

Abstract
Numerous studies have attributed the psychopathology of anxiety and stress disorders to maladaptive behavioral responses such as an inability to extinguish fear. Therefore, understanding neural substrates of fear extinction is imperative for developing more effective therapies for anxiety and stress disorders. Although several studies indicated a role for cholinergic transmission and nicotinic acetylcholine receptors (nAChRs) in anxiety and stress disorder symptomatology, very little is known about the specific contribution of nAChRs in the fear extinction process. In the present study, we first examined the involvement of several brain regions essential for fear extinction (i.e., dorsal and ventral hippocampus, dHPC and vHPC; infralimbic, IL, and prelimbic, PL of the medial prefrontal cortex, mPFC; basolateral nucleus of the amygdala, BLA) in the impairing effects of a nAChR agonist, nicotine, on contextual fear extinction in mice. Our results showed that systemic administration of nicotine during contextual fear extinction increased c-fos expression in the vHPC and BLA while not affecting dHPC, IL or PL. In line with these results, local nicotine infusions into the vHPC, but not dHPC, resulted in impaired contextual fear extinction. Interestingly, we found that local nicotine infusions into the PL also resulted in impairment of contextual fear extinction. Second, we measured the protein levels of the GABA synthesizing enzymes GAD65 and GAD67 in the dHPC and vHPC during contextual fear extinction. Our results showed that in the group that received acute nicotine, both GAD65 and GAD67 protein levels were downregulated in the vHPC, but not in dHPC. This effect was negatively correlated with the level of freezing response during fear extinction suggesting that the downregulated GAD65/67 levels were associated with disrupted fear extinction. Finally, using c-fos/GAD65/67 double immunofluorescence, we showed that nicotine mainly increased c-fos expression in non-GABAergic ventral hippocampal cells, indicating that acute nicotine increases vHPC excitability. Overall, our results suggest that acute nicotine’s impairing effects on fear extinction is associated with ventral hippocampal disinhibition. Therefore, these results further our understanding of the interaction between nicotine addiction and anxiety and stress disorders by describing novel neural mechanisms mediating fear extinction.
Keywords: Nicotinic receptors, GABA, fear extinction, hippocampus, PTSD
Nicotine dependence is highly comorbid with anxiety and stress disorders such as Post-traumatic Stress Disorder (PTSD, Breslau et al, 2004)), which is characterized by inability to inhibit and extinguish learned fear (Rothbaum and Davis, 2003; VanElzakker et al, 2014).There is a bidirectional relationship between nicotine dependence and PTSD where nicotine increases one’s risk for developing PTSD and one may be inclined to increase his/her nicotine intake after experiencing an event that triggers PTSD (Breslau et al, 2003, 2004; Koenen et al, 2005). Consequently, nicotine dependence is nearly 23% higher among PTSD patients (Ziedonis et al, 2008). Importantly, in parallel with these statistics, a growing body of literature suggests that nicotine may directly impact PTSD symptoms. Recent studies from our lab and others show that smokers have difficulty in learning cues associated with safety (Kutlu et al, 2018a) and nicotine intake increases the amount of intrusive memories associated with a traumatic event (Hawkins and Cougle, 2013). Therefore, nicotine exposure should be considered a contributing factor to PTSD symptoms in humans.
Animal studies have demonstrated that nicotine can alter fear memories via modulation of nicotinic acetylcholine receptors (nAChRs, Kutlu and Gould, 2015). Both hippocampus-dependent contextual and trace fear conditioning are enhanced by an acute dose of nicotine and these effects are dependent on high-affinity α4β2 nAChRs (Davis et al, 2005, 2006; Davis and Gould, 2006; Gould, 2003; Gould et al, 2004; Gould and Higgins, 2003; Gould and Lommock, 2003; Gould and Wehner, 1999). However, the effects of nicotine are not limited to enhancing fear memories. Previous studies from our laboratory have demonstrated that nicotine administration impaired fear extinction and safety learning in mice (Connor et al, 2017; Kutlu et al, 2014, 2016a, 2017a, 2017b, 2018a; Kutlu and Gould, 2014). Specifically, acute nicotine administration was shown to cause deficits in encoding, consolidation, and retrieval of contextual fear extinction memories (Kutlu et al, 2016a, 2016b; Kutlu and Gould, 2014; Oliver et al, 2018), as well as deficits in contextual safety discrimination (Kutlu and Gould, 2014). These effects were specific to extinction and safety learning, as nicotine did not affect general freezing behavior (Kutlu and Gould, 2014). These results clearly suggest that nicotine is a strong modulator of fear extinction and inhibitory learning and may have negative consequences for psychopathologies associated with deficits in these processes.
There are several hippocampal processes that may be involved in the acute nicotine-induced deficits in fear extinction. Similar to the effects of acute nicotine on fear acquisition, we previously showed that high affinity α4β2 nAChRs, which are expressed in the hippocampus (Séguéla et al, 1993; Wada et al, 1989), were required for the impairing effects of acute nicotine on contextual fear extinction (Kutlu et al, 2016a). Further, we recently showed that hippocampal cell signaling cascades, such as the mitogen-activated protein kinase (MAPK) pathway, were altered by acute nicotine administration during fear extinction (Kutlu et al, 2017a). Nevertheless, the neural mechanism underlying the acute nicotine-induced deficits in fear extinction is still unknown. Therefore, in the present study, we defined how acute nicotine changes the activity of brain regions associated with fear extinction, including the dorsal (dHPC) and ventral (vHPC) hippocampus, infralimbic (IL) and prelimbic (PL) cortices, and basolateral amygdala (BLA), using c-fos immunohistochemistry. Then, we tested how nicotine affects fear extinction when locally infused into the dHPC, vHPC, IL and PL to isolate the main target of nicotine. Finally, we examined how acute nicotine alters GABA-synthesizing enzyme protein levels and spatial distribution of GABAergic activity in the hippocampus. Our results demonstrate that acute nicotine augments ventral hippocampal activity during fear extinction by reducing GABAergic activity in this brain region. Together these results define for the first time how acute nicotine controls hippocampal inhibitory/excitatory balance during fear extinction and provides a mechanism for its impairing effects.
Methods
Subjects
Eight-week-old male C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME) were group-housed in a colony room and maintained on a 12 hour light/dark cycle. Subjects had ad libitum access to food and water. Training and testing took place between 9:00 am and 6:00 pm. All behavioral procedures were approved by the Temple University and Penn State Institutional Animal Care and Use Committees.
Apparatus
Contextual fear conditioning training, testing, and extinction took place in 4 identical chambers (18.8 × 20 ×18.3 cm) within sound-attenuating boxes (MED Associates, St. Albans, VT). A ventilation fan located at the back of each box produced background noise (65 dB). A white noise conditioned stimulus (CS, 85 dB) was produced by a speaker within the right wall of the chambers. Each chamber was composed of Plexiglas walls and ceiling with metal grid floors (0.20 cm and 1.0 cm apart) connected to a shock generator, which produced a 2 s long, 0.57 mA foot-shock unconditioned stimulus (US). Both the CS and US were controlled by an IBM-PC compatible computer running MED-PC software. The open field testing took place within a Plexiglas arena (49.5 cm × 59.7 cm). Between subjects, the fear conditioning chambers and open field arena were cleaned with 70% ethanol.
