

Abstract
Objective:
Lymphatic vessel dysfunction and increased lymph leakage have been directly associated with several metabolic diseases. However, the underlying cellular mechanisms causing lymphatic dysfunction have not been determined. Aberrant insulin signaling affects the metabolic function of cells, and consequently impairs tissue function. We hypothesized that insulin resistance in lymphatic endothelial cells (LECs) decreases eNOS activity, disrupts barrier integrity increases permeability, and activates mitochondrial dysfunction and pro-inflammatory signaling pathways.
Methods:
LECs were treated with insulin and/or glucose to determine the mechanisms leading to insulin resistance.
Results:
Acute insulin treatment increased eNOS phosphorylation and NO production in LECs via activation of the PI3K/Akt signaling pathway. Prolonged hyperglycemia and hyperinsulinemia induced insulin resistance in LECs. Insulin resistant LECs produced less NO due to a decrease in eNOS phosphorylation and showed a significant decrease in impedance across an LEC monolayer that was associated with disruption in the adherence junctional proteins. Additionally, insulin resistance in LECs impaired mitochondrial function by decreasing basal-, maximal-, and ATP-linked-oxygen consumption rates and activated NF-κB nuclear translocation coupled with increased pro-inflammatory signaling.
Conclusion:
Our data provide the first evidence that insulin resistance disrupts endothelial barrier integrity, decreases eNOS phosphorylation and mitochondrial function, and activates inflammation in LECs.
Keywords: Insulin resistance, inflammation, lymphatic endothelial cells, mitochondria, permeability
Introduction
Insulin resistance, defined as the inability of insulin to optimally stimulate the transport of glucose into the body’s cell, is one of the major risk factors for the development of metabolic diseases [1]. In addition to its target tissues, such as liver, skeletal muscle, and adipose tissue, the vascular endothelium has emerged as an early responder to the development and progression of metabolic insulin resistance [2,3]. It has been shown that vascular endothelial cells respond more rapidly to insulin compared to other insulin target tissues in the obesity model induced by a high-fat-diet regimen [2,4]. Hence, the vasculature is believed to be the most susceptible to nutrient overload and changes in insulin, which significantly contributes to the progression of pathology [4]. Although a wealth of evidence exists for the role of blood vascular endothelium and its response to high insulin conditions [5–7], there is almost nothing known about how lymphatic endothelial cells (LECs) respond under similar conditions. In addition, LECs are central players in the process of inflammation and immune responses and participate in multiple signaling processes [8,9], so LEC dysfunction could have important consequences for health.
It has been shown that lymphatic dysfunction and increased vessel permeability are associated with metabolic disorders and obesity [10,11]. Mice heterozygous for Prox1, a key lymphatic endothelial transcription factor, showed leakage of lymph due to impaired endothelial tight junctions and exhibited late-onset obesity [10]. Apolipoprotein E deficient mice (ApoE−/−) on a high-fat diet regimen exhibited leaky and dilated lymphatic vessels leading to tissue swelling [12]. Further, decreased lymphatic vessel integrity and permeability in a type 2 diabetic mouse model suggested the importance of lymphatic endothelial barrier function in the progression of metabolic diseases [13]. We previously demonstrated that lymphatic vessels exhibit impaired intrinsic contractile activity and reduced force generation during conditions of metabolic syndrome (MetSyn) [14–16]. Further, the lymphatic vessels from MetSyn rats showed a significant decrease in the relative levels of endothelial nitric oxide synthase (eNOS) that could be associated with endothelial dysfunction [14]. We also showed that insulin resistance directly altered cellular bioenergetics and inflammatory signaling, coupled with impairment of a key contractile regulatory molecule in lymphatic muscle cells (LMCs) [17]. We hypothesized that insulin resistance in LECs would decrease eNOS activity and would activate mitochondrial dysfunction and pro-inflammatory signaling pathways, consequently disrupting barrier integrity and increasing permeability. To test this hypothesis, we induced insulin resistance in LECs by prolonging hyperglycemic and hyperinsulinemic culture conditions and determined the effects of insulin or insulin resistance on: a) eNOS activity and NO bioavailability, b) endothelial barrier function, C) mitochondrial function and d) inflammatory signaling pathways in LECs.
MATERIALS AND METHODS
Materials
Phospho-eNOS (Ser1177), eNOS, Phospho-Akt (Thr308), Phospho-Akt (Ser473), Akt, and β-actin antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). A β-catenin antibody was purchased from Santa Cruz Biotechnology (Dallas, TX, USA). LY294002 and L-N-Nitroarginine methylester (L-NAME) were purchased from EMD Millipore (Billerica, MA, USA). The 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl) amino]-2-deoxyglucose (2-NBDG, 11046), DAN Reagent (780070), NaOH (780068), Nitrite Standard (780016), cell-based assay buffer, and Akt inhibitor XI (902779–59-3) were purchased from Cayman Chemical (Ann Arbor, MI, USA). Endothelial Cell Growth Medium (EGM2 MV) and phenol red-free Endothelial Basal Medium (EBM) were purchased from Lonza (Basel, Switzerland). Human dermal lymphatic endothelial cells (HDLECs) were purchased from Promo Cell (Heidelberg, Germany). High pore density PET track-etched membrane cell culture inserts for permeable supports were purchased from Corning (#353495, Corning, NY, USA). ProLong™ Gold Anti-fade mounting medium with DAPI was purchased from ThermoFisher Scientific (Waltham, MA, USA). S961, an insulin receptor antagonist, was purchased from Phoenix Pharmaceuticals, Inc. (Burlingame, CA, USA). All other chemicals and reagents were from Sigma-Aldrich (St. Louis, MO, USA).
Cell culture and treatments
LECs were grown directly on cell culture dishes in EGM2 MV medium containing 5% FBS, (Lonza) and maintained at 37°C in a 5% CO2 incubator, as described previously [9]. LECs were plated in 24-well culture plates and grown to confluence. The cells were then treated with different doses of insulin (1, 10, 100, 150, 200 and 300nM) for various periods of time (5 min, 20 min, 1, 12, 48 and 72 h) to determine the effect of insulin on Akt and eNOS phosphorylation in LECs. Cells were also treated with an insulin receptor antagonist, S961 (100nM), a PI3K inhibitor, LY294002 (20μg/ml), and an Akt inhibitor, Akt inhibitor XI (20μM), to identify insulin signaling pathways in LECs. LECs were pretreated for 2 h with inhibitors and then treated in the presence or absence of insulin for 20 mins. LECs were also treated with high glucose (HG, 25mM) or mannose (25mM) for various periods of time to examine the effect of high glucose or osmolality on LECs. Osmolality was measured for EGM and complete EGM media with or without glucose, mannose, or insulin using a micro-osmometer, Model 3300 (Advanced Instruments Inc. Norwood, MA, USA). TNF-α (20ng/ml) treatment was utilized in some experiments as a positive control for inflammatory conditions in LECs.
Glucose transport assay
Insulin sensitivity in LECs was assessed by measuring glucose uptake using 2-NBDG, as described previously [17]. To determine the optimal incubation time, LECs were grown in the presence of 2-NBDG and the 2-NBDG uptake was measured at various time points from 10min to 6 h. Results showed that 2NBDG fluorescence intensity reached a maximal level in LECs at the 1-h incubation time point. Hence, we used 1-h 2-NBDG incubation for subsequent glucose transport assays. Following completion of the HG, insulin or insulin + HG treatments, cells were incubated with 2-NBDG (150μg/ml) in phenol red-free EBM medium for 1 h and the plate was centrifuged for 5 min at 400g at room temperature. The supernatants were removed and the cells were washed twice with the cell-based assay buffer (Cayman Chemical). Buffer was added to each well and the plate was centrifuged for 5 min at 400g at room temperature, and the supernatants were removed. After the final wash, the cell-based assay buffer was added to the cells and the amount of 2-NBDG taken up by LECs was measured at 485/535 nm wavelengths using a plate reader. All experiments were repeated a minimum of three times. Data were normalized to corresponding controls and the data presented as mean fold change ± SEM.
Trans-endothelial electrical resistance (TEER) measurement
TEER was measured as described [18]. LECs were seeded on 6.4mm polyethylene terephthalate Transwell™ inserts with a 0.4μm pore size (Corning Inc. Corning, NY, USA). Cells were incubated for at least 48 h in order to ensure a completely confluent monolayer. TEER measurements were made with an Epithelial Volt/Ohm Meter (EVOM2) and STX2 chopstick electrode (World Precision Instruments, Sarasota, FL, USA), just before and after the addition of treatments (20 min, 1h, 12h, 24h, and 48h). Resistance (ΔTEER, Ω × cm2) of the LEC monolayers under the different conditions was calculated by subtracting the mean resistance of control inserts. To compare independent experiments, normalized ΔTEER values were calculated in relation to the ΔTEER in basal conditions before the treatment. Data were presented as mean fold change ± SEM.
LEC permeability assay
The LEC monolayer barrier function assay was performed as previously described [19]. LECs were grown to confluent monolayers on the 6.4mm Transwell™ inserts and then treated with EBM containing either insulin (100nM), high glucose (25mM), mannose (25mM), TNFα (20ng/ml) or left untreated. EBM2 medium was then added to the upper and lower chambers. FITC-labeled bovine serum albumin (10 mg/ml) was added to the upper chamber. Cells were incubated at 37 °C for 30 min and then aliquots of medium were removed from the lower chamber, diluted with Milli-Q water and fluorescence measured at 494nm/518nm wavelengths using a plate reader. All experiments were repeated three times. Data were corrected for background and reported as the percent of control. Data were presented as mean fold changes ± SEM.
Nitric oxide production
The production of NO was assessed by measuring the concentration of nitrite in LEC-conditioned culture medium [20] using a sensitive nitrite fluorometric assay kit (Cayman Chemical). Briefly, LECs were plated and grown to completely confluent monolayers in 48-well cell culture plates and then treated with HG, insulin, mannose, L-NAME (a NOS inhibitor, 10μM, 24h), or TNF-α (24h), as described earlier [19]. Nitrite measurement was carried out according to the manufacturer’s instructions. The fluorescence was monitored at dual wavelengths of 375 and 415 nm. Experiments were repeated nine times and data were presented as mean ± SEM.
Mitochondrial bioenergetics
Mitochondrial function in LECs was assessed using the XFe96 Extracellular Flux Analyzer (Seahorse Bioscience, North Billerica, MA, USA), by measuring the rate changes in the extracellular flux of dissolved oxygen and protons, as previously described [17]. The LECs were seeded in 96-well XFe microplates at 50 × 103 cells/well in EGM2 MV at 370C in a 5% CO2 incubator and grown to confluence. The LECs were treated as described above (n=4/group). The medium was replaced with the assay medium and corresponding treatments (i.e., glucose, insulin) for 1 h before beginning the assay at 37°C. The oxygen consumption rates (OCRs) were measured with the following sequential reagents: oligomycin (5μM, mitochondrial complex V inhibitor), FCCP (1μM, an uncoupling agent), and rotenone (5μM, mitochondrial complex I inhibitor). On completion of the assay, the number of cells was counted for normalization of the values. All the OCRs are expressed in picomoles per minute of oxygen consumed. Reserve capacity was calculated as the difference between basal OCR and that obtained in the presence of FCCP. Maximum respiratory capacity was calculated as the difference in OCR between rotenone-treated and FCCP-treated cells.
