

Abstract
Adaptive resistance to MEK inhibitors (MEK-Is) typically occurs via induction of genes for different receptor tyrosine kinases (RTKs) and/or their ligands, even in tumors of the same histotype, making combination strategies challenging. SHP2 (PTPN11) is required for RAS/ERK pathway activation by most RTKs, and might provide a common resistance node. We found that combining the SHP2 inhibitor SHP099 with a MEK-I inhibited the proliferation of multiple cancer cell lines in vitro. PTPN11 knockdown/MEK-I treatment had similar effects, while expressing SHP099 binding-defective PTPN11 mutants conferred resistance, demonstrating that SHP099 is on-target. SHP099/trametinib was highly efficacious in xenograft and/or genetically engineered models of KRAS-mutant pancreas, lung, and ovarian cancer and in wild type RAS-expressing triple negative breast cancer. SHP099 inhibited activation of KRAS mutants with residual GTPase activity, impeded SOS/RAS/MEK/ERK1/2 reactivation in response to MEK-Is and blocked ERK1/2-dependent transcriptional programs. We conclude that SHP099/MEK-I combinations could have therapeutic utility in multiple malignancies.
Keywords: SHP2, SHP099, RAS, MEK inhibitor, Adaptive resistance
INTRODUCTION
The RAS/ERK mitogen-activated protein kinase (MAPK) pathway is one of the most commonly affected signaling pathways in human cancer (1–3). Mutations in genes encoding pathway components, including those for receptor tyrosine kinases (RTKs), SHP2, NF1, RAS or RAF, cause inappropriate pathway activation and promote oncogenesis. Attempts have been made to target the ERK pathway in different cancer types, and can lead to initial responses. Unfortunately, a form of intrinsic resistance termed “adaptive resistance” occurs frequently, resulting in lack of efficacy, recurrence or progression (4).
KRAS is the most frequently mutated RAS/ERK pathway gene (1–3). Approaches to target KRAS-mutant cancers with MEK-inhibitors (MEK-Is) have failed, often due to the induction of RTK genes and/or their ligands. For example, FGFR1 is activated in MEK-I-treated KRAS-mutant lung cancers, leading to increased upstream signaling and ERK reactivation (5). Another group found that MEK-I resistance can be mediated through ERBB3 in KRAS-mutant lung and colon cancers (6), whereas a third reported that MEK-I treatment leads to EGFR activation in KRAS mutant pancreatic cancer lines (7). Malignancies that lack mutations in pathway genes but nonetheless hyperactivate ERK also show adaptive resistance in response to MEK-Is. For example, MEK-I-treated triple negative breast cancer (TNBC) cells induce the expression of genes encoding AXL, DDR1, FGFR2, IGF1R, KIT, PDGFRB and VEGFRB (8,9).
Because resistance to MEK-Is can be mediated by multiple RTKs, combining MEK and RTK inhibition is probably not a viable therapeutic approach. However, a strategy that efficiently blocks signals from multiple activated RTKs might prevent adaptive resistance. The protein-tyrosine phosphatase SHP2 is a positive (i.e., signal-enhancing) signal transducer, acting between RTKs and RAS (10,11). A potent, highly specific inhibitor targeting SHP2, SHP099, has been developed, and blocks ERK activation and proliferation of cancer cells driven by over-expressed, hyperactivated RTKs (12,13). We hypothesized that SHP099 would inhibit signals from RTKs activated following MEK inhibition, and thereby block adaptive resistance. This idea comports with the previous finding that PTPN11 shRNA or CRISPR/Cas9-mediated deletion prevents adaptive resistance to vemurafenib in BRAF-mutant colon cancer (14).
Here, we test this hypothesis in multiple KRAS-mutant and wild type cancer cells from different histotypes. Our results suggest that SHP2 inhibition could provide a general strategy for preventing MEK-I resistance in a wide range of malignancies and might also have single agent efficacy against KRAS mutants that retain significant GTPase activity.
RESULTS
SHP099 abrogates adaptive resistance to MEK-inhibitors in vitro
Previous work showed that several cancer models develop adaptive resistance to MEK-Is through RTK upregulation. We analyzed RTKs/RTK ligand gene expression by qRT-PCR in pancreatic ductal adenocarcinoma (PDAC) cell lines treated with AZD6244, a well-established MEK-I (Fig. 1A). Consistent with earlier findings, several—but different—RTKs were induced by MEK-I treatment, including EGFR, FGFR3, IGFR1, MET and PDGFRB in MIAPaCa-2 cells, ERBB2/3, FGFR2/3 and IGFR1 in Capan-2 cells, and ERBB2/3 and FGFR3 in CFPAC-1 cells. The same lines variably induced EGF, FGF2, PDGFB, PDGFC, PDGFD and/or VEGFA/B. These observations make it difficult, if not impossible, to design an efficient combination therapy with MEK-Is by targeting RTKs directly.
Figure 1: Combined SHP2 and MEK inhibition abrogates adaptive resistance in PDAC cell lines.
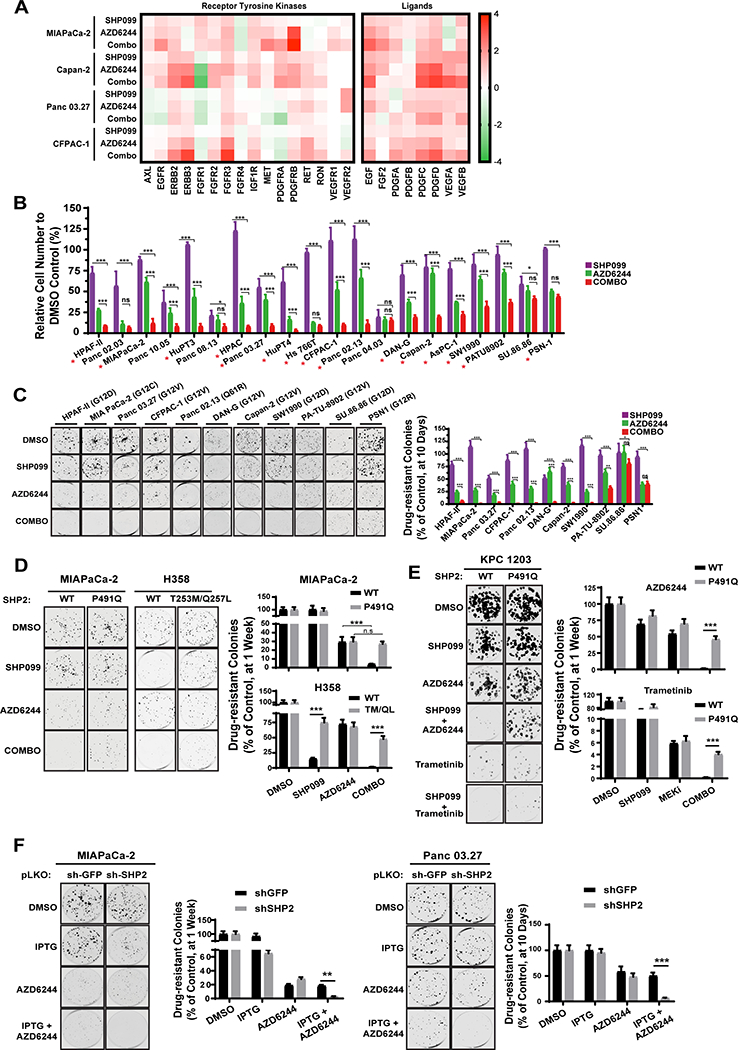
A, Time-dependent increase in in RTK (left) and RTK ligand (right) gene expression in PDAC cells after DMSO, SHP099, AZD6244, or SHP099/AZD6244 (Combo) treatment, determined by qRT-PCR. B-C, PDAC cell lines were treated with DMSO, SHP099, AZD6244, or both drugs (Combo). Cell viability, by PrestoBlue assay (B), and colony formation (C) were assessed at seven or ten days, respectively (*P < 0.05, **P < 0.01, ***P < 0.001, two-tailed t test). Representative results from a minimum of three biological replicates are shown per condition. Red asterisks indicate synergistic interaction between the two drugs by BLISS independent analysis. D, Colony formation assay (one week) in MiaPaCa-2 cells either expressing an SHP099-resistant PTPN11 mutant (P491Q) or wild-type PTPN11 (WT) and H358 NSCLC cells expressing an SHP099-resistant PTPN11 mutant (T253M/Q257L) or wild-type PTPN11 (WT) (***P < 0.001, two-sided t test). E, Colony formation assay (one week) in KPC 1203 cells either expressing an SHP099-resistant PTPN11 mutant (P491Q) or wild-type PTPN11 (WT). F, Colony formation assay (one week) in MiaPaCa-2 (left) and Panc 03.27 (right) cells expressing IPTG-inducible PTPN11 (sh-SHP2) or CTRL (sh-GFP) shRNAs. Representative results from a minimum of three biological replicates are shown per condition. For all experiments, drug doses were: SHP099 10 μM, AZD6244 1 μM, Combo= SHP099 10 μM + AZD6244 1μM. Trametinib (10 nM) was used where indicated.