Drugs and Administration
For all experiments, nicotine hydrogen tartrate salt (0.18mg/kg freebase, Fisher Scientific) dissolved in 0.9% physiological saline (saline) or saline alone was injected intraperitoneally (i.p.) 2–4 mins prior to each fear extinction session. Injection volumes were 10mL/kg as in previous studies (Kutlu and Gould, 2014). The 0.18mg/kg dose of acute nicotine was chosen based on our previous reports showing impaired contextual fear extinction at this dose (Kutlu et al, 2016a; Kutlu and Gould, 2014).
Behavioral Procedures
Following Kutlu and Gould (2014), mice were placed in the fear conditioning chambers and after a 120s baseline period, 2 conditioned stimulus-unconditioned stimulus pairings were delivered. A 30s white noise (85dB) served as the CS, and a 2s 0.57-mA foot shock served as the US. Twenty-four hours after training, the subjects were again placed into the fear conditioning chambers to assess their freezing behavior to the context (retention test). Both the fear conditioning training and retention test were 5min and 30s. Each day for the next five days, mice were placed into the chambers for 5min to assess their freezing behavior during contextual fear extinction. Prior to each extinction session, they received an i.p. injection of nicotine (0.18 mg/kg). Freezing behavior was defined as the absence of voluntary movement except respiration (Blanchard and Blanchard, 1969). Scores were then converted to percent freezing. Experimenters were blinded to drug conditions when scoring.
Immunohistochemistry and Double Immunofluorescence
To examine the effects of nicotine on the expression of c-fos in the dHPC, vHPC, IL, PL, and BLA, a cohort of male C57BL/6J mice underwent the same drug and behavioral procedures outlined previously and were placed in one of six groups. A group of nicotine and saline mice underwent training and extinction (Nic-Extinction and Sal-Extinction). To control for any effects of nicotine or injections, a group of mice received nicotine and saline injections in their homecages and did not receive any behavioral training (Nic-Homecage and Sal-Homecage groups). Mice were perfused using sterile phosphate buffered saline (PBS) and 4% paraformaldehyde (PFA) 1 hour following the last extinction session. After perfusion, brains were collected, stored in 4% PFA for 24 hours, and then incubated in a 30% sucrose solution for an additional 24 hours. Fixed brains were frozen and sectioned using a cryostat and coronal sections were collected.
The c-fos immunohistochemistry was performed using floating sections as described previously (Kutlu et al, 2016b). Briefly, sections were incubated in a rabbit-host polyclonal antibody (c-fos, 1:1000 in PBS-TX-BSA with Sodium Azide; Santa Cruz Biotechnology) for two nights at room temperature. Following PBS-TX-BSA washes (3 × 10 min), the sections were incubated in the secondary antibody (biotinylated goat–anti rabbit, 1:200 in PBS-TX-BSA; Jackson Immuno-Research) for 90 min and incubated in avidin–biotin complex (1:600 in PBS; Vector ABC kit) for 30 min at room temperature. An oxidase–diaminobenzidine– nickel (DAB) method was used for the immunostaining reaction (5 min, diaminobenzidine, DAB Nickel Substrate Kit, Vector Laboratories). The sections were then mounted on slides and cover slipped using Permount (Fisher Chemicals).
The dorsal hippocampus (dHPC; CA1, CA3, and dentate gyrus, DG), ventral hippocampus (vHPC; CA1, CA3, and dentate gyrus), medial prefrontal cortex (prelimbic [PL] and infralimbic [IL] cortices), and basolateral nucleus of the amygdala (BLA) were selected as the regions of interest (ROI). The ROI coordinates were based on Paxinos and Franklin (2001), dHPC, 21.58 to 22.54 mm posterior to bregma; vHPC, 22.70 to 23.64mm posterior to bregma; PL, 1.54 to 1.98mm anterior to bregma; IL, 1.54 to 1.98mm anterior to bregma; and BLA, 20.82 to 21.70mm posterior to bregma). Sections were visualized using a brightfield microscope (Leica), 20× images were taken for each brain regio n and processed using the ImageJ (1.48v) software. A custom ImageJ macro was used to count the c-fos immunoreactive (IR) neurons based on pixel intensity. Only c-fos signals that were above threshold were counted as c-fos-positive cells following previous studies (e.g., Zhao and Li, 2010). Three measurements from the given anterior–posterior coordinates for each brain region (every fourth section for IL and PL and every eighth section for dHPC, vHPC, and BLA) were averaged (Martinez et al. 2013) and expressed as the number of c-fos-positive cells per millimeter square (mm2).
Another group of mice were trained in fear extinction following saline and nicotine administrations and the brain tissue was collected following perfusion as described above. The sections from this cohort were stained for c-fos/GAD65/67 double immunofluorescence. Briefly, sections were incubated in 4 mL of 10% goat serum for one hour. Sections were incubated at room temperature with the primary antibodies, (anti-c-fos mouse mAb, Santa-Cruz, 1:100; anti-GAD65/67 rabbit pAb, Abcam, 1:100) for 48 hours in 2 mL of 1% goat serum. After incubation, the sections were washed three times in 4 mL of PBS-Triton X for 5 minutes per wash. In a dark room, sections were incubated in 2 mL of 1% goat serum and secondary antibodies (Alexa Fluor 488, anti-mouse, Invitrogen, 1:250; Alexa Fluor 647, anti-rabbit, Invitrogen, 1:250), for 3 hours. Sections were washed four times in PBS-Triton-X for 10 minutes per wash. Sections were then mounted onto slides using PBS and allowed to dry. A drop of Prolong Gold anti-fade was placed on each section prior to placing the coverslip on the slide. An epifluorescence microscope (Olympus) was used to take fluorescent images. C-fos and GAD65/67 IR cells in dHPC and vHPC ROIs as listed above were manually counted by 3 experimenters blinded to the experimental groups (see Supp Fig1 for counts from all 3 experimenters).
Cannula surgeries
Mice were placed in a stereotaxic surgical apparatus after being anesthetized with isoflurane (5% induction, 2.5% maintenance). Bilateral guide cannulae were implanted into the dHPC (A/P −1.7, M/L ±3.0, D/V −2.3 mm), vHPC (A/P −2.8, M/L ±3.0, D/V −4.0 mm), IL (A/P +1.7, M/L ±0.5, D/V −3.8 mm) or PL (A/P +1.7, M/L ± 0.5, D/V −2.5 mm). All region coordinates were based on Paxinos and Franklin (2001). Mice were allowed to recover for at least five days prior to initiation of behavioral experimental procedures. Infusion sites were confirmed through cresyl violet staining and subjects with placements determined to fall outside of the target regions (8 mice total) were excluded from all analyses (see Supp Fig2&3 for placements).
Local Drug Infusions
We locally infused 0.18 ug/side of nicotine or saline into the dHPC, vHPC, IL and PL. All mice in this experiment were trained in fear conditioning and fear extinction. Nicotine or saline was administered prior to each extinction session. Due to potential risks of tissue damage after repeated drug administration, we employed 4 instead of 5 fear extinction sessions. Mice were momentarily restrained and the dummy cannula was removed. Using a 22 gauge internal cannula (dHPC and vHPC) and a 33 gauge internal cannula (IL and PL) attached to PE50 polyethylene tubing, the drug was infused directly at a 0.5 uL/min rate with a dosing volume of 0.5 uL per side. A microinfusion pump (KD Scientific, New Hope, PA, USA) was utilized to control the infusion with a 10 uL Hamilton syringe (Reno, NV, USA). To allow diffusion of the drug, the internal cannulae were in place for one minute after infusion. Previous studies from our lab have shown that this local infusion procedure yielded a spread of infusion about 1 mm3 (Davis et al, 2007; Lewis and Gould, 2007). Mice were placed in chambers immediately after infusion.
