

Abstract
CNS inflammatory responses are linked to cognitive impairment in humans. Research in animal models supports this connection by showing that inflammatory cytokines suppress long-term potentiation (LTP), the best-known cellular correlate of memory. Cytokine-induced modulation of LTP has been previously studied in vivo or in brain slices, two experimental approaches containing multiple cell populations responsive to cytokines. In their target cells, cytokines commonly increase the expression of multiple cytokines, thus increasing the complexity of brain cytokine networks even after single-cytokine challenges. Whether cytokines suppress LTP by direct effects on neurons or by indirect mechanisms is still an open question. Here, we evaluated the effect of a major set of inflammatory cytokines including TNFα (tumor necrosis factor-α), IL-1β (interleukin-1β) and IL-18 (interleukin-18) on chemically induced LTP (cLTP) in isolated hippocampal synaptosomes of mice, using Fluorescence Analysis of Single-Synapse Long-Term Potentiation (FASS-LTP). We found that TNFα and IL-1β suppress synaptosomal cLTP. In contrast, cLTP was not affected by IL-18, at a concentration previously shown to block LTP in hippocampal slices. We also found that IL-18 does not impair cLTP or BDNF (brain-derived neurotrophic factor) signaling in primary hippocampal neuronal cultures. Thus, using both synaptosomes and neuron cultures, our data suggest that IL-18 impairs LTP by indirect mechanisms, which may depend on non-neuronal cells, such as glia. Notably, our results demonstrate that TNFα and IL-1β directly suppress hippocampal plasticity via neuron-specific mechanisms. A better understanding of the brain’s cytokine networks and their final molecular effectors is crucial to identify specific targets for intervention.
Keywords: inflammation, cytokines, hippocampus, synaptosomes, cLTP, FASS-LTP
Introduction
Brain inflammation comprises a complex response orchestrated by multiple cells and modulated by molecular networks of soluble factors, mainly cytokines [1]. Observations in both humans and animals indicate a link between brain inflammation and cognitive alterations [2–4], which might be reflecting suppression of neuronal functions by cytokines. However, elucidating neuron-specific effects of cytokines has been challenging because most brain cells express cytokine receptors [5, 6]. Moreover, cytokines commonly increase the expression of multiple cytokines in their target cells, thus increasing the complexity of brain cytokine networks even after single-cytokine challenges [6]. A better understanding of these cascades and their final neuron-specific effectors is crucial to identify specific targets for intervention.
Relevant for cytokine-cell maps, data on peripheral and brain levels of cytokines provide information reflecting steady-state levels of cytokine networks in health and disease. Notably, interleukin-1β (IL-1β), interleukin-18 (IL-18), and tumor necrosis factor-α (TNFα) belong to a major set of cytokines driving inflammatory states in the brain in both acute (e.g., infection and surgery) and chronic diseases (e.g., depression and Alzheimer’s disease (AD)) [7–9]. In particular, changes in peripheral and cerebrospinal fluid (CSF) levels of IL-1β, TNFα and IL-18 accompany AD [10–12]. A comprehensive analysis of 118 research articles to compare levels of 66 cytokines in blood or CSF obtained from mild cognitive impairment (MCI) and AD patients suggested that IL-1β and TNFα increase slowly during disease progression, while IL-18 transiently increases at time of MCI to AD conversion [10]. Moreover, in both humans and animals, IL-1β [10, 13–18], IL-18 [18, 19], and TNFα [20–24] have been associated with cognitive impairment, suggesting that these cytokines can impair functional properties of neuronal circuitries.
The hippocampus, a region containing key neuronal circuitries for memory, is a brain target for inflammation. Accordingly, inflammatory states induced by infection [15], stress [25], brain pathology [26] and aging [13] impair hippocampal-dependent memory in animal models. Consistent with the evidence that memory deficits arise from synaptic dysfunction [27–30], inflammatory states suppress long-term potentiation (LTP), a crucial synaptic mechanism defined as a rapid and remarkably persistent increase in synaptic transmission elicited by brief patterns of afferent activity [31]. Evidence from several laboratories using in vivo [32, 33] and in vitro [34–38] electrophysiological recordings have demonstrated that IL-1β suppresses LTP in the hippocampus. Similarly, IL-18 [39–41] and TNFα [36, 41–43] suppress LTP in hippocampal slices from rodents. Using a single-synapse approach to study LTP, we have recently reported that IL-1β can suppress chemically-induced LTP (cLTP) directly in mouse synaptosomes [44], a preparation containing presynaptic terminals attached to post-synaptic dendritic spines [45], providing intact synaptic units for biochemical, structural [46] and functional [47–50] analysis. However, whether TNFα or IL-18 similarly acts as final effectors directly targeting synapses to suppress LTP remains to be investigated. Alternatively, these cytokines may impair LTP by activating indirect mechanisms driven by cellular-molecular cascades involving microglia and astrocytes. To test the possibility that TNFα and IL-18 suppress LTP directly at synapses, we used Fluorescence Analysis of Single-Synapse Long-Term Potentiation (FASS-LTP), our novel method to study cLTP in isolated synaptosomes [44, 51]. Synaptosomal cLTP is based on application of the NMDA receptor co-agonist glycine, which facilitates NMDA receptor activation [52, 53]. Previous work in hippocampal slices using cLTP and electrical-stimulated LTP has confirmed that glycine-induced cLTP and classical LTP approaches share underlying cellular processes [53]. In contrast to current electrophysiological approaches however, FASS-LTP allows for the analysis of synapses in isolation (i.e., in the absence of microglia and astrocytes) and thus for identifying neuron-specific mechanisms. To further focus on neuron-specific mechanisms, we used hippocampal neuronal cultures to evaluate cytokines’ effects on cLTP and BDNF signaling, which facilitates hippocampal LTP [54–56]. Overall, our data support the idea that inflammatory cytokines differentially suppress LTP via direct and indirect mechanisms.