To explore whether SHP2 inhibition could suppress MEK-I adaptive resistance, we performed in vitro viability (PrestoBlue) and colony formation assays on a panel of KRAS-mutant PDAC lines (Fig. 1B and C). Resistant cell populations and drug-resistant colonies were observed after one or two weeks, respectively, of AZD6244 treatment. AZD6244 itself had variable effects, leading to 30–90% reduction in proliferation/colony formation compared with control DMSO treatment; nevertheless, nearly all lines showed significant resistance. Consistent with a previous report (12), KRAS-mutant cell lines exhibited low sensitivity to SHP099 alone. By contrast, all but two of the lines had markedly reduced cell numbers and few or no detectable colonies after SHP099/MEK-I combination treatment; in most cases, the combination was synergistic (Fig. 1B, red asterisks, Table S1). Similar effects were seen in growth curve assays (Fig. S1A), with the more potent MEK-I, trametinib (Fig. S1B), on short-term cultures of cells from patient-derived xenografts (PDXs), and in KRAS-mutant non-small cell lung cancer (NSCLC) lines (Fig S1C-E). The drug combination decreased cell cycle progression and, in some lines, enhanced cell death (measured at 48h and 6 days of treatment, respectively), compared with either single agent alone (Fig. S1F).
Expression of a mutant (PTPN11P491Q) predicted to lack SHP099 binding in MIAPaCa-2 cells (which are quite sensitive to SHP099/MEK-I) and in KPC 1203, a cell line derived from induced LSL-KrasG12DTrp53R172H (KPC) mice (15), eliminated the effects of SHP099 in combination-treated cells (Fig. 1D and E). Another drug-resistant mutant, PTPN11T253M/Q257L (12), rescued the effects of the combination on H358 NSCLC cells (Fig. 1D). Moreover, combining MEK inhibition and PTPN11 shRNA expression had similar effects to SHP099/MEK-I treatment (Fig. 1F). These data indicate that SHP099 is “on-target” and that SHP2 inhibition diminishes adaptive resistance to MEK-Is in multiple KRAS-mutant cancer cell lines, arising from two distinct tissues.
SHP099 impedes MEK inhibitor-induced reactivation of the ERK MAPK pathway
We assessed the biochemical effects of each single agent and the drug combination on RAS/ERK pathway activity after short (1h)- and longer-term (48h) treatments. Short-term AZD6244 exposure had no detectable effect on RAS. At 48h, however, RAS activation (monitored by RAF-RBD assay) was enhanced, consistent with signaling from the induced RTKs/RTK ligands (Fig. 2A). Isoform-specific antibodies revealed increased activation of KRAS and NRAS in response to 48h MEK-I treatment of MIAPaCa-2 cells (Fig. 2B). SHP2 acts upstream of RAS, but whether it promotes RAS exchange (e.g., via SOS), inhibits RAS-GAP, or both, has been less clear (10,11). MIAPaCa-2 cells have no WT KRAS (mutant allele frequency=0.99) (16,17), so the increase in KRAS activation following MEK-I treatment must reflect enhanced cycling of KRASG12C. As KRAS(G12C) is highly resistant to RAS-GAP (18), the decreased KRAS-GTP in SHP099/AZD6244-treated MIAPaCa-2 cells indicates that residual KRAS(G12C) GTPase activity in contributes significantly to the steady state level of KRAS-GTP in these cells and that SHP2 must promote RAS exchange. Increased NRAS-GTP in response to SHP099 reflects activation of normal, endogenous NRAS.
Figure 2: SHP2 inhibition acts upstream of RAS to abrogate MEK-I-evoked ERK MAPK pathway reactivation.
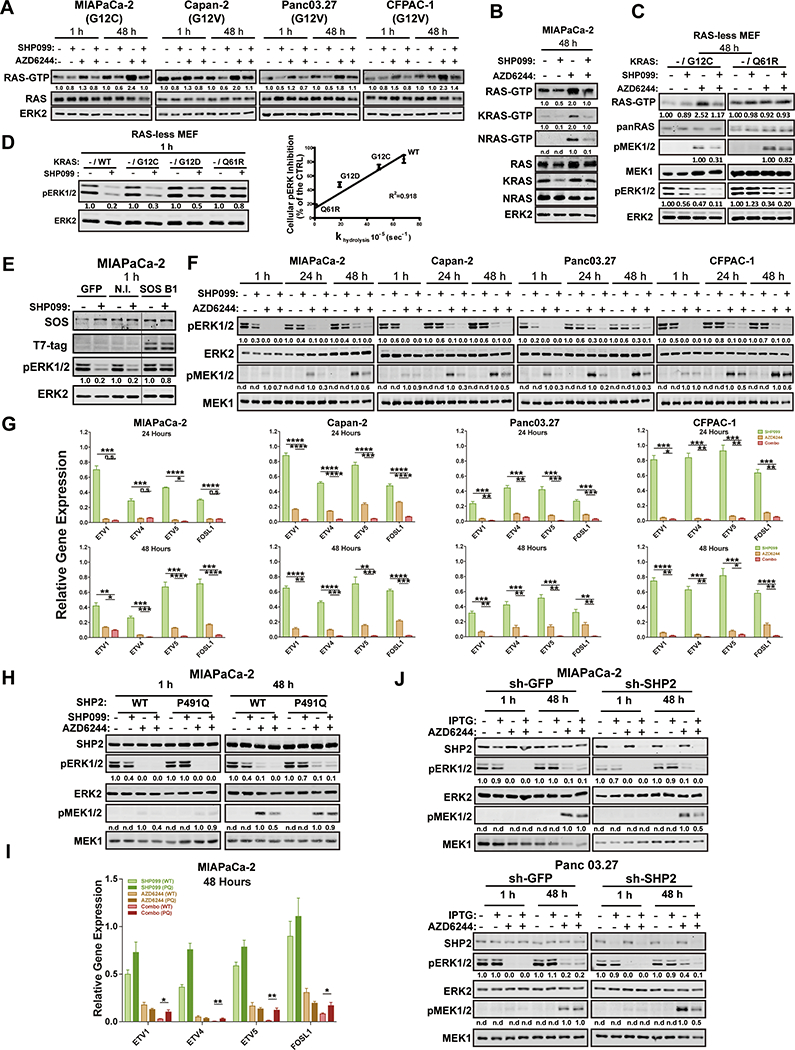
A-B, Immunoblots of whole cell lysates or GST-RBD-precipitated (RAS-GTP, KRAS-GTP and NRAS-GTP) lysates from PDAC cells treated with DMSO, SHP099 10 μM, AZD6244 1 μM, or both drugs for the times indicated. The images shown are representative of at least two independent biological replicates. C, GST-RBD pulldown assay in RAS-less MEFs reconstituted with RASG12C or RASQ61R. Total RAS, p-ERK/ERK and p-MEK/MEK were also detected in whole cell lysates prepared in modified RIPA buffer from the same cells. D, Immunoblots of whole cell lysates in RAS-less MEFs reconstituted with KRASWT, KRASG12C, KRASG12D or KRASQ61R, treated with or without 10 μM SHP099 (left). Linear regression of SHP099-induced p-ERK inhibition compared with intrinsic GTPase activity of the different KRAS mutants (from Ref. 17) in RAS-less MEFs (right). E, Effect of SHP099 on p-ERK levels in MiaPaCa-2 cells expressing a SOS1 mutant (SOS B1) that targets the SOS1 catalytic domain constitutively to the plasma membrane. Cells were incubated for 1 hour with SHP099, and lysates were immunoblotted for p-ERK and total ERK (as a loading control). F, Immunoblots of lysates from PDAC lines treated as indicated. Image shown is representative of three independent biological replicates. G, ERK-dependent gene expression (ETV1,4, 5 and FOSL1), assessed by qRT-PCR, in PDAC lines treated as indicated (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, two-tailed t test). H, Immunoblots of SHP2, p-ERK, ERK, p-MEK and MEK from MiaPaCa-2 cells ectopically-expressing wild-type SHP2 (WT) or an SHP099- resistant mutant (P491Q), treated as indicated. I, ERK-dependent gene expression in MIAPaCa-2 cells ectopically expressing wild-type SHP2 (WT) or an SHP099-resistant mutant (P491Q), treated as in F (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.001, two-tailed t test). J, Immunoblot of lysates from MIAPaCa-2 (upper panel) and Panc 03.27 (lower panel) cells expressing IPTG-inducible PTPN11 (sh-SHP2) or CTRL (sh-GFP) shRNA, subjected to the indicated drugs. Numbers under blots indicate relative intensities, compared with untreated controls, quantified by LICOR.
