siRNA (NC1) and siRNA for five SFKs. Fold expression targeting/non-targeting is depicted as geometric mean +/−geometric standard deviation (n=3). *p<0.05 (D) Relative cell proliferation in HuCCT-1 cell lines transfected with targeting and non-targeting siRNA for five SFKs, quantitation. *p<0.05 (E) HuCCT-1 cell lines transfected with a doxycycline inducible CRISPR/Cas9 construct targeting LCK were incubated with doxycycline (7.5 µg/mL) for the noted time. Cell lysates were subjected to immunoblot for pYAPY357 (total YAP as loading control) and LCK (GAPDH as loading control). (F) HuCCT-1 cell lines transfected with a doxycycline inducible CRISPR/Cas9 construct targeting LCK or empty vector were incubated with doxycycline (7.5 µg/mL) for the noted time. Immunofluorescence for YAP was undertaken and representative images are displayed (upper panel) with quantification of YAP localization (lower panel). Original magnification 40×. Scale bar = 10 microns. (G) Phosphorylation of recombinant YAP in a cell free system by recombinant LCK was evaluated by luminescence and immunoblot (inset). (H) Heat map representation of FPKM for LCK and ARID3a from PDX derived from patients undergoing curative intent liver resection with recurrence in less than 10 months (L10) versus recurrence in greater than 10 months (G10).
YAP tyrosine phosphorylation regulates nuclear retention, independent of S127 serine/threonine phosphorylation
We next sought to confirm the role of tyrosine phosphorylation in regulating YAP subcellular localization. The SB1 cells express an epitope-tagged YAP containing the S127A mutation, rendering S127A-YAP insensitive to serine/threonine kinase phosphorylation, providing a useful reagent to determine if YAP subcellular localization may be controlled independent of the canonical serine/threonine kinase cascades. We examined S127A-YAP subcellular localization in SB1 cells at baseline and following incubation with dasatinib. Following treatment with dasatinib, S127A-YAP translocated from the nucleus to the cytoplasm (Figure 4A). Next, we examined potential S127A-YAP binding partners in the presence and absence of dasatinib by immunoprecipitation of the FLAG epitope followed by mass spectroscopy. We identified multiple tyrosine phosphatases associated with S127A-YAP and this association increased following incubation with dasatinib (Table 1). In keeping with these findings we observed that incubation of HuCCT-1 cells with the tyrosine phosphatase inhibitor sodium orthovanadate (Na3VO4) abolished the dasatinib-induced nuclear to cytosplasmic redistribution of YAP (Figure 4B). Similarly, S127A-YAP cellular localization in the SB1 cells incubated with Na3VO4 plus and dasatinib remained nuclear (Figure 4A). These data suggest that YAP nuclear retention in CCA cells is likely independent of S127 phosphorylation by serine/threonine kinases and that YAP nuclear retention is due, in part, to a balance between tyrosine kinase and tyrosine phosphatase activity. To further validate this nuclear retention pathway and rule out the possibility that the Hippo kinase complex could be regulating YAP localization through phosphorylation of serine residues other than S127, we next overexpressed a FLAG-tagged YAP in which eight serine residues are mutated to alanine (FLAG-5SA-YAP), in HuCCT-1 cells. The localization of this 5SA-YAP as determined by immunofluorescence of the epitope-tag was examined following dasatinib incubation. Confirming our previous findings, 5SA -YAP also redistributed from the nucleus to the cytosol in dasatinib treated cells and this redistribution was abrogated by pre-incubation with Na3VO4 (Figure 4C). To exclude an effect of dasatinib on the actin cytoskeleton, which is known to regulate YAP cellular localization, we also examined wild-type YAP localization after pretreatment with the actin inhibitor latrunculin-A. Following incubation with latrunculin-A, the cells rounded as expected, however YAP was still localized to the nucleus and subsequently redistributed to the cytosol only after dasatinib treatment (Figure 4D). Recent work has also suggested a role for tyrosine phosphorylation in regulating LATS1 activity.