

Abstract
KRAS mutation is present in approximately 30% of human lung adenocarcinomas. Although recent advances in targeted therapy have shown great promise, effective targeting of KRAS remains elusive, and concurrent alterations in tumor suppressors render KRAS-mutant tumors even more resistant to existing therapies. Contributing to the refractoriness of KRAS-mutant tumors are immunosuppressive mechanisms, such as increased presence of suppressive regulatory T cells (Tregs) in tumors and elevated expression of the inhibitory receptor PD-1 on tumor-infiltrating T cells. Treatment with BET bromodomain inhibitors is beneficial for hematologic malignancies, and they have Treg-disruptive effects in a non-small cell lung cancer (NSCLC) model. Targeting PD-1 inhibitory signals through PD-1 antibody blockade also has substantial therapeutic impact in lung cancer, although these outcomes are limited to a minority of patients. We hypothesized that the BET bromodomain inhibitor JQ1 would synergize with PD-1 blockade to promote a robust antitumor response in lung cancer. In the present study, using Kras+/LSL-G12D; Trp53L/L (KP) mouse models of NSCLC, we identified cooperative effects between JQ1 and PD-1 antibody. The numbers of tumor-infiltrating Tregs were reduced and activation of tumor-infiltrating T cells, which had a T-helper type 1 (Th1) cytokine profile, was enhanced, underlying their improved effector function. Furthermore, lung tumor–bearing mice treated with this combination showed robust and long-lasting antitumor responses compared to either agent alone, culminating in substantial improvement in the overall survival of treated mice. Thus, combining BET bromodomain inhibition with immune checkpoint blockade offers a promising therapeutic approach for solid malignancies such as lung adenocarcinoma.
Keywords: bromodomain inhibitor, epigenetic, immunotherapy, KRAS, NSCLC, Tregs
Introduction
Non-small cell lung cancer (NSCLC), comprising lung adenocarcinoma, squamous-cell lung carcinoma, and large cell carcinoma, makes up 85–90% of lung cancers, and the different subtypes differ in genomic features and clinical histories (1). Mutation of KRAS, encoding a small GTPase linking growth factor signaling and downstream MAPK signaling, is present in approximately 30% of lung adenocarcinoma and associates with poor prognosis in NSCLC (2,3). Although drugs such as MEK inhibitors and PI3K inhibitors are under investigation in NSCLC, there is no approved therapy directly targeting this oncogene (2,3). Furthermore, KRAS mutation concurrent with other genetic alterations provokes differential responses to current therapeutics and therapeutic resistance (4,5). For example, TP53 or LKB1 co-mutation makes KRAS-mutant tumors more resistant to chemotherapy (4). These concurrent genetic lesions are also associated with lung tumors that are characterized by distinct immune cells dynamics within the tumor microenvironment (5). In a preclinical study of Kras+/LSL-G12D; Trp53L/L (KP) genetically engineered mice (GEM), the concurrent p53 deficiency rendered KP tumors more chemoresistant, compared with either Kras alone or Kras with concurrent Lkb1 mutation (4). Considering the high rate of p53 deficiency in KRAS-mutant NSCLCs, an urgent need exists to explore new therapeutic modalities targeting this NSCLC subset.
Epigenetic mechanisms such as acetylation, methylation, and phosphorylation regulate gene transcription without altering the genetic code (6,7). Proteins with epigenetic functions can be categorized into three main types: writers (such as DNA methyl transferases; DNMTs), readers (such as bromodomain proteins) and erasers (such as histone deacetylases; HDACs) (8–10). As a consequence of their activities, these epigenetic enzymes modulate many biological processes and their hyperactivity or overexpression has been reported in a number of cancers (8). Accumulating evidence demonstrates that a number of pharmacological agents targeting these proteins elicit cytostatic and/or cytotoxic effects in tumor cells (9). Given reported contributions of immune cells to durable antitumor responses (11,12), caution should be exercised in utilization of these agents for oncologic therapeutic applications as deleterious effects on tumor-associated immune cells may impede immune-orchestrated antitumor responses. In this vein, JQ1, an inhibitor of the BET family of bromodomain-containing proteins, may be a promising epigenetics-targeting drug for the treatment of NSCLC due to its reported efficacy in hematologic malignancies such as acute myeloid leukemia and multiple myeloma, as well as in other investigational solid cancer models (13–16). Our previous study demonstrated antitumor activity of JQ1 in KRAS-mutant lung cancers, either as a single agent or in combination with HDAC inhibitors (17,18). However, the therapeutic efficacy of this combination in these settings is still somewhat limited.
Immunotherapy, in particular, the rapid development of multiple immune checkpoint blockade molecules, is among the most exciting recent breakthroughs in cancer treatment (12,19). Immune therapies such as PD-1/PD-L1 and CTLA-4 blocking antibodies have been approved in multiple advanced and metastatic cancers, including NSCLC (11,20). Immunotherapy promotes greater duration of survival in some patients than conventional chemotherapies in some lung cancer subtypes (21,22). However, with the exception of melanoma, the majority of cancer patients do not exhibit clinical benefit from anti-PD-1 therapy due to a variety of resistance mechanisms stemming from lack of presentation of effective antigens, JAK1/2 mutations, and other immune response alterations (23–25). Thus, a critical unmet need remains to develop combination therapies to expand the group of immunotherapy responders. This notion is supported by our previous studies in which anti-PD-1 showed efficacy in certain subtypes of Egfr-mutant lung cancer GEM (26), and when coupled with radiotherapy in Kras-mutant tumors (27).
Immunosuppressive mechanisms, such as PD-1 expression on tumor-infiltrating T cells, and accumulation of suppressive T cells, such as CD4+Foxp3+ regulatory T (Treg) cells, in the tumor microenvironment impede antitumor immune responses. Several lines of evidence demonstrate that eliminating such inhibitory mechanisms in tumors can pave the way for a more productive antitumor response, thereby facilitating a tumor microenvironment where immune stimulatory signals prevail over inhibitory ones. In the present study, we hypothesized that combining JQ1, which we previously demonstrated disrupts tumor-Treg function (18), with anti-PD-1 blockade will alleviate potential tumor-reactive effector T cells of the inhibitory barriers posed by PD-1 signaling and Treg presence to favor enhanced T cell function within the tumor milieu. We tested this hypothesis by evaluating the effects of both agents on phenotypic and functional features of tumor-infiltrating T cells, upon their administration in a genetically engineered mouse model (GEMM) of NSCLC in which lung tumor development is driven by activating Kras mutation and p53 deficiency (KP) . Furthermore, the therapeutic potential of this unique combinatorial regimen in this NSCLC model was explored.
Materials and Methods
Mice
All breeding and treatment experiments were performed with the approval of DFCI Animal Care and Use Committee. Mice were maintained under specific-pathogen-free conditions in an AAALAC-accredited facility. All animal work was conducted in accordance with ARRIVE guidelines (28).