Materials and Methods
Animals
Both mice and rats were housed with food and water ad libitum. Lights were maintained on a 12:12 light/dark cycle. All procedures used in the present study followed the Principles of Laboratory Animal Care from NIH and were approved by the University of California, Irvine, Institutional Animal Care and Use Committee.
Hippocampal Cell Cultures
Primary cultures were prepared from E18 Sprague-Dawley rats as described previously [57]. Cells were maintained at 37°C, O2/CO2 (95%/5%) in Neurobasal medium supplemented with B27, GlutaMAX, and penicillin/streptomycin (all culture reagents from Life Sciences). After 5–7 DIV, neurons were treated at 37 °C with 50 ng/mL BDNF, 100 ng/ml IL-18 and 50 ng/ml IL-1β (PeproTech), with control neurons receiving equal volumes of vehicle. For cLTP, 7–10 DIV neurons were treated as described previously [52, 58]. Briefly, hippocampal neurons were changed to normal pre-warmed (37°C) external solution (125 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 33 mM glucose, 5 mM HEPES, pH = 7.4) for 15 min. Next, cLTP was induced by changing the external buffer to Mg2+-free external solution (125 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 33 mM glucose, 5 mM HEPES, pH = 7.4) supplemented with 0.2 mM glycine, 0.02 mM bicuculline and 0.003 mM strychnine, for 10 min. After cLTP stimulation, the incubation solution was changed back to external solution for 20 min. Neurons were treated with vehicle or cytokines before (15 min), during (10 min) and after cLTP stimulation (20 min). It should be noted that we used E18 rats for culture studies and mice for FASS-LTP experiments to open the possibility of follow up studies in transgenic models. Rat cultures provided great quantities of tissue for signaling pathway studies.
FASS-LTP
Synaptosomes isolation
Fresh crude synaptosome P2 fractions were obtained from whole mouse hippocampus using our long-standing protocol [47]. All the steps for synaptosome P2 fraction isolation were carried out at 4°C; sucrose buffer, grinder, pestle and microfuge tubes were all pre-cooled on ice. Hippocampi were rapidly dissected form a single mouse and homogenized in 320 mM sucrose (1.5 ml) containing HEPES [10 mM] and protease/phosphatase inhibitors cocktail (Pierce), pH 7.4. Homogenization consisted of 6–8 manual strokes in a Glass-Teflon grinder, clearance (between plunger and glass): 0.15–0.25mm. Plunger was gently rotated during strokes while the grinder was kept on ice. The homogenate was centrifuged at 1200 × g for 10 min. Supernatant (S1, containing mitochondria and synaptosomes) was transferred into two clean microfuge tubes and centrifuged at 12,000 × g for 20 min. Supernatants (S2) were carefully removed using a plastic tip and vacuum. Pellets (P2, corresponding to the crude synaptosome fraction) were resuspended by gently pipetting up and down (10–20 times) in 1.5 ml of extracellular (tube 1) or cLTP (tube 2) solutions. Extracellular solution contains (in mM): 120 NaCl, 3 KCl, 2 CaCl2, 2 MgCl2, 15 glucose, 15 HEPES, pH=7.4; whereas cLTP solution is Mg2+-free and contains (in mM): 125 NaCl, 2 CaCl2, 5 KCl, 10 HEPES, 30 glucose, pH=7.4 [59]. Synaptosome P2 fractions were filtered with a 40-μm-pore cell strainer (BD Biosciences) and incubated in a cell culture dish (30 mm) with agitation at RT (room temperature) for 10–15 min for recovery. A P2 fraction aliquot was used to determine protein concentration (BCA assay using BSA as a standard, Thermo). To prevent synaptosome damage, Finntip™ pipette tips (Thermo) were used in all steps.