Western Blotting
Prior to each extinction session, subjects were given either nicotine (0.18 mg/kg, i.p.) or saline as described above. One hour following the 1st or 3rd extinction session, the hippocampal tissue was collected and dissected into dorsal and ventral sections (in a 1:1 ratio). Sections were frozen on dry ice immediately after dissection. Using a sonic dismembrator (Fisher Scientific), dorsal and ventral hippocampi were homogenized in RIPA buffer (Sigma) containing 1X HALT Protease and Phosphate Inhibitor Cocktail (Thermo Scientific). DC™ Protein Assay (Bio-Rad) was used to determine each sample’s protein concentration. Samples were diluted with RIPA buffer to obtain 15 ug total protein. Each sample was diluted 1:1 using Laemmeli sample buffer (Bio-Rad) containing 5% β-mercaptoethanol. Samples were loaded into TGX 4–20% gradient gels (Bio-Rad). Proteins were separated using electrophoresis conducted at 100v constant voltage for 80 min in 1X Tris-Glycine buffer (Bio-Rad). Proteins were transferred onto nitrocellulose membranes (Bio-Rad) for 2hr at 400mA constant current in ice-cold 1X Tris-Glycine buffer (Bio-Rad) containing 20% methanol. Membranes were then washed with Tris buffered solution (TBS) and blocked with 5% bovine serum albumin (BSA, fraction V, Omnipur) in 1X TBS with 0.1% Tween-20 (TBS-T) for 1hr at room temperature. Membranes were incubated in primary antibodies (anti-GAD67 rabbit pAb, Cell Signaling, 1:1000; anti-GAD65 rabbit pAb, Cell Signaling, 1:1000; anti-ChAT goat pAb, Sigma-Aldrich, 1:5000; anti-β-Actin mouse mAb, Sigma–Aldrich, 1:5000) in 5% BSA overnight at 4°C. The following day, blots were incubated in HRP-conjugated secondary antibodies (Vector Laboratories, anti-rabbit 1:1000 for GAD65 and GAD67; anti-goat 1:5000 for ChAT; anti-mouse 1:10,000 for β –actin in 5%BSA) for 1h in room temperature and enhanced chemiluminescence (SuperSignal West Pico Chemiluminescent Substrate; Thermo Scientific) was applied for 5min. To image the blots, a 60s exposure on a Kodak camera (Gel Logic 1500 Imaging System) was utilized. Using ImageJ software, bands were quantified as optical density values. Levels of GAD65, GAD67, and ChAT were normalized to β-actin loading control protein levels for final quantification (see Supp Fig4 for whole blot images).
Statistical Analysis
Behavioral fear extinction data following systemic and local nicotine administration was analyzed using a two-way mixed-design ANOVA that examined 2 levels of Drug across extinction sessions for each compound. To eliminate potential between-group baseline differences in contextual freezing, which may affect subsequent fear extinction curves, the dependent variable was percent freezing to the context normalized to the individual freezing levels at the initial testing session (freezing × 100/ initial freezing; Kutlu et al, 2016a; Tian et al, 2008). For c-fos immunohistochemistry, the number of c-fos IR cells was converted to number of cells per mm2 (number of c-fos IR cells per area of the count; 0.31952 mm2). A multivariate ANOVA was run for all ROIs together to detect Drug (nicotine versus saline) × Training (extinction versus homecage) interaction, which was followed by Tukey post hoc tests to examine group differences. For the western blotting experiments, the optical density values from western blot experiments were represented as fold-changes relative to the respective saline control group and analyzed using a 2 × 2 × 2 ANOVA to test the interaction between Extinction Day, Brain Region and Drug as well as one-way ANOVAs for each brain region and protein. Finally, for the c-fos/GAD65/67 immunofluorescence experiments, we analyzed the total c-fos IR numbers as well as the c-fos+GAD65/7 to c-fos IR cell number ratios (c-fos+GAD/c-fos) for each brain region using one-way ANOVAs. We also analyzed correlations of c-fos IR cell numbers for all brain regions as well as normalized optical densities (OD) for GAD65, GAD67, and ChAT with normalized freezing responses using Pearson’s r. Group sizes were indicated in figure captions. All statistical analyses were run using SPSS 21.
Results
Acute nicotine increases c-fos expression in the ventral hippocampus during contextual fear extinction
First, using c-fos immunohistochemistry, we examined general neural activity levels in the dHPC, vHPC, PL, IL, and BLA during fear extinction following systemic nicotine and saline administration. For the behavioral experiment, 3 contextual fear extinction sessions resulted in significant Drug × Trial interaction (Figure 1A, F(3,30)=4.973, p<0.01). Our c-fos immunohistochemistry results showed that the Drug x Training interaction was significant for each subregion of the vHPC (Figure 1B; total vHPC: F(3,20)=34.451, p<0.01; vCA1: F(3,20)=17.369, p<0.01; vCA3: F(3,20)=6.795, p<0.05; vDG: F(3,20)=7.755, p<0.05) as well as for BLA (Figure 1C; F(3,20)=7.489, p<0.05). Further, Tukey post hoc analysis showed that the number of Nic-Extinction c-fos IR cells was significantly higher than Sal-Extinction, Nic-Homecage, and Saline-Homecage controls for all vHPC subregions (ps<0.05). None of the mPFC (Figure 1D) or dHPC (Figure 1B) subregions showed significant Drug x Training interactions (ps>0.05). We also showed that individual number of c-fos IR cells and normalized %freezing response were correlated for the vHPC (r= 0.69, p<0.5), but not for dHPC, PL, IL, or BLA (Figure 1E; ps>0.05) suggesting that vHPC activation is the strongest predictor of the acute nicotine-induced impairment of contextual fear extinction. Overall, these results demonstrate that acute nicotine increases c-fos expression in the vHPC and BLA while not affecting mPFC subregions PL and IL or dHPC. This suggests systemic nicotine administration augments specific targets of the limbic system associated with fear expression and anxiety such as the vHPC and BLA, which may contribute to disrupted fear extinction observed following acute nicotine administration.
Figure 1. Acute nicotine administration during contextual fear extinction increases vHPC and BLA c-fos expression.