Kras+/LSL-G12D; Trp53L/L (KP) GEMs and treatment studies
KP mice were induced with adeno-Cre intranasally, and lung tumors were confirmed and monitored by magnetic resonance imaging (MRI ) with BioSpec USR70/30 horizontal bore system (Bruker) (4) 3D Slicer software was used to quantify the tumor volume (4) . After MRI-confirmation of tumors, KP mice were treated with JQ1 (50 mg/kg I.P. daily), anti-PD-1 (clone 29F.1A12; 200 μg/mouse I.P. three times per week), or in combination, and tumor growth was monitored by MRI every two weeks. For depleting antibody treatments, anti-CD4 (GK1.5) and anti-CD8 (53–6.72) were purchased from Bio X Cell (Lebanon, NH). Mice in each group were given two consecutive doses (400 μg/mouse) of antibodies at day −2 and day −1 and twice per week thereafter together with JQ1/α-PD-1 combination treatment.
Adoptive T cell transfer and tumor inoculation studies
For adoptive transfer and tumor inoculation studies in athymic nude mice, trans-thoracic injection of KP cell line (2×106) was first performed. Upon establishment of lung tumors as confirmed by MRI, total CD4+ or CD4+CD25- T cells (2.5×106) isolated from KP mice were transferred i.v. into these tumor-bearing mice. Two weeks later, the phenotype of the transferred CD4+ T cells present in tumors were analyzed. KP cell lines were established in our laboratory using lung tumor nodules of genetically engineered Kras+/LSL-G12D;Trp53L/L (KP) mice. All cell lines were authenticated by DNA fingerprinting and verified as Mycoplasma-free using Universal Mycoplasma Detection Kit (ATCC).
Immune profiling with multicolor flow cytometry
Tumor-bearing mouse lungs were collected from KP mice after which tumor nodules were excised and cut into about 1 mm pieces before placement under Hank’s Balanced Salt Solution (HBSS) containing 100 U/mL Collagenase D from Clostridium histolyticum (Sigma Aldrich) and 50 μg/mL DNase I grade II from bovine pancreas (Sigma Aldrich) for 40 minutes at 37°C. After digestion, cells were passed through a 70 µm strainer to remove clumps, and treated with ACK Lysing Buffer (Life Technologies). Cells were resuspended in FACS buffer (PBS + 2% fetal bovine serum) for flow cytometry. For multicolor flow cytometry, cells were first stained with LIVE/DEAD Fixable Aqua Dead Cell Stain Kit, for 405 nm excitation (Life Technologies) for 30 minutes at 4 °C and washed twice with FACS buffer. Cells were treated with purified anti-mouse CD16/32 (BioLegend) for 15 minutes, and then antibody mixture was added. Thirty minutes later, the cells were washed twice with FACS buffer and fixed in 1% formalin or further processed for intracellular staining. For intracellular staining, cells were fixed/permeabilized with Foxp3/Transcription Factor Staining Buffer Set Kit (eBioscience) before antibodies were added. After two washes, samples were resuspended in FACS buffer before acquisition using BD LSR Fortessa or BD FACS Canto (BD Biosciences)].
Antibodies
All antibodies used for flow cytometry analysis were purchased from BD Biosciences (San Jose, CA), Biolegend (San Diego, CA), or eBioscience (Santa Clara, CA) and are listed in supplementary table 1.
Ex vivo CD8+ T cell activation assay
Leukocytes from lungs of treated mice were isolated using Ficoll gradient separation after single cell disassociation. Then, 106 isolated cells were stimulated at 37°C with Leukocyte Activation Cocktail for 6 hours with FITC-CD107a (Biolegend) and BD GolgiPlug™ (BD Pharmingen™) added in the last 5 hours. Cells were washed and stained for intracellular cytokines using BD cytofix/cytoperm kit (BD Pharmingen) according to the manufacturer’s instructions.
Ex vivo tumor cell death assay
EpCAM+ tumor cells and CD3+Foxp3- T cells were sorted from the tumors of treated mice. Cells were rested in complete media for 1 hour, after which 1×105 T cells were added to 1×103 tumor cells in 96-well plate and incubated at 37°C for 24 hours in the presence of 5 IU/ml of IL2. Tumor cell death was assessed by FACS staining using fixable live/dead dye.
T-cell proliferation/Treg-suppression assay
For mouse Treg-suppression assays, CD4+CD25hi Tregs in tumor cell suspensions were sorted into KLRG1+ or KLRG1- cells and cultured at 1:2, 1:4, and 1:8 ratios with Carboxyfluorescein diacetate succinimidyl ester (CFSE)-labelled CD4+CD25- cells (0.25×105) isolated from the spleen of the same mouse. T cell–depleted, mitomycin C–treated splenocytes (0.25×105) were added as antigen presenting cells (APCs) and cultures were stimulated with soluble anti-CD3 (clone 2C11, ebioscience) at 0.5 µg/ml concentration for 3 days.
Cytokine analysis
Bronchoalveolar lavage (BAL) fluid was collected from the tumor-bearing lungs of KP mice by injecting 05ml of 1x PBS into the lungs with repeated flushing before dispensing into collection tubes. BAL fluids were then immediately centrifuged for 5 minutes at 1200 rpm at 4°C and subsequently stored at −80°C and thawed on ice prior to analysis. Each assay used 50 μl of each sample. A multiplex magnetic bead-based sandwich ELISA assay was utilized to simultaneously measure 23 secreted proteins, including IL4, RANTES, KC, G-CSF, IL17, IL5, IL1α, IL9, MIP-1β, MIP-1α, MCP-1, IL13, IL12 (p70), IL12 (p40), IL3, Eotaxin, GM-CSF, IFNɣ, IL10, IL1β, IL2, IL6, TNFα were tested in a single assay format (Bio-rad: Bioplex 23x ms panel – Cat# M60009RDPD). Each bead is encoded with a ratiometric concentration of 2 IR dyes and act as a barcode, unique to each protein. The final detection complex is formed with the addition of streptavidin-phycoerythrin conjugate, where the phycoerythrin serves as the fluorescent reporter. Concentration levels, measured in pg/ml, for each cytokine are derived from 5-parameter curve fitting models. Fold change relative to control are calculated and plotted as log2FC to establish biological significance.
RNA-seq and data analysis
A small portion of snap frozen lung tumor from each mouse was pulverized using the Covaris T-prep method on dry ice (cat # 520097). The Agencourt RNAadvance kit for isolation of nucleic acids from frozen tissue (cat # A32645) was implemented on a Biomek® FXP Laboratory Automation Workstation (Dual arm system with multichannel pipette and span-8 pipetters, cat# A31844). The automated protocol is based upon Solid Phase Reversible Immobilization (SPRI) paramagnetic bead-based technology, which does not require vacuum filtration, centrifugation, or organic solvents such as phenol or xylene associated with traditional methods. Briefly the tissue is lysed, after which the RNA is immobilized onto the magnetic particles. The RNA is then treated with DNase and the contaminants rinsed away using a simple wash procedure. RNA isolates were eluted in a 55 µl volume of RNase/DNase-free H2O. All RNA was stored at −80°C. RNA quality was assessed by Agilent Bioanalyzer using the RNA 6000 pico kit (cat# 5067–1513). 50 ng RNA was utilized as input for RNA-Seq library preparation utilizing the TruSeq RNA Access Library Prep Kit (cat# RS-301–2001). The method was automated on the Biomek® FXP Laboratory Automation Workstation. This method facilitates enrichment of the coding regions of the transcriptome that are captured using sequence-specific probes to create the final library. cDNA libraries were quantified utilizing the Quanti-iT PicoGreen assay (Life Technologies, cat# P7589). One µl of cDNA was required for quantification. Concentration is measured as ng/ul. Libraries were excited at 480 nm and the fluorescence emission intensity was measured at 520 nm using a Victor X3 spectrophotometer (Perkin Elmer cat# 2030–0030). Fluorescence intensity was plotted versus concentration over the low calibration range, 0–50 ng/μl. Libraries were also quality checked by Agilent Bioanalyzer using the High Sensitivity DNA kit (cat# 5067–4626). cDNA libraries were then sequenced on the Illumina NextSeq500 platform as 75bp paired end reads. The STAR RNA sequencing alignment tool (Spliced Transcripts Alignment to a Reference) aligner [STAR_2.5.0a]) was utilized to align the data to the mouse genome, (RefSeq gene annotations). DeSeq2 (29) was utilized to perform differential expression analysis. We conducted pathway enrichment analysis utilizing the GeneGo MetaCore+MetaDrug™ tool (Thompson Reuters, version 6.31 build 68930). RNA sequencing data have been deposited in the NCBI GEO public data repository (reference series accession GSE114601).