Stimulation
After recovery, 180 μl synaptosomes (50–200 μg protein, BCA assay) maintained in cLTP solution were transferred to cytometry tubes. As control, an equal volume (180 μl) of synaptosome maintained in external solution was also transferred to a cytometry tube. Synaptosomes in external solution were used to determine basal levels of potentiated synaptosomes (see below). All cytometry tubes were pre-warmed in a 37°C bath (5 min) before stimulation. External, glycine and KCl solutions were also pre-warmed at 37°C. Next, 20 μl of external solution was added to control synaptosomes (in external solution), whereas 20 μl of glycine ([5 mM] in cLTP solution freshly supplemented with 0.001 mM strychnine and 0.02 mM bicuculline methiodide) was added to synaptosomes in cLTP solution (final [glycine]=500 μM[60]). Glycine was incubated for 15 min to prime synaptic NMDAR. After glycine treatment, synaptosomes were depolarized with 100 μl of a [high] KCl solution consisting of (in mM): 50 NaCl, 2 CaCl2, 100 KCl, 10 HEPES, 30 glucose, 0.5 glycine 0.001 strychnine, 0.02 bicuculline methiodide, pH 7.4 (final [KCl]=37 mM), and incubated for 30 min; 100 μl of external solution were added to control synaptosomes. After KCl incubation, stimulation was stopped by sequential addition of 0.5 ml of ice-cold 0.1 mM EDTA-PBS (pH 7.4) and 4 ml of ice-cold blocking buffer (5% FBS in PBS). Tubes were chilled on ice and immediately centrifuged at 2,500 × g for 6 min at 4°C (9000 rpm, Sorvall RT6000B). After centrifugation, the pellet was resuspended by gentle finger agitation (no vortex) and kept on ice.
Immunolabeling
Primary antibody solution (400 μl) was added to the resuspended pellet and incubated for 30 min on ice with agitation. Primary antibody solution contained rabbit anti-GluA1 (Cell signaling #13185; 1:400) and mouse anti-Nrx1β (UC Davis/NIH NeuroMab Facility, 75–216) antibodies, both at 2.5 μg/ml in blocking buffer (5% FBS in PBS). After incubation synaptosomes were washed with 4 ml of ice-cold blocking buffer and centrifuged (2,500 × g/6min/4°C). Supernatant was discarded and pellet gently resuspended as previously described. Secondary antibody solution (400 μl) was added to each tube and incubated for 30 min on ice with agitation and protected from light. After incubation synaptosomes were washed as previously described. Secondary antibody solution contained anti-rabbit-Alexa-488 and anti-mouse-Alexa 647 antibodies (Life Sciences), both at 2.5 μl/ml. Endogenous/non-specific background fluorescence for each marker were determined using secondary antibody staining only in a tube containing synaptosomes maintained in external solution (37°C, 45 min); no differences in background fluorescence was found when comparing synaptosomes in external solution (basal state) or following cLTP stimulation. After the last wash, the pellet was resuspended as previously described and 400 μl of 0.25% paraformaldehyde in PBS was added to each tube. Samples were protected from light, maintained at 4°C and run on a flow cytometer within 6 h.
Flow cytometry
Samples were acquired using a Becton Dickinson FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA) equipped with argon 488 nm and helium-neon 635 nm lasers. Relative size and granularity was determined by forward (FSC) and side scatter (SSC) properties. FSC, SSC and fluorescence (FL1 [530±15 nm] and FL4 [650±25 nm]) signals were collected using log amplification. FSC-SSC plots were used to select particles matching the size of synaptosomes (~1.0 μm) using calibrated beads (Polysciences, Inc.) (Fig. 1a), as previously described [51]. Identical FSC settings were used for acquiring data on bead standards and samples. Small fragments and debris were excluded by establishing a FSC-H threshold to exclude particles smaller than 0.5 μm. Settings for fluorescence amplification on FL1 and FL4 photomultiplier tube detectors were based on the emission detected on size-based gated particles. Alexa 488 and Alexa 647 fluorochromes were detected by the FL1 and FL4 detectors, respectively. Ten thousand size-gated particles were collected and analyzed for each sample; event rate: approximately 500/sec. Analysis was performed using the CellQuest Pro software (BD Biosciences). Gates are set based on standard immunostaining protocols for flow cytometry [61–63]. Briefly, we use staining controls to discriminate background fluorescence due to both sample itself and unspecific binding of fluorescence-labeled antibodies. These controls are synaptosomes incubated only with secondary antibodies (no primary antibodies, Fig. 1c).
Fig. 1.