The other PDAC lines tested express KRAS mutants with less intrinsic GTPase activity than KRAS(G12C) (18) and retain WT-KRAS. Hence, it was not clear whether SHP099 can also block activation of these RAS mutants in response to MEK-I treatment or affects WT-KRAS or the other RAS isoforms (Fig. 2A). To more directly interrogate the effects of SHP2 inhibition on other KRAS mutants, we used RAS-less mouse embryonic fibroblasts (RAS-less MEFs) (19). As in MIAPaCa-2 cells, KRAS(G12C)-reconstituted RAS-less cells showed increased KRAS-GTP after 48h of MEK-I treatment, and this increase was prevented by SHP099. By contrast, SHP099 had no effect on KRAS(Q61R)-GTP levels (Fig. 2C). The ability of single agent SHP099 to inhibit ERK activation in RAS-less MEFs reconstituted with different KRAS mutants was linearly related to their reported GTPase activity (17) (Fig. 2D). These results confirm that SHP2 is required for RAS exchange, most likely acting upstream of SOS1/2. Indeed, expressing the SOS1 catalytic domain tagged with a C-terminal CAAX BOX of RAS (20) rescued the effects of SHP099 on ERK activation in MIAPaCa-2 cells (Fig. 2E).
Single agent AZD6244 blocked MEK and ERK1/2 phosphorylation after 1h, but these effects were successively abolished after 24h and 48h of treatment, respectively, and MEK and ERK activity rebounded (Fig. 2F and Fig. S2A). Trametinib also caused MEK/ERK rebound, although to a lesser extent (Fig. S2B). Consistent with its effects on RAS, SHP099 co-administration blocked the adaptive increase in MEK and ERK phosphorylation in response to either MEK-I (Fig. 2F and S2A and B). ERK-dependent gene expression can provide a more sensitive assessment of pathway output than p-ERK levels (21), so we measured FOS-like 1 (FOSL1) and ETS variants 1, 4, 5 (ETV1, 4 and 5) RNA by qRT-PCR. Compared with the effects of either single agent, SHP099/AZD6244 or SHP099/trametinib combination caused greater suppression of ERK-dependent transcription (Fig. 2G and Fig. S2C). Other RTK-evoked pathways (e.g., PI3K/AKT, STAT, JNK/p38) showed no consistent effects of either single agent or the drug combination (Fig. S2D and data not shown). These findings confirm that ERK reactivation is a key component of the adaptive program activated in KRAS-mutant cancer cells treated with MEK-Is, and show that SHP099 blocks this adaptive response. Importantly, the biochemical effects of SHP099 (like its effects on colony formation; Fig. 1D) were reversed in MIAPaCa-2 (Fig. 2H), H358 (Fig. S2E) and KPC 1203 cells expressing SHP099-resistant SHP2. PTPN11 depletion had similar biochemical effects as SHP2 inhibition (Fig. 2J), confirming on-target effects of SHP099.
We also explored the mechanism of resistance of two KRAS-mutant PDAC lines to SHP099/MEK-I. PSN1 cells failed to suppress MEK-ERK reactivation or ERK-dependent gene expression (Fig. S3A and S3B). In SU.86.86 cells, MEK/ERK and ERK-dependent genes were inhibited to an extent similar to sensitive cells, consistent with a downstream escape mechanism (Fig. S3C and D). Further investigation will be required to uncover the precise molecular explanation for resistance in these cell lines.
Combined SHP2/MEK inhibition suppresses KRAS-mutant tumor growth in vivo
We next established Capan-2, MIAPaCa-2, and H358 xenografts, and treated them with vehicle control (methyl-cellulose+Tween80), trametinib alone, SHP099 alone, or SHP099/trametinib. We used trametinib because of its favorable mouse pharmacokinetic properties (t½ = 33h (22)), which enables single daily dosing, as does SHP099 (13). In initial experiments, mice were treated daily with trametinib (1mg/kg), a dose used commonly in mouse tumor studies (6,23,24), SHP099 (75mg/kg), or both. Each single agent was well tolerated, but mice receiving the combination lost weight (>10%), exhibited lassitude, and began dying at day 7 of treatment. Some showed gross GI bleeding (Fig. S4A), and histology revealed multi-focal GI tract ulceration, acute esophagitis and gastritis, and villus blunting, which could explain malabsorption, diarrhea, and weight loss (Fig. S4B).
Although trametinib is usually administered to mice at this dose or even at doses as high as 3mg/kg (5,25), the mouse allometric equivalent of the maximum tolerated dose (MTD) in humans is ~0.25mg/kg (26). We treated a small group of mice with this dose of trametinib and SHP099 (75mg/kg) daily (QD). Although treated mice lived longer than with the higher trametinib dose, this combination also led to weight loss and death (data not shown). Exploratory dose finding resulted in a tolerable schedule, in which trametinib is delivered at 0.25mg/kg, with SHP099 (75mg/kg) every other day (QOD). These mice showed no observable histopathology (Fig. S4C). A few developed mild, self-limited, non-bloody diarrhea, but all showed stable weight, normal behavior and appeared healthy for up to 37 days of continuous treatment (Fig. S4D and S4E and data not shown).