Mouse-derived organotypic tumor (MDOT) culture
Mouse-derived organotypic tumor (MDOT) cultures were established from gently dissociated KP tumors, after which the generated tumor spheroids were mixed with supporting collagen matrix and loaded into three-dimensional AIM glass chambers (AIM Biotech, Singapore) in the presence of complete media containing JQ1 (250nm) and anti-PD-1 (10ug/ml). DMSO served as control. After three days in culture, media was aspirated and chambers were gently washed with 1X PBS prior to immunofluorescent staining. Briefly, purified CD16/32 FCγR blocking reagent was added to chambers for 15 minutes at room temperature followed by Alexa fluor 488 anti-mouse CD3 and Alexa fluor 594 anti-mouse EpCAM for one hour. Then, chambers were washed gently with 1X PBS/2% FBS twice and DAPI (Invitrogen, Carlsbad, CA; 1:1000X) was added and incubated for five minutes followed by two washes with 1X PBS/2%FBS. Images were captured on a Nikon Eclipse 80i fluorescence microscope equipped with CoolSNAP CCD camera and merged images created with NIS elements imaging software. Quantification of CD3+ T cells under each treatment condition was determined from an average of 10–11 fields of view as evaluated by fluorescent microscopy.
Statistical analysis
Data were analyzed using mean ± standard error of the mean (SEM). Unpaired two-tailed Student t test was used for comparisons between two groups using GraphPad Prism software. P values < 0.05 were considered statistically significant (*); P values < 0.01 are marked **, and P values < 0.001 are marked ***.
RESULTS
PD-1 blockade and BET bromodomain inhibition attenuates PD-1 expression and Treg proportions
In previous studies, we demonstrated that JQ1, an inhibitor of BET family of bromodomain (BRD) proteins, disrupts Treg phenotype and function in tumors of GEMM of NSCLC (18). Existing reports also show that, although clinical responses seen in patients treated with PD-1 blockade can be remarkable, these therapeutic benefits are limited to a subset of patients (20,23,24). Consistent with this, only two of the six GEM-harboring Kras-driven lung adenocarcinomas showed a delay in tumor growth following PD-1 treatment (27), Supplementary Fig. S1). We hypothesized that combining JQ1 with PD-1 blockade would dampen the cellular and molecular inhibitory mechanisms ascribed to Treg function and PD-1 signaling, respectively, to favor improved T cell activation and function within the tumor-immune microenvironment. To test this hypothesis, we employed the use of GEM harboring activating Kras mutations with concurrent p53 deficiency (KP; Fig. 1A), with the rationale that this model mirrors clinical presentations of aggressive lung adenocarcinomas driven by KRAS mutations and/or p53 deficiency. First, we evaluated KP mice treated with JQ1/anti-PD-1 for two weeks in order to characterize changes to immunological parameters accompanying these treatments (Fig. 1A). Although the numbers of cells in most of the tumor CD45+ leukocyte sub-populations that we evaluated did not significantly change (Supplementary Table. S2, Supplementary Fig. S2), the proportion of CD4+Foxp3+ Tregs decreased about 60% in JQ1 and JQ1/anti-PD-1-treated mice, relative to vehicle controls (Fig. 1B). Comprehensive profiling of tumor-infiltrating CD4+ and CD8+ T cells revealed that among several immune checkpoint receptors evaluated, the frequency of PD-1–expressing T cells, which is more prominently expressed than CTLA-4 on tumor-infiltrating T cells (Supplementary Fig. S3), was diminished after JQ1 treatment with or without anti-PD-1 but not with anti-PD-1 treatment alone (CD8+ T cells in Fig. 1C, CD4+ T cells in Supplementary Fig. S4A). A modest decrease in T cell expression of CTLA-4 was also observed upon PD-1 treatment which was significant when combined with JQ1 administration (Fig. 1D, Supplementary Fig. S4B). These changes were not observed in peripheral (splenic) T cells where PD-1 and CTLA-4 were only expressed at basal levels (Supplementary Fig. S3). Taken together, these results demonstrate quantitative and phenotypic changes on Tregs and potential effector T cells, respectively, that is consistent with reduced cellular and molecular inhibitory entities in tumor-infiltrating T cells upon combined administration of JQ1 and anti-PD-1 in NSCLC-bearing mice.
Fig 1.
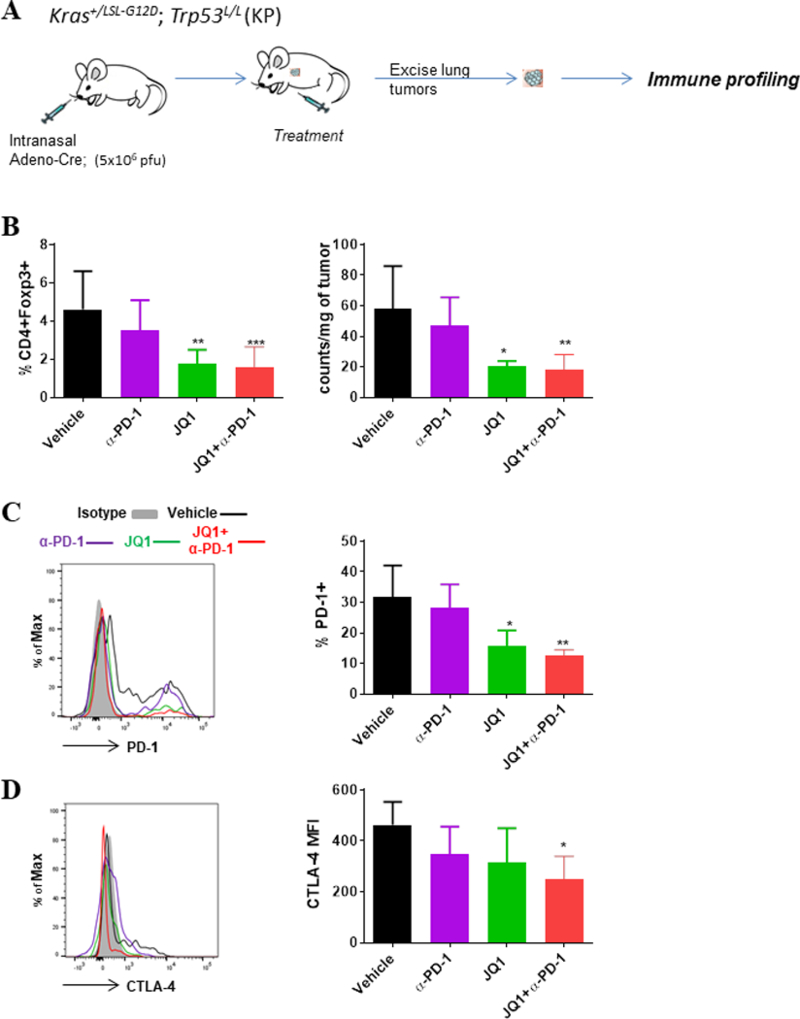
PD-1 blockade and BET bromodomain inhibition promotes reduced Treg proportions and expression of inhibitory receptors on tumor-infiltrating T cells in GEMM of NSCLC. (A) Schematics of treatment study and immune analysis in KP GEM. KP mice were induced with adeno-Cre intranasally, and treatment was started upon tumor establishment as confirmed by MRI. After two weeks of treatment, tumor nodules were excised from the lungs of JQ1 and/or α-PD-1-treated mice, and multiparameter flow cytometric analysis was conducted on single cell suspensions to assess the frequencies and phenotype of tumor-infiltrating T cell subsets. (B) Percent CD4+Foxp3+ Tregs within CD45+ leukocytes (left) and absolute Treg counts within the tumors of KP mice treated as indicated. (C, D) Representative histograms (left) and summary (right) of expression levels for (C) PD-1 and (D) CTLA-4 on tumor-infiltrating CD8+ T cells. Data are mean ± SEM for 4–5 mice per group. *P < 0.05; **P < 0.01; ***P < 0.001.