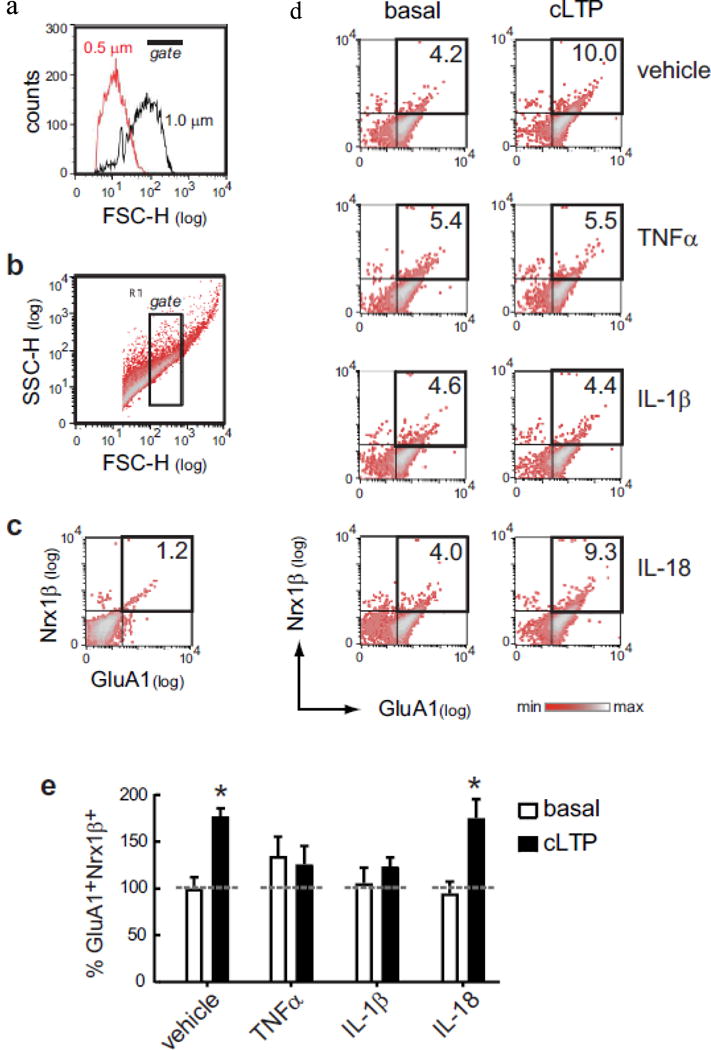
Cytokine-induced modulation of cLTP in synaptosomes. a Flow cytometry analysis includes size-based identification of synaptosomes. We exclude small particles having a size equivalent to 0.5 μm calibrated beads, while select particles of ~ 1.0 μm calibrated beads. b Based on calibrated beads, we set a threshold in the Forward scatter (FSC) channel. Forward-Side (FSC-SSC) density plot shows the size-complexity profile of particles in the synaptosomal P2 fraction isolated from fresh mouse hippocampus. The inside rectangle (gate) selects putative synaptosomes according with their size (~ 1.0 μm = size-gated synaptosomes), relative to calibrated beads. In size-gated synaptosomes, FASS-LTP identifies potentiated synapses by tracking GluA1 and Nrx1β surface staining. c Thresholds for endogenous/non-specific fluorescence for each marker were set by staining with secondary antibodies only. d Synaptosomal P2 fractions maintained either in external or Mg2+-free external solution were treated with vehicle or cytokines (50 ng/ml TNFα, 50 ng/ml IL-1β or 100 ng/ml IL-18) for 15 min. After cytokine treatment, cLTP was induced by sequential stimulation using 500 μM glycine (15 min) and 37 mM KCl (30 min). As control, equivalent volumes of external solution were added to a parallel set of synaptosomal fractions maintained in external solution (basal). Representative two-color parameter density plots show GluA1 (x-axis) and Nrx1β (y-axis) surface levels in size-gated synaptosomes following experimental treatments. e Values normalized to basal state of vehicle-treated synaptosomes, mean ± SEM, P = 0.003, two-way ANOVA, effect of cLTP stimulation, F1,38 = 10.0, *P < 0.05 Bonferroni posttest, n = 8 independent experiments (8 mice, 9–10 months)
Western blot
Homogenates from neuronal cultures and immunoblotting were prepared as described previously [57]. After treatment, cells were washed in ice-cold PBS, then lysed in RIPA/Nonidet P-40 buffer containing protease and phosphatase inhibitor mixtures (Pierce, Thermo Fisher Scientific), and immediately frozen. Cells were harvested in Laemmli buffer, boiled, run on Criterion Tris-HCl gels, and then transferred to PVDF membranes according to the manufacturer’s instructions (BD Biosciences). Membranes were blocked in 5% BSA for 1 h and then probed with the primary antibody anti-p-Akt (Cell Signaling, #4060, dil=1:2000) (4 °C, overnight). The membranes were washed (4 × 10 min in TBS with 0.1% Tween-20, vol/vol) and probed with HRP-conjugated secondary antibody for 1 h. The membranes were washed and developed by using Pierce Chemiluminescent Substrate (Pierce; 32106). Blots were washed, stripped, and reprobed with antibodies to Akt (Cell Signaling, #2920, dil=1:1000) and beta-actin (Sigma, #A2066, dil=1:3000). Membranes were incubated in stripping buffer according to the manufacturer’s instructions (Pierce, 46430). ImageJ software was used for densitometry analysis.