Glycolytic capacity analysis
Cellular glycolytic rates in LECs were estimated by measuring extracellular acidification rate (ECAR), as described in our previous study [17]. The LECs were treated as described above (n=4/group). The extracellular acidification (milli-pH per minute) was measured at basal and maximal levels by injection of oligomycin (5μM). Values were normalized by cell number. Glycolytic capacity was determined as the difference between basal ECAR and that obtained in the presence of oligomycin.
Cytokine analysis in LECs using real-time PCR
LECs were plated in 24-well culture plates and serum starved for one hr. Cells were then treated with glucose (25mM) or insulin (100nM) for 48h, as described previously. Total RNA was extracted using the RNeasy kit (Qiagen, Valencia, CA, USA), according to the manufacturer’s instructions. The quality and quantity of RNAs were determined using a NanoDrop (ThermoFisher Scientific, Wilmington, DE, USA). RNA was converted to cDNA using a Superscript III cDNA synthesis kit (ThermoFisher Scientific). The cDNA was mixed with SYBR Green PCR master mix (ThermoFisher Scientific). Primers specific to IL-1β, IL-4, IL-6, iNOS, TNFα, monocyte chemoattractant protein (MCP)-1, and macrophage inflammatory protein (MIP)-2, CXCR2, CFOS, c-Jun were designed and used for real-time PCR [9,17]. RPL19 amplification was used as a control for all samples and the differences in the relative expression of cytokines between groups was calculated using the 2-ΔΔCt method.
Immunofluorescence
Immunofluorescence experiments were performed as described earlier to determine β-catenin and NF-κB localization [9,17]. In brief, the treated LECs were fixed with 2% paraformaldehyde. Cells were then permeabilized and incubated with the corresponding primary antibodies, β-catenin or NF-κB. Corresponding normal IgG was used as a control. After washing off the primary antibody, cells were incubated with a fluorescence-conjugated secondary antibody, followed by washing, and cells were then mounted in ProLong™ Gold Antifade solution containing DAPI (ThermoFisher Scientific). Images were taken with an Olympus BX41 fluorescence microscope using an UPlan Apo x20 (NA=0.7) or Uplan FLN × 40 objective (NA=1.3). NIH Image J edge finder and plug-in co-localization were used to quantify adherent junction fluorescence signal intensity and nuclear localization, respectively [21].
Protein isolation and western blot analysis
Protein expression was quantified by western blot analysis [9,22]. Protein lysates from LECs were separated on a 4–20% gradient SDS-polyacrylamide gel, and western blot analysis was performed with antibodies for the following: p-eNOS (Ser1177), total eNOS, p-Akt (Ser473), p-Akt (Thr308), total Akt, β-catenin, and β-actin. Appropriate secondary antibodies were used and protein detection was carried out using a chemiluminescence system (ThermoFisher) followed by using the Fuji LAS-4000 Mini image processor (GE Healthcare Bio-Science, Pittsburgh, PA, USA). Densitometry analyses were carried out with Image J (National Institutes of Health, Bethesda, MD, USA) [21]. All experiments were repeated in triplicate and mean fold change ± SEM was calculated.
Statistical analysis
Time-dependent and dose-dependent effects of insulin, glucose uptake assays, permeability assays, NO measurements and TEER measurements were analyzed using one-way ANOVA. Mean differences were compared to control LECs or basal levels using the Dunnett’s post hoc test (SPSS19.0; IBM Corp., Armonk, New York, USA). Effects of S961, Akt inhibitor XI, L-NAME, and prolonged HG and insulin treatments were analyzed using two-way ANOVA, followed by Bonferroni’s post hoc test. If there were any significant interactions or main effects between treatments, mean differences between test groups were assessed using one-way ANOVA with Fisher’s Least Significant Difference (LSD) post hoc test. Mitochondrial OCR data were analyzed using a two-tailed t-test. NF-κB co-localization data were analyzed using one-way ANOVA with Fisher’s LSD post hoc test. All values are expressed as mean ± SEM; p=0.05 was considered as significant. All graphs were generated with Prism 5 (GraphPad Software, La Jolla, CA, USA).
Results
Acute insulin treatment activates Akt/eNOS phosphorylation in LECs.
Insulin regulates endothelium-derived NO production via Akt signaling in blood endothelium [23,24]. To determine whether insulin also stimulates eNOS phosphorylation in LECs via the Akt pathway, we treated LECs with various concentrations of insulin and quantified Akt and eNOS phosphorylation. Results showed a dose-dependent increase in Akt serine and threonine phosphorylation in LECs treated with insulin (Figures 1A & B). While 10nM of insulin increased Akt (Ser473) phosphorylation (+2.34 fold vs. Control, p<0.002), 100nM of insulin had significant effects on Akt (Thr308) phosphorylation (+2.37 fold vs. Control, p<0.034). Additionally, 100nM or higher doses of insulin significantly induced eNOS (Ser1177) phosphorylation (+4.58 fold vs. Control, p<0.0001, Figure 1C). We next examined the effect of insulin (100nM) on Akt and eNOS phosphorylation at various time points (5 min, 20 min, 1 h, 12 h, 24 h, 48 h, 72 h) (Figures 1D, E, F). As shown in Figure 1D, Akt (Ser473) phosphorylation was significantly elevated following 20 min of insulin treatment (+1.09 fold vs. Control, p<0.03) and decreased to the basal level within 24 hours. Phosphorylation of Akt (Thr308) was upregulated after 5 min of insulin treatment (+0.57 fold vs. Control, p<0.014) and downregulated to the basal level within 12 hours (Figure 1E). Insulin treatment also significantly increased eNOS phosphorylation after 20 min (+0.83-fold vs Control, p<0.004). Compared to the basal level, insulin treatment longer than 24h did not induce any significant differences in eNOS phosphorylation (Figure 1F).
Figure 1.
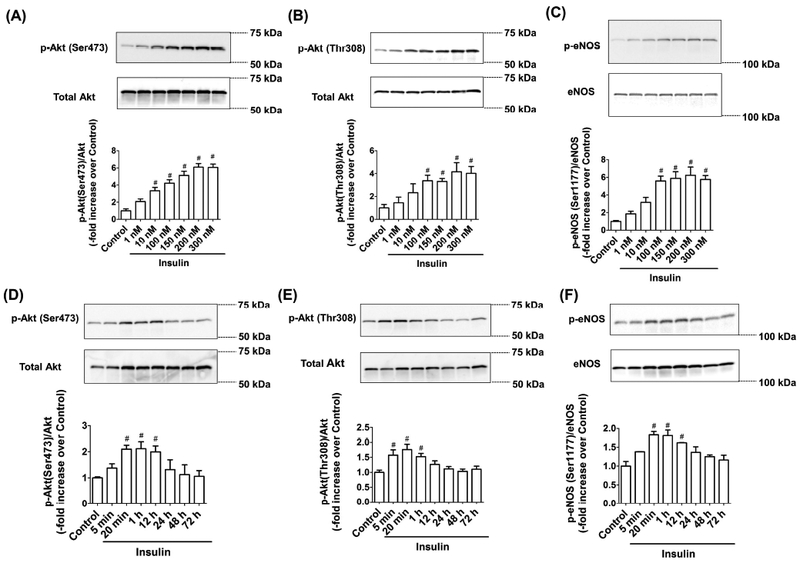
Acute insulin treatment activates Akt/eNOS phosphorylation in LEC. LECs were treated with different doses of insulin (1–300nM) for 20 minutes. Representative western blots are shown: (A) Akt (Ser473) phosphorylation, (B) Akt (Thr308) phosphorylation, and (C) eNOS (Ser1177) phosphorylation. The relative expression of p-Akt (Ser473)/Akt, p-Akt (Thr308)/Akt, and p-eNOS (Ser1177)/eNOS were quantified and plotted (n=4/group). LECs were treated with insulin (100nM) for varying time periods (5 min-72 h). Representative western blots are shown: (D) Akt (Ser473) phosphorylation, (E) Akt (Thr308) phosphorylation, and (F) eNOS (Ser1177) phosphorylation. The relative expression of p-Akt (Ser473)/Akt, p-Akt (Thr308)/Akt, and p-eNOS (Ser1177)/eNOS were quantified and plotted (n=4/group). Data represent mean fold changes ± SEM. *p<0.05 vs. control.
Insulin-mediated eNOS phosphorylation is regulated by PI3K/Akt pathways.
To ascertain whether insulin-induced eNOS phosphorylation is mediated via the insulin receptor (IR), LECs were treated with S961, an insulin receptor antagonist, and evaluated for Akt/eNOS phosphorylation. In the presence of this IR inhibitor, insulin-induced Akt and eNOS phosphorylation levels remained at basal levels (Figure 2A). To elucidate the specific roles of PI3K and Akt in modulating eNOS phosphorylation as a downstream signaling pathway of insulin, we also used a PI3K inhibitor (LY294002) or Akt inhibitor (Akt Inhibitor XI) in conjunction with insulin treatment of LECs. Inhibition of PI3K blunted insulin-mediated eNOS phosphorylation (−1.22 fold vs. Insulin, p<0.001). Treatment with PI3K inhibitor alone did not influence eNOS phosphorylation compared to untreated controls (Figure 2B). Similarly, Akt inhibitor caused a significant reduction of insulin-induced eNOS phosphorylation (−0.54 fold vs. Insulin, p<0.005), while Akt inhibitor alone did not alter the eNOS phosphorylation level compared to the control group (Figure 2C). These results suggest that insulin activates eNOS phosphorylation via the PI3K/Akt signaling pathway in the LECs.
Figure 2.
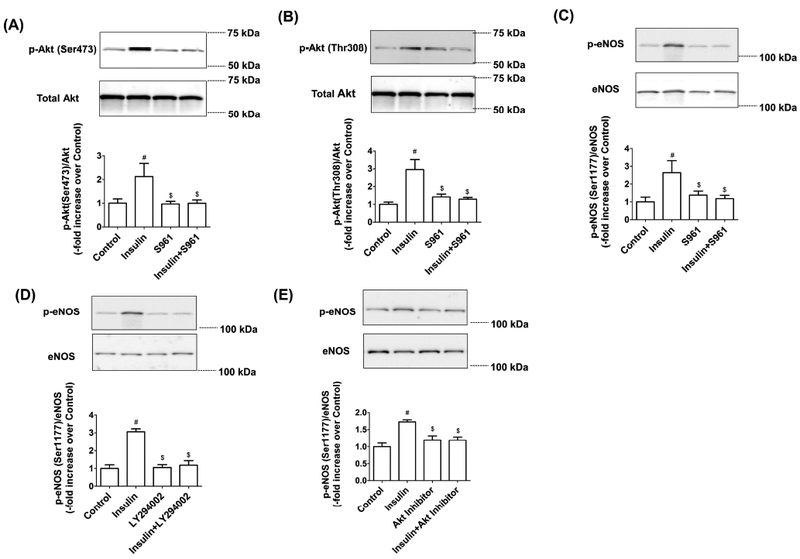
Insulin-mediated eNOS phosphorylation is regulated by PI3K/Akt pathways. LECs were treated with insulin (100nM) and S961(100nM), as described in the Materials and Methods, and representative western blots are shown: (A) Akt (Ser473) phosphorylation, (B) Akt (Thr308) phosphorylation, and (C) eNOS (Ser1177) phosphorylation. The relative expression of p-Akt (Ser473)/Akt, p-Akt (Thr308)/Akt, and p-eNOS/eNOS was quantified and plotted (n=3/group). Data represent mean ± SEM. #p<0.05 vs. Control; $p<0.05 vs. insulin. LECs were treated with insulin (100nM) and (D) LY294002 (20μg/ml) or (E) Akt inhibitor XI (20μM), as described in the Materials and Methods, and representative western blots are shown. The p-eNOS/eNOS expression was quantified and plotted (n=3/group). Data represent mean fold changes ± SEM. #p<0.05 vs. control; $p<0.05 vs. insulin.