PD-1 blockade combined with BET bromodomain inhibition enhances T cell-activation and effector activity
Given that T cell activation and function is most permissive under reduced expression of molecular inhibitors and/or hindrance by suppressive cells, we tested whether our findings above correlate with effector activity by evaluating the activation status and functional capacity of tumor-infiltrating T cells in the treated GEM. Phenotypic assessments showed that tumor-infiltrating CD4+ and CD8+ T cells exhibited an increased activation profile under anti-PD-1 or JQ1 treatment, given by increased frequencies of CD69+ T cells, an effect that was further amplified upon their combination (Fig. 2A, B). Upon ex vivo stimulation, tumor-infiltrating CD8+ T cells under single agent treatment exhibited an augmented ability to secrete the effector cytokine IFNγ and the expression of degranulation-associated membrane protein CD107a. This increased effector profile was most pronounced upon combination of JQ1 and anti-PD-1 (Fig. 2C, D), indicating that both agents cooperatively promote enhanced activation and effector function of CD8+ T cells in the tumor bed. Similar findings for IFNγ were also noted for tumor-CD4+ T cells (Supplementary Fig. S4C). This re-invigoration in T cell effector function likely stems in part from the diminished PD-1 expression on tumor-T cells, as their capacity to secrete the effector cytokine IFNγ inversely correlated with frequency of PD-1+ T cells (Supplementary Fig. S4D). Although analysis of myeloid cells (CD45+CD11b+ / CD45+CD11c+) did not reveal broad phenotypic changes, treatment with JQ1 alone or with anti-PD-1 was accompanied by increased expression of MHC class I on tumor-associated macrophages (TAMs), which also exhibited modest decrease in PD-L1 expression (Supplementary Fig. S5A-C). Of note, JQ1 treatment also evoked measurable effects on tumor cells, such as downregulation of genes encoding tumor growth and survival proteins, as well as concurrent upregulation of a number of tumor suppressors (Supplementary Fig. S6 Fig. S11, Supplementary Table. S3)
Fig 2.
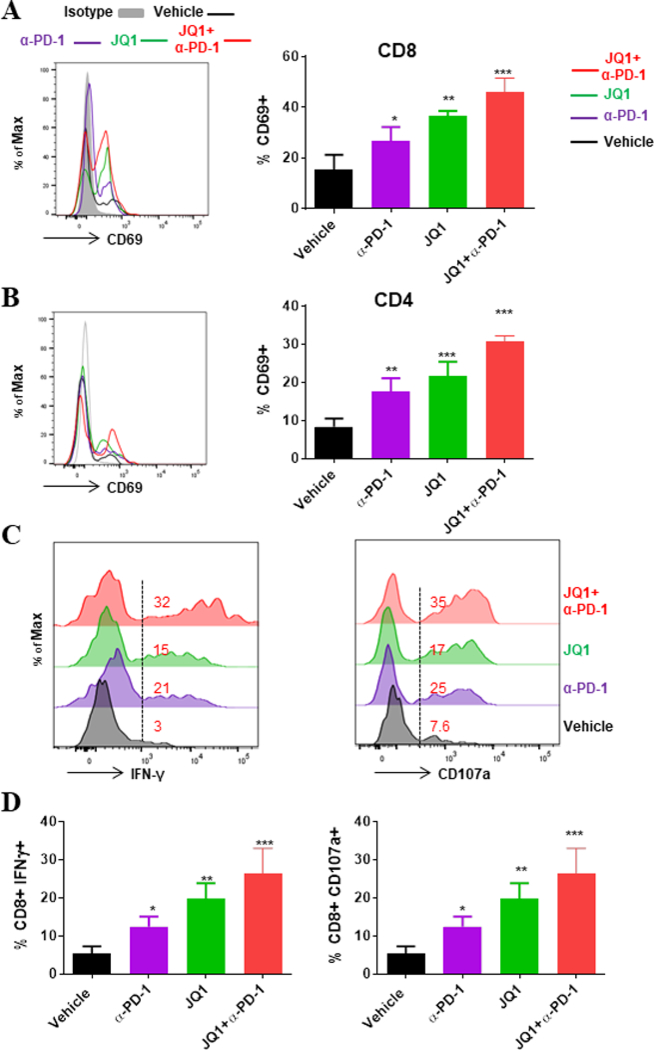
PD-1 blockade and BET bromodomain inhibition promote increased activation and effector capacity of tumor-infiltrating T cells in GEMM of NSCLC. Phenotypic assessments were conducted by flow cytometry of tumor cell suspensions to evaluate activation status of T cells. (A, B) Representative histograms (left) and summary (right) of CD69 expression on tumor-infiltrating (A) CD8+ and (B) CD4+ T cells. (C, D) Immune cells from KP tumors treated with vehicle or indicated agents were isolated by Ficoll gradient and stimulated for 6 hours with leukocyte activation cocktail (PMA and Ionomycin); golgi plug and CD107a antibody were added in the last 5 hours of culture. (C) Representative histograms with corresponding percentages and (D) summary of CD8+ T cells secreting IFNγ after intracellular cytokine staining. Data are mean ± SEM for 3–4 independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001.