Statistical analyses
Sample sizes were chosen on the basis of previous experience with FASS-LTP [44, 51] and cell cultures [57]. As assumptions of normality (Shapiro-Wilk test) and equal variance (Bartlett’s test) were met, for mean comparisons of three or more groups one-way ANOVA was followed by post hoc Tukey’s test. Two-way ANOVA was followed by Bonferroni’s post hoc test. Statistical tests were performed using GraphPad Prism 5.0. Data are presented as mean ± SEM. A P value < 0.05 was considered significant.
Results
We used our flow cytometry-based method FASS-LTP to evaluate cLTP in hippocampal synaptosomes of adult mice (9–10 month-old). To induce cLTP, synaptosomes were primed with glycine (500 μM, 15 min) in Mg2+-free solution followed by depolarization using KCl (37 mM) [44, 51]. Using flow cytometry and size calibrated beads (Fig. 1a), FASS-LTP selectively and reliably identified synaptosomal particles from the crude P2 fraction [44, 51], which can be rapidly obtained (~30 min) and is enriched in synaptosomes [47, 64, 65]. We refer to these particles as size-gated synaptosomes. Using flow cytometry analysis on the crude P2 fraction, we have previously shown that the subset of particles of ~1.0 μm is enriched in viable synaptosomes: a high proportion of these size-gated particles stain positive for calcein-AM (> 90%), and express extra- and intra-cellular synaptic markers (e.g., synaptophysin, synapsin-I, PSD95) but no histone-H3 (nuclear) [44, 51, 66, 67]. Once gated by size (Fig. 1b), FASS-LTP identifies potentiated synaptosomes by extracellular labeling (without permeabilization) using antibodies specific for extracellular epitopes on the AMPA receptor subunit GluA1 and neurexin-1β (Nrx1β) (Fig. 1c, d). Nrx1β is a presynaptic adhesion molecule stabilized at the membrane surface by synaptic activity [68] which captures postsynaptic surface GluA1 via PSD95 [69]. GluA1 and Nrx1β double labeling ensures that we are analyzing intact synaptosomes that contain both pre- and post-synaptic elements.
A useful advantage of FASS-LTP is the possibility of testing multiple samples in parallel using minimal amount of tissue [51, 70]. In particular, we simultaneously tested the effect of TNFα, IL-1β and IL-18 by incubating hippocampal synaptosomes with each cytokine or vehicle for 15 min before cLTP stimulation. In synaptosomes treated with vehicle we detected an increase in the level of GluA1+Nrx1β+ synaptosomes following cLTP, relative to non-stimulated synaptosomes (basal condition: synaptosomes maintained in Mg2+-containing external solution) (P < 0.05, basal vs cLTP, Bonfferoni post hoc, two-way ANOVA; Fig. 1d, e). In contrast, in TNFα-treated synaptosomes levels of potentiated GluA1+Nrx1β+ synaptosomes were found to be similar comparing basal and cLTP conditions (Fig. 1d, e), thus indicating that TNFα suppresses cLTP directly at the synapse. It is noteworthy that, by itself, TNFα slightly increased GluA1+Nrx1β+ levels in the absence of cLTP stimulation (135 ± 20% GluA1+Nrx1β+ in basal condition, Fig. 1d, e). This result resembles previous studies showing that TNFα slightly and rapidly increases basal neurotransmission [43], as well as surface GluA1-containing AMPA receptors at postsynaptic spines to control neuronal firing rate via homeostatic plasticity (scaling) mechanisms [71]. In IL-1β-treated synaptosomes, analysis of GluA1+Nrx1β+ double-labeled events showed that this cytokine suppresses cLTP (Fig. 1d, e). Notably, we found that IL-1β specifically impairs activity-dependent plasticity, but has no detectable effect in non-stimulated synaptosomes (106 ± 16% GluA1+Nrx1β+ in basal condition, Fig. 1d, e). In contrast to TNFα and IL-1β effects on plasticity, IL-18 treatment did not affect synaptosomal cLTP, which displayed a significant increase in GluA1+Nrx1β+ size-gated synaptosomes in the presence of IL-18 (P < 0.05, basal vs cLTP, Bonfferoni post hoc, two-way ANOVA; Fig. 1d, e). IL-18 also did not affect basal levels of potentiated synapses (94 ± 13% GluA1+Nrx1β+ in basal condition, Fig. 1d, e).