Capan-2, MIAPaCa-2 or H358 xenografts were allowed to grow to 500mm3 (Fig. 3A), and then mice were treated with vehicle, trametinib (0.25mg/kg QD), SHP099 (75 mg/kg QD), or trametinib (0.25mg/kg QD)/SHP099 (75mg/kg QOD). Remarkably, the combination caused substantial regressions in all mice (Fig. 3A and Fig. S4F). Tumor shrinkage averaged >65% in the three models, well above Response Evaluation Criteria for Solid Tumors (RECIST) criteria (27). The decrease in tumor size probably underestimates the anti-neoplastic effect, as the ratio of tumor cells/area also decreased, with the residual area occupied by fibroblasts (Fig. 3B and C). Strikingly, Capan-2 xenografts treated for 37 days with SHP099/trametinib failed to regrow after 40 days of drug withdrawal (Fig. 3D). The residual tumor appeared to have undergone cellular senescence, as shown by βgal staining (Fig. 3E) and elevated expression of the senescence-associated cytokine interleukin-6 (Fig. 3F).
Figure 3: Combined MEK/SHP2 inhibition is efficacious in PDAC models in vivo.
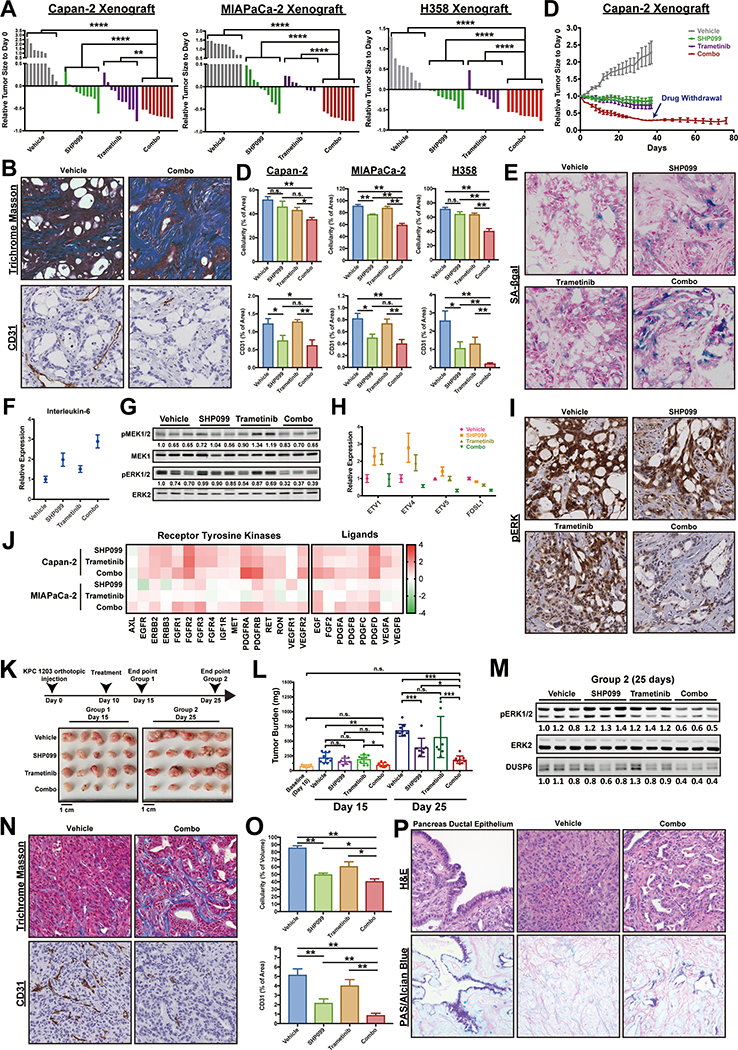
A, Response of Capan-2, MIAPaCa-2 and H358 subcutaneous xenografts to treatment with SHP099 (75 mg/kg body weight, daily), trametinib (0.25 mg/kg QD) or both drugs (trametinib 0.25 mg/kg QD; SHP099 75 mg/kg QOD). Waterfall plot shows response of each tumor after 37 days (Capan-2), 19 days (MIAPaCa-2) and 21 days (H358) of treatment; n = 8–10 mice per group. (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, two-tailed Mann Whitney test). B, Masson Trichrome (collagen) and CD31 (blood vessels) staining in treated Capan-2 tumors showing reduced tumor cellularity and vascularity, respectively. C, Quantification of tumor cellularity (Masson Trichrome stain) and vascularity (CD31) of treated Capan-2, MIAPaCa-2 and H358 xenografts (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, Masson Trichrome: two-tailed t test, CD31: one-tailed t test). D, Tumor growth curve of treated Capan-2 xenografts (drug withdrawal after 37 days of combo treatment). E, SA-β gal staining on treated Capan-2 tumors following 37 days of treatment. F, qRT-PCR of senescence-associated cytokine interleukin-6 in treated Capan-2 tumors. G, Immunoblot showing p-ERK and p-MEK levels in treated Capan-2 tumors. H, ERK-dependent gene expression (ETV1,4, 5 and FOSL1), assessed by qRT-PCR, in Capan-2 tumors. I, Immunohistochemical stain for p-ERK in treated Capan-2 tumors. J, qRT-PCR of RTK and RTK ligand genes in treated Capan-2 and MIAPaCa-2 tumors. K-L, Syngeneic mice injected orthotopically with KPC 1203 cells were treated with vehicle, SHP099 (75 mg/kg QD), trametinib (0.25 mg/kg QD) or both drugs (trametinib 0.25 mg/kg QD; SHP099 75 mg/kg QOD), as depicted in the scheme. Tumor size was measured at day 15 and 25 (*P < 0.05, **P < 0.01, ***P < 0.001, one-way ANOVA with Tukey’s multiple comparison test). M, Immunoblot showing p-ERK and DUSP6 levels in KPC 1203 tumors from K. N-O Masson Trichrome and CD31 staining and quantification in treated KPC tumors. P, H&E and PAS/Alcian Blue staining of treated KPC tumors. Numbers under blots indicate relative intensities, compared with untreated controls, quantified by LICOR.
Single agent effects were more variable both within each treatment group and in mice bearing MIAPaCa-2 versus Capan-2-/H358-derived tumors. Trametinib had minimal effects on MIAPaCa-2 tumors, although it caused significant shrinkage of about half of the Capan-2 and H358 xenografts (Fig. 3A and Fig. S4F). Only a few trametinib-treated mice met RECIST criteria (>30%), however, and the SHP099/MEK-I combination was more effective (Fig. 3A and Fig. S4F). Surprisingly, in contrast to its lack of effect on proliferation in cell culture or on colony formation, SHP099 alone caused tumor shrinkage in ~80% of Capan-2, ~60% of MIAPaCa-2, and ~70% of H358 xenografts. It is not clear whether this discrepancy reflects effects of SHP099 on normal RAS/ERK signaling in cells within the tumor microenvironment (e.g., fibroblasts, blood vessels), effects on the malignant cells themselves with secondary consequences for stroma, or both. Consistent with at least some non-autonomous effects, SHP099 decreased tumor vascularity as monitored by CD31 immunostaining (Fig. 3B and C), without major effects on VEGF or FGF mRNA levels (see Fig. 3J). Nevertheless, single agent SHP099 was, like trametinib alone, inferior to the drug combination (Fig. 3A and Fig. S4F). Comporting with these biological effects, immunoblotting (Fig. 3G and Fig. S4G), qRT-PCT (Fig. 3H) and immunohistochemical (IHC) analysis (Fig. 3I) revealed greater p-ERK inhibition in combination-, than in single agent-, treated tumors. Notably, tumors induced RTK and RTK ligand expression following MEK-I treatment, confirming that adaptive resistance via RTK over-activation occurs in vivo (Fig. 3J).