(25) In order to exclude an effect of dasatinib on LATS1 activity as a mechanism of regulating YAP subcellular compartmentation in CCA cells, we examined the phosphorylation status of serine 909 on LATS1 (LATS1S909) in the presence and absence of dasatinib. LATS1S909 phosphorylation signifies an enzymatically active LATS1. Incubation of HuCCT-1 cells with dasatinib led to the expected decrease in YAPY357 phosphorylation with no appreciable effect on LATS1S909 phosphorylation (Figure 4E). Previous work has demonstrated that SFK may also phosphorylate YAP on tyrosines 341, 357, and 394(26). Therefore, we also examined the role of these tyrosine phospho sites in regulating YAP cellular compartmentation by expressing an eGFP-fusion protein in which these three tyrosine residues were mutated to phenylalanine (Y341F, Y357F, Y394F-YAP-eGFP). Compared to the wild-type YAP-eGFP, which was located in both the nucleus and cytosol, Y341,357,394F-YAP-eGFP was enriched in the cytosol and was excluded from the nucleus (Figure 4F). We next examined whether mutation of the Y357 site was sufficient to limit YAP localization to the cytosol. Indeed, Y357F-YAP-eGFP was also excluded from the nucleus; suggesting that phosphorylation of the Y357 residue was sufficient to regulate nuclear retention of YAP in cholangiocarcinoma cells (Figure 4F). Importantly, incubation of the wild-type YAP-eGFP HuCCT-1 cells with dasatinib lead to rapid redistribution of the YAP to the cytosol, suggesting transport kinetics for the GFP-tagged protein were similar to untagged protein (Figure 4G). Taken together, these data suggest a nuclear retention pathway for YAP regulated by tyrosine phosphorylation of Y357 and independent of YAP serine phosphorylation and therefore LATS1 activity.
Figure 4. Phosphorylation of YAPY357 regulates nuclear retention in cholangiocarcnoma.

(A) Representative images of FLAG immunofluorescence in the SB1 CCA cell line incubated with vehicle, dasatinib (100 nM, 1 hour), or dasatinib (100 nM, 1 hour) and sodium orthovanadate (10 mM, 1 hour) (upper panel) and quantification of YAP nuclear localization (lower panel). (B) Representative images of YAP immunofluorescence in HuCCT-1 cells incubated with vehicle, dasatinib (100 nM, 1 hour), or dasatinib (100 nM, 1 hour) and sodium orthovanadate (10 mM, 1 hour) (upper panel), quantification of YAP nuclear localization (lower left panel), and cell fractionation evaluated by immunoblot (lower right panel). Lamin B1 and GAPDH were used as nuclear and cytoplasmic markers respectively. (C) Representative images of FLAG immunofluorescence in HuCCT-1 cells transfected with FLAG-5SA-YAP incubated with vehicle, dasatinib (100 nM, 1 hour), or dasatinib (100 nM, 1 hour) and sodium orthovanadate (10 mM, 1 hour) (upper panel) and quantification of YAP nuclear localization (lower panel). (D) Representative images of YAP immunofluorescence in HuCCT-1 cells incubated with vehicle, latrunculin A (1 µM, 1 hour), or latrunculin A (1 µM, 1 hour) and dasatinib (100nM, 1 hour) (upper panel) and quantification of YAP nuclear localization (lower panel). (E) Cell lysates from HuCCT-1 cells treated with dasatinib (1µM, 2 hours) were subjected to immunoblot for pYAPY357, pYAPS127 (total YAP as a loading control) and pLATS1S909 (total LATS1 as a loading control). Actin was used as a loading control. (F) Representative images of fluorescent microscopy in HuCCT-1 cells expressing wild-type or mutated YAP-eGFP. (G) Representative images of time lapse fluorescent microscopy in HuCCT-1 cells expressing wild-type YAP-eGFP. Original magnification 40×. Scale bar = 10 microns for all images.
Table 1.
Tyrosine Phosphatases Associated with YAP in Murine SB1 Cells: Ratio of Total Spectrum Count(TSC) in Dasatinib Treated/Untreated Controls
| Phosphatase | Ratio TSC Treated/Control |
|---|---|
| PTPN11 | 3.5 |
| PTPRK | 3.0 |
| PTPN1 | 2.0 |
| PTPN23 | 2.0 |
The SFK inhibitor dasatinib is therapeutic in vitro and in vivo
Incubation of HuCCT1 and SB1 cells with dasatinib lead to an increased cell death (Figure 5A), and loss of cell number in a concentration dependent manner with significant effects noted at clinically achievable concentrations (≤ 1 µM) (Figure 5B). Consistent with prior observations that the pro-survival protein MCL-1 is a YAP target gene, dasatinib treatment of the CCA cells lead to a decrease in MCL-1 protein and gene expression (Figure 2B, Figure 5C). Next, we examined dasatinib therapeutic efficacy in NOD/SCID mice bearing CCA PDX. Patient-derived xenografts from three different patients (PDX 42, 115, 135) were implanted bilaterally in the mice and tumor-bearing animals treated with daily gavage of dasatinib (15 mg/kg) or vehicle; initiated once the tumors were palpable (~125 mm3). Following 14 days of treatment there was a dramatic difference in the size of tumors from dasatinib treated animals as compared to vehicle treated (Figure 5D). This was accompanied by a marked increase in cell death as indicated by TUNEL staining. (Figure 5E) The increase in cell death was an early process and present even after short course treatment (3 days). Collectively, SFK inhibition with dasatinib was tumor suppressive in vitro and in vivo suggesting the LCK-mediated YAP tyrosine phosphorylation pathway in CCA is potentially therapeutically targetable.