Prolonged hyperinsulinemia and hyperglycemia cause insulin resistance in LECs.
One of the key pathological determinants of insulin resistance is impairment of insulin sensitivity that reduces glucose uptake into peripheral tissues or cells, including endothelial cells [25,26]. We measured glucose uptake level using fluorescently labeled 2-NBDG to determine whether prolonged hyperinsulinemia and/or hyperglycemia induces insulin resistance in LECs. Acute insulin treatment significantly increased LEC glucose uptake at 5 minutes (+2.47 fold vs. Control, p<0.001), 20 minutes (+2.83 fold vs. Control, p<0.001) and 12 hrs (+0.56 fold vs. Control, p<0.008). After 12 hrs there was no significant change in glucose uptake in LECs treated with insulin (Figure 3A). In contrast to insulin, hyperglycemia did not alter glucose uptake at any of the time points (Figure 3B). Previous studies showed both hyperglycemia and hyperinsulinemia were necessary to develop insulin resistance in various cell types, including LMCs [17,27,28]. In the presence of insulin and HG, LECs showed an increase in glucose uptake within 5 minutes (+2.46 fold vs. Control, p<0.001). This effect was found to last until 12 h (+0.65 fold vs. Control, p<0.05) and was found to decrease to the basal level at 24 h (Figure 3C). LECs that were exposed to hyperglycemic and hyperinsulinemic conditions for 48h showed a significant reduction (−0.52 fold vs. Control, p<0.022) in glucose uptake (Figure 3C). Therefore, we chose 100nM insulin with 25mM glucose treatments for 48h to investigate the effects of insulin resistance in LECs. Hyperglycemia- and hyperinsulinemia-induced insulin resistance in LECs was reversed by removing glucose and insulin from the culture medium. Insulin resistant LECs returned to basal LEC culture medium for 48h showed increased glucose uptake (+0.15 fold vs. HG+Ins 48h, p<0.05), but it was still lower than control LECs (−0.17 fold vs. Control, p<0.05, Figure 3D). Glucose uptake was similar to control LECs in insulin resistant LECs after returning to LEC culture medium for 72 h (Figure 3D).
Figure 3.
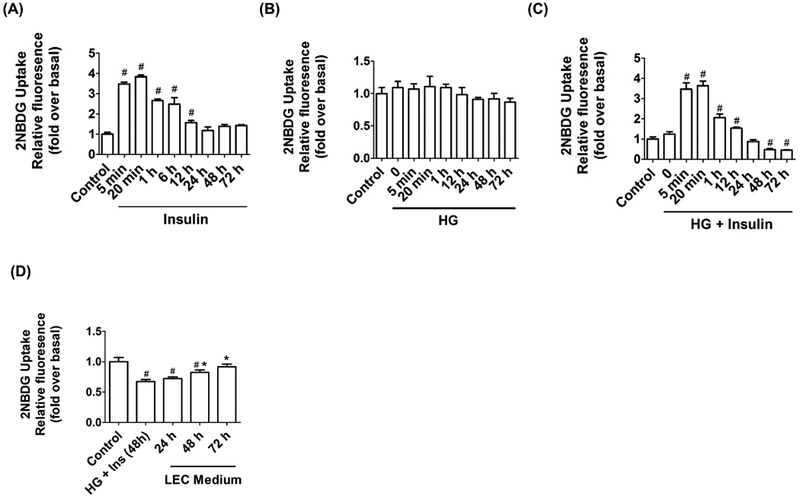
Prolonged hyperinsulinemia and hyperglycemia conditions induced insulin resistance in LECs. Glucose uptake was measured to test insulin sensitivity using 2NBDG in LECs at different time points (5 min-72 h): (A) insulin (100nM) treatment in low glucose (5mM), (B) HG (25mM), and (C) insulin (100nM) and HG (25mM) combined (n=6/group). (D) Glucose uptake was also measured after returning cells to standard growth medium following 48h of HG and insulin treatment of LECs. Data represent mean fold changes ± SEM (n=4/group). #p<0.05 vs. control.
Hyperglycemia- and hyperinsulinemia-induced insulin resistance impairs insulin-dependent PI3K/Akt/eNOS signaling in LECs.
We previously demonstrated that MetSyn rats showed a decrease in eNOS expression coupled with impaired flow responses in thoracic duct lymphatics [14]. Therefore, we investigated the mechanisms by which insulin resistance impairs eNOS regulation in LECs. Phosphorylation of both Akt (Ser473) and Akt (Thr308) was significantly reduced by 48 h HG and insulin treatments (−0.39 fold vs. Control, p<0.003 and −0.43-fold vs. Control, p<0.05, respectively) compared to all other groups (Figures 4A and B). Additionally, eNOS phosphorylation (Ser1177) was significantly decreased in insulin resistant LECs (−0.43 fold vs. Control, p<0.01) (Figure 4C). To confirm whether the decreased eNOS phosphorylation impaired NO production in insulin resistant LECs, we measured levels of nitrite, a stable metabolite of NO, in conditioned medium as a direct index of NO production in LECs. When eNOS was blocked by a NOS inhibitor (i.e., L-NAME), nitrite content in LECs was significantly reduced (0.63 ± 0.1nM/hour/105 cells, p<0.05 vs. Control) compared to the control group (2.27 ± 0.29nM/hour/105 cells, Figure 4D). Acute insulin treatment elevated the medium nitrite content (3.57 ± 0.32nM/hour/105 cells, p<0.01 vs. Control), and insulin-induced nitrite production was glucose-independent (3.63 ± 0.33nM/hour/105 cells, p<0.01 vs. Control). In contrast, insulin resistant LECs showed a significantly diminished nitrite level (1.45 ± 0.08nM/hour/105 cells, p<0.05 vs. Control). To rule out possible effects of osmolality on NO production, we treated LECs with 25mM mannose-containing medium, with or without insulin. The osmolality of each solution (mOsmol/Kg) was measured: control, 277.1±2.91; HG, 302.1±2.81; Mannose, 302.2±2.74. The mannose-treated LECs did not show any significant difference in nitrite level (2.05 ± 0.24nM/hour/105 cells) compared to control LECs, ruling out an effect of osmolality.
Figure 4.

Hyperglycemia- and hyperinsulinemia-induced insulin resistance impaired insulin-dependent PI3K/Akt/eNOS signaling in LECs. LECs were treated with HG (25mM) and insulin (100nM) for 48h and representative western blots are shown: (A) Akt (Ser473) phosphorylation, (B) Akt (Thr308) phosphorylation, and (C) eNOS (Ser1177) phosphorylation. The relative expression of p-Akt (Ser473)/Akt, p-Akt(Thr308)/Akt, and p-eNOS/eNOS were quantified and plotted (n=3/group). Data represent mean fold changes ± SEM. #p<0.05 vs. Control; $p<0.05 vs. insulin; *p<0.05 vs. HG. (D) Nitrite level was measured in LECs treated with HG (25mM), insulin (100nM), and L-NAME (10μM) as described (n=7–9/group). Data represent mean ± SEM. #p<0.05 vs. control.
Insulin resistance mechanisms impair mitochondrial function and cellular energetics in LECs.
Mitochondria play an important role in maintaining endothelial function by producing essential cellular energy and by regulating cellular Ca2+ homeostasis. Unlike blood endothelial cells (BECs), LECs are more dependent on mitochondria as a source of energy than on glycolysis [29]. However, the mechanisms regulating LEC metabolism and mitochondrial function during insulin resistance conditions have not been previously investigated. We first compared the energetic phenotypes of LECs with the highly energetic LMCs. LECs showed significantly lower basal- (19.96 ± 0.82 vs. 2.11±0.20pmol/min/104 cells, LMCs vs. LECs, p<0.05), maximal- (37.18 ± 2.77 vs. 6.06 ± 0.27pmol/min/104 cells, LMCs vs. LECs, p<0.05) and ATP-linked OCR values when compared to LMCs (14.7± 1.32 vs. 1.87± 0.12pmol/min/104 cells, LMCs vs. LECs, p<0.05, Figure 5B–C). When compared to control LECs, insulin resistant LECs exhibited significant decreases in the basal (2.368 ± 0.12 vs. 1.392 ± 0.07pmol/min/104 cells, p<0.001), maximal (3.65 ± 0.65 vs. 1.734 ± 0.04pmol/min/104 cells, p<0.021), and ATP-linked OCR (1.72 ± 0.09 vs. 0.803 ± 0.19pmol/min/104 cells, p<0.001) values (Figure 5E–F); no significant differences were observed between HG- or insulin-treated LECs. Additionally, there were no significant differences between groups in maximal glycolytic capacity (Figure 5G).
Figure 5.
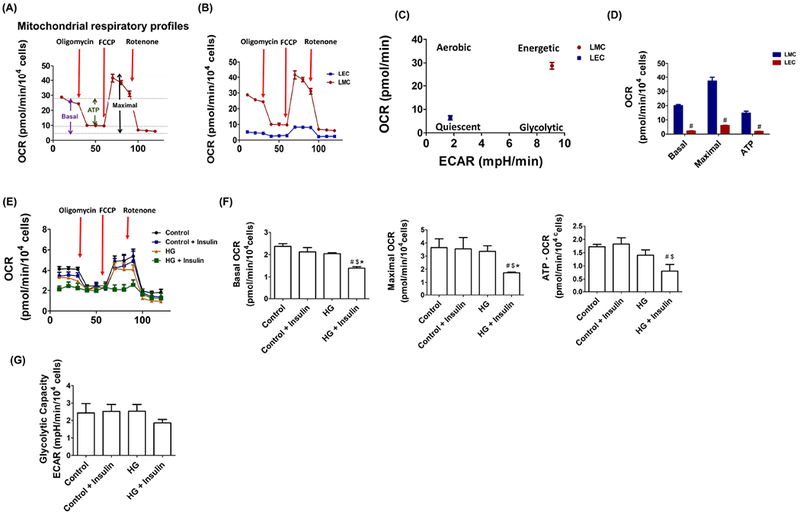
Insulin resistance conditions impaired mitochondrial function in LEC. A) Graph of the mitochondrial bioenergetics test detailing the three key parameters of mitochondrial function (basal respiration, ATP-respiration, and maximal respiration) with the sequential treatment of oligomycin (ATP synthesis inhibitor), FCCP (mitochondrial uncoupler), and rotenone (mitochondrial complex I inhibitor). B) Graphic representation showing real-time analysis of mitochondrial OCRs in LMCs and LECs (n=4/group). Arrows point to where oligomycin, FCCP, or rotenone were applied to demonstrate basal, maximal, and ATP-linked OCR. C) Metabolic phenogram. Basal OCR and ECAR rates were plotted in LMCs and LECs. D) Basal, maximal, and ATP-linked OCR were quantified and plotted. # p<0.05 vs. LMCs. E) Real-time analysis of OCR in LECs after 48 h HG and insulin treatments (n=4/group). Arrows; oligomycin, FCCP, and rotenone application. F) Basal, maximal, and ATP-linked OCR were quantified and plotted (n=4/group). G) Glycolytic capacity was quantified and plotted (n=4/group from triplicate). Data represent mean ± SEM. # p<0.05 vs. control, $ p<0.05 vs. control + insulin, *p<0.05 vs. HG.