We also tested syngeneic mice injected orthotopically with KPC 1203 cells. Tumors were allowed to grow for 10 days, 5 mice were sacrificed to obtain baseline tumor sizes, and the rest were treated with single agent or SHP099/trametinib for 5 or 15 days, respectively. Again, mice in the combination arms showed markedly inhibited tumor growth, compared with trametinib-treated mice (Fig. 3K and L). Although the combination was superior (Day 15, Fig. 3L), SHP099 also had significant effects, even though KRAS(G12D) has significantly less residual GTPase activity than KRAS(G12C) (18). Immunoblot analysis revealed greater inhibition of p-ERK and DUSP6 (an ERK target gene product) in combination-treated tumors than in those treated with trametinib or SHP099 alone (Fig. 3M).
As in the xenografts, tumor vascularity and overall tumor cellularity was reduced in combination-treated GEMMs (Fig. 3N and O). Furthermore, residual tumor cells in combination-treated mice showed ductal differentiation, compared with vehicle- or single agent-treated mice (Fig. 3P). PAS/Alcian Blue staining revealed secretory activity (Fig. 3P), whereas qRT-PCR analysis showed induction of ductal and, more prominently, endocrine markers (Fig. S4H). All SHP099/trametinib-treated xenografts and syngeneic tumors showed decreased proliferation and increased apoptotic cell death (Fig. S5 and Fig. S6).
SHP099/MEK-I combination is also effective in TNBC and serous ovarian cancer models
Genetic (28,29) and functional genomic (30,31) analyses reveal striking similarities between TNBC and high-grade serous ovarian cancer (HGSC). These malignancies typically express WT RAS, and in some TNBC models, MEK inhibition results in RTK upregulation and adaptive resistance (8). To explore the potential generality of combination MEK/SHP2 inhibition as a therapeutic strategy (and the utility of this combination in adaptive resistance to MEK-I in WT RAS-expressing cells), we tested SHP099/MEK-I combinations in TNBC and HGSC models.
Similar to its effects on KRAS-mutant cells, MEK-I treatment increased RTK and RTK ligand gene expression in TNBC and HGSC lines (Fig. S7A). SHP099 (10μM) alone had little effect on cell number or colony formation (Fig. 4A-B, Fig. S7B). The MEK-Is AZD6244 or UO126 had variable single agent effects, often (but not always) causing reduced cell proliferation compared with controls. Nevertheless, resistant cell populations were seen in almost all cell lines. The SHP099/MEK-I combination showed increased efficacy, with additive to synergistic effects (Fig. 4A, Fig. S7B and Table S1). As in KRAS-mutant cell models (above), combination treatment (for 48h) slowed cell cycle progression and enhanced cell death (Fig. S7C and D). After 48h of single-agent treatment, SHP099 had little or no effect on RAS activation in any of the models (Fig. 4D). After MEK-I treatment, however, RAS was hyper-activated to varying degrees in MDA-MB-468, HCC1954, CAL-120, and OVCAR-8 cells. In SHP099/MEK-I-treated cells, RAS-GTP decreased to normal levels in randomly growing cells. These findings indicate that RAS activation is largely SHP2-independent under normal serum growth conditions, but is essential for the increase in RAS-GTP evoked by the adaptive (RTK-driven) program evoked by MEK-I treatment. As expected, 48h AZD6244 treatment caused increased MEK1/2 and ERK phosphorylation; consistent with its effects on RAS, SHP099 suppressed this increase (Fig. 4E) as well as ERK-dependent gene expression (Fig. 4F).
Figure 4: Combined MEK/SHP2 inhibition is also effective in TNBC and HGSC models.
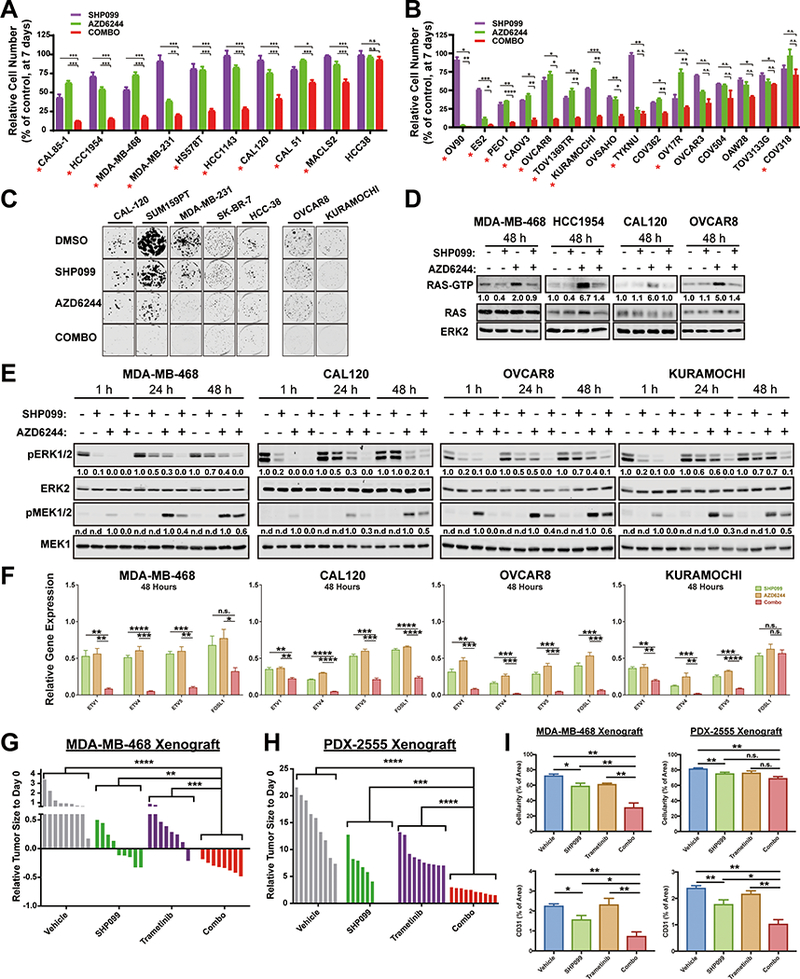
A-C, TNBC (A) and HGSC (B) cell lines were treated with DMSO, SHP099, AZD6244, or both (COMBO). PrestoBlue intensity (A) or cell number (B) were assessed at one week. Colony formation (C) was quantified at two weeks. Representative results from a minimum of three biological replicates are shown per condition: SHP099 10 μM, AZD6244 (1 μM), Combo=SHP099 10 μM +AZD6244 1 μM (*P < 0.05, **P < 0.01, ***P < 0.001, two-tailed t test). Red asterisks indicate synergistic interaction between the two drugs by BLISS independent analysis. D, GST-RBD pull down assay from TNBC and HGSC cell line lysates treated with DMSO, SHP099 10 μM, AZD6244 1 μM, or both for 48h. Image is representative of at least two independent experiments. E, Immunoblots of lysates from TNBC and HGSC lines, treated as indicated. Image is representative of three independent experiments. F, ERK-dependent gene expression (ETV1,4, 5 and FOSL1), assessed by qRT-PCR, in TNBC and HGSC lines treated for 48h with the indicated drugs (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, two-tailed t test). G-H, MDA-MB-468 (G) and PDX-2555 (B) mammary fat pad xenografts following treatment with SHP099 (75 mg/kg QD), trametinib (0.25 mg/kg QD) or both (trametinib 0.25 mg/kg QD; SHP099 75 mg/kg QOD). Waterfall plots of tumor response after 29 days (MDA-MB-468) and 9 days (PDX-2555) of treatment are shown; n = 8–10 mice per group. (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, two-tailed Mann Whitney test). I, Quantification of Masson Trichrome and CD31 staining of treated MDA-MB-468 and PDX-2555 tumor sections (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, Masson Trichrome: two-tailed t test, CD31: one-tailed t test). Numbers under blots indicate relative intensities, compared with untreated controls, quantified by LICOR.