Figure 5. The SFK inhibitor dasatinib is therapeutic both in vitro and in vivo.

(A) Cell death was evaluated after staining with propidium iodide in HuCCT1 and SB1 cells treated with dasatinib (1 µM, 24hrs). *p<0.05 (B) HuCCT-1 and SB1 cells were incubated with vehicle or the indicated concentration of dasatinib for 3 days. Cell proliferation was evaluated by cell counting after staining with Hoechst 33342 and was normalized to vehicle treatment. *p<0.01 (C) Cell lysates from HuCCT-1 cell lines incubated +/− dasatinib (1µM, 2hrs) were subjected to immunoblot for MCL-1. GAPDH were used as loading control. (D) Tumor bearing NOD/SCID mice from PDX42, PDX115, and PDX 135 were treated with daily gavage of vehicle or dasatinib (15 mg/kg, 14 days)(n=7/treatment) and tumor volumes recorded daily. Day 14 tumor volumes are displayed for all three PDX. *p<0.05 ***p<0.001 (E) Representative images following TUNEL staining in tumors from vehicle and dasatinib treated animals (upper panel), with quantification of TUNEL positive cells (lower panel). Original magnification 10×. Scale bar = 50 microns.
DISCUSSION
This study delineates a regulatory role for YAP tyrosine phosphorylation in cholangiocarcinoma cell lines and patient-derived xenografts (Figure 6). These data indicate that: (1) tyrosine phosphorylation regulates YAP nuclear retention independent of the serine/threonine cytosolic retention pathway, (2) the SRC-family kinase LCK mediates YAPY357 phosphorylation and subsequent activity, and (3) SFK inhibition is therapeutic in vitro and in vivo. These findings are discussed in greater detail below.
Figure 6. YAP nuclear and cytosolic retention pathways model.
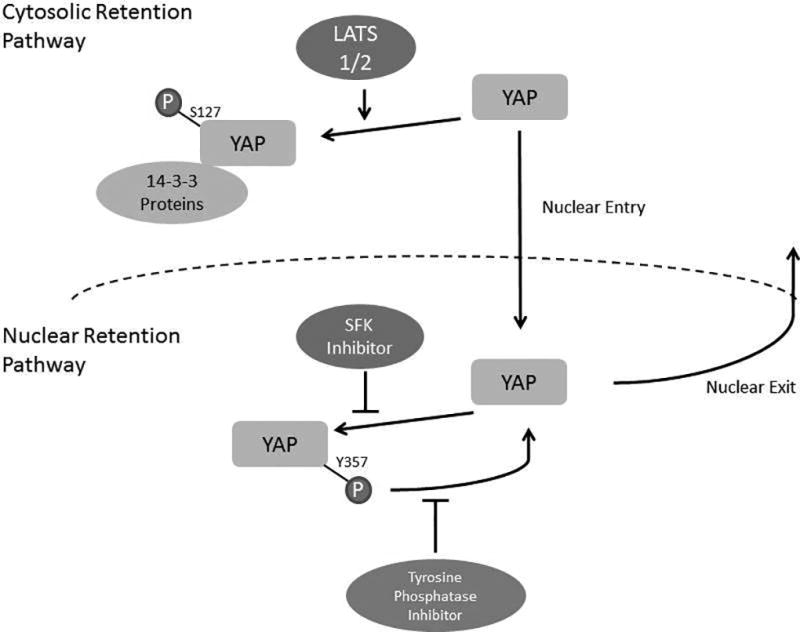
The canonical regulatory pathway described for YAP involves a core Hippo serine/threonine kinase relay module involving MST1/2 and LATS1/2. This serine/threonine kinase relay culminates in phosphorylation of regulatory serine residues on YAP; regulatory serine phosphorylation promotes YAP cytosolic sequestration and/or proteasomal degradation.(4, 27) This pathway, which we have termed the cytosolic retention pathway, restrains the activity of YAP; and consistent with these concepts we and others have demonstrated complete exclusion of phosphorylated YAPS127 from the nucleus.(2, 28) However, accumulating evidence also implicates receptor tyrosine kinases in the regulation of YAP cellular compartmentation.(2, 8) In the current study, we further examined the role of tyrosine phosphorylation in regulating YAP nuclear localization, a so-called nuclear retention pathway. We were able to separate the serine/threonine and tyrosine phospho-regulation effects by utilizing our CCA cell line expressing an epitope-tagged S127A-YAP and subsequently by enforced expression of an epitope tagged 5SA-YAP in a human CCA cell line. S127A-YAP is predominantly compartmentalized to the nucleus. Utilizing an inhibitory paradigm to inform the regulatory pathways, we noted redistribution of S127A-YAP from the nucleus into the cytosol following SFK inhibition. This redistribution of S127A-YAP out of the nucleus was accompanied by its association with tyrosine phosphatases and was blocked by a tyrosine phosphatase inhibitor. These data suggest a regulatory process whereby YAP tyrosine phosphorylation promotes nuclear retention. Conversely, dephosphorylation, either through decreased tyrosine kinase activity or via tyrosine phosphatase activity, enhances transport of YAP out of the nucleus. While S127 is the most comprehensively studied serine residue regulating YAP cellular compartmentation, other potential regulatory serine phosphorylation sites exist in YAP, and one possibility is that the Hippo kinase module could be regulating the subcellular localization by phosphorylation at serines other than S127. Moreover, LATS1 tyrosine phosphorylation may also negatively regulate LATS1 serine kinase activity.