Lymphatic permeability is increased in insulin resistant LECs.
Metabolic diseases are accompanied by disrupted lymphatic barrier function and increased lymph leakage [10,11,30]. Since LECs are actively involved in the regulation of lymphatic permeability [19,30–32], we wanted to determine whether insulin resistance brought about by hyperglycemic and hyperinsulinemic conditions in LECs directly impaired LEC barrier integrity. Glucose or acute/prolonged insulin treatment did not affect permeability in LECs (Figure 6A). Notably, hyperglycemic and hyperinsulinemic conditions caused a significant increase in permeability in LECs (+0.96 fold vs. Control, p<0.05). TNF-α, a well-known pro-inflammatory mediator that increases lymphatic endothelial permeability [19,32], was used as a positive control and it significantly increased lymphatic permeability (+2.36 fold vs. Control, p<0.0001).
Figure 6.

Insulin resistance affects LEC permeability. (A) Permeability of LEC monolayers to FITC-labeled bovine serum albumin was measured in the LECs treated with insulin (100nM), HG (25mM), or TNF-α (20ng/ml) as described. Data represent mean fold changes ± SEM. #p<0.05 vs. control (n=9/group). (B) LEC were treated with insulin (100nM), HG (25mM), and TNF-α (20 ng/ml) and time-dependent TEER was measured. Data represent mean fold changes ± SEM. #p<0.05 vs. basal level (n=3/group). (C) Representative image of β-catenin in LEC treated with high glucose (25mM) and insulin (100nM) for 48h. Images were obtained using a 20× (NA=0.7) UPlan Apo objective (upper panel) and 40× (NA=1.3) Uplan FLN objective (lower panel). Bar indicates 100μM and 50μM, respectively. A minimum of 9 fields was quantified from triplicate experiments (n=9/group). (D) Quantification of β-catenin in the junctional area is plotted. Data represent mean fold changes ± SEM. #p<0.05 vs. control; $p<0.05 vs. insulin; *p<0.05 vs. HG. (E) LECs were treated with HG (25mM) and insulin (100nM) for 48h and representative western blots for β-catenin are shown (n=4/group). Data represent mean fold changes ± SEM.
To further determine at what time point hyperglycemia and hyperinsulinemia begin to disrupt the LEC barrier, we employed trans-endothelial electrical resistance (TEER) measurements to assess barrier integrity. We found that insulin or glucose alone did not affect TEER throughout the time periods tested (Figure 6B). High glucose and insulin treated-LECs exhibited significantly decreased TEER at 48h (−0.32 fold vs. Basal, p<0.05), matching the time point at which LECs displayed insulin resistance. TNF-α-treated LECs showed a steady decline in TEER with a significant decrease at the 24h time point (−0.56-fold vs Basal, p<0.001, Figure 6B). In order to determine whether changes occur in the adherens junction, we examined the localization of β-catenin in the insulin resistant LECs. Insulin resistant LECs showed a decrease in β-catenin expression at the junctional regions (−0.48 fold vs. Control, p<0.05) when compared to control LECs, while insulin- or glucose-treated LECs did not show any significant differences (Figures 6C and D). No significant changes were observed in the total amount of β-catenin in the LECs from the different groups (Figure 6E).
Insulin resistance causes activation of NF-κB and increased cytokine gene expression in LECs.
Obesity and insulin resistance are associated with chronic systemic inflammation [33]. Our previous data demonstrated an increase in inflammatory signaling in insulin resistant LMCs [17] and in the lymphatic vessels from MetSyn animals [15,16]. Therefore, we investigated whether insulin resistance also promotes pro-inflammatory signaling in LECs. TNF-α-treated LECs showed robust NF-κB nuclear translocation (Figures 7A and B), indicating activation of inflammation, as we have shown previously [9]. HG and high insulin conditions also led to a significant increase in NF-κB nuclear translocation in LECs (+0.85 fold vs. Control, p<0.05). ICAM-1, one of the key attractants for inflammatory leukocytes, was significantly upregulated in the HG- and insulin-treated LECs (+0.74 fold vs. Control, p<0.05) compared to all other groups, while there was no significant difference between control, insulin-, and glucose-treated LECs (Figure 7C). Furthermore, we analyzed gene expression for the inflammatory cytokines MCP1, IL1β, IL6, TNF-α, iNOS, MIF, MMP2, CXCR2, CFOS, CJUN, and MIP2 in the LECs under the different treatment conditions described above. MCP1 and MIP2 showed significant increases in expression in insulin resistant LECs, while no other cytokines showed a significant increase in expression (Figure 7D).
Figure 7.
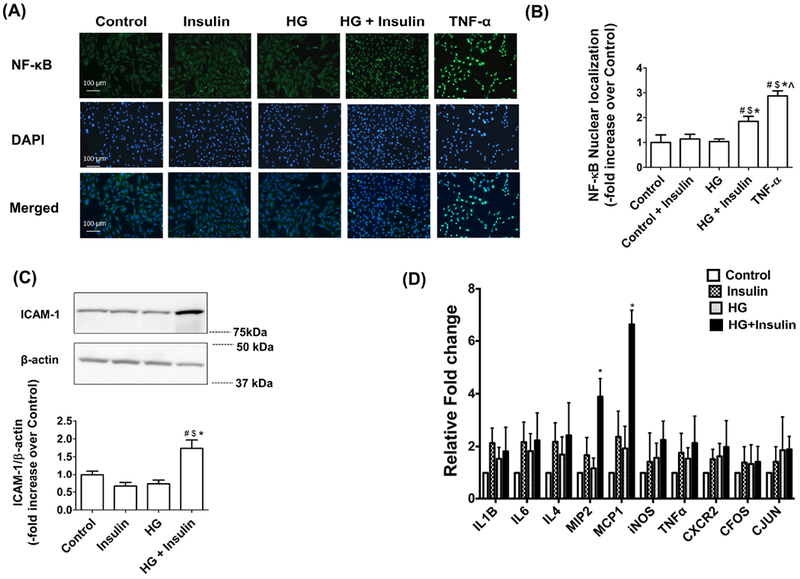
Insulin resistance activated NF-κB inflammatory cell adhesion molecules and cytokines. (A) Immunofluorescence images of activated NF-κB (green) translocation into the nucleus (DAPI: blue) in LEC treated with HG (25mM), insulin (100nM), and TNF-α (20ng/ml) as described using 20X objective (NA=0.7). (B) Relative nuclear signal intensity in each cell was quantified. Nine fields of view were used for data quantification from triplicate experiments (n=9/group). Data represent mean fold changes ± SEM. #p<0.05 vs. control; $p<0.05 vs. insulin; *p<0.05 vs. HG; and ^p<0.001 vs. TNF-α. (C) LECs were treated with HG (25mM) and insulin (100nM) for 48h and representative western blots are shown for ICAM-1 expression. Data represent mean fold changes ± SEM. #p<0.01 vs. control; $p<0.01 vs. insulin; *p<0.01 vs. HG. (D) Analyses of inflammatory cytokine mRNA expression in insulin resistant LECs. RNA was isolated from LECs treated with HG (25mM), insulin (100nM) or HG and insulin combined as described. Expression levels of various cytokines were quantified by real time PCR. Fold change relative to untreated control was calculated. RPL19 was used as endogenous control (n=4/group). Data represent mean fold changes ± SEM. #p<0.05 vs. control.
Discussion
The mechanistic relationship between insulin resistance and lymphatic endothelial dysfunction is not clearly understood. Lymphatic vascular integrity has been shown to be disrupted in diabetic db/db mice, and these conditions were associated with low NO bioavailability and increased permeability [13]. However, the specific molecular mechanisms governing this physiological response have not been investigated and is the highlight of the present study. In this study, we provide the first evidence to our knowledge that while acute insulin exposure promotes eNOS phosphorylation and consequently increases NO production via PI3K/AKT signaling in LECs, insulin resistance activates proinflammatory signaling in LECs, decreases cellular bioenergetics, and decreases NO production, thereby impacting lymphatic barrier function, junctional integrity and increasing LEC permeability (Figure 8). In the following section, we justify the concentrations of glucose and insulin used in this study and correlat our data with the available literature on insulin resistance in BECs.
Figure 8.
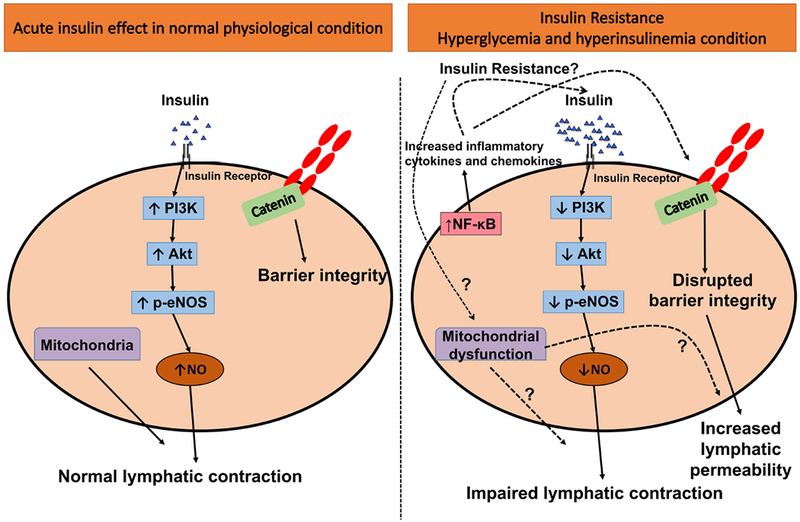
Schematic representation of insulin resistance-mediated mechanisms in LECs. Under normal physiologic conditions, insulin stimulates eNOS phosphorylation via PI3K/Akt signaling that regulates NO production. LEC junctions are well regulated via adherens junctions in the normal state, thus maintaining lymphatic function. However, the onset of insulin resistance impairs the PI3K/Akt/eNOS pathway, diminishing NO production, and impairs mitochondrial function in LECs. These mechanisms further disrupt the endothelial membrane integrity by disrupting the adherens junction and thus increase LEC permeability and activate proinflammatory signaling, together leading to impaired lymphatic function.
The level of glucose used as our control (5mM) is similar to the physiological glucose level, while our HG level (25mM) is higher than the published in vivo pathological value (e.g.,8.1 ± 2.5mM). The insulin concentration (100nM) used in our study has been widely used in several in vitro studies [34–37] and is also within the range of insulin concentrations used to treat various types of BECs (Tables 1 and 2).
Table 1.