Finally, we treated mice bearing mammary fat pad xenografts derived from MDA-MB-468 or an extremely aggressive HGSC patient-derived xenograft (PDX) with SHP099, trametinib or both drugs. Single agents did not produce consistent regressions of MDA-MB-468 xenografts, and the ovarian PDX was highly resistant to both drugs. However, SHP099/trametinib caused substantial regression of MDA-MB-468 xenografts and markedly inhibited the growth of the HGSC PDX (Fig. 4G, H and Fig. S4F), while significantly reducing tumor angiogenesis and cellularity in both models (Fig. 4I). Decreased tumor cell proliferation and increased apoptosis were observed in combination-treated tumors (Fig. S6).
DISCUSSION
Tumors evade targeted cancer therapies via an extensive repertoire of resistance mechanisms. One common theme involves activation of RTKs by inducing their expression and/or the expression of their ligands, which reactivates the inhibited pathway (5–9,24,32). Indeed, multiple, distinct sets of RTKs/RTK ligands were activated in response to MEK-I treatment in the models that we tested. The heterogeneity of this adaptive response renders unfeasible combination therapies with MEK-Is and RTK inhibitors. However, targeting a common downstream component of RTK signaling in combination with MEK-Is might yield substantial efficacy. SHP2 has long been known to signal downstream of normal RTKs (10,11), and cancer cells dependent on RTK activity are susceptible to SHP099 monotherapy (12,13). We find that combined MEK/SHP2 inhibition blocks cell proliferation and promotes shrinkage of tumors with increased RAS/ERK pathway activation, including those that typically have WT RAS (TNBC, HGSC), as well as those driven by mutant KRAS (PDAC, NSCLC). Unexpectedly, our studies also shed new light on the long elusive effect of SHP2 on RAS.
Although SHP099 reportedly has off-target effects in some cells (33), it is clearly “on-target” in our experiments. We observed its expected biochemical effects on RAS/ERK pathway activation in the multiple lines tested. Moreover, two different drug-resistant mutants (PTPN11P491Q, PTPN11T253M/Q257L) rescue the effects of SHP099 in PDAC and NSCLC cells, respectively, whereas PTPN11 shRNA expression has similar biological and biochemical effects to SHP099. The MEK-inhibitors employed here also are highly validated, giving us confidence that the effects we observed reflect dual SHP2/MEK inhibition.
We expected SHP099 to block the adaptive increase in normal RAS activation that accompanies increased RTK signaling in MEK-I-treated cells. Surprisingly, SHP099 also decreased mutant KRAS activation in MIAPaCA-2 cells, which only express KRAS(G12C), yet show clearly decreased KRAS activation following SHP099 treatment. KRAS(G12C) has significant intrinsic GTPase activity (18), as exemplified by covalent RAS inhibitors that target KRAS(G12C)-GDP (34). Hence, even though KRAS(G12C) is largely refractory to RAS-GAPs, significant conversion to RAS-GDP must occur in cells via this intrinsic GTPase activity, and ongoing GDP/GTP exchange is required to maintain steady state levels of KRAS(G12C)-GTP. Other RAS mutants (except Q61 alleles) also retain some intrinsic GTPase activity, although less than does KRAS(G12C). SHP099 also led to decreased p-ERK levels in RAS-less MEFs expressing KRAS(G12D) and KRAS(G12V) in a manner linearly related to residual KRAS-GTPase activity. Recently, Nichols et al. (35) also reported variable effects of a new allosteric SHP2 inhibitor on mutant KRAS, although its chemical matter was not reported and its specificity was not established using drug-resistant mutants.
Genetic and biochemical analyses have firmly established that SHP2 acts upstream of RAS (10,11), but whether it promotes exchange or inhibits GAP activity, or both, has been controversial. Early work showed that SHP2, via its C-terminal tyrosine phosphorylation sites, can recruit GRB2/SOS (36,37). A subsequent study reported diminished RAS exchange in lysates from cells expressing a GAB1 mutant that cannot bind SHP2 (38). However, multiple other reports claim that SHP2 antagonizes RAS-GAP by dephosphorylating its binding sites on RTKs or on SHP2-binding scaffolding adapters (39–41). Studies of Drosophila embryogenesis also argue for actions of the SHP2 ortholog, CSW, on the GAP binding sites in TORSO (42). Our findings, and those of Nichols et al. (35), show clearly that SHP2 acts upstream of SOS, and SHP2 inhibition can be bypassed by SOS, although we cannot exclude additional effects on RAS-GAP.
Single agent SHP099 had little effect on 2D proliferation or colony formation by cancer cells, but it significantly affected some xenografts and the KPC GEMM. There are several potential, non-mutually exclusive explanations for this apparent discrepancy. First, tumors occupy a hypoxic, nutrient-challenged, and potentially growth factor-deficient microenvironment; under such conditions, SHP2 might be essential for proliferation. Second, SHP2 might affect stromal support functions (e.g., growth factor production by cancer-associated fibroblasts, tumor angiogenesis). Tumor vascularity was decreased in all SHP099-treated xenografts, although whether this reflects direct inhibition of tumor angiogenesis or indirect effects on the tumor, with secondary effects on vessels, remains unclear. Third, SHP2 might affect the anti-tumor immune response, at least in the GEMM. Interestingly, SHP099 and the drug combination had greater inhibitory effects in syngeneic, than in nude, mice (data not shown).
Our results comport with, and extend, previous studies of the effects of SHP2 modulation on other ERK pathway inhibitors. Prahallad et al. (14) found that SHP2 depletion (via PTPN11 shRNA or deletion) blocked adaptive resistance to the BRAFV600E inhibitor vemurafenib. They claimed that an SHP2 catalytic domain inhibitor (GS493) had similar effects, but that agent has off-target effects on tyrosine kinases (43). While this manuscript was in revision, 3 independent groups reported that inhibition of SHP2 can sensitize KRAS mutant or amplified cancers to MEK inhibitors (44–46). Our results are in general agreement with their findings, although these reports either used the non-specific SHP2 inhibitor GS493(45), the earlier generation MEK-I AZD6244, which has a very short half-life in vivo and is not approved for cancer therapy (44), or mouse trametinib doses 4 times higher than the human MTD (45,46). We show that SHP099 is “on-target” using drug-resistant SHP2 mutants, and provide new evidence that combination therapy affects the tumor microenvironment (angiogenesis and stroma), can, at least in some models, promote differentiation of highly anaplastic tumor cells, and when delivered for sufficient time, can prevent tumor regrowth after drug withdrawal. Taken together, all of these studies suggest that SHP2 inhibition might be a broadly applicable strategy to prevent or overcome adaptive resistance to kinase inhibition in a wide array of malignancies.
METHODS
Cell Lines and Reagents
Cells were maintained in 5% CO2 at 37C° under the conditions described by the vendor or the source laboratory; details are available from CF or KHT upon request. Cells were tested at least every 3 months for mycoplasma contamination by PCR (47), and genotyped by STR analysis at IDEXX Bioresearch. See Supplementary Methods for details. SHP099 (HY-100388A) was purchased from MedChemExpress. Selumetinib-AZD6244 (S1008), UO126 (S1102) and trametinib (GSK1120212-S2673) were purchased from Selleckchem.