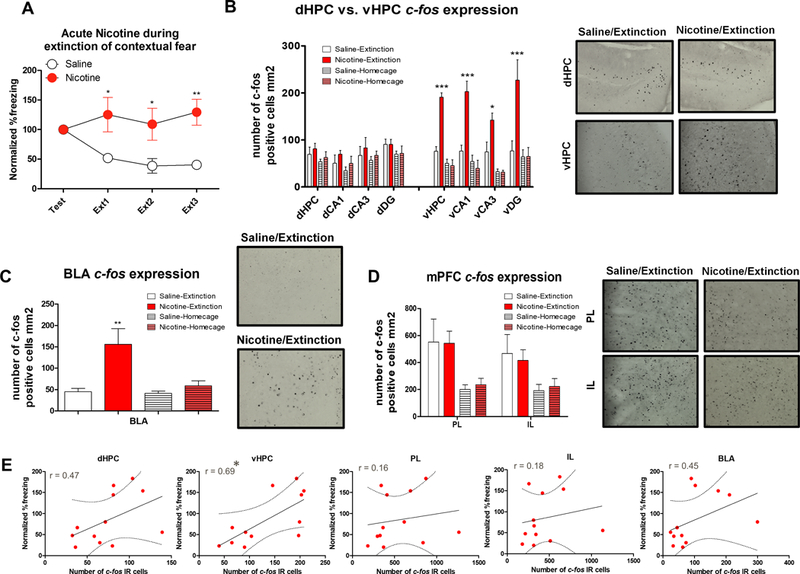
A. Normalized %freezing scores across initial testing and 3 extinctions sessions (n=6 per group). B. Acute nicotine increased the number of c-fos IR cells within the vHPC and vCA1, VCA3, and vDG but did not affect c-fos expression in the dHPC and its subregions. Right panel shows representative c-fos immunohistochemistry images from dHPC and vHPC of the Saline-Extinction and Nicotine-Extinction groups. C. BLA c-fos expression was increased in the group that received acute nicotine during contextual fear extinction. Right panel shows representative c-fos immunohistochemistry images from BLA of the Saline-Extinction and Nicotine-Extinction groups. D. Acute nicotine did not affect IL or PL c-fos expression, but extinction training increased c-fos expression in these brain regions. Right panel shows representative c-fos immunohistochemistry images from IL and PL of the Saline-Extinction and Nicotine-Extinction groups. E. Correlation plots between dHPC, vHPC, PL, IL, and BLA c-fos IR cell numbers and normalized %freezing scores for each subject. Error bars show standard error of the mean. *p<0.05; **p<0.01; ***p<0.001 compared to Saline-Extinction controls.
Local infusions of nicotine into the ventral hippocampus and mPFC impair contextual fear extinction
Following our c-fos expression experiment, we aimed to understand whether nicotine administration into the vHPC is sufficient to produce the fear extinction deficits we observed following systemic injections. Therefore, we infused nicotine directly into the vHPC. We also locally infused nicotine into the dHPC and mPFC (i.e, IL and PL) as our control targets. Separate 2-way ANOVAs yielded that the Drug × Trial interaction was not significant in the groups that received local nicotine infusions into the dHPC (Figure 2A; F(4,48)=0.224, p>0.05), IL (Figure 2C; F(4,32)=1.709, p>0.05), or PL (Figure 2D; F(4,32)=1.948, p>0.05). However, Drug × Trial interaction was significant following vHPC local nicotine infusions (Figure 2B; F(4,48)=5.953, p<0.05). Further, Drug main effect was significant for vHPC (F(1,12)=21.180, p<0.001) and PL (F(1,8)=21.070, p<0.001) but not for dHPC or IL (ps>0.05). Bonferroni corrected t-tests showed that the difference between Saline and Nicotine normalized % freezing was significant in Ext1, Ext2, Ext,3 and Ext4 for vHPC and in Ext3 for PL (ps<0.05). Therefore, nicotine infusion into the vHPC had the strongest impairment of fear extinction while dHPC or IL infusions did not produce an effect on extinction of contextual fear. These results parallel our c-fos immunohistochemistry experiment showing that the vHPC is critically involved in the impairing effects of nicotine on fear extinction. Interestingly, although c-fos expression was not increased in the mPFC as a response to nicotine injections prior to contextual fear extinction, local infusion of nicotine into the PL in isolation disrupted fear extinction. This suggests that activation of PL nAChRs may also be sufficient to produce impaired fear extinction, but they may not be necessary for the systemic effect.
Figure 2. Local nicotine infusion into the vHPC and PL result in contextual fear extinction deficits.
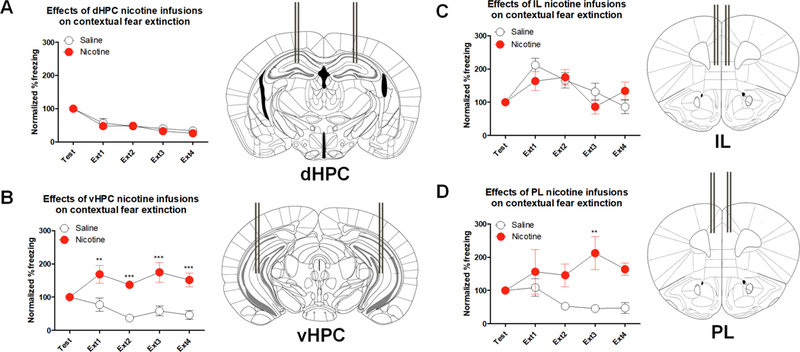
A. Local nicotine infusion into the dHPC did not affect normalized %freezing scores across 4 extinctions sessions (n=7 per group). B. vHPC local nicotine infusions disrupted contextual fear extinction (n=7 per group). C. IL nicotine infusions had no effect on contextual fear extinction (n=4–6 per group). D. Local nicotine infusions into the PL also resulted in impaired extinction of contextual fear. (n=5 per group). Right panels show placement of guide cannulas. Error bars show standard error of the mean. *p<0.05; **p<0.01; ***p<0.001 compared to Saline controls.
Systemic administration of acute nicotine during contextual fear extinction decreases protein levels of GABA-synthesizing enzymes in the ventral hippocampus
Our initial immunohistochemistry results suggested that systemic nicotine injections increased overall activity in the vHPC as indicated by enhanced c-fos expression during contextual fear extinction. One possibility is that the nicotine-induced hyperactivation of the vHPC may be a result of decreased inhibitory signaling within the vHPC. Therefore, to test whether inhibitory GABAergic function is affected by nicotine, we measured the protein levels of GABA-synthesizing enzymes GAD65 and GAD67 in the dHPC and vHPC during fear extinction. We also tested ChAT protein levels to control for potential effects of nicotine on cholinergic function. For the immunoblotting data, we analyzed lysates collected for a previously published behavioral data showing acute nicotine-induced impaired fear extinction following 3 extinction, but not 1 extinction, session (Kutlu et al, 2017a).
Three 2 × 2 × 2 repeated measures ANOVAs showed that the interaction between Extinction Day (1 vs. 3 days), Drug (Nicotine vs. Saline), and Brain Region (dHPC vs. vHPC) was not significant for GAD65 (F(1,25)=2.562, p>0.05), GAD67 (F(1,25)=3.224, p>0.05), or ChAT (F(1,25)=0.173, p>0.05). However, separate one-way ANOVAs yielded that after the 1st Extinction Day there was no significant Drug main effects for GAD65 (Figure 3A), GAD67 (Figure 3B), or ChAT (Figure 3C) in the dHPC or vHPC (ps>0.05). However, after the 3rd Extinction Day, Drug main effect was significant for vHPC GAD65 (Figure 3D; F(1,12)=6.382, p<0.05) and GAD67 (Figure 3E; F(1,12)=4.828, p<0.05) but not ChAT (Figure 3F; F(1,12)=1.512, p>0.05) fold-changes. There were no significant Drug main effects for dHPC for any of the proteins (ps>0.05). Together, these results show that nicotine administration during contextual fear extinction reduces GAD65 and GAD67 levels in the vHPC without affecting dHPC GAD65 and GAD67. ChAT protein levels were not affected by acute nicotine in either the dHPC or vHPC. Further, the effects of acute nicotine on contextual fear extinction and vHPC GAD65 and GAD67 levels only appeared on the 3rd day of extinction, but not on the 1st day of extinction. Finally, we found that vHPC GAD67 (r= −0.65, p<0.5) and GAD65 (r= −0.65, p<0.5), but not ChAT (r= −0.35, p>0.5), optical density values after the 3rd extinctions session were negatively correlated with the individual normalized %freezing responses (Figure 3G). This suggests that decreased levels of vHPC GABA synthesis may be a contributing factor for the increased activity patterns we observed in the vHPC. Further, the negative correlation between vHPC GAD65/67 levels and freezing response during fear extinction suggests that the decreased GAD65 and GAD67 protein levels may be associated with disrupted fear extinction.