Insulin activates Akt/eNOS phosphorylation in BECs
| Cell types | Insulin treatment | Results | Reference |
|---|---|---|---|
| Bovine aortic endothelial cells (BAEC) | 500nM for 2 mins | Increased eNOS and NO production | [89] |
| Human umbilical vein EC (HUVEC) | 500nM (<10 mins) | Increased NO production | [24] |
| Human umbilical vein EC (HUVEC) | 100nM for (5 to 180 mins) | Increased Akt phosphorylation | [90] |
| Mouse aortic endothelial cells | 100nM for 10 mins | Increased Akt phosphorylation and eNOS phosphorylation | [91] |
| Human umbilical vein EC (HUVEC) | 1μM for 10 mins | Increased Akt phosphorylation and eNOS phosphorylation | [53] |
| Bovine aortic endothelial cells (BAEC) | 100nM for 5 mins | Increased Akt phosphorylation and eNOS phosphorylation | [92] |
| Human umbilical vein EC (HUVEC) | 500nM for 2 mins | Increased Akt phosphorylation, eNOS phosphorylation, and NO production. | [93] |
| Bovine aortic endothelial cells (BAEC) | 100nM for 10 mins | Increased Akt phosphorylation, eNOS phosphorylation, and NO production. | [47] |
| Bovine aortic endothelial cells (BAEC) | 500nM for3, 5, and 10 mins[1] | Increased Akt phosphorylation, eNOS phosphorylation, and NOS activity. | [7] |
| Human umbilical vein EC (HUVEC) | 100nM for 30 mins | Increased eNOS phosphorylation and NO production | [94] |
| Human umbilical vein EC (HUVEC) | 50nM for 10 mins | Increased Akt phosphorylation, eNOS phosphorylation, and NO production. | [95] |
| Human umbilical vein EC (HUVEC) | 1–10nM for 10 mins | Increased Akt phosphorylation and eNOS phosphorylation | [96] |
Table 2.
Insulin stimulates glucose uptake in BECs
| Cell types | Reagents/Tracers | Insulin treatment | Result | Reference |
|---|---|---|---|---|
| Human coronary artery endothelial cells (HCAEC) | 2NBDG | 17.4pM for 20 min | Increased 2NBDG uptake by 64% | [26] |
| Human umbilical vein EC (HUVEC) | 2-3HDG | 100nM for 90 min | Increased 2-3HDG uptake | [97] |
| Bovine aortic endothelial cells (BAEC) | 2-deoxyglucose | 17pM to 17.4μM | Increased 2DG uptake at 1.7nM insulin | [98] |
| Rabbit cardiac muscle endothelial cells (RCME) | 2-deoxyglucose | 2nM for 90 min | Increased 2DG uptake | [99] |
| Rat capillary endothelial cell | 2-deoxyglucose | 0.16nM – 160nM for 30 min | Increased glucose uptake from 0.16nM of insulin | [100] |
| Human aortic endothelial cells (HAEC) | 2-deoxy[14C]glucose | 347.3pM for 24h | Increased glucose uptake | [101] |
| Human umbilical vein EC (HUVEC) | 2-deoxyglucose | 100nM insulin for 30 min | Increased glucose uptake | [94] |
| Bovine pulmonary artery (PAEC) and aortic endothelial cells (AEC) | Glucose | No difference between PAEC vs. AEC | [102] |
Previous studies have shown comparable levels of gastrointestinal hormones, including insulin, within lymph and plasma in canine and rodent models [38,39]. Other studies have shown higher glucose and lower insulin levels in the lymph compared to blood plasma [40–42] (Tables 3 and 4). The insulin levels in human lymph and plasma are similar under basal conditions, and there is a strong positive correlation between whole-body glucose uptake and lymph insulin, supporting comparable levels of insulin in the lymph and systemic circulation [43]. Insulin sensitivity was higher in the lymphatic vessel (i.e., thoracic duct) compared to right jugular vein, while plasma insulin level achieved at a steady state faster than the lymph insulin following insulin infusion [39]. However, this temporal difference could be due to the spatial gap between venous and lymphatic vessels since the insulin was infused via the hind limb vein.
Table 3.
Insulin levels in Plasma and lymph
| Study model | Plasma | Insulin level | Reference |
|---|---|---|---|
| Sprague-Dawley rats (150–225g) | Not applicable | 8.023 ± 0.19nM | [103] |
| Mongrel dogs (17.9–25.4 kg) | 36 ± 6pM | 30 ± 6pM (Thoracic) 36 ± 6pM (Hindlimb) | [104] |
| Mongrel dogs (23–28kg) | 129.15 ± 2pM | 86.1 ± 7.18pm | [41] |
| Mongrel dogs (23.2–34.5kg) | 108 ± 12pM | 84 ± 8.6pM (Thoracic) | [39] |
| Bulldogs (18–42 kg) | Not applicable | 93.28 ± 11.48pM | [105] |
| Mongrel dogs | 70.75 ± 10.98pM (Femoral artery) 57.18 ± 8.32pM (Femoral vein) |
30.49 ± 5.17pM | [40] |
| Sprague-Dawley | 77 ± 14pM (portal vein) | 55 ± 10pM (mesentery) | [38] |
| Mongrel dogs (19.1–30 kg) | 44 ± 7pM (Hind limb artery) | 24 ± 3pM (Hind limb) | [106] |
Table 4.
Glucose level in plasma and lymph
| Study model | Plasma Glucose level (mM) | Lymph glucose (mM) | Reference |
|---|---|---|---|
| Mongrel dogs (17.9–25.4 kg) | 6.6 ± 0.4 | 7.1± 0.4 (Thoracic) 6.7 ± 0.5 (hind limb) | [104] |
| Mongrel dogs (23–28kg) | 5.4 ± 1 | 5.7 ± 1 | [41] |
| Mongrel dogs (23.2–34.5kg) | 5.4 ± 0.1 | 5.7 ± 0.1 (Thoracic) | [39] |
| Mongrel dogs | 5.05 ± 0.1 (Femoral artery) 3.86 ± 0.8 (Femoral vein) |
5.58 ± 0.9 (mg/dl) | [40] |
| Sprague-Daley | 5.1 ± 2 (portal vein) | 6.4 ± 0.2 (mesentery) | [38] |
| Mongrel dogs (19.1–30 kg) | 6.6 ± 0.5 (Hind limb artery) | Not applicable | [106] |
We previously showed that the collecting lymphatic vessels from MetSyn animals demonstrated increased constriction with reduced contractile frequency, resulting in decreased lymph transport [15,16]. In addition, the lymphatic thoracic duct from MetSyn animals exhibited insensitivity to shear stress due to downregulated eNOS expression [14]. NO plays a critical role in the flow/shear-mediated regulation of lymphatic contractility [44–46]. The data presented in this study demonstrate that insulin increases eNOS and NO production in LECs via activation of the PI3K/Akt signaling pathway (Figures 1 and 2). However, under insulin resistance conditions, eNOS phosphorylation and NO production were reduced significantly compared to the basal condition (Figure 4). Hence, we proposed that the reduced lymphatic contractile activity and the smaller diameter lymphatic vessels observed in MetSyn rats could be caused by the diminished eNOS phosphorylation and NO production in the insulin resistant LECs of MetSyn animals. Table 1 provides the data from various studies showing the effects of insulin on eNOS phosphorylation in BECs from different sources [6,7,47,48]. Though these studies used different insulin concentrations on various BECs, the results of Akt signaling and eNOS phosphorylation are similar to the present study with LECs (Table 1 and Figures 1 and 2).
BECs predominantly rely on glycolysis to regulate bioenergetics due to easy access to glucose [49,50]. Although insulin plays a vital role in endothelial function by regulating eNOS phosphorylation [23,24,51], it is not directly linked with energy metabolism in BECs [52,53]. Table 2 indicates the differential effect of insulin on glucose uptake in diverse BECs. In contrast to BECs, very little is known about metabolic pathways in LECs [2]. Wong et al [29] had shown that fatty acid β-oxidation was higher in LECs than in arterial, venous and microvascular ECs, whereas glycolytic flux was lower, suggesting the important role of mitochondrial oxidation in LEC metabolism. Studies have also shown that mitogen activated protein kinase kinase kinase kinase 4 (Map4k4) impairs energy metabolism in ECs and promotes insulin resistance during high-fat diet-induced obesity and its systemic loss improves insulin sensitivity [54–56]. Primary ECs that were a mixture of lymphatic and blood ECs showed enhanced glycolytic and mitochondrial respiration in the absence of MAP4K4. However, specific signaling mechanisms and metabolic effects in the blood and lymphatic endothelium that may be contributing to the observed effects were not delineated [55]. In this study, we have shown that while insulin resistance promotes mitochondrial dysfunction in LECs (Figures 5E and F), glycolytic capacity (Figure 5G) is not altered, suggesting that LECs may utilize mitochondria as a predominant energy source. LECs exhibit a reduced cellular energetics status when compared to LMCs (Figures 5A–C), which might be due to the higher mitochondrial content in LMCs. LEC mitochondria might play a prominent role in cellular signaling responses to environmental cues [57], as well as in regulating NO bioavailability [58,59]. Further studies are warranted to determine the mechanisms by which insulin resistance impairs mitochondrial function more severely than glycolytic capacity in LECs and to identify the source and mechanisms of energy metabolism in LECs under physiological and pathological conditions.
Maintaining lymphatic endothelial barrier function is crucial for maintenance of lymphatic functions and limiting inflammation [13,19,31,60,61]. The lymphatic endothelial barrier is controlled by the cell-cell junction that controls physiological lymphatic function [61]. While BECs are impermeable to albumin and other large molecules in a steady state condition [62], lymphatics constitutively leak a portion of the fluid and solute [63,64]. Elevated lymph leakage due to disrupted LEC junctions aids development of obesity with hyperinsulinemia and with increased leptin levels [10]. Further, diabetic mice exhibited increased permeability in mesenteric collecting lymphatic vessels, suggesting that compromised lymphatic barrier function would lead to severe leakage of lymph into the tissue [13]. In the present study, our results demonstrate that there is a significant increase in permeability in insulin resistant LECs (Figure 6A). This is consistent with the decreased TEER across LECs in response to prolonged hyperglycemia and hyperinsulinemia conditions (Figure 6B). In addition, the expression of the adherence junction protein, β-catenin, which is one of the essential molecules for endothelial integrity and the stabilization of cadherin [32,65], is significantly lower in the junctional area of the insulin resistant LECs compared to control cells (Figures 6C–E). Thus, our data suggest that the decreased impedance across the LECs and increased LEC permeability under insulin resistance conditions may be due to the disruption of β-catenin at the junctional areas of the cells. In the current study, we were not able to assess the initial step of increase in solute flux that reflects more quantitative permeability. In addition, the increased permeability may be due to changes in active albumin transport as the FITC-albumin assay is reflective of the rate of transport of albumin across the endothelial monolayer via passive diffusion through cell junction and active ATP-dependent transcytosis [66,67].
NO is also known as a crucial regulator for permeability in endothelium. Studies have provided evidence that NO increases or decreases the blood microvascular or lymphatic permeability [68–71]. For instance, inhibition of NO increased blood vascular permeability in the rat [72] and cat [73]. In contrast, eNOS-derived NO was necessary for VEGF-induced permeability in endothelial cells [74]. The role of NO in the lymphatic endothelial barrier integrity is poorly understood. iNOS- derived NO brought about by pro-inflammatory cytokines increased permeability in rat mesenteric LECs [19]. Scallan et al. [13] performed a more direct assessment of the role of NO and permeability using the mouse lymphatic vessel, and the results were a paradox. In healthy lymphatics, NO increased permeability. In contrast, inducing NO synthesis using L-arginine supplementation decreased permeability in the lymphatics of diabetic animals [13]. Our data presented in this study show that insulin induces NO in LECs but does not decrease permeability; whereas NO is decreased in insulin resistant LECs and those cells show an increase in permeability (Figure 6).