Plasmids, Retro and Lentiviral production
Lenti- and retro-viral constructs were generated by standard methods (see Supplementary Methods). Viruses were produced by co-transfecting HEK293T cells with lentiviral or retroviral constructs and packaging vectors. Stable pools of infected cells were selected by using the appropriate antibiotic or by fluorescence activated cell sorting (FACS) for EGFP.
Cell Assays
Cell number was monitored by PrestoBlue assay (Thermo Fisher). Potential drug synergy was determined by Bliss analysis as: Yab,P = Ya + Yb – YaYb, where Ya stands for percentage inhibition of drug a and Yb stands for percentage inhibition of drug b (48). For colony assays, cells (100–500) were seeded in six-well plates, and after 24 hr, treated with DMSO or the indicated drugs. Colonies were stained with crystal violet, visualized by using the Odyssey Imaging System (LICOR) and quantified with the ImageJ Colony Area PlugIn (49). For details, see Supplemental Methods. Cell cycle distribution was monitored by flow cytometry using 7AAD and analyzed by ModFit LT software (Versity Software House). Apoptosis was quantified by using the PE Annexin V Apoptosis Detection Kit (BD).
Biochemical Assays
RAS activity was assessed by GST-RBD pulldown, followed by immunoblotting with pan-RAS or RAS isoform-specific antibodies. Whole cell lysates were resolved by SDS-PAGE, followed by transfer to Nylon membranes. Immunoblots were performed with the indicated primary antibodies, followed by IRDye-conjugated secondary antibodies and visualization by LICOR. For details, see Supplementary Methods.
Immunohistochemistry
p-ERK (Cell Signaling, 4370), CD31 (Cell Signaling, D8V9E), Cleaved Caspase 3 (Cell Signaling, D3E9), and Ki67 (Spring Biosciences, SP6) staining was performed on paraffin sections. OCT frozen sections were used for SA-βgal staining. H&E, Masson Trichrome and PAS/Alcian Blue staining were performed by the Experimental Pathology Shared Resource at Perlmutter Cancer Center (PCC).
Animal Experiments
All animal experiments were approved by the NYU Langone Institutional Animal Care and Use Committee (IACUC). Pancreas and lung cell line xenografts were established by sub-cutaneous injection of 5 × 106 cells in 50% Matrigel (Corning) into nude mice (nu/nu, #088 Charles River) MDA-MB-468 xenografts were established by injecting 5 × 106 cells in 50% Matrigel into the right lower mammary pad. Ovarian PDXs were established by injecting 5 × 105 cells in 50% Matrigel into the right lower mammary pad of NSG mice (Jackson Lab). KPC 1203 cells (1 × 105 in Matrigel) were implanted into the pancreata of syngeneic male mice. Single agents and drug combinations were administered and tumor size and body weight were monitored. For details, see Supplemental Methods.
qRT-PCR
Total RNA was isolated by the Qiagen RNeasy kit. cDNA was generated by using the SuperScript IV First Strand Synthesis System (Invitrogen). qRT-PCR was performed with Fast SYBR™ Green Master Mix (Applied Biosystems), following the manufacturer’s protocol, in 384-well format in C1000 Touch Thermal Cycler (Biorad). Differential gene expression analysis was performed with CFX Manager (Biorad) and normalized to GAPDH expression. Primers used are listed in Supplementary Table 2.
Statistical Analysis
Data are expressed as mean ± standard deviation. Statistical significance was determined using Student t test, Mann–Whitney U test, or one-way ANOVA. Statistical analyses were performed in Prism 7 (GraphPad Software). Significance was set at P = 0.05.
Supplementary Material
SIGNIFICANCE:
MEK inhibitors show limited efficacy as single agents, in part because of the rapid development of adaptive resistance. We find that SHP2/MEK inhibitor combinations prevent adaptive resistance in multiple cancer models expressing mutant and wild-type KRAS.
Acknowledgements:
We thank Drs. Alec Kimmelman, Dafna Bar-Sagi, Douglas A. Levine, Gottfried E. Konecny, Robert Rottapel, and Kwok-Kin Wong for cell lines, Drs. Dafna Bar-Sagi, Jason Moffat, and David Root for plasmids, and the PCC Experimental Pathology and Precision Immunology shared resources for technical support. We also thank Dr. Toshiyuki Araki for advice and discussion on this project. This work was supported by R01CA49152 to B.G.N., and R01CA131045 to D.M.S..
Financial Support: This work was supported by NIH Research Project Grant Program R01 CA49152 and CA131045.
Abbreviations:
- HGSC
high-grade serous ovarian cancer
- MAPK
mitogen-activated protein kinase
- MEK-Is
MEK-inhibitors
- NSCLC
non-small cell lung cancer, PDAC, pancreatic ductal adenocarcinoma
- TNBC
triple negative breast cancer
Footnotes
Conflicts of Interest: B.G.N. is a co-founder, chair of the Scientific Advisory Board, and holds equity in Navire Pharmaceuticals, which is developing SHP2 inhibitors for cancer therapy.
REFERENCES
- 1.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature 2013;502(7471):333–9 doi 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet 2013;45(10):1127–33 doi 10.1038/ng.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 2017;23(6):703–13 doi 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caunt CJ, Sale MJ, Smith PD, Cook SJ. MEK1 and MEK2 inhibitors and cancer therapy: the long and winding road. Nat Rev Cancer 2015;15(10):577–92 doi 10.1038/nrc4000. [DOI] [PubMed] [Google Scholar]
- 5.Manchado E, Weissmueller S, Morris JPt, Chen CC, Wullenkord R, Lujambio A, et al. A combinatorial strategy for treating KRAS-mutant lung cancer. Nature 2016;534(7609):647–51 doi 10.1038/nature18600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun C, Hobor S, Bertotti A, Zecchin D, Huang S, Galimi F, et al. Intrinsic resistance to MEK inhibition in KRAS mutant lung and colon cancer through transcriptional induction of ERBB3. Cell Rep 2014;7(1):86–93 doi 10.1016/j.celrep.2014.02.045. [DOI] [PubMed] [Google Scholar]
- 7.Anderson GR, Winter PS, Lin KH, Nussbaum DP, Cakir M, Stein EM, et al. A Landscape of Therapeutic Cooperativity in KRAS Mutant Cancers Reveals Principles for Controlling Tumor Evolution. Cell Rep 2017;20(4):999–1015 doi 10.1016/j.celrep.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duncan JS, Whittle MC, Nakamura K, Abell AN, Midland AA, Zawistowski JS, et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell 2012;149(2):307–21 doi 10.1016/j.cell.2012.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zawistowski JS, Bevill SM, Goulet DR, Stuhlmiller TJ, Beltran AS, Olivares-Quintero JF, et al. Enhancer Remodeling during Adaptive Bypass to MEK Inhibition Is Attenuated by Pharmacologic Targeting of the P-TEFb Complex. Cancer Discov 2017;7(3):302–21 doi 10.1158/2159-8290.CD-16-0653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ran H, Tsutsumi R, Araki T, Neel BG. Sticking It to Cancer with Molecular Glue for SHP2. Cancer Cell 2016;30(2):194–6 doi 10.1016/j.ccell.2016.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan G, Neel BG. Role of PTPN11 (SHP2) in Cancer Protein Tyrosine Phosphatases in Cancer. New York: Springer; 2016. p 115–43. [Google Scholar]
- 12.Chen YN, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG, et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 2016;535(7610):148–52 doi 10.1038/nature18621. [DOI] [PubMed] [Google Scholar]
- 13.Garcia Fortanet J, Chen CH, Chen YN, Chen Z, Deng Z, Firestone B, et al. Allosteric Inhibition of SHP2: Identification of a Potent, Selective, and Orally Efficacious Phosphatase Inhibitor. J Med Chem 2016;59(17):7773–82 doi 10.1021/acs.jmedchem.6b00680. [DOI] [PubMed] [Google Scholar]