Figure 3. Acute nicotine decreased protein levels of vHPC GAD65 and GAD67 following the 3rd extinction session.
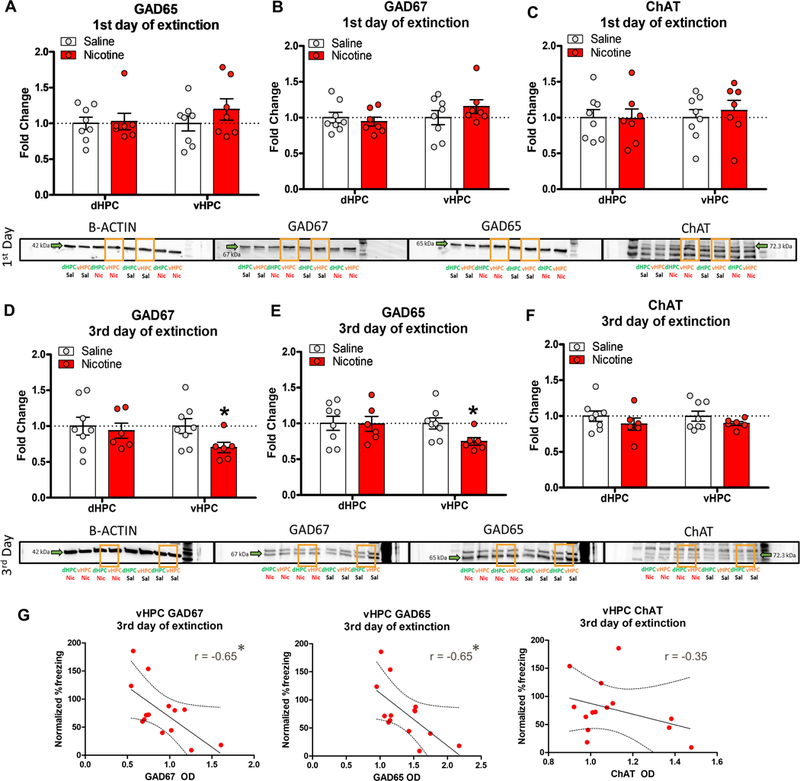
A. GAD65, B. GAD67, and C. ChAT levels in the dHPC and vHPC were not altered by acute nicotine administration after the 1st contextual fear extinction session (n=7–8 per group). Acute nicotine decreased vHPC levels of D. GAD65 and E. GAD67, but not F. ChAT (n=6–8 per group). dHPC levels of these proteins were not altered. Lower panels show representative immunoblots for each target protein and brain region in the Nicotine and Saline treated groups. G. Correlation plots between vHPC GAD67, GAD65, ChAT optical density values and normalized %freezing scores after the 3rd extinction session for each subject. Error bars show standard error of the mean. *p<0.05 compared to Saline controls.
Acute nicotine reduces active GAD to non-GAD ratio in vHPC during contextual fear extinction
Our results show that nicotine increases c-fos expression and decreases the GABA synthesizing enzyme in the vHPC during fear extinction. Therefore, we hypothesized that nicotine may decrease the GABAergic to non-GABAergic cell activation ratio within the vHPC. To test this hypothesis, we ran a c-fos/GAD65/67 double immunofluorescence and identified the c-fos expressing vHPC cells following contextual fear extinction (Figure 4A&B). For total c-fos IR counts, we found no changes in c-fos IR counts in the dHPC subregions (Figure 4C; ps>0.05). However, the Drug main effect was significant for all vHPC subregions (Figure 4D; vHPC total: F(1,11)=15.387, p<0.05; vCA1: F(1,11)=20.404, p<0.05; vCA3: F(1,11)=20.351, p<0.05; vDG: F(1,11)=6.351, p<0.05). This result replicates our findings using c-fos immunohistochemistry described above. Similarly, the dHPC subregions showed no significant Drug main effects for c-fos+GAD/c-fos ratios (Figure 4E; ps>0.05). The main effect of Drug for c-fos+GAD/c-fos ratios was significant for all vHPC subregions (Figure 4F; vHPC total: F(1,11)=15.387, p<0.05; vCA1: F(1,11)=20.404, p<0.05; vCA3: F(1,11)=20.351, p<0.05; vDG: F(1,11)=6.351, p<0.05). Interestingly, we found no significant main effects of Drug for the number of c-fos expressing GAD65/67 cells for the dHPC and vHPC or any of the subregions (ps<0.05). This demonstrates that acute nicotine did not alter the number of active GAD65/67 cells but increased c-fos expression in the non-GAD65/67 cells in the vHPC. Overall, our results show that although the number of active vHPC GABAergic cells were not decreased, acute nicotine increased the number of active excitatory neurons. Together with our immunoblotting results showing decreased GAD65/67 synthesis, these results suggest that nicotine increases vHPC activation during fear extinction by decreasing GABAergic transmission. Thus, acute nicotine may disinhibit excitatory vHPC neuronal populations, which may result in disrupted fear extinction learning.
Figure 4. vHPC c-fos expression in non-GAD cells was enhanced by acute nicotine administration during contextual fear extinction.