The MetSyn condition is characterized by a mild, but chronic inflammatory state [33]. We found that conditions of high glucose and high insulin caused NF-κB nuclear trans-localization in LECs (Figure 7A–B), which is also associated with the induction of inflammatory chemoattractant molecules, such as MCP1 and MIP2, that play an important role in recruitment of inflammatory M1 macrophages [75,76]. In addition, insulin resistant LECs showed increased ICAM-1 expression, which is involved in recruiting innate and adaptive immune cells and mediating LEC permeability [77–79]. In LECs, ICAM-1 is one of the critical adhesion molecules facilitating dendritic cell transmigration in response to inflammatory cytokines [78]. ICAM-1 blockage inhibited flow-dependent dendritic cell migration and permeability, suggesting an important role of ICAM-1 in both immune cell regulation and lymphatic permeability [77]. Inflammation increased ICAM-1 expression in LECs [19,80] and increased permeability [19,32]. We have previously shown that the dietary endotoxin, LPS, increased innate immune cell recruitment that was associated with an ICAM-1 upregulation [22]. Therefore, we speculate that increased ICAM-1 by insulin resistance in LECs would promote immune cell recruitment into the collecting lymphatic vessels, similar to what we observed in the mesenteric collecting lymphatic vessels of MetSyn rats [15]. Our data did not show significant changes in several other key inflammatory molecules, such as IL-1β, IL-6, and IL-4, indicating a mild inflammatory activation under insulin resistance conditions in LECs. Therefore, it is plausible that insulin resistant LECs exhibit an increase in permeability due to decreased lymphatic endothelial barrier integrity caused by mild chronic inflammatory signaling. Indeed, animals with MetSyn or related diseases (i.e. Type 2 diabetes) exhibited leaky lymphatic vessels [13] and impaired pumping activity with reduced intrinsic force generation [16], altered inflammatory signaling [15] and decreased eNOS expression [14].
In conclusion, this study provides the first direct evidence that prolonged hyperglycemia and hyperinsulinemia conditions induce insulin resistance in LECs. As seen in Figure 8, insulin resistance impairs the PI3K/Akt pathway that dysregulates eNOS phosphorylation and decreases NO production. These changes are also associated with alterations in the mitochondrial function of the LECs. Disruption of LEC barrier function during insulin resistance increases permeability. Insulin resistance also activates pro-inflammatory signaling in LECs (Figure 8). We acknowledge that the LECs in the present study were grown directly on cell culture plastic. Numerous cell types are affected by their microenvironment, including types of extracellular matrix (ECM) composition and physical properties [81]. BECs showed different vascular structure properties and angiogenic characteristics in response to various extracellular microenvironments [82–87]. A recent study showed that decreased ECM stiffness mediates lymphangiogenesis both in vivo and in vitro [88]. Therefore, further studies are warranted to understand how the remodeling of ECM and other changes in the microenvironment occurring in the in vivo conditions influence the molecular mechanisms of the complex inflammatory, mitochondrial and metabolic signaling in the lymphatic endothelium in response to nutrient overload.
Perspectives
Insulin promotes nitric oxide production via the PI3K/Akt/eNOS pathway in lymphatic endothelial cells. In insulin resistance conditions, nitric oxide production is diminished due to impaired signaling via the PI3K/Akt pathway in the lymphatic endothelial cell. Additionally, insulin resistance disrupts lymphatic endothelial barrier integrity and induces inflammation. Thus, impairments in the insulin resistant lymphatic endothelial cell functional mechanisms would weaken lymphatic vessel pumping activity, and consequently, lymph flow in metabolic syndrome or other related metabolic diseases.
Acknowledgement
This work was supported by NIH RO1 DK99221 (to M.M.) and the Department of Medical Physiology Lymphatic Graduate Student Fellowship (to Y.L). The authors declare no conflicts of interest. The authors wish to thank Se-Hoon Jeong, Department of Biomedical Engineering, Texas A&M University for technical help with TEER measurements.
Abbreviations
- ANOVA
Analysis of variance
- eNOS
Endothelial nitric oxide synthase/nitric oxide synthase 3
- ECAR
Extracellular acidification rate
- FCCP
Carbonyl cyanide-p-trifluoromethoxyphenylhydrazone 2
- HG
High glucose
- ICAM-1
Intercellular adhesion molecule 1
- IL-1β
Interleukin 1 beta
- IL-4
Interleukin 4
- IL-6
Interleukin 6
- iNOS
Inducible nitric oxide synthase/nitric oxide synthase
- LECs
Lymphatic endothelial cells
- L-NAME
L-NG-Nitroarginine methylester
- LPS
Lipopolysaccharide
- MCP1
Monocyte chemoattractant protein-1
- MetSyn
Metabolic syndrome
- MIF
Macrophage migration inhibitory factor/glycosylation-inhibiting factor
- MIP
Macrophage inflammatory proteins
- NF-κB
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NO
Nitric oxide
- OCR
Oxygen consumption rate
- PI3K
Phosphatidylinositol-4,5-bisphosphate-3-kinase
- SDS
Sodium dodecyl sulphate
- TEER
Transendothelial electrical resistance
- TNF-α
Tumor necrosis factor alpha
References
- 1.Roberts CK, Hevener AL, Barnard RJ. Metabolic syndrome and insulin resistance: underlying causes and modification by exercise training. Comprehensive Physiology 3: 1–58, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barrett EJ, Liu Z. The endothelial cell: an “early responder” in the development of insulin resistance. Reviews in endocrine & metabolic disorders 14: 21–27, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barrett EJ, Eggleston EM, Inyard AC, Wang H, Li G, Chai W, Liu Z. The vascular actions of insulin control its delivery to muscle and regulate the rate-limiting step in skeletal muscle insulin action. Diabetologia 52: 752–764, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim F, Pham M, Maloney E, Rizzo NO, Morton GJ, Wisse BE, Kirk EA, Chait A, Schwartz MW. Vascular inflammation, insulin resistance, and reduced nitric oxide production precede the onset of peripheral insulin resistance. Arteriosclerosis, thrombosis, and vascular biology 28: 1982–1988, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cubbon RM, Rajwani A, Wheatcroft SB. The impact of insulin resistance on endothelial function, progenitor cells and repair. Diabetes & vascular disease research 4: 103–111, 2007. [DOI] [PubMed] [Google Scholar]
- 6.Duncan ER, Crossey PA, Walker S, Anilkumar N, Poston L, Douglas G, Ezzat VA, Wheatcroft SB, Shah AM, Kearney MT. Effect of endothelium-specific insulin resistance on endothelial function in vivo. Diabetes 57: 3307–3314, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tanigaki K, Mineo C, Yuhanna IS, Chambliss KL, Quon MJ, Bonvini E, Shaul PW. C-reactive protein inhibits insulin activation of endothelial nitric oxide synthase via the immunoreceptor tyrosine-based inhibition motif of FcgammaRIIB and SHIP-1. Circulation research 104: 1275–1282, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huggenberger R, Siddiqui SS, Brander D, Ullmann S, Zimmermann K, Antsiferova M, Werner S, Alitalo K, Detmar M. An important role of lymphatic vessel activation in limiting acute inflammation. Blood 117: 4667–4678, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chakraborty S, Zawieja DC, Davis MJ, Muthuchamy M. MicroRNA signature of inflamed lymphatic endothelium and role of miR-9 in lymphangiogenesis and inflammation. American journal of physiology Cell physiology 309: C680–692, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Escobedo N, Proulx ST, Karaman S, Dillard ME, Johnson N, Detmar M, Oliver G. Restoration of lymphatic function rescues obesity in Prox1-haploinsufficient mice. JCI insight 1, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harvey NL, Srinivasan RS, Dillard ME, Johnson NC, Witte MH, Boyd K, Sleeman MW, Oliver G. Lymphatic vascular defects promoted by Prox1 haploinsufficiency cause adult-onset obesity. Nature genetics 37: 1072–1081, 2005. [DOI] [PubMed] [Google Scholar]
- 12.Lim HY, Rutkowski JM, Helft J, Reddy ST, Swartz MA, Randolph GJ, Angeli V. Hypercholesterolemic mice exhibit lymphatic vessel dysfunction and degeneration. The American journal of pathology 175: 1328–1337, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scallan JP, Hill MA, Davis MJ. Lymphatic vascular integrity is disrupted in type 2 diabetes due to impaired nitric oxide signalling. Cardiovascular research 107: 89–97, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zawieja SD, Gasheva O, Zawieja DC, Muthuchamy M. Blunted flow-mediated responses and diminished nitric oxide synthase expression in lymphatic thoracic ducts of a rat model of metabolic syndrome. American journal of physiology Heart and circulatory physiology 310: H385–393, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zawieja SD, Wang W, Chakraborty S, Zawieja DC, Muthuchamy M. Macrophage alterations within the mesenteric lymphatic tissue are associated with impairment of lymphatic pump in metabolic syndrome. Microcirculation (New York, NY : 1994) 23: 558–570, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zawieja SD, Wang W, Wu X, Nepiyushchikh ZV, Zawieja DC, Muthuchamy M. Impairments in the intrinsic contractility of mesenteric collecting lymphatics in a rat model of metabolic syndrome. American journal of physiology Heart and circulatory physiology 302: H643–653, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee Y, Fluckey JD, Chakraborty S, Muthuchamy M. Hyperglycemia- and hyperinsulinemia-induced insulin resistance causes alterations in cellular bioenergetics and activation of inflammatory signaling in lymphatic muscle. FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Srinivasan B, Kolli AR, Esch MB, Abaci HE, Shuler ML, Hickman JJ. TEER measurement techniques for in vitro barrier model systems. Journal of laboratory automation 20: 107–126, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cromer WE, Zawieja SD, Tharakan B, Childs EW, Newell MK, Zawieja DC. The effects of inflammatory cytokines on lymphatic endothelial barrier function. Angiogenesis 17: 395–406, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bowers MC, Hargrove LA, Kelly KA, Wu G, Meininger CJ. Tetrahydrobiopterin attenuates superoxide-induced reduction in nitric oxide. Frontiers in bioscience (Scholar edition) 3: 1263–1272, 2011. [DOI] [PubMed] [Google Scholar]