(25) In this scenario, SFK inhibition may increase LATS1 activity and subsequent YAP phosphorylation on multiple serine residues. Our data argue against this mechanism in CCA cells as YAPS127 phosphorylation did not increase in HuCCT-1 cells treated with dasatinib, the 5SA-YAP (in which 8 serine residues have been mutated to alanine) still redistributed to the cytosol with dasatinib treatment, and p-LATS1S909 was not altered. Additional studies will be required to fully understand the interplay between the cytosolic and nuclear retention pathways and potential cross regulation, however our data suggest functions for both serine and tyrosine phospho-regulation of YAP and its cellular compartmentation.
Based on our previous observations linking SFK activity to PDGF regulation of YAP activity and localization, we sought to determine more precisely which SFK may be involved in this pathway. In IDH1/2 mutated CCA, Saha et al, demonstrated a central role for SRC itself.(29) In our studies we did not find a significant effect on YAP tyrosine phosphorylation with downregulation of SRC levels. We did however note a decrease in YAP tyrosine phosphorylation and transcriptional activity with siRNA targeted knockdown of LCK. This association between LCK and YAP phosphorylation has not been previously reported. In fact, LCK is most comprehensively described in its role in signal transduction from the T-cell receptor to downstream intracellular signaling molecules.(30) We found LCK is expressed in not only normal human cholangiocytes, but both of the CCA cell lines we utilized, and the CCA PDX tumors examined. Interestingly, when dasatinib was undergoing development as a pan-SRC kinase inhibitor it was LCK that was utilized as the binding partner for the compound screen and subsequent optimization strategies.(31) It is possible that other SFK are involved in YAP regulation in a redundant fashion, but that thresholds for expression may be different; such that more complete knockdown of the other expressed family members may be necessary to see effects on YAP phosphorylation and localization. The degree of homology between the family members makes pharmacologic inhibition of individual members challenging; however, other strategies such as CRISPR/Cas9 constructs would further delineate the roles of the individual SFK.
The tyrosine phosphorylation of YAP is an attractive therapeutic target as it represents an activating phosphorylation (versus the inhibitory serine phosphorylation). SFK inhibition can be achieved with the pan-SRC inhibitor dasatinib and efficacy has previously been demonstrated in IDH-mutated intrahepatic cholangiocarcinoma models.(29) We found dramatic sensitivity to dasatinib in all of our models, cell lines and PDX. The effect appears to be, at least partially, due to downregulation of the pro-survival YAP target gene MCL-1. Further studies are underway to determine if targeted downregulation of LCK has a similar effect on tumor survival, however the accumulated preclinical data suggest SFK inhibition as a therapeutic strategy in intrahepatic cholangiocarcinoma.