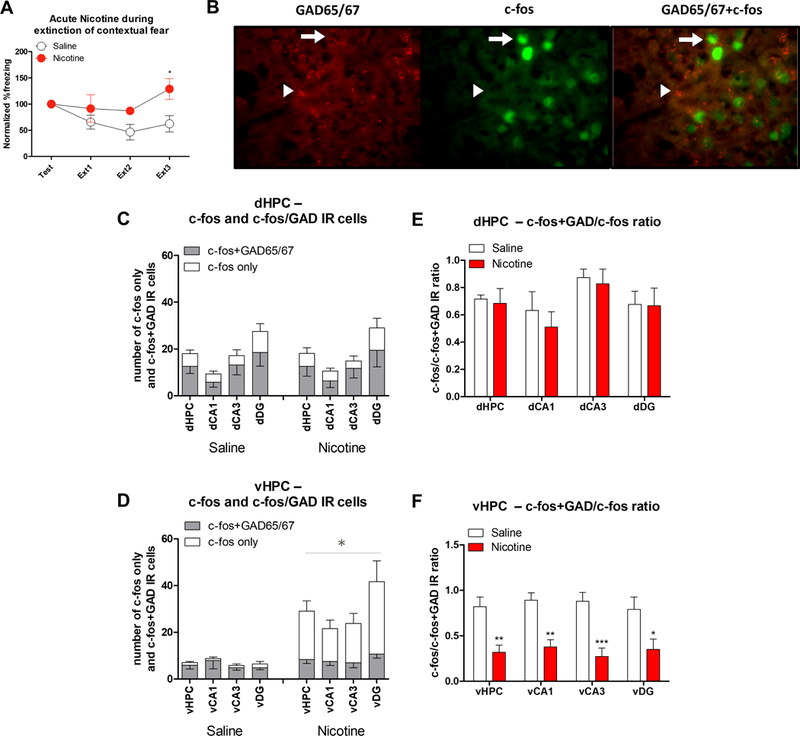
A. Behavioral results showing that acute nicotine injections increased normalized %freezing response to the context and impaired contextual fear extinction. B. Representative image showing c-fos-GAD65/67 double immunofluorescent labeled cells in the hippocampus. Arrows show cells double labeled for GAD65/67 and c-fos, while arrowheads show cells labeled for only GAD65/67. C. Number of dHPC c-fos and c-fos+GAD65/67 labeled cells was not altered by acute nicotine during contextual fear extinction (n=6–7 per group). D. Acute nicotine increased non-GAD65/67 c-fos expression in the vHPC without altering c-fos expression in the GAD65/67 cells. E. dHPC c-fos+GAD/c-fos ratios were not affected by acute nicotine. F. Acute nicotine decreased c-fos+GAD/c-fos ratios in the vHPC during contextual fear extinction. Error bars show standard error of the mean. *p<0.05; **p<0.01; ***p<0.001 compared to Saline controls.
Discussion
The results of the present study demonstrate that systemic injections of acute nicotine prior to contextual fear extinction increases neural activity in the limbic brain regions critical for fear learning and anxiety. Specifically, acute nicotine increased c-fos expression in the vHPC and BLA while not affecting dHPC, IL or PL subregions of the mPFC. Further, local infusions of nicotine into the vHPC and PL, but not into the dPHC and IL, prior to contextual fear extinction also impaired fear extinction. These results suggest that vHPC and PL nAChR activation is sufficient for the acute nicotine-induced impairment of contextual fear extinction. We also showed that the acute nicotine administration during fear extinction reduced vHPC GAD65 and GAD67 protein levels. This effect was accompanied by increased c-fos activation in non-GABAergic vHPC cells. Overall, our results suggest a novel mechanism that underlies the acute nicotine-induced impairment of contextual fear extinction. That is, acute nicotine augments activity in the brain regions that regulate fear expression and anxiety by decreasing the protein levels of GABA-synthesizing enzymes and disinhibiting excitatory neuronal populations.
Previous literature on fear extinction supports an important role of the ventral hippocampus in fear-related behaviors. Importantly, in contrast to the dorsal hippocampus, the ventral hippocampus directly innervates the amygdala, which has a role as a general mediator of fear conditioning (Pitkänenet al, 2000). This way the ventral hippocampus is mainly involved in emotional processing as opposed to the dorsal hippocampus’ role in cognitive function (Fanselow and Dong, 2010). For example, the projections from the ventral hippocampus to the amygdala are necessary for the formation and retrieval of successful context-shock associations (Hunsaker and Kesner, 2008; Maren and Fanselow, 1995). In contrast to fear conditioning, only a few studies have directly investigated the role of the ventral hippocampus in fear extinction. Nevertheless, evidence from those studies suggests that the ventral hippocampus may be critically involved in several stages of extinction learning (Hobin et al, 2006; Kutlu et al, 2016b; Orsini et al, 2011). Specifically, failure to retrieve extinction memories was associated with increased activation in the ventral hippocampus (Kutlu et al, 2016b; Orsini et al, 2011). Moreover, ventral hippocampal inactivation enhanced encoding (Sierra-Mercado et al, 2011) and retrieval (Hobin et al, 2006) of fear extinction memories. We recently showed that retrieval of previously acquired contextual fear extinction memories induced c-fos expression in the vHPC and acute nicotine increased c-fos expression and enhanced spontaneous recovery of extinguished contextual fear (Kutlu et al, 2016b). Importantly, the enhancing effects of acute nicotine on spontaneous recovery was also accompanied by decreased c-fos expression in the IL, an effect we did not observe for the encoding of contextual fear extinction in the present study. The differential IL activation pattern in the acute nicotine-induced impairment of encoding and retrieval of fear extinction memories are in line with previous results suggesting that IL is mainly involved in fear extinction retrieval (Milad and Quirk, 2002). Overall, these studies support our conclusion that the ventral hippocampus is a key region for the nAChR-mediated effects on contextual fear extinction.
In addition to identifying the vHPC as the critical site for the impairing effects of nicotine on contextual fear extinction, our results suggest that the nicotine-induced enhancement of vHPC activation may result from decreased GABAergic transmission in this region. Specifically, acute nicotine administration during contextual fear extinction downregulated GABA-synthesizing enzymes GAD67 and GAD65 in the vHPC, but not in dHPC. In support, multiple studies showed that GABA is required for contextual fear extinction (Harris and Westbrook, 1998; Hobin et al, 2006; Shumyatsky et al, 2002). Importantly, nAChRs modulate several aspects of GABAergic transmission. For example, nicotine leads to GABA release in the hippocampus (Alkondon et al, 1999), reverses GABAergic inhibition of hippocampal long-term potentiation (Fujii et al, 2000), a process that may underlie formation of long-term memory, and impairs LTP of GABAergic neurons (Niehaus et al, 2010). Thus, nAChRs may both downregulate and upregulate GABA signaling in the hippocampus. In support of this hypothesis, several studies have shown that nAChR-induced increase in the inhibitory GABAergic transmission may result in both inhibition and disinhibition of the excitatory pyramidal neurons in the hippocampus depending on the activated population of interneurons (Alkondon et al, 2000; Ji et al, 2001; Ji and Dani, 2000). That is, if nicotine activates inhibitory interneurons that form synapses with pyramidal neurons, it results in inhibition of those neurons, whereas if nicotine activates interneurons that form synapses with other inhibitory interneurons, it leads to disinhibition of the pyramidal neurons. Therefore, it is possible that nicotine may cause disinhibition of the hippocampal network resulting in over-activation of the ventral hippocampus and thus impair contextual fear extinction. Nevertheless, it is important to note that other mechanisms may be in play to compensate for the nicotine-induced changes we have observed in GABAergic function, which requires further investigation of this effect.
One possible way for nAChRs to modulate GABA-synthesizing enzymes during contextual fear extinction is altering cell signaling cascades responsible for inhibitory plasticity in the ventral hippocampal interneurons. We recently found that the impairing effects of nicotine on fear extinction are absent in α4 and β2 knockout mice (Kutlu et al, 2016a) and inactivation and desensitization of high-affinity α4β2 nAChRs via systemic injections of antagonists and partial agonists prior to each extinction session enhanced fear extinction learning in mice (Kutlu et al, 2018b). Importantly, activation of nAChRs triggers GABA release in pre-synaptic interneurons (Alkondon et al, 2000). The nAChR-induced GABA release can be mediated by phosphorylation of protein kinase A (PKA) and extracellular signal regulated kinases (ERK)1/2 through calcium (Ca2+) influx into the cell. In support, an interaction between the mitogen-activated protein kinase (MAPK) cascade and GABA receptors has been documented (Cui et al, 2008; Matsumoto et al, 2006). Specifically, these results showed that inhibition of ERK1/2 resulted in a decrease in GABA release (Cui et al, 2008). However, it is possible that the nAChR-induced GABA release from the pre-synaptic interneurons leads to a decrease in intercellular Ca2+ levels through GABA receptor activation. Consequently, the decreased Ca2+ levels will result in downregulation of PKA and ERK1/2, which in turn, will reduce GABA release and disinhibit the excitatory pyramidal neurons. In line with this model, recent studies from our lab showed that acute nicotine administration prior to contextual fear extinction sessions downregulated ERK1/2 levels in the ventral but not in dorsal hippocampus compared to saline controls (Kutlu et al, 2017a). Together with our results showing that acute nicotine decreases GAD67 and GAD65 protein levels and enhances ventral hippocampal activity, the downregulation of ERK1/2 in the ventral hippocampus may explain the nicotine-induced hyperactivity observed in the ventral hippocampus during contextual fear extinction.