- 14.Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012;483(7387):100–3 doi 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 15.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005;7(5):469–83 doi 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 16.Moore PS, Sipos B, Orlandini S, Sorio C, Real FX, Lemoine NR, et al. Genetic profile of 22 pancreatic carcinoma cell lines. Analysis of K-ras, p53, p16 and DPC4/Smad4. Virchows Arch 2001;439(6):798–802. [DOI] [PubMed] [Google Scholar]
- 17.Sun C, Yamato T, Furukawa T, Ohnishi Y, Kijima H, Horii A. Characterization of the mutations of the K-ras, p53, p16, and SMAD4 genes in 15 human pancreatic cancer cell lines. Oncol Rep 2001;8(1):89–92. [DOI] [PubMed] [Google Scholar]
- 18.Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, Westover KD. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol Cancer Res 2015;13(9):1325–35 doi 10.1158/1541-7786.MCR-15-0203. [DOI] [PubMed] [Google Scholar]
- 19.Drosten M, Dhawahir A, Sum EY, Urosevic J, Lechuga CG, Esteban LM, et al. Genetic analysis of Ras signalling pathways in cell proliferation, migration and survival. EMBO J 2010;29(6):1091–104 doi 10.1038/emboj.2010.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patgiri A, Yadav KK, Arora PS, Bar-Sagi D. An orthosteric inhibitor of the Ras-Sos interaction. Nat Chem Biol 2011;7(9):585–7 doi 10.1038/nchembio.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pratilas CA, Taylor BS, Ye Q, Viale A, Sander C, Solit DB, et al. (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc Natl Acad Sci U S A 2009;106(11):4519–24 doi 10.1073/pnas.0900780106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gilmartin AG, Bleam MR, Groy A, Moss KG, Minthorn EA, Kulkarni SG, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res 2011;17(5):989–1000 doi 10.1158/1078-0432.CCR-10-2200. [DOI] [PubMed] [Google Scholar]
- 23.Yao Z, Yaeger R, Rodrik-Outmezguine VS, Tao A, Torres NM, Chang MT, et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 2017;548(7666):234–8 doi 10.1038/nature23291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin L, Sabnis AJ, Chan E, Olivas V, Cade L, Pazarentzos E, et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat Genet 2015;47(3):250–6 doi 10.1038/ng.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xue Y, Martelotto L, Baslan T, Vides A, Solomon M, Mai TT, et al. An approach to suppress the evolution of resistance in BRAF(V600E)-mutant cancer. Nat Med 2017;23(8):929–37 doi 10.1038/nm.4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm 2016;7(2):27–31 doi 10.4103/0976-0105.177703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45(2):228–47 doi 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 28.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature 2012;490(7418):61–70 doi 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature 2011;474(7353):609–15 doi 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marcotte R, Sayad A, Brown KR, Sanchez-Garcia F, Reimand J, Haider M, et al. Functional Genomic Landscape of Human Breast Cancer Drivers, Vulnerabilities, and Resistance. Cell 2016;164(1–2):293–309 doi 10.1016/j.cell.2015.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marcotte R, Brown KR, Suarez F, Sayad A, Karamboulas K, Krzyzanowski PM, et al. Essential gene profiles in breast, pancreatic, and ovarian cancer cells. Cancer Discov 2012;2(2):172–89 doi 10.1158/2159-8290.CD-11-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010;468(7326):973–7 doi 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fulcher LJ, Hutchinson LD, Macartney TJ, Turnbull C, Sapkota GP. Targeting endogenous proteins for degradation through the affinity-directed protein missile system. Open Biol 2017;7(5) doi 10.1098/rsob.170066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dang CV, Reddy EP, Shokat KM, Soucek L. Drugging the ‘undruggable’ cancer targets. Nat Rev Cancer 2017;17(8):502–8 doi 10.1038/nrc.2017.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nichols RJ, Haderk F, Stahlhut C, Schulze CJ, Hemmati G, Wildes D, et al. Efficacy of SHP2 phosphatase inhibition in cancers with nucleotide-cycling oncogenic RAS, RAS-GTP dependent oncogenic BRAF and NF1 loss. bioRxiv 2017. [Google Scholar]
- 36.Bennett AM, Tang TL, Sugimoto S, Walsh CT, Neel BG. Protein-tyrosine-phosphatase SHPTP2 couples platelet-derived growth factor receptor beta to Ras. Proc Natl Acad Sci U S A 1994;91(15):7335–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li W, Nishimura R, Kashishian A, Batzer AG, Kim WJ, Cooper JA, et al. A new function for a phosphotyrosine phosphatase: linking GRB2-Sos to a receptor tyrosine kinase. Mol Cell Biol 1994;14(1):509–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamasaki S, Nishida K, Yoshida Y, Itoh M, Hibi M, Hirano T. Gab1 is required for EGF receptor signaling and the transformation by activated ErbB2. Oncogene 2003;22(10):1546–56 doi 10.1038/sj.onc.1206284. [DOI] [PubMed] [Google Scholar]
- 39.Agazie YM, Hayman MJ. Molecular mechanism for a role of SHP2 in epidermal growth factor receptor signaling. Mol Cell Biol 2003;23(21):7875–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Montagner A, Yart A, Dance M, Perret B, Salles JP, Raynal P. A novel role for Gab1 and SHP2 in epidermal growth factor-induced Ras activation. J Biol Chem 2005;280(7):5350–60 doi 10.1074/jbc.M410012200. [DOI] [PubMed] [Google Scholar]
- 41.Fambrough D, McClure K, Kazlauskas A, Lander ES. Diverse signaling pathways activated by growth factor receptors induce broadly overlapping, rather than independent, sets of genes. Cell 1999;97(6):727–41. [DOI] [PubMed] [Google Scholar]
- 42.Cleghon V, Feldmann P, Ghiglione C, Copeland TD, Perrimon N, Hughes DA, et al. Opposing actions of CSW and RasGAP modulate the strength of Torso RTK signaling in the Drosophila terminal pathway. Mol Cell 1998;2(6):719–27. [DOI] [PubMed] [Google Scholar]
- 43.Tsutsumi R, Ran H, Neel BG. Off-target inhibition by active site-targeting SHP2 inhibitors. FEBS Open Bio 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mainardi S, Mulero-Sánchez A, Prahallad A, Germano G, Bosma A, Krimpenfort P, et al. SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nature Medicine 2018. [DOI] [PubMed] [Google Scholar]
- 45.Ruess DA, Heynen GJ, Ciecielski KJ, Ai J, Berninger A, Kabacaoglu D, et al. Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nature Medicine 2018. [DOI] [PubMed] [Google Scholar]
- 46.Wong GS, Zhou J, Liu JB, Wu Z, Xu X, Li T, et al. Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nature Medicine 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Young L, Sung J, Stacey G, Masters JR. Detection of Mycoplasma in cell cultures. Nat Protoc 2010;5(5):929–34 doi 10.1038/nprot.2010.43. [DOI] [PubMed] [Google Scholar]
- 48.Zhao W, Sachsenmeier K, Zhang L, Sult E, Hollingsworth RE, Yang H. A New Bliss Independence Model to Analyze Drug Combination Data. J Biomol Screen 2014;19(5):817–21 doi 10.1177/1087057114521867. [DOI] [PubMed] [Google Scholar]
- 49.Guzman C, Bagga M, Kaur A, Westermarck J, Abankwa D. ColonyArea: an ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS One 2014;9(3):e92444 doi 10.1371/journal.pone.0092444. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.