To validate FASS-LTP data showing no IL-18 effect on synaptosomal cLTP, we evaluated whether IL-18 influences activity-dependent signaling in primary neuronal cultures, a widely used approach to study neuron-specific mechanisms. In particular, we tested whether IL-18 modulates cLTP or BDNF signaling in rat hippocampal neuronal cultures. We used a hippocampal neuron culture model of cLTP based on a brief application of the NMDA receptor co-agonist glycine [52, 53]. In this experimental model, glycine-induced cLTP facilitates insertion of AMPA receptors into the postsynaptic surface and spine remodeling [52, 58, 72, 73]. As activation of PI3K-Akt pathway is crucial for insertion of AMPA receptors into the postsynaptic surface after LTP [74–77], we evaluated whether IL-18 influences cLTP-induced Akt activation. Consistent with FASS-LTP data (Fig. 1), we found that IL-18 does not affect cLTP-induced Akt activation, as reflected by its phosphorylation at serine-473 (p-Akt) (Fig. 2). In contrast, IL-1β treatment significantly reduced cLTP-induced increase in p-Akt levels (Fig. 2). These results confirm FASS-LTP data and further support previous reports on the impairment of synaptic plasticity by IL-1β [32–37].
Fig. 2.
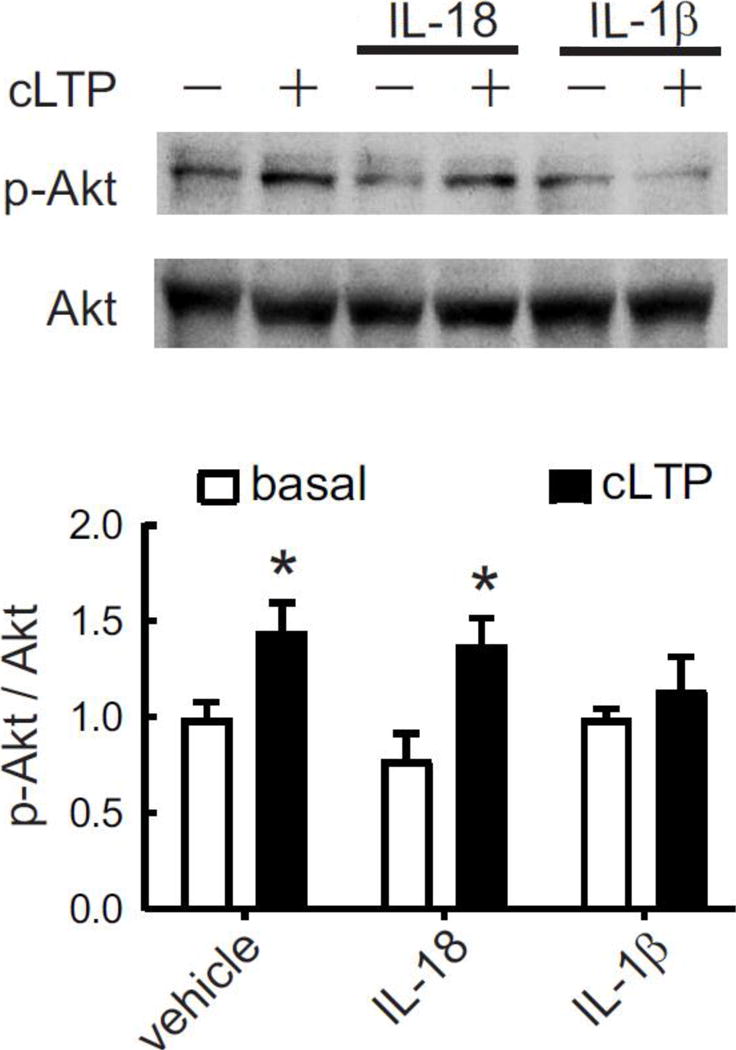
Cytokine-induced modulation of cLTP in neurons. After 20 min of cLTP stimulation, phosphorylated and total levels of Akt were evaluated by Western blot in primary rat hippocampal neurons (7–10 DIV) pretreated for 15 min with IL-18 (100 ng/ml), IL-1β (50 ng/ml) or vehicle, as control. Densitometry values were normalized to non-stimulated, vehicle-treated neurons. For p-Akt/Akt ratio: P = 0.003, two-way ANOVA, effect of cLTP stimulation, F1,41 = 9.85, *P < 0.05 Bonferroni posttest vs respective controls, n = 9 independent experiments (neurons from different embryo litters). Data are presented as mean ± SEM.
BDNF is crucial to hippocampal LTP [54–56]. While inhibition of BDNF signaling by IL-1β in cortical [78] and hippocampal [44, 57, 67] neurons has been previously reported, to the best of our knowledge no previous report had tested whether IL-18 modulates neuronal BNDF signaling. In particular, we evaluated whether IL-18 or IL-1β (as positive control) modulates BDNF-induced Akt activation in cultures of hippocampal neurons, which express both IL-1 receptor-1 (IL-1R1) [44] and IL-18 receptor [79]. We treated neurons with IL-18, IL-1β or vehicle for 2 h before BDNF stimulation. Data showed that IL-1β but not IL-18 suppresses BDNF-induced Akt phosphorylation (P < 0.05, Bonfferoni post hoc, two-way ANOVA; Fig. 3a). Next, considering that IL-18 signaling in neurons activates multiple pathways that follow differential kinetics (e.g., early MAPK but late STAT3 modulation [79]), we pretreated neurons with IL-18 for 0.5, 1 and 2 h, before BDNF stimulation for 1h. Data showed no inhibition of BDNF signaling by IL-18 at any time point tested (Fig. 3b). Altogether, our results indicate that IL-18 does not reduce activity-dependent signaling either in synaptosomes or in hippocampal neuron cultures.