Our results have several implications for anxiety and stress disorders in humans. In conjunction with the results of our previous studies (e.g., (Connor et al, 2017; Kutlu et al, 2014, 2016a, 2016b; Kutlu and Gould, 2014), the results of the present study strongly suggest that nicotine exposure might be a contributing factor to fundamental maladaptive behaviors such as inability to inhibit and extinguish fear observed in these psychopathologies. In line with this hypothesis, we recently showed that chronic nicotine exposure both in mice and humans is associated with a decreased ability to learn cues signaling safety (Kutlu et al, 2018a). Nevertheless, the nicotinic action on GABAergic signaling and fear extinction may also offer novel ways to treat fear-related symptoms in anxiety and stress disorders. For example, we found that acute nicotine requires high-affinity α4β2 subtype of nAChRs to exert its effects on fear extinction (Kutlu et al, 2016a), and inactivation of these receptors via direct antagonists or partial-agonists enhances fear extinction in mice (Kutlu et al, 2018a). Therefore, in future clinical trials, hippocampal nAChR mechanisms may be utilized to alleviate deficits in fear extinction in anxiety and stress disorder patients.
Supplementary Material
Highlights:
Systemic administration of nicotine during contextual fear extinction increased c-fos expression in the vHPC and BLA
Local nicotine infusions into the vHPC, but not dHPC, resulted in impaired contextual fear extinction
Acute nicotine downregulated both GAD65 and GAD67 protein levels in the vHPC, but not in dHPC
Acknowledgements
This work was funded with grant support from the National Institute on Drug Abuse (T.J.G., DA017949; 1U01DA041632), Jean Phillips Shibley Endowment, and Penn State Biobehavioral Health Department.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
We declare no potential conflict of interest.
References
- Alkondon M, Pereira EF, Eisenberg HM, Albuquerque EX (1999). Choline and selective antagonists identify two subtypes of nicotinic acetylcholine receptors that modulate GABA release from CA1 interneurons in rat hippocampal slices. J Neurosci 19: 2693–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkondon M, Pereira EF, Eisenberg HM, Albuquerque EX (2000). Nicotinic receptor activation in human cerebral cortical interneurons: a mechanism for inhibition and disinhibition of neuronal networks. J Neurosci 20: 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard RJ, Blanchard DC (1969). Crouching as an index of fear. J Comp Physiol Psychol 67: 370–375. [DOI] [PubMed] [Google Scholar]
- Breslau N, Davis GC, Schultz LR (2003). Posttraumatic stress disorder and the incidence of nicotine, alcohol, and other drug disorders in persons who have experienced trauma. Arch Gen Psychiatry 60: 289–294. [DOI] [PubMed] [Google Scholar]
- Breslau N, Novak SP, Kessler RC (2004). Psychiatric disorders and stages of smoking. Biol Psychiatry 55: 69–76. [DOI] [PubMed] [Google Scholar]
- Connor DA, Kutlu MG, Gould TJ (2017). Nicotine disrupts safety learning by enhancing fear associated with a safety cue via the dorsal hippocampus. J Psychopharmacol 31: 934–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Costa RM, Murphy GG, Elgersma Y, Zhu Y, Gutmann DH, et al. (2008). Neurofibromin Regulation of ERK Signaling Modulates GABA Release and Learning. Cell 135: 549–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JA, Gould TJ (2006). The effects of DHBE and MLA on nicotine-induced enhancement of contextual fear conditioning in C57BL/6 mice. Psychopharmacology (Berl) 184: 345–352. [DOI] [PubMed] [Google Scholar]
- Davis JA, James JR, Siegel SJ, Gould TJ (2005). Withdrawal from chronic nicotine administration impairs contextual fear conditioning in C57BL/6 mice. J Neurosci 25: 8708–8713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JA, Kenney JW, Gould TJ (2007). Hippocampal α4β2 nicotinic acetylcholine receptor involvement in the enhancing effect of acute nicotine on contextual fear conditioning. J Neurosci 27: 10870–10877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JA, Porter J, Gould TJ (2006). Nicotine enhances both foreground and background contextual fear conditioning. Neurosci Lett 394: 202–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanselow MS, Dong H-W (2010). Are the Dorsal and Ventral Hippocampus Functionally Distinct Structures? Neuron 65: 7–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii S, Jia Y, Yang A, Sumikawa K (2000). Nicotine reverses GABAergic inhibition of long-term potentiation induction in the hippocampal CA1 region. Brain Res 863: 259–265. [DOI] [PubMed] [Google Scholar]
- Gould TJ (2003). Nicotine produces a within-subject enhancement of contextual fear conditioning in C57BL/6 mice independent of sex. Integr Physiol Behav Sci 38: 124–132. [DOI] [PubMed] [Google Scholar]
- Gould TJ, Feiro O, Moore D (2004). Nicotine enhances trace cued fear conditioning but not delay cued fear conditioning in C57BL/6 mice. Behav Brain Res 155: 167–173. [DOI] [PubMed] [Google Scholar]
- Gould TJ, Higgins JS (2003). Nicotine enhances contextual fear conditioning in C57BL/6J mice at 1 and 7 days post-training. Neurobiol Learn Mem 80: 147–157. [DOI] [PubMed] [Google Scholar]
- Gould TJ, Lommock JA (2003). Nicotine enhances contextual fear conditioning and ameliorates ethanol-induced deficits in contextual fear conditioning. Behav Neurosci 117: 1276. [DOI] [PubMed] [Google Scholar]
- Gould TJ, Wehner JM (1999). Nicotine enhancement of contextual fear conditioning. Behav Brain Res 102: 31–39. [DOI] [PubMed] [Google Scholar]
- Harris JA, Westbrook RF (1998). Evidence that GABA transmission mediates context-specific extinction of learned fear. Psychopharmacology (Berl) 140: 105–115. [DOI] [PubMed] [Google Scholar]
- Hawkins KA, Cougle JR (2013). The effects of nicotine on intrusive memories in nonsmokers. Exp Clin Psychopharmacol 21: 434–442. [DOI] [PubMed] [Google Scholar]
- Hobin JA, Ji J, Maren S (2006). Ventral hippocampal muscimol disrupts context-specific fear memory retrieval after extinction in rats. Hippocampus 16: 174–182. [DOI] [PubMed] [Google Scholar]
- Hunsaker MR, Kesner RP (2008). Dissociations across the dorsal-ventral axis of CA3 and CA1 for encoding and retrieval of contextual and auditory-cued fear. Neurobiol Learn Mem 89: 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji D, Dani J a (2000). Inhibition and disinhibition of pyramidal neurons by activation of nicotinic receptors on hippocampal interneurons. J Neurophysiol 83: 2682–2690. [DOI] [PubMed] [Google Scholar]
- Ji D, Lape R, Dani JA (2001). Timing and location of nicotinic activity enhances or depresses hippocampal synaptic plasticity. Neuron 31: 131–141. [DOI] [PubMed] [Google Scholar]