Overall our study contributes to the understanding of YAP function and regulation in cancer. Our observations extend the concept of phospho-regulation of YAP to include a role for tyrosine phosphorylation independent of LATS 1/2 serine phosphorylation and a separation of the cytosolic and nuclear retention signals. Additionally we have identified a potentially novel role for LCK in the regulation of YAP in CCA. Finally we have generated additional pre-clinical data supporting the role of SFK inhibition in the treatment of intrahepatic cholangiocarcinoma.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Grant Support: This work was supported by National Institutes of Health Grants R01DK59427 (to G.J.G), Optical Microscopy Core services and a Pilot and Feasibility Award (to R.L.S) provided through the Mayo Clinic Center for Cell Signaling in Gastroenterology (P30DK084567), the Mayo Clinic Center for Biomedical Discovery Pilot Award (to R.L.S), Department of Defense CDMRP Career Development Award CA171017 (to R.L.S), the Mayo Clinic Medical Genome Facility - Proteomics Core, a supplement for liver cancer infrastructure (NCI Cancer Center Support Grant 5P30 CA15083-43C1), MH CZ - DRO (UHHK, 00179906) (to PH), and by the Mayo Clinic.
Footnotes
The authors declare no potential conflicts of interest.
References
- 1.Pei T, Li Y, Wang J, Wang H, Liang Y, Shi H, Sun B, Yin D, Sun J, Song R, Pan S, Sun Y, Jiang H, Zheng T, Liu L. YAP is a critical oncogene in human cholangiocarcinoma. Oncotarget. 2015;6(19):17206–20. doi: 10.18632/oncotarget.4043. PubMed PMID: 26015398; PMCID: PMC4627302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smoot RL, Werneburg NW, Sugihara T, Hernandez MC, Yang L, Mehner C, Graham RP, Bronk SF, Truty MJ, Gores GJ. Platelet-derived growth factor regulates YAP transcriptional activity via Src family kinase dependent tyrosine phosphorylation. J Cell Biochem. 2017 doi: 10.1002/jcb.26246. PubMed PMID: 28661054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamada D, Rizvi S, Razumilava N, Bronk SF, Davila JI, Champion MD, Borad MJ, Bezerra JA, Chen X, Gores GJ. IL-33 facilitates oncogene-induced cholangiocarcinoma in mice by an interleukin-6-sensitive mechanism. Hepatology. 2015;61(5):1627–42. doi: 10.1002/hep.27687. PubMed PMID: 25580681; PMCID: PMC4406813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol Rev. 2014;94(4):1287–312. doi: 10.1152/physrev.00005.2014. PubMed PMID: 25287865. [DOI] [PubMed] [Google Scholar]
- 5.Farshidfar F, Zheng S, Gingras MC, Newton Y, Shih J, Robertson AG, Hinoue T, Hoadley KA, Gibb EA, Roszik J, Covington KR, Wu CC, Shinbrot E, Stransky N, Hegde A, Yang JD, Reznik E, Sadeghi S, Pedamallu CS, Ojesina AI, Hess JM, Auman JT, Rhie SK, Bowlby R, Borad MJ, Cancer Genome Atlas N. Zhu AX, Stuart JM, Sander C, Akbani R, Cherniack AD, Deshpande V, Mounajjed T, Foo WC, Torbenson MS, Kleiner DE, Laird PW, Wheeler DA, McRee AJ, Bathe OF, Andersen JB, Bardeesy N, Roberts LR, Kwong LN. Integrative Genomic Analysis of Cholangiocarcinoma Identifies Distinct IDH-Mutant Molecular Profiles. Cell Rep. 2017;19(13):2878–80. doi: 10.1016/j.celrep.2017.06.008. PubMed PMID: 28658632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li H, Wolfe A, Septer S, Edwards G, Zhong X, Abdulkarim AB, Ranganathan S, Apte U. Deregulation of Hippo kinase signalling in human hepatic malignancies. Liver Int. 2012;32(1):38–47. doi: 10.1111/j.1478-3231.2011.02646.x. PubMed PMID: 22098159; PMCID: PMC4175712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moroishi T, Hansen CG, Guan KL. The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer. 2015;15(2):73–9. doi: 10.1038/nrc3876. PubMed PMID: 25592648; PMCID: PMC4562315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rizvi S, Yamada D, Hirsova P, Bronk SF, Werneburg NW, Krishnan A, Salim W, Zhang L, Trushina E, Truty MJ, Gores GJ. A Hippo and Fibroblast Growth Factor Receptor Autocrine Pathway in Cholangiocarcinoma. J Biol Chem. 2016;291(15):8031–47. doi: 10.1074/jbc.M115.698472. PubMed PMID: 26826125; PMCID: PMC4825008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenbluh J, Nijhawan D, Cox AG, Li X, Neal JT, Schafer EJ, Zack TI, Wang X, Tsherniak A, Schinzel AC, Shao DD, Schumacher SE, Weir BA, Vazquez F, Cowley GS, Root DE, Mesirov JP, Beroukhim R, Kuo CJ, Goessling W, Hahn WC. beta-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell. 2012;151(7):1457–73. doi: 10.1016/j.cell.2012.11.026. PubMed PMID: 23245941; PMCID: PMC3530160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fan R, Kim NG, Gumbiner BM. Regulation of Hippo pathway by mitogenic growth factors via phosphoinositide 3-kinase and phosphoinositide-dependent kinase-1. Proc Natl Acad Sci U S A. 2013;110(7):2569–74. doi: 10.1073/pnas.1216462110. PubMed PMID: 23359693; PMCID: PMC3574943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taniguchi K, Wu LW, Grivennikov SI, de Jong PR, Lian I, Yu FX, Wang K, Ho SB, Boland BS, Chang JT, Sandborn WJ, Hardiman G, Raz E, Maehara Y, Yoshimura A, Zucman-Rossi J, Guan KL, Karin M. A gp130-Src-YAP module links inflammation to epithelial regeneration. Nature. 2015;519(7541):57–62. doi: 10.1038/nature14228. PubMed PMID: 25731159; PMCID: PMC4447318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O'Hara SP, Splinter PL, Trussoni CE, Pisarello MJ, Loarca L, Splinter NS, Schutte BF, LaRusso NF. ETS Proto-oncogene 1 Transcriptionally Up-regulates the Cholangiocyte Senescence-associated Protein Cyclin-dependent Kinase Inhibitor 2A. The Journal of biological chemistry. 2017;292(12):4833–46. doi: 10.1074/jbc.M117.777409. Epub 2017/02/12. PubMed PMID: 28184004; PMCID: 5377799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rizvi S, Fischbach SR, Bronk SF, Hirsova P, Krishnan A, Dhanasekaran R, Smadbeck JB, Smoot RL, Vasmatzis G, Gores GJ. YAP-associated chromosomal instability and cholangiocarcinoma in mice. Oncotarget. 2018;9(5):5892–905. doi: 10.18632/oncotarget.23638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3(6):1101–8. doi: 10.1038/nprot.2008.73. Epub 2008/06/13. PubMed PMID: 18546601. [DOI] [PubMed] [Google Scholar]
- 15.Aubrey BJ, Kelly GL, Kueh AJ, Brennan MS, O'Connor L, Milla L, Wilcox S, Tai L, Strasser A, Herold MJ. An inducible lentiviral guide RNA platform enables the identification of tumor-essential genes and tumor-promoting mutations in vivo. Cell Rep. 2015;10(8):1422–32. doi: 10.1016/j.celrep.2015.02.002. PubMed PMID: 25732831. [DOI] [PubMed] [Google Scholar]
- 16.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36. doi: 10.1186/gb-2013-14-4-r36. PubMed PMID: 23618408; PMCID: PMC4053844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–9. doi: 10.1038/nmeth.1923. PubMed PMID: 22388286; PMCID: PMC3322381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roberts A, Pimentel H, Trapnell C, Pachter L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics. 2011;27(17):2325–9. doi: 10.1093/bioinformatics/btr355. PubMed PMID: 21697122. [DOI] [PubMed] [Google Scholar]