Fig. 3.
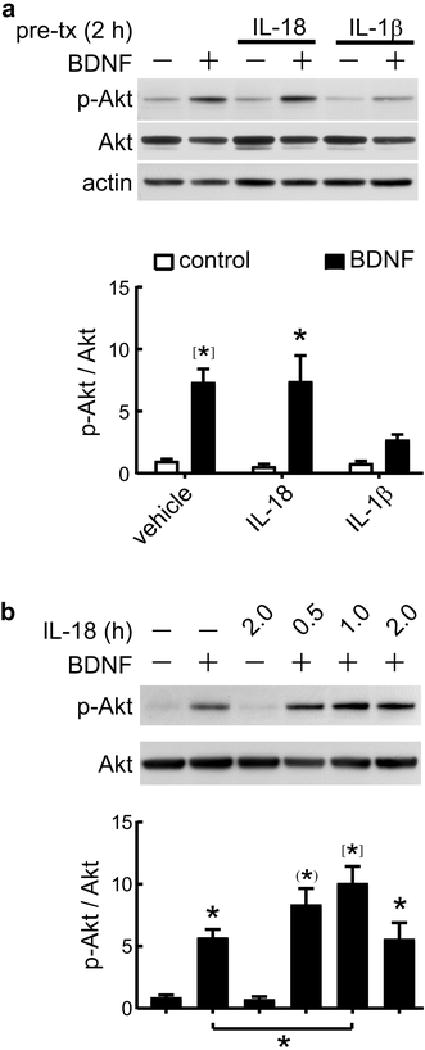
Cytokine-induced modulation of BDNF signaling in neurons. a Primary rat hippocampal neurons (5–7 DIV) were preincubated for 2 hr with IL-18 (100 ng/ml), IL-1β (50 ng/ml) or vehicle, as control. After cytokine treatment, neurons were stimulated with BDNF (50 ng/ml) for 1hr. Image shows representative Western blot of phosphorylated and total levels of Akt, as well as beta-actin, as loading control. Relative levels of p-Akt (Ser-473)/Akt were normalized with vehicle-treated neurons (control). P < 0.0001, two-way ANOVA, effect of BDNF stimulation, F1,26 = 24.53, *P < 0.05, [*]P < 0.001, Bonferroni posttest vs respective controls, n = 6. b Neurons (5–7 DIV) were preincubated with 100 ng/ml IL-18 for the indicated times. After cytokine treatment, neurons were stimulated with BDNF (50 ng/ml) for 1hr. Image shows representative Western blot of phosphorylated and total levels of Akt. Relative p-Akt/Akt levels were normalized with vehicle-treated neurons, P < 0.0001, ANOVA, *P < 0.05; (*), P < 0.01; [*]P < 0.001, Tukey’s post hoc test vs control, n = 5. Data are presented as mean ± SEM
Surprisingly, compared to neurons treated with BDNF, p-Akt levels were higher in neurons treated with BDNF (1 h) and preincubated with IL-18 for 1 h (total time IL-18 treatment = 2 h) (P < 0.05, Fig. 3b), a finding resembling the transient activation of Akt by IL-18 treatment for 2 h but not 3 h in cortical neurons [80]. It is noteworthy that we did not detect additive effects on p-Akt levels by BDNF (1 h) plus IL-18 pretreatment (2 h) (total time IL-18 = 3 h) (Fig. 3b), thus supporting the idea that IL-18 signaling, in a time-dependent manner, may be additive to BDNF signaling at the PI3K-Akt pathway.
Discussion
The idea that brain inflammatory signaling contributes to cognitive impairment is increasingly being supported by clinical and basic research. A key molecular signature of inflammation comprises sets of cytokines released by multiple brain cells including astrocytes and microglia. While cytokine networks are complex, including feedback loops, pleiotropy and crosstalk, we propose that dissecting final factors directly impairing synaptic plasticity may establish crucial endpoints in the cytokines-cell map. Identifying endpoint effectors could significantly contribute for developing novel therapeutics, designed to target selectively deleterious effects of inflammation on neurons and cognition, while preserving the benefits of brain immune responses. In this study, we report for first time that TNFα and IL-1β, but not IL-18, can suppress cLTP directly at synapses of adult mice. These data further exemplifies unique roles of members of the IL-1 family, a major set of 11 cytokines including IL-1β and IL-18.