- Koenen KC, Hitsman B, Lyons MJ, Niaura R, McCaffery J, Goldberg J, et al. (2005). A twin registry study of the relationship between posttraumatic stress disorder and nicotine dependence in men. Arch Gen Psychiatry 62: 1258–1265. [DOI] [PubMed] [Google Scholar]
- Kutlu MG, Garrett B, Gadiwalla S, Tumolo JM, Gould TJ (2017a). Acute nicotine disrupts consolidation of contextual fear extinction and alters long-term memory-associated hippocampal kinase activity. Neurobiol Learn Mem 145: 143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutlu MG, Gould TJ (2014). Acute nicotine delays extinction of contextual fear in mice. Behav Brain Res 263: 133–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutlu MG, Gould TJ (2015). Nicotine modulation of fear memories and anxiety: Implications for learning and anxiety disorders. Biochem Pharmacol 97: 498–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutlu MG, Holliday E, Gould TJ (2016a). High-affinity α4β2 nicotinic receptors mediate the impairing effects of acute nicotine on contextual fear extinction. Neurobiol Learn Mem 128: 17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutlu MG, Marin M-F, Tumolo JM, Kaur N, VanElzakker MB, Shin LM, et al. (2018a). Nicotine exposure leads to deficits in differential cued fear conditioning in mice and humans: a potential role of the anterior cingulate cortex. Neurosci Lett . [DOI] [PMC free article] [PubMed]
- Kutlu MG, Oliver C, Gould TJ (2014). The effects of acute nicotine on contextual safety discrimination. J Psychopharmacol 28: 1064–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutlu MG, Tumolo JM, Cann C, Gould TJ (2018b). Differential effects of α4β2 nicotinic receptor antagonists and partial-agonists on contextual fear extinction in male C57BL/6 mice. Psychopharmacology (Berl) 235: 1211–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutlu MG, Tumolo JM, Holliday E, Garrett B, Gould TJ (2016b). Acute nicotine enhances spontaneous recovery of contextual fear and changes c-fos early gene expression in infralimbic cortex, hippocampus, and amygdala. Learn Mem 23: 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutlu MG, Zeid D, Tumolo JM, Gould TJ (2017b). Pre-adolescent and adolescent mice are less sensitive to the effects of acute nicotine on extinction and spontaneous recovery. Brain Res Bull . [DOI] [PMC free article] [PubMed]
- Lewis MC, Gould TJ (2007). Reversible inactivation of the entorhinal cortex disrupts the establishment and expression of latent inhibition of cued fear conditioning in C57BL/6 mice. Hippocampus 17: 462–470. [DOI] [PubMed] [Google Scholar]
- Maren S, Fanselow MS (1995). Synaptic plasticity in the basolateral amygdala induced by hippocampal formation stimulation in vivo. J Neurosci 15: 7548–7564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto T, Numakawa T, Yokomaku D, Adachi N, Yamagishi S, Numakawa Y, et al. (2006). Brain-derived neurotrophic factor-induced potentiation of glutamate and GABA release: different dependency on signaling pathways and neuronal activity. Mol Cell Neurosci 31: 70–84. [DOI] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ (2002). Neurons in medial prefrontal cortex signal memory for fear extinction. Nature 420: 70. [DOI] [PubMed] [Google Scholar]
- Niehaus JL, Murali M, Kauer JA (2010). Drugs of abuse and stress impair LTP at inhibitory synapses in the ventral tegmental area. Eur J Neurosci 32: 108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver CF, Kutlu MG, Zeid D, Gould TJ (2018). Sex differences in the effects of nicotine on contextual fear extinction. Pharmacol Biochem Behav 165: 25–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsini CA, Kim JH, Knapska E, Maren S (2011). Hippocampal and Prefrontal Projections to the Basal Amygdala Mediate Contextual Regulation of Fear after Extinction. J Neurosci 31: 17269–17277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ (2001). The mouse brain in stereotaxic coordinates. 2001. This opens a new W to Investig impact Neuropsychiatr Cond such as drug, alcohol Addict fear or Depress like Behav Connect As case amygdala, thalamus, another gray matter Reg sends axonal Proj 2: . [Google Scholar]
- Pitkänen A, Pikkarainen M, Nurminen N, Ylinen A (2000). Reciprocal connections between the amygdala and the hippocampal formation, perirhinal cortex, and postrhinal cortex in rat: a review. Ann N Y Acad Sci 911: 369–391. [DOI] [PubMed] [Google Scholar]
- Rothbaum BO, Davis M (2003). Applying Learning Principles to the Treatment of Post-Trauma Reactions. Ann N Y Acad Sci 1008: 112–121. [DOI] [PubMed] [Google Scholar]
- Séguéla P, Wadiche J, Dineley-Miller K, Dani J a, Patrick JW (1993). Molecular cloning, functional properties, and distribution of rat brain alpha 7: a nicotinic cation channel highly permeable to calcium. J Neurosci 13: 596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shumyatsky GP, Tsvetkov E, Malleret G, Vronskaya S, Hatton M, Hampton L, et al. (2002). Identification of a signaling network in lateral nucleus of amygdala important for inhibiting memory specifically related to learned fear. Cell 111: 905–918. [DOI] [PubMed] [Google Scholar]
- Sierra-Mercado D, Padilla-Coreano N, Quirk GJ (2011). Dissociable roles of prelimbic and infralimbic cortices, ventral hippocampus, and basolateral amygdala in the expression and extinction of conditioned fear. Neuropsychopharmacology 36: 529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian S, Gao J, Han L, Fu J, Li C, Li Z (2008). Prior chronic nicotine impairs cued fear extinction but enhances contextual fear conditioning in rats. Neuroscience 153: 935–943. [DOI] [PubMed] [Google Scholar]
- VanElzakker MB, Kathryn Dahlgren M, Caroline Davis F, Dubois S, Shin LM (2014). From Pavlov to PTSD: The extinction of conditioned fear in rodents, humans, and anxiety disorders. Neurobiol Learn Mem 113: 3–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada E, Wada K, Boulter J, Deneris E, Heinemann S, Patrick J, et al. (1989). Distribution of alpha2, alpha3, alpha4, and beta2 neuronal nicotinic receptor subunit mRNAs in the central nervous system: A hybridization histochemical study in the rat. J Comp Neurol 284: 314–335. [DOI] [PubMed] [Google Scholar]
- Zhao C, Li M (2010). c-Fos identification of neuroanatomical sites associated with haloperidol and clozapine disruption of maternal behavior in the rat. Neuroscience 166: 1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziedonis D, Hitsman B, Beckham JC, Zvolensky M, Adler LE, Audrain-McGovern J, et al. (2008). Tobacco use and cessation in psychiatric disorders: National Institute of Mental Health report. Nicotine Tob Res 10: 1691–1715. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.