- 21.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature methods 9: 671–675, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chakraborty S, Zawieja SD, Wang W, Lee Y, Wang YJ, von der Weid PY, Zawieja DC, Muthuchamy M. Lipopolysaccharide modulates neutrophil recruitment and macrophage polarization on lymphatic vessels and impairs lymphatic function in rat mesentery. American journal of physiology Heart and circulatory physiology: ajpheart.00467.02015, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 399: 597–601, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zeng G, Quon MJ. Insulin-stimulated production of nitric oxide is inhibited by wortmannin. Direct measurement in vascular endothelial cells. The Journal of clinical investigation 98: 894–898, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jais A, Solas M, Backes H, Chaurasia B, Kleinridders A, Theurich S, Mauer J, Steculorum SM, Hampel B, Goldau J, Alber J, Forster CY, Eming SA, Schwaninger M, Ferrara N, Karsenty G, Bruning JC. Myeloid-Cell-Derived VEGF Maintains Brain Glucose Uptake and Limits Cognitive Impairment in Obesity. Cell 165: 882–895, 2016. [DOI] [PubMed] [Google Scholar]
- 26.Gaudreault N, Scriven DR, Laher I, Moore ED. Subcellular characterization of glucose uptake in coronary endothelial cells. Microvascular research 75: 73–82, 2008. [DOI] [PubMed] [Google Scholar]
- 27.Chen R, Peng X, Du W, Wu Y, Huang B, Xue L, Wu Q, Qiu H, Jiang Q. Curcumin attenuates cardiomyocyte hypertrophy induced by high glucose and insulin via the PPARgamma/Akt/NO signaling pathway. Diabetes research and clinical practice 108: 235–242, 2015. [DOI] [PubMed] [Google Scholar]
- 28.Niu P, Zhang Y, Shi D, Chen Y, Deng J. Cardamonin ameliorates insulin resistance induced by high insulin and high glucose through the mTOR and signal pathway. Planta medica 79: 452–458, 2013. [DOI] [PubMed] [Google Scholar]
- 29.Wong BW, Wang X, Zecchin A, Thienpont B, Cornelissen I, Kalucka J, Garcia-Caballero M, Missiaen R, Huang H, Bruning U, Blacher S, Vinckier S, Goveia J, Knobloch M, Zhao H, Dierkes C, Shi C, Hagerling R, Moral-Darde V, Wyns S, Lippens M, Jessberger S, Fendt SM, Luttun A, Noel A, Kiefer F, Ghesquiere B, Moons L, Schoonjans L, Dewerchin M, Eelen G, Lambrechts D, Carmeliet P. The role of fatty acid beta-oxidation in lymphangiogenesis. Nature, 2016. [DOI] [PubMed] [Google Scholar]
- 30.Sawane M, Kajiya K, Kidoya H, Takagi M, Muramatsu F, Takakura N. Apelin inhibits diet-induced obesity by enhancing lymphatic and blood vessel integrity. Diabetes 62: 1970–1980, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dejana E, Orsenigo F, Molendini C, Baluk P, McDonald DM. Organization and signaling of endothelial cell-to-cell junctions in various regions of the blood and lymphatic vascular trees. Cell and tissue research 335: 17–25, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kakei Y, Akashi M, Shigeta T, Hasegawa T, Komori T. Alteration of cell-cell junctions in cultured human lymphatic endothelial cells with inflammatory cytokine stimulation. Lymphatic research and biology 12: 136–143, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. The Journal of clinical investigation 115: 1111–1119, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mellor KM, Varma U, Stapleton DI, Delbridge LM. Cardiomyocyte glycophagy is regulated by insulin and exposure to high extracellular glucose. American journal of physiology Heart and circulatory physiology 306: H1240–1245, 2014. [DOI] [PubMed] [Google Scholar]
- 35.Zhang M, Niu X, Hu J, Yuan Y, Sun S, Wang J, Yu W, Wang C, Sun D, Wang H. Lin28a protects against hypoxia/reoxygenation induced cardiomyocytes apoptosis by alleviating mitochondrial dysfunction under high glucose/high fat conditions. PloS one 9: e110580, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khodabukus A, Baar K. Glucose concentration and streptomycin alter in vitro muscle function and metabolism. Journal of cellular physiology 230: 1226–1234, 2015. [DOI] [PubMed] [Google Scholar]
- 37.Wang K, Wen L, Peng W, Li H, Zhuang J, Lu Y, Liu B, Li X, Li W, Xu Y. Vinpocetine attenuates neointimal hyperplasia in diabetic rat carotid arteries after balloon injury. PloS one 9: e96894, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.D’Alessio D, Lu W, Sun W, Zheng S, Yang Q, Seeley R, Woods SC, Tso P. Fasting and postprandial concentrations of GLP-1 in intestinal lymph and portal plasma: evidence for selective release of GLP-1 in the lymph system. American journal of physiology Regulatory, integrative and comparative physiology 293: R2163–2169, 2007. [DOI] [PubMed] [Google Scholar]
- 39.Yang YJ, Hope I, Ader M, Poulin RA, Bergman RN. Dose-response relationship between lymph insulin and glucose uptake reveals enhanced insulin sensitivity of peripheral tissues. Diabetes 41: 241–253, 1992. [DOI] [PubMed] [Google Scholar]
- 40.Chiu JD, Richey JM, Harrison LN, Zuniga E, Kolka CM, Kirkman E, Ellmerer M, Bergman RN. Direct administration of insulin into skeletal muscle reveals that the transport of insulin across the capillary endothelium limits the time course of insulin to activate glucose disposal. Diabetes 57: 828–835, 2008. [DOI] [PubMed] [Google Scholar]
- 41.Yang YJ, Hope ID, Ader M, Bergman RN. Insulin transport across capillaries is rate limiting for insulin action in dogs. The Journal of clinical investigation 84: 1620–1628, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dea MK, Hamilton-Wessler M, Ader M, Moore D, Schaffer L, Loftager M, Volund A, Bergman RN. Albumin binding of acylated insulin (NN304) does not deter action to stimulate glucose uptake. Diabetes 51: 762–769, 2002. [DOI] [PubMed] [Google Scholar]
- 43.Castillo C, Bogardus C, Bergman R, Thuillez P, Lillioja S. Interstitial insulin concentrations determine glucose uptake rates but not insulin resistance in lean and obese men. The Journal of clinical investigation 93: 10–16, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bohlen HG, Gasheva OY, Zawieja DC. Nitric oxide formation by lymphatic bulb and valves is a major regulatory component of lymphatic pumping. American journal of physiology Heart and circulatory physiology 301: H1897–1906, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bohlen HG, Wang W, Gashev A, Gasheva O, Zawieja D. Phasic contractions of rat mesenteric lymphatics increase basal and phasic nitric oxide generation in vivo. American journal of physiology Heart and circulatory physiology 297: H1319–1328, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gasheva OY, Zawieja DC, Gashev AA. Contraction-initiated NO-dependent lymphatic relaxation: a self-regulatory mechanism in rat thoracic duct. The Journal of physiology 575: 821–832, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Symons JD, McMillin SL, Riehle C, Tanner J, Palionyte M, Hillas E, Jones D, Cooksey RC, Birnbaum MJ, McClain DA, Zhang QJ, Gale D, Wilson LJ, Abel ED. Contribution of insulin and Akt1 signaling to endothelial nitric oxide synthase in the regulation of endothelial function and blood pressure. Circulation research 104: 1085–1094, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vicent D, Ilany J, Kondo T, Naruse K, Fisher SJ, Kisanuki YY, Bursell S, Yanagisawa M, King GL, Kahn CR. The role of endothelial insulin signaling in the regulation of vascular tone and insulin resistance. The Journal of clinical investigation 111: 1373–1380, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, Quaegebeur A, Ghesquiere B, Cauwenberghs S, Eelen G, Phng LK, Betz I, Tembuyser B, Brepoels K, Welti J, Geudens I, Segura I, Cruys B, Bifari F, Decimo I, Blanco R, Wyns S, Vangindertael J, Rocha S, Collins RT, Munck S, Daelemans D, Imamura H, Devlieger R, Rider M, Van Veldhoven PP, Schuit F, Bartrons R, Hofkens J, Fraisl P, Telang S, Deberardinis RJ, Schoonjans L, Vinckier S, Chesney J, Gerhardt H, Dewerchin M, Carmeliet P. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 154: 651–663, 2013. [DOI] [PubMed] [Google Scholar]
- 50.Krutzfeldt A, Spahr R, Mertens S, Siegmund B, Piper HM. Metabolism of exogenous substrates by coronary endothelial cells in culture. Journal of molecular and cellular cardiology 22: 1393–1404, 1990. [DOI] [PubMed] [Google Scholar]
- 51.Zeng G, Nystrom FH, Ravichandran LV, Cong LN, Kirby M, Mostowski H, Quon MJ. Roles for insulin receptor, PI3-kinase, and Akt in insulin-signaling pathways related to production of nitric oxide in human vascular endothelial cells. Circulation 101: 1539–1545, 2000. [DOI] [PubMed] [Google Scholar]
- 52.Pekala P, Marlow M, Heuvelman D, Connolly D. Regulation of hexose transport in aortic endothelial cells by vascular permeability factor and tumor necrosis factor-alpha, but not by insulin. The Journal of biological chemistry 265: 18051–18054, 1990. [PubMed] [Google Scholar]
- 53.Artwohl M, Brunmair B, Furnsinn C, Holzenbein T, Rainer G, Freudenthaler A, Porod EM, Huttary N, Baumgartner-Parzer SM. Insulin does not regulate glucose transport and metabolism in human endothelium. European journal of clinical investigation 37: 643–650, 2007. [DOI] [PubMed] [Google Scholar]
- 54.Roth Flach RJ, Danai LV, DiStefano MT, Kelly M, Menendez LG, Jurczyk A, Sharma RB, Jung DY, Kim JH, Kim JK, Bortell R, Alonso LC, Czech MP. Protein Kinase Mitogen-activated Protein Kinase Kinase Kinase Kinase 4 (MAP4K4) Promotes Obesity-induced Hyperinsulinemia. The Journal of biological chemistry 291: 16221–16230, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roth Flach RJ, DiStefano MT, Danai LV, Senol-Cosar O, Yawe JC, Kelly M, Garcia Menendez L, Czech MP. Map4k4 impairs energy metabolism in endothelial cells and promotes insulin resistance in obesity. American journal of physiology Endocrinology and metabolism 313: E303–e313, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Danai LV, Flach RJ, Virbasius JV, Menendez LG, Jung DY, Kim JH, Kim JK, Czech MP. Inducible Deletion of Protein Kinase Map4k4 in Obese Mice Improves Insulin Sensitivity in Liver and Adipose Tissues. Molecular and cellular biology 35: 2356–2365, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Darley-Usmar V. The powerhouse takes control of the cell; the role of mitochondria in signal transduction. Free radical biology & medicine 37: 753–754, 2004. [DOI] [PubMed] [Google Scholar]
- 58.Kluge MA, Fetterman JL, Vita JA. Mitochondria and endothelial function. Circulation research 112: 1171–1188, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kadlec AO, Beyer AM, Ait-Aissa K, Gutterman DD. Mitochondrial signaling in the vascular endothelium: beyond reactive oxygen species. Basic research in cardiology 111: 26, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baluk P, Fuxe J, Hashizume H, Romano T, Lashnits E, Butz S, Vestweber D, Corada M, Molendini C, Dejana E, McDonald DM. Functionally specialized junctions between endothelial cells of lymphatic vessels. The Journal of experimental medicine 204: 2349–2362, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yao LC, Baluk P, Srinivasan RS, Oliver G, McDonald DM. Plasticity of button-like junctions in the endothelium of airway lymphatics in development and inflammation. The American journal of pathology 180: 2561–2575, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Predescu D, Vogel SM, Malik AB. Functional and morphological studies of protein transcytosis in continuous endothelia. American journal of physiology Lung cellular and molecular physiology 287: L895–901, 2004. [DOI] [PubMed] [Google Scholar]
- 63.Patterson RM, Ballard CL, Wasserman K, Mayerson HS. Lymphatic permeability to albumin. The American journal of physiology 194: 120–124, 1958. [DOI] [PubMed] [Google Scholar]
- 64.Mayerson HS, Patterson RM, Mc KA, Lebrie SJ, Mayerson P. Permeability of lymphatic vessels. The American journal of physiology 203: 98–106, 1962. [DOI] [PubMed] [Google Scholar]