Combination of anti-PD-1 and JQ1 evokes a Th1 cytokine signature in the local tumor niche
The cytokine milieu within or around tumor microenvironment plays crucial roles in regulating cell-mediated immunity (30–32). We therefore asked how the cytokine composition associated with the tumor niche is impacted by anti-PD-1 and JQ1 treatment. We analyzed the BAL fluid within the local tumor-bearing lung of treated KP mice. Relative to the vehicle treatments, a number of cytokines that regulate T lymphocyte differentiation or signify their functional tendencies, including IL2, IL12, IFNγ, and TNFα, were increased in the BAL fluid of JQ1 or anti-PD-1-treated mice whereas IL4, IL5, IL6, IL10, and IL13 were either decreased or marginally increased. These patterns appeared somewhat additive under combined JQ1/anti-PD-1 treatments, resulting in greater changes than associated with either agent alone (Fig. 3A). Dissecting the pattern of the T cell-related cytokines further, we found that as compared to vehicle control treatment, a Th1 cytokine profile, including elevated IL12 (p40), TNFα, and IFNγ, was more dominant in the BAL fluid associated with tumors of single agent-treated KP mice when assessed against Th2 cytokinesIL4, IL5, IL10, and IL13. As with previous phenotypic changes, this bias in favor of Th1 cytokines was most striking with the combination of JQ1 and PD-1 (Fig. 3B, C), demonstrating that an overall effector cytokine profile is evoked under the combinatorial regimen. A number of cytokines implicated in myeloid cell trafficking and recruitment (e.g. MCP-1, MIP-1, RANTES, Eotaxin) were also substantially elevated in the combination treatment (Fig. 3A).
Fig 3.
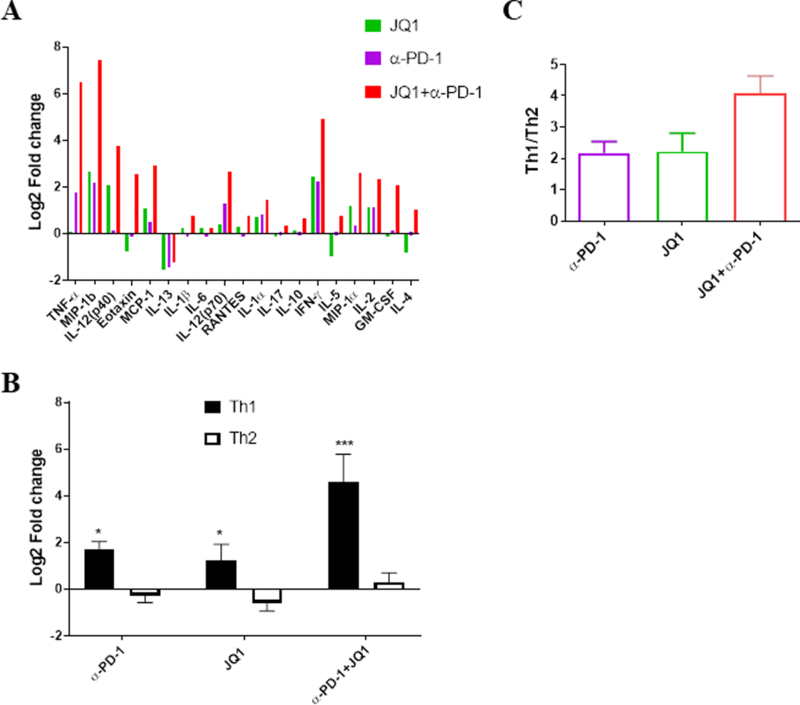
PD-1 blockade and BET bromodomain inhibition is associated with a Th1 cytokine skewing in the BAL fluid of treated GEMM of NSCLC. BAL fluid obtained from the lungs of KP-tumor-bearing mice treated with α-PD-1, JQ1 or the combination were subjected to multiplex cytokine evaluation. (A) Summary of listed cytokines detected in the BAL fluid of mice that were treated as indicated shown as log2 fold-change relative to samples from vehicle-treated mice. (B) Mean fold change for Th1 (IL2, IFNγ, TNFα) and Th2 (IL4, IL5, IL6, IL10, IL13) cytokines relative to the vehicle controls in BAL fluids. (C) Ratio of the mean values for Th1 versus Th2 cytokines concentrations. Data in panel A are the average value for 4 replicates expressed as Log2 fold change over vehicle for each cytokine. Data shown in panels B and C represent mean ± SEM for 4 replicates/group.
Combination of JQ1 and anti-PD-1 delays tumor growth and improves survival of KP mice
Based on the enhanced effector profile of tumor-infiltrating T cells following JQ1 and anti-PD-1 treatment, we hypothesized that the combination of both agents should potentiate an improved antitumor response in long-term treatment studies. Thus, to evaluate the therapeutic potential of this drug combination, KP mice were assessed by magnetic resonance imaging (MRI) every two weeks during the course of single and dual agent treatment, and tumor volume was quantified. Although anti-PD-1 treatment led to moderate delay in tumor progression (change in tumor volume of ~10–30%) relative to vehicle treatment, JQ1 administration resulted in a more robust tumor growth arrest and shrinkage (change in tumor volume of −10–10%) at two weeks post treatment initiation. Tumors regressed in all mice with combination therapy (Fig. 4A, B). Although PD-1 alone promoted a modest improved overall survival of tumor-bearing mice relative to control treatment [median survival time (MST) 36 days vs 16 days], JQ1 treatment resulted in a significantly prolonged survival that was further increased under the combination therapy (Fig. 4C; MST 69 and 87, respectively). In separate cohorts, we found that addition of CD8- or CD4-depleting antibodies to the JQ1/anti-PD-1 combination regimen led to substantial or modest loss of antitumor response and survival benefits, respectively, suggesting that the protection from tumor progression under this drug combination is largely immune (T cell) mediated (Fig. 4A, C). In support of this notion, treatment of immunodeficient mice bearing similar lung tumors as the GEM showed only marginal or slightly modest improvement in their overall survival when treated with anti-PD-1 or JQ1, respectively (Supplementary Fig. S7). Furthermore, and consistent with the in vivo therapeutic outcomes, CD8+ T cells that were isolated from tumors of treated KP mice showed enhanced degranulation and cytotoxic activity in vitro that paralleled increased tumor cell death. This effect was most robust with JQ1/anti-PD-1 combination therapy, providing evidence linking the augmented activation/effector profile evoked in this treatment setting with antitumor activity (Supplementary Fig. S8A-C). Underscoring the relatively safe profiles of this JQ1/anti-PD-1 combination was the lack of deleterious effect on the overall proportions of tumor-associated T cells (Supplementary Fig. S9). Collectively, these data demonstrate that partnering immune checkpoint blockade with BET bromodomain inhibition improved antitumor T cell function and led to robust and long-lasting therapeutic outcomes in the aggressive Kras/p53-mutant NSCLC.
Fig 4.
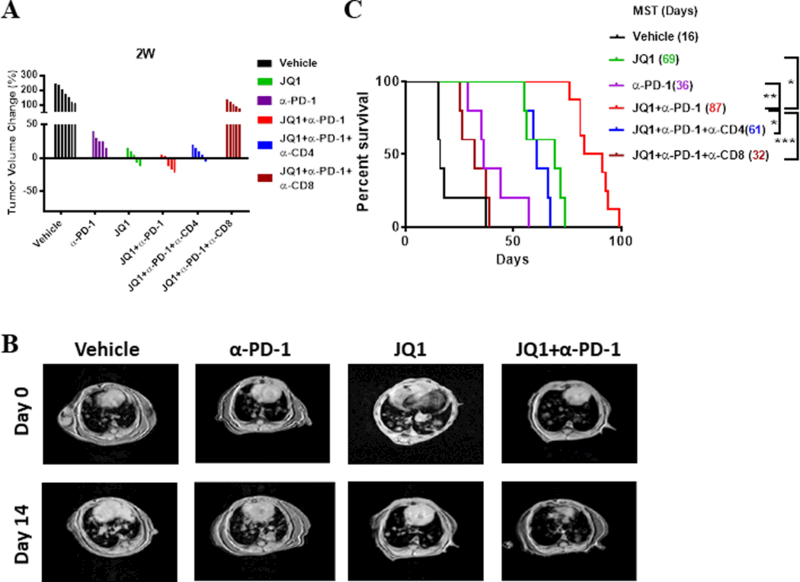
Combination of BET bromodomain inhibitor and anti-PD-1 promotes long-lasting therapeutic outcomes in KP-driven lung adenocarcinomas. (A) Waterfall plots showing tumor volume change (%) under indicated treatments after two weeks compared to pre-treatment tumor burden. Each column represents one individual mouse under each treatment (B) Representative MRI images of lung tumors in KP mice on day zero and day 14 after vehicle (control), JQ1, and/or α-PD-1 treatments. (C) Overall survival curve of KP mice under each indicated treatment with corresponding median survival time in days. Data are from 5–7 mice/treatment group.