- 19.Roberts A, Trapnell C, Donaghey J, Rinn JL, Pachter L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011;12(3):R22. doi: 10.1186/gb-2011-12-3-r22. PubMed PMID: 21410973; PMCID: PMC3129672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562–78. doi: 10.1038/nprot.2012.016. PubMed PMID: 22383036; PMCID: PMC3334321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–5. doi: 10.1038/nbt.1621. PubMed PMID: 20436464; PMCID: PMC3146043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31(1):46–53. doi: 10.1038/nbt.2450. PubMed PMID: 23222703; PMCID: PMC3869392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giridhar KV, Sosa CP, Hillman DW, Sanhueza C, Dalpiaz CL, Costello BA, Quevedo FJ, Pitot HC, Dronca RS, Ertz D, Cheville JC, Donkena KV, Kohli M. Whole Blood mRNA Expression-Based Prognosis of Metastatic Renal Cell Carcinoma. Int J Mol Sci. 2017;18(11) doi: 10.3390/ijms18112326. PubMed PMID: 29099775; PMCID: PMC5713295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zimmerman EI, Turner DC, Buaboonnam J, Hu S, Orwick S, Roberts MS, Janke LJ, Ramachandran A, Stewart CF, Inaba H, Baker SD. Crenolanib is active against models of drug-resistant FLT3-ITD-positive acute myeloid leukemia. Blood. 2013;122(22):3607–15. doi: 10.1182/blood-2013-07-513044. Epub 2013/09/21. PubMed PMID: 24046014; PMCID: 3837509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Si Y, Ji X, Cao X, Dai X, Xu L, Zhao H, Guo X, Yan H, Zhang H, Zhu C, Zhou Q, Tang M, Xia Z, Li L, Cong YS, Ye S, Liang T, Feng XH, Zhao B. Src Inhibits the Hippo Tumor Suppressor Pathway through Tyrosine Phosphorylation of Lats1. Cancer Res. 2017;77(18):4868–80. doi: 10.1158/0008-5472.CAN-17-0391. PubMed PMID: 28754671. [DOI] [PubMed] [Google Scholar]
- 26.Li P, Silvis MR, Honaker Y, Lien WH, Arron ST, Vasioukhin V. alphaE-catenin inhibits a Src-YAP1 oncogenic module that couples tyrosine kinases and the effector of Hippo signaling pathway. Genes Dev. 2016;30(7):798–811. doi: 10.1101/gad.274951.115. PubMed PMID: 27013234; PMCID: PMC4826396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ehmer U, Sage J. Control of Proliferation and Cancer Growth by the Hippo Signaling Pathway. Mol Cancer Res. 2016;14(2):127–40. doi: 10.1158/1541-7786.MCR-15-0305. PubMed PMID: 26432795; PMCID: PMC4755889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lian I, Kim J, Okazawa H, Zhao J, Zhao B, Yu J, Chinnaiyan A, Israel MA, Goldstein LS, Abujarour R, Ding S, Guan KL. The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev. 2010;24(11):1106–18. doi: 10.1101/gad.1903310. PubMed PMID: 20516196; PMCID: PMC2878649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saha SK, Gordan JD, Kleinstiver BP, Vu P, Najem MS, Yeo JC, Shi L, Kato Y, Levin RS, Webber JT, Damon LJ, Egan RK, Greninger P, McDermott U, Garnett MJ, Jenkins RL, Rieger-Christ KM, Sullivan TB, Hezel AF, Liss AS, Mizukami Y, Goyal L, Ferrone CR, Zhu AX, Joung JK, Shokat KM, Benes CH, Bardeesy N. Isocitrate Dehydrogenase Mutations Confer Dasatinib Hypersensitivity and SRC Dependence in Intrahepatic Cholangiocarcinoma. Cancer Discov. 2016;6(7):727–39. doi: 10.1158/2159-8290.CD-15-1442. PubMed PMID: 27231123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palacios EH, Weiss A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene. 2004;23(48):7990–8000. doi: 10.1038/sj.onc.1208074. PubMed PMID: 15489916. [DOI] [PubMed] [Google Scholar]
- 31.Das J, Chen P, Norris D, Padmanabha R, Lin J, Moquin RV, Shen Z, Cook LS, Doweyko AM, Pitt S, Pang S, Shen DR, Fang Q, de Fex HF, McIntyre KW, Shuster DJ, Gillooly KM, Behnia K, Schieven GL, Wityak J, Barrish JC. 2-aminothiazole as a novel kinase inhibitor template. Structure-activity relationship studies toward the discovery of N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1- piperazinyl)]-2-methyl-4-pyrimidinyl]amino)]-1,3-thiazole-5-carboxamide (dasatinib, BMS-354825) as a potent pan-Src kinase inhibitor. J Med Chem. 2006;49(23):6819–32. doi: 10.1021/jm060727j. PubMed PMID: 17154512. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.