- 65.Cattelino A, Liebner S, Gallini R, Zanetti A, Balconi G, Corsi A, Bianco P, Wolburg H, Moore R, Oreda B, Kemler R, Dejana E. The conditional inactivation of the beta-catenin gene in endothelial cells causes a defective vascular pattern and increased vascular fragility. The Journal of cell biology 162: 1111–1122, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reed AL, Rowson SA, Dixon JB. Demonstration of ATP-dependent, transcellular transport of lipid across the lymphatic endothelium using an in vitro model of the lacteal. Pharmaceutical research 30: 3271–3280, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Triacca V, Guc E, Kilarski WW, Pisano M, Swartz MA. Transcellular Pathways in Lymphatic Endothelial Cells Regulate Changes in Solute Transport by Fluid Stress. Circulation research, 2017. [DOI] [PubMed] [Google Scholar]
- 68.Yuan Y, Granger HJ, Zawieja DC, Chilian WM. Flow modulates coronary venular permeability by a nitric oxide-related mechanism. The American journal of physiology 263: H641–646, 1992. [DOI] [PubMed] [Google Scholar]
- 69.Scallan JP, Huxley VH. In vivo determination of collecting lymphatic vessel permeability to albumin: a role for lymphatics in exchange. The Journal of physiology 588: 243–254, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Scallan JP, Davis MJ, Huxley VH. Permeability and contractile responses of collecting lymphatic vessels elicited by atrial and brain natriuretic peptides. The Journal of physiology 591: 5071–5081, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yuan SY. New insights into eNOS signaling in microvascular permeability. American journal of physiology Heart and circulatory physiology 291: H1029–1031, 2006. [DOI] [PubMed] [Google Scholar]
- 72.Kurose I, Kubes P, Wolf R, Anderson DC, Paulson J, Miyasaka M, Granger DN. Inhibition of nitric oxide production. Mechanisms of vascular albumin leakage. Circulation research 73: 164–171, 1993. [DOI] [PubMed] [Google Scholar]
- 73.Kubes P, Granger DN. Nitric oxide modulates microvascular permeability. The American journal of physiology 262: H611–615, 1992. [DOI] [PubMed] [Google Scholar]
- 74.Di Lorenzo A, Lin MI, Murata T, Landskroner-Eiger S, Schleicher M, Kothiya M, Iwakiri Y, Yu J, Huang PL, Sessa WC. eNOS-derived nitric oxide regulates endothelial barrier function through VE-cadherin and Rho GTPases. Journal of cell science 126: 5541–5552, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Patsouris D, Cao JJ, Vial G, Bravard A, Lefai E, Durand A, Durand C, Chauvin MA, Laugerette F, Debard C, Michalski MC, Laville M, Vidal H, Rieusset J. Insulin resistance is associated with MCP1-mediated macrophage accumulation in skeletal muscle in mice and humans. PloS one 9: e110653, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sartipy P, Loskutoff DJ. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proceedings of the National Academy of Sciences of the United States of America 100: 7265–7270, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Miteva DO, Rutkowski JM, Dixon JB, Kilarski W, Shields JD, Swartz MA. Transmural flow modulates cell and fluid transport functions of lymphatic endothelium. Circulation research 106: 920–931, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Johnson LA, Clasper S, Holt AP, Lalor PF, Baban D, Jackson DG. An inflammation-induced mechanism for leukocyte transmigration across lymphatic vessel endothelium. The Journal of experimental medicine 203: 2763–2777, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ledgerwood LG, Lal G, Zhang N, Garin A, Esses SJ, Ginhoux F, Merad M, Peche H, Lira SA, Ding Y, Yang Y, He X, Schuchman EH, Allende ML, Ochando JC, Bromberg JS. The sphingosine 1-phosphate receptor 1 causes tissue retention by inhibiting the entry of peripheral tissue T lymphocytes into afferent lymphatics. Nature immunology 9: 42–53, 2008. [DOI] [PubMed] [Google Scholar]
- 80.Podgrabinska S, Kamalu O, Mayer L, Shimaoka M, Snoeck H, Randolph GJ, Skobe M. Inflamed lymphatic endothelium suppresses dendritic cell maturation and function via Mac-1/ICAM-1-dependent mechanism. Journal of immunology (Baltimore, Md : 1950) 183: 1767–1779, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell 126: 677–689, 2006. [DOI] [PubMed] [Google Scholar]
- 82.Astrof S, Hynes RO. Fibronectins in vascular morphogenesis. Angiogenesis 12: 165–175, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Davis GE, Senger DR. Endothelial extracellular matrix: biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circulation research 97: 1093–1107, 2005. [DOI] [PubMed] [Google Scholar]
- 84.Francis SE, Goh KL, Hodivala-Dilke K, Bader BL, Stark M, Davidson D, Hynes RO. Central roles of alpha5beta1 integrin and fibronectin in vascular development in mouse embryos and embryoid bodies. Arteriosclerosis, thrombosis, and vascular biology 22: 927–933, 2002. [DOI] [PubMed] [Google Scholar]
- 85.Reynolds LE, Wyder L, Lively JC, Taverna D, Robinson SD, Huang X, Sheppard D, Hynes RO, Hodivala-Dilke KM. Enhanced pathological angiogenesis in mice lacking beta3 integrin or beta3 and beta5 integrins. Nature medicine 8: 27–34, 2002. [DOI] [PubMed] [Google Scholar]
- 86.Stenzel D, Lundkvist A, Sauvaget D, Busse M, Graupera M, van der Flier A, Wijelath ES, Murray J, Sobel M, Costell M, Takahashi S, Fassler R, Yamaguchi Y, Gutmann DH, Hynes RO, Gerhardt H. Integrin-dependent and -independent functions of astrocytic fibronectin in retinal angiogenesis. Development (Cambridge, England) 138: 4451–4463, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.van der Flier A, Badu-Nkansah K, Whittaker CA, Crowley D, Bronson RT, Lacy-Hulbert A, Hynes RO. Endothelial alpha5 and alphav integrins cooperate in remodeling of the vasculature during development. Development (Cambridge, England) 137: 2439–2449, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Frye M, Taddei A, Dierkes C, Martinez-Corral I, Fielden M, Ortsater H, Kazenwadel J, Calado DP, Ostergaard P, Salminen M, He L, Harvey NL, Kiefer F, Makinen T. Matrix stiffness controls lymphatic vessel formation through regulation of a GATA2-dependent transcriptional program. Nature communications 9: 1511, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Montagnani M, Ravichandran LV, Chen H, Esposito DL, Quon MJ. Insulin receptor substrate-1 and phosphoinositide-dependent kinase-1 are required for insulin-stimulated production of nitric oxide in endothelial cells. Molecular endocrinology (Baltimore, Md) 16: 1931–1942, 2002. [DOI] [PubMed] [Google Scholar]
- 90.Hermann C, Assmus B, Urbich C, Zeiher AM, Dimmeler S. Insulin-mediated stimulation of protein kinase Akt: A potent survival signaling cascade for endothelial cells. Arteriosclerosis, thrombosis, and vascular biology 20: 402–409, 2000. [DOI] [PubMed] [Google Scholar]
- 91.Akakabe Y, Koide M, Kitamura Y, Matsuo K, Ueyama T, Matoba S, Yamada H, Miyata K, Oike Y, Ikeda K. Ecscr regulates insulin sensitivity and predisposition to obesity by modulating endothelial cell functions. Nature communications 4: 2389, 2013. [DOI] [PubMed] [Google Scholar]
- 92.Kim JA, Montagnani M, Koh KK, Quon MJ. Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation 113: 1888–1904, 2006. [DOI] [PubMed] [Google Scholar]
- 93.Montagnani M, Chen H, Barr VA, Quon MJ. Insulin-stimulated activation of eNOS is independent of Ca2+ but requires phosphorylation by Akt at Ser(1179). The Journal of biological chemistry 276: 30392–30398, 2001. [DOI] [PubMed] [Google Scholar]
- 94.Jeong SO, Son Y, Lee JH, Cheong YK, Park SH, Chung HT, Pae HO. Resveratrol analog piceatannol restores the palmitic acid-induced impairment of insulin signaling and production of endothelial nitric oxide via activation of anti-inflammatory and antioxidative heme oxygenase-1 in human endothelial cells. Molecular medicine reports 12: 937–944, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang Y, Cheng KK, Lam KS, Wu D, Wang Y, Huang Y, Vanhoutte PM, Sweeney G, Li Y, Xu A. APPL1 counteracts obesity-induced vascular insulin resistance and endothelial dysfunction by modulating the endothelial production of nitric oxide and endothelin-1 in mice. Diabetes 60: 3044–3054, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.De Nigris V, Pujadas G, La Sala L, Testa R, Genovese S, Ceriello A. Short-term high glucose exposure impairs insulin signaling in endothelial cells. Cardiovascular diabetology 14: 114, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Huang Y, Lei L, Liu D, Jovin I, Russell R, Johnson RS, Di Lorenzo A, Giordano FJ. Normal glucose uptake in the brain and heart requires an endothelial cell-specific HIF-1alpha-dependent function. Proceedings of the National Academy of Sciences of the United States of America 109: 17478–17483, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bar RS, Siddle K, Dolash S, Boes M, Dake B. Actions of insulin and insulinlike growth factors I and II in cultured microvessel endothelial cells from bovine adipose tissue. Metabolism: clinical and experimental 37: 714–720, 1988. [DOI] [PubMed] [Google Scholar]
- 99.Gerritsen ME, Burke TM, Allen LA. Glucose starvation is required for insulin stimulation of glucose uptake and metabolism in cultured microvascular endothelial cells. Microvascular research 35: 153–166, 1988. [DOI] [PubMed] [Google Scholar]
- 100.Kwok CF, Goldstein BJ, Muller-Wieland D, Lee TS, Kahn CR, King GL. Identification of persistent defects in insulin receptor structure and function capillary endothelial cells from diabetic rats. The Journal of clinical investigation 83: 127–136, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gosmanov AR, Stentz FB, Kitabchi AE. De novo emergence of insulin-stimulated glucose uptake in human aortic endothelial cells incubated with high glucose. American journal of physiology Endocrinology and metabolism 290: E516–522, 2006. [DOI] [PubMed] [Google Scholar]
- 102.Viator RJ, Khader H, Hingorani N, Long S, Solodushko V, Fouty B. Hypoxia-induced increases in glucose uptake do not cause oxidative injury or advanced glycation end-product (AGE) formation in vascular endothelial cells. Physiological reports 3, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.King MJ, Michel D, Foldvari M. Evidence for lymphatic transport of insulin by topically applied biphasic vesicles. The Journal of pharmacy and pharmacology 55: 1339–1344, 2003. [DOI] [PubMed] [Google Scholar]
- 104.Poulin RA, Steil GM, Moore DM, Ader M, Bergman RN. Dynamics of glucose production and uptake are more closely related to insulin in hindlimb lymph than in thoracic duct lymph. Diabetes 43: 180–190, 1994. [DOI] [PubMed] [Google Scholar]
- 105.Camu F, Rasio E. Peripheral glucose uptake in relation to physiological levels of plasma and lymph insulin. European journal of clinical investigation 2: 188–194, 1972. [DOI] [PubMed] [Google Scholar]
- 106.Hamilton-Wessler M, Ader M, Dea MK, Moore D, Loftager M, Markussen J, Bergman RN. Mode of transcapillary transport of insulin and insulin analog NN304 in dog hindlimb: evidence for passive diffusion. Diabetes 51: 574–582, 2002. [DOI] [PubMed] [Google Scholar]