Diminished KLRG1+ Tregs associate with increased survival following JQ1/anti-PD-1 treatment
Unlike in the spleen, a prominent proportion of CD4+Foxp3+ Tregs infiltrating the tumors of KP mice express KLRG1(Fig. 5A), a cell surface protein that has been linked to terminally-differentiated T cells (33). Although there is generally an accumulation of tumor-infiltrating Tregs with increasing tumor burden, we found that the KLRG1+ Treg subset (Fig. 5B), showed a direct correlation with tumor size. We hypothesized that this KLRG1-expressing, tumor-infiltrating Treg subset represents a distinct Treg fraction, the characterization of which could provide clues to their molecular features and functional activities. To test this hypothesis, further phenotypic and functional studies were conducted. Analysis of these KLRG1+ Treg subset revealed that they are more activated, as indicated by the vast majority being CD69+, CD44hi, and CD62Llo, when compared to their KLRG1– counterparts or peripheral pool of splenic Tregs (Supplementary Fig. S10A). Despite this distinction, their high expression of Helios and Neuropilin-1, two proteins that are highly expressed on natural Tregs, mirrored that of the KLRG1– cells and splenic Tregs, suggesting that the KLRG1+ Tregs likely arise from the peripheral pool of naturally-occurring Tregs of thymic origin (Supplementary Fig. S10B). We next employed an orthotopic tumor model by injecting KP tumor cells transthoracically and subsequently performed adoptive transfer of CD4+ T cells, which were isolated from spleens of KP mice (Supplementary Fig. S11A). Analyses of the input cells, which were either bulk CD4+ T cells or a subset depleted of CD25+, showed that the major Foxp3+ population within the CD25+ subset, or the minute Foxp3+ fraction within the CD25– subset did not express KLRG1 at time of transfer (Supplementary Fig. S11B). Two weeks after transfer of either the bulk CD4+ T cells or CD25–CD4+ population, the lung tumors were collected and the T cells were analyzed. A sizable pool of tumor-infiltrating KLRG1+ CD25+Foxp3+ Tregs were readily detected in the lung tumors of mice that received CD4+ T cells containing a largely KLRG1– Treg pool. In contrast, CD4+CD25– conventional T cells that were transferred into parallel cohorts of tumor-bearing mice largely retained their Foxp3– phenotype. The few Foxp3+ cells that were present in the tumors were CD25dim/– and did not generate a detectable KLRG1-expressing sub-population (Supplementary Fig. S11C). These data demonstrate that the KLRG1+ Treg pool is a derivative of CD25+Foxp3+ Tregs and not a peripherally-induced subset converted from CD25– conventional CD4+ T cells.
Fig 5.

Reduced proportions of KLRG1+ Tregs is associated with low tumor burden and improved survival and response to treatment in KP mice. Following identification of a subset of tumor-infiltrating KLRG1+ CD4+Foxp3+ Tregs , their proportions were further assessed. (A) Representative histograms for the expression of KLRG1 on splenic versus tumor-Tregs. (B) Percent of KLRG1+ Tregs in the tumors of KP mice as a function of tumor volume. (C) Representative histograms with corresponding percentages and (D) summary of KLRG1+ Tregs in the tumors of vehicle and JQ1/anti-PD-1-treated KP mice. (E) Percent of KLRG1+ Tregs in the tumors of KP mice as a function of survival time in days. Data in D are mean ± SEM for 4–5 mice/group. *P < 0.05; **P < 0.01.
As CTLA-4 is critical for Treg function, we also found that the tumor-associated KLRG1+ Tregs exhibit higher CTLA-4 levels that parallels their superior suppressive function compared to their KLRG1– counterparts (Supplementary Fig. S12A, B). Given our observations in vivo, we then examined whether there is any correlation between the presence of these cells and the therapeutic responses observed. Whereas a moderate decrease in proportion of KLRG1+ Tregs was observed in the tumors of anti-PD-1-treated mice, a significant reduction was observed following JQ1 monotherapy, and was further reduced by 5–10% following treatment in combination with anti-PD-1 (Fig. 5C, D). Consistent with their decline under JQ1 treatment (Fig. 5D), the KLRG1+ Treg fraction exhibited diminished expression of Foxp3, CTLA-4, GITR and Bcl-2 compared to the negative subset, suggesting that JQ1 evokes a greater disruption to their molecular features, compared with anti-PD-1, along with a propensity towards reduced survival (Supplementary Fig. S13). Lastly, the inverse relationship between the KLRG1+ Treg frequency and overall survival in the treatment settings (Fig. 5E) suggest that terminally-differentiated KLRG1-expressing Treg proportions in the tumor may be a surrogate for severity of disease and their reduction associated with therapeutic response.
DISCUSSION
Although multiple strategies have been under development against KRAS-mutant NSCLC, KRAS remains an untargetable driver oncogene. Concurrent gene alterations make KRAS-driven lung cancers even more heterogeneous and usually more resistant to treatment. Our present study demonstrates that the immune-enhancing properties of agents that disrupt the activity of epigenetics-regulating bromodomain proteins could be harnessed to enhance the therapeutic benefits of immune checkpoint blockade in NSCLC.
Previously, we reported that inhibition of BET bromodomain proteins by JQ1 promoted downregulation of Foxp3 and CTLA-4 in lung tumor-infiltrating Tregs and disrupted their suppressive function (18). Our finding that Treg numbers declined in the presence of JQ1 treatment with or without PD-1 blockade, in the present study, is consistent with this observation, as these signature proteins are crucial for Treg maintenance and survival (34,35). Whether JQ1 preferentially targets a sub-population of tumor-infiltrating Tregs warrants further investigation, although our finding that the KLRG1+ Treg subset was diminished with this treatment supports this possibility and the idea that the prevalence of this subset may associate with tumor severity. As their frequency correlates with tumor burden, we deduce that their increasing proportions is likely causative, rather than a consequence of tumor growth. We surmise that the activated status of this KLRG1+ subset relative to the KLRG1– cells may be a key feature rendering them more vulnerable to epigenetic modulation, including propensity towards cell death as observed under JQ1 treatment. Their reduced presence under this treatment therefore likely emanates from a gradual disruption in molecular programming that is normally operative in Treg maintenance, which is supported by the more dramatic downregulation of key Treg signature proteins in this subset upon JQ1 administration. Although it is tempting to also speculate that the functions of tumor-associated Tregs may be partially impaired following PD-1 blockade, our data do not provide evidence in support of this. Besides, the role of PD-1 in Treg function remains to be deduced, as indicated by evidence from conflicting reports (36,37).
Cytokines regulate T cell differentiation and function (38,39). The substantially elevated levels of IL12/IFNγ, compared with IL4, IL6, IL10, and IL13, in the presence of JQ1 and anti-PD-1 therapy supports the premise that this combination likely promotes Th1 cell differentiation and the observed Th1 cytokines, which are implicated in antitumor immunity (40–42). Future work is planned to interrogate additional conundrums, such as the status of macrophages recruited to the tumors in the presence of this drug combination (i.e. M1 or M2), which may explain potential sources of these T cell differentiation–modulating cytokines. Nonetheless, the enhanced activation and improved effector activity of tumor-infiltrating T cells, coupled with a Th1 cytokine profile, all of which were significantly potentiated with the combination treatment, aligns well with the durable antitumor response seen with this novel therapy combination.
A number of reports demonstrate that BET bromodomain proteins regulate PD-L1, and their inhibition by JQ1 downmodulate its expression (43–45). Thus, one could argue that the therapeutic effect of JQ1 as shown in this study emanates from this effect on PD-L1. Our findings do not support this premise, as we found only a modest decrease in PD-L1 expression on TAMs, whereas PD-1 expression was predominantly impacted in tumor-infiltrating T cells. This result is consistent with the idea that the therapeutic efficacy of JQ1 in this model can be attributed mostly to its Treg-disruptive effect along with PD1 downregulation in potential effector T cells. We emphasize that JQ1-mediated reduction in PD-1 levels is only partial on conventional T cells, raising the possibility that the residual PD-1-mediated negative signals may contribute to impediment of antitumor responses. Further blocking of remaining PD-1 promotes a more sustained delay of tumor growth. This is consistent with the notion that aggressive attenuation of negative signals induced by this inhibitory axis through a multipronged approach is projected to support enhanced antitumor immunity.
The cooperative nature of both drugs likely stems from a number of plausible arguments. The increased activation profile and effector function associated with JQ1 treatment could be attributed in part to a reduction in tumor-Treg proportions, which are reported to inhibit T-cell activation, proliferation and effector function in various inflammatory settings (34). The reduced levels of molecular checkpoints, such as PD-1 on tumor-associated conventional T cells, is another plausible explanation for these phenotypic changes upon JQ1 treatment. Engagement of these inhibitory receptors blunt T cell priming, a series of events that include T cell activation (46–48). In the case of PD-1 blockade, a reinvigoration of partially exhausted tumor-T cells is one of the likely mechanisms accounting for the enhancement in T cell effector function as demonstrated by ex vivo functional studies. Thus, we believe the mechanism for the cooperative effect between JQ1 and anti-PD-1 in the present study is a multifaceted targeting of inhibitory signals in the tumor microenvironment: JQ1-mediated reduction of Tregs, especially a highly suppressive KLRG1+ subset, and the dampening of the negative TCR signaling that is normally operative in the presence of un-perturbed PD-1-PD-L1 signaling.
The idea that JQ1 promotes a reduction in PD-1 expression on tumor-CD8+ T cells, whereas anti-PD-1 blocks residual PD-1 signaling, is an attractive one for immunotherapy design, as existing reports demonstrate only a subset of patients derive therapeutic benefits from anti-PD-1 therapy. Our data highlight the notion that tumor immunity is a balancing act (Fig. 6), in which inhibitory mechanisms must be reversed or attenuated sufficiently to allow stimulatory mechanisms to prevail. This notion is supported by the finding that the most robust effector activity by tumor-CD8+ T cells (as measured by IFNγ secretion) coincided with the lowest proportion of PD-1-expressing cells. This introduces the possibility that the differential responses to anti-PD-1 therapy observed in the clinic is a reflection of the degree of success to which PD-1 signals are dampened in patients. As more data emerge from clinical use of anti-PD-1 therapy, this hypothesis may be supported if pre-treatment PD-1 levels correlate with therapeutic response to anti-PD-1 therapy. Nevertheless, targeting inhibitory immune checkpoint receptors on multiple fronts in addition to dampening suppressive T cell presence, as was done here with BET bromodomain inhibition and PD-1 blockade, promotes a tumor immune microenvironment whereby potential tumor-reactive T cells can better exert their antitumor functions with less impedance.
Fig. 6.
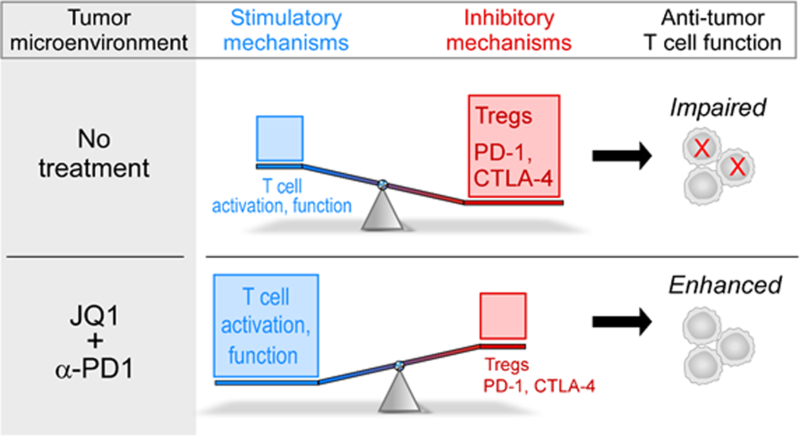
Proposed model for JQ1/α-PD-1 therapeutic effects in the KP model of NSCLC. In the absence of therapeutic intervention, inhibitory mechanisms including increased Treg presence and PD-1/CTLA-4 expression on tumor-infiltrating T cells outweigh stimulatory mechanisms leading to impairment in antitumor T cell function. Treatment with JQ1 and α-PD-1 cooperatively reduce these inhibitory mechanisms in distinct ways to allow stimulatory signals to prevail and tip the scale in favor of enhanced T cell function.
We hereby propose that BET inhibitors like JQ1 are a double-edged swords that will likely complement the therapeutic efficacy of immune checkpoint blockade. On the one hand, they evoke tumor cell-intrinsic effects, such as changes in the transcriptional landscape characterized by downregulation of genes encoding tumor growth and survival proteins, as well as concurrent upregulation of a number of tumor suppressors . On the other hand, their immunostimulatory effect is projected to favor increased immune reactivity in tumor settings. Despite JQ1’s antiproliferative effect on tumor cells (15–17,49), the long-lasting therapeutic outcomes following treatment with JQ1 or when coupled with anti-PD-1 therapy seen in the present study was not reproduced in immunodeficient mice treated with either drug suggesting that immune (T) cells predominantly contribute to the antitumor benefits derived from this dual agent treatment regimen. The possibility that JQ1 may have deleterious effects on non-Tregs, i.e. potential effector T cells, in humans is a point of consideration; however, in the present study, we found that mice tolerated the combination quite well. There was also no evidence for deleterious or toxic effects of JQ1/anti-PD-1 on T conventional cells in vitro, suggesting a relatively safe profile in the context of tumor-associated T cells.
With PD-1-blocking antibodies already approved by the FDA for management of advanced/metastatic lung cancer, and JQ1-like drugs in clinical testing, our proof-of-concept study demonstrates promising therapeutic efficacy by combining these drugs to improve antitumor responses that exceed the potential of either agent alone. As immunotherapeutic agents evolve beyond prototypical agents such as antibodies, rationally selected combinatorial approaches, such as the approach taken in this study warrant further investigation and future evaluation in the clinic.
Supplementary Material
Acknowledgements:
We would like to thank Zach Herbert (Dana-Farber Molecular biology core facility) for RNA sequencing and data assistance, and Ting Chen and Yanxi Zhang for technical assistance. This work was supported by the National Cancer Institute R01 CA216188, CA205150, CA166480, CA213333, CA098101 and CA154303 to K.K. Wong.
Footnotes
Conflict of Interest: The authors declare no potential conflict of interest.
References
- 1.Swanton C, Govindan R. Clinical Implications of Genomic Discoveries in Lung Cancer. N Engl J Med 2016;374(19):1864–73. [DOI] [PubMed] [Google Scholar]
- 2.Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511(7511):543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roberts PJ, Stinchcombe TE. KRAS mutation: should we test for it, and does it matter? J Clin Oncol 2013;31(8):1112–21. [DOI] [PubMed] [Google Scholar]
- 4.Chen Z, Cheng K, Walton Z, Wang Y, Ebi H, Shimamura T, et al. A murine lung cancer co-clinical trial identifies genetic modifiers of therapeutic response. Nature 2012;483(7391):613–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov 2015;5(8):860–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet 2016;17(8):487–500. [DOI] [PubMed] [Google Scholar]
- 7.Liu F, Wang L, Perna F, Nimer SD. Beyond transcription factors: how oncogenic signalling reshapes the epigenetic landscape. Nat Rev Cancer 2016;16(6):359–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahuja N, Sharma AR, Baylin SB. Epigenetic Therapeutics: A New Weapon in the War Against Cancer. Annu Rev Med 2016;67:73–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Copeland RA, Olhava EJ, Scott MP. Targeting epigenetic enzymes for drug discovery. Curr Opin Chem Biol 2010;14(4):505–10. [DOI] [PubMed] [Google Scholar]
- 10.Tough DF, Tak PP, Tarakhovsky A, Prinjha RK. Epigenetic drug discovery: breaking through the immune barrier. Nat Rev Drug Discov 2016;15(12):835–53. [DOI] [PubMed] [Google Scholar]
- 11.Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol 2016;13(6):394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palucka AK, Coussens LM. The Basis of Oncoimmunology. Cell 2016;164(6):1233–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011;478(7370):524–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature 2010;468(7327):1067–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011;146(6):904–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov 2013;3(3):308–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shimamura T, Chen Z, Soucheray M, Carretero J, Kikuchi E, Tchaicha JH, et al. Efficacy of BET bromodomain inhibition in Kras-mutant non-small cell lung cancer. Clin Cancer Res 2013;19(22):6183–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adeegbe D, Liu Y, Lizotte PH, Kamihara Y, Aref AR, Almonte C, et al. Synergistic Immunostimulatory Effects and Therapeutic Benefit of Combined Histone Deacetylase and Bromodomain Inhibition in Non-small Cell Lung Cancer. Cancer Discov 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 2015;161(2):205–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest 2015;125(9):3384–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N Engl J Med 2016;375(19):1823–33. [DOI] [PubMed] [Google Scholar]
- 22.Langer CJ, Gadgeel SM, Borghaei H, Papadimitrakopoulou VA, Patnaik A, Powell SF, et al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: a randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol 2016;17(11):1497–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med 2016;375(9):819–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov 2017;7(2):188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015;348(6230):124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov 2013;3(12):1355–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herter-Sprie GS, Koyama S, Korideck H, Hai J, Deng J, Li YY, et al. Synergy of radiotherapy and PD-1 blockade in Kras-mutant lung cancer. JCI Insight 2016;1(9):e87415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. J Pharmacol Pharmacother 2010;1(2):94–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015;527(7577):249–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med 2013;19(11):1423–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008;27(45):5904–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bin Dhuban K, Kornete M, E SM, Piccirillo CA. Functional dynamics of Foxp3(+) regulatory T cells in mice and humans. Immunol Rev 2014;259(1):140–58. [DOI] [PubMed] [Google Scholar]
- 34.Ohkura N, Kitagawa Y, Sakaguchi S. Development and maintenance of regulatory T cells. Immunity 2013;38(3):414–23. [DOI] [PubMed] [Google Scholar]
- 35.Friedline RH, Brown DS, Nguyen H, Kornfeld H, Lee J, Zhang Y, et al. CD4+ regulatory T cells require CTLA-4 for the maintenance of systemic tolerance. J Exp Med 2009;206(2):421–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang B, Chikuma S, Hori S, Fagarasan S, Honjo T. Nonoverlapping roles of PD-1 and FoxP3 in maintaining immune tolerance in a novel autoimmune pancreatitis mouse model. Proc Natl Acad Sci U S A 2016;113(30):8490–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Penaloza-MacMaster P, Kamphorst AO, Wieland A, Araki K, Iyer SS, West EE, et al. Interplay between regulatory T cells and PD-1 in modulating T cell exhaustion and viral control during chronic LCMV infection. J Exp Med 2014;211(9):1905–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol 2010;28:445–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cox MA, Harrington LE, Zajac AJ. Cytokines and the inception of CD8 T cell responses. Trends Immunol 2011;32(4):180–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tugues S, Burkhard SH, Ohs I, Vrohlings M, Nussbaum K, Vom Berg J, et al. New insights into IL-12-mediated tumor suppression. Cell Death Differ 2015;22(2):237–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Disis ML. Immune regulation of cancer. J Clin Oncol 2010;28(29):4531–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zanetti M Tapping CD4 T cells for cancer immunotherapy: the choice of personalized genomics. J Immunol 2015;194(5):2049–56. [DOI] [PubMed] [Google Scholar]
- 43.Zhu H, Bengsch F, Svoronos N, Rutkowski MR, Bitler BG, Allegrezza MJ, et al. BET Bromodomain Inhibition Promotes Antitumor Immunity by Suppressing PD-L1 Expression. Cell Rep 2016;16(11):2829–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Melaiu O, Mina M, Chierici M, Boldrini R, Jurman G, Romania P, et al. PD-L1 Is a Therapeutic Target of the Bromodomain Inhibitor JQ1 and, Combined with HLA Class I, a Promising Prognostic Biomarker in Neuroblastoma. Clin Cancer Res 2017;23(15):4462–72. [DOI] [PubMed] [Google Scholar]
- 45.Hogg SJ, Vervoort SJ, Deswal S, Ott CJ, Li J, Cluse LA, et al. BET-Bromodomain Inhibitors Engage the Host Immune System and Regulate Expression of the Immune Checkpoint Ligand PD-L1. Cell Rep 2017;18(9):2162–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boussiotis VA. Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. N Engl J Med 2016;375(18):1767–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev 2009;229(1):12–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAS, Andrews MC, et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell 2017;170(6):1120–33 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bhadury J, Nilsson LM, Muralidharan SV, Green LC, Li Z, Gesner EM, et al. BET and HDAC inhibitors induce similar genes and biological effects and synergize to kill in Myc-induced murine lymphoma. Proc Natl Acad Sci U S A 2014;111(26):E2721–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.