Brain expression of IL-1 (α or β) and TNFα genes is upregulated in at least eight major diseases/disorders of the brain (e.g., Alzheimer’s, Down’s, Parkinson’s, major depressive disorder, multiple sclerosis, ischemia, trauma and epilepsy) [9, 81]. Elevated levels of IL-1 and TNFα in the brain may contribute to disease symptoms by affecting basic neuron functions. For instance, IL-1β and TNFα may lead to dementia by affecting synaptic plasticity. In support of this idea, research in animal models has shown that elevated signaling of both IL-1β [32–34, 36, 37, 82] and TNFα [20, 36, 42, 43] impairs LTP and memory. In agreement with our previous report that IL-1β impairs cLTP in hippocampal synaptosomes of middle-aged mice (12–15 month-old) [44], the present study shows that IL-1β similarly suppresses synaptosomal cLTP in adult mice (9–10 month-old), thus providing unequivocal proof that IL-1β can impair synaptic plasticity via neuron-specific mechanisms. We further extended FASS-LTP analysis to TNFα, and demonstrated that this cytokine suppresses cLTP in hippocampal synaptosomes of adult mice. Thus, by showing that both IL-1β and TNFα suppress cLTP directly in synaptosomes, our data support the evolving concept that neuronflammation negatively affects cognition directly at the synapse, and may lead to dementia [8]. It is worth mentioning that we treated synaptosomes with IL-1β and TNFα at 50 ng/ml, a high concentration compared to CSF levels of IL-1β and TNFα in AD (< 300 pg/ml) [83, 84]. However, in line with previous reports testing the effects of IL-1β and TNFα at 1–100 ng/ml in hippocampal slices [34, 36–38, 42, 43], our data provide valuable in vitro information supporting a role of high-possible saturating-concentrations of inflammatory cytokines on synaptic dysfunction. In AD, the most common dementia, neuroinflammation interacts with key pathological and genetic risk factors (e.g., amyloid beta, tau, and APOE4). Studies in humans [85] and in transgenic mice [86, 87] have demonstrated that ApoE4, the strongest genetic risk factor for late-onset AD, is associated with higher innate immune reactivity than ApoE2 and ApoE3. Either whole blood from healthy APOE4 carriers or microglia derived from APOE4 targeted replacement mice demonstrate higher production of IL-1β [85, 87] and TNFα [85–87], as compared to APOE3 carriers or microglia derived from APOE3 targeted replacement mice. Altogether, these evidences position IL-1β and TNFα systems in the group of promising targets to alleviate memory loss in AD.
The heterodimeric IL-1β receptor comprises the ligand-binding subunit, IL-1 receptor type 1 (IL-1R1), and the accessory protein subunits AcP and AcPb, which function as coreceptors [88]; whereas TNFα signaling is transduced via either TNFR1 and TNFR2 receptors. Hippocampal neurons express high levels of IL-1R1 [89, 90], AcP and AcPb [91], as well as TNFR1 and TNFR2 [92]. Our finding that IL-1β and TNFα directly modulate cLTP in isolated synaptosomes strongly suggests that essential signal transduction components of these cytokines are locally present in synapses. Supporting this possibility, functional analysis of synaptosomes [93–95], as well as biochemical and imaging studies in cultured neurons [96] provide evidence that synapses contain IL-1β and TNFα receptors along with their common downstream effector p38, a stress kinase by which IL-1β [34] and TNFα [42] suppress electrically stimulated LTP in hippocampal slices. Thus, hippocampal synapses are well positioned to be modulated by both IL-1β and TNFα, rendering hippocampus vulnerable to age- and disease-associated inflammation. Alterations in the expression of receptors for IL-1β and TNFα may further aggravate the vulnerability of hippocampus. We have previously reported that aging increases IL-1R1-AcP receptor levels in hippocampal synaptosomes of mice. This age-dependent IL-1R1-AcP upregulation potentiates IL-1β signaling at the synapse, rendering cLTP vulnerable even to a low IL-1β concentration (3 pM) [44]. Similarly, binding affinity of TNFR1 increases whereas TNFR2 levels decreases in AD brains compared to healthy brains [97]. As it is thought that TNFR1 promotes inflammatory neurodegeneration whereas TNFR2 is neuroprotective [98, 99], overall these data suggest that brain inflammation can be accompanied by reconfigurations in IL-1β and TNFα systems.
A highly promising therapeutic target for neurodegeneration is the Nlrp3 inflammasome [100, 101], a macromolecular complex that controls systemic low-grade age-related inflammation in both periphery and brain [102, 103]. Activation of the Nlrp3 inflammasome leads to caspase-1-dependent cleavage of IL-1β and IL-18 precursors, converting these cytokines into mature forms. That caspase-1 processes both IL-1β and IL-18 suggests a possible crosstalk between these two cytokines in brain inflammation. Adding IL-18 to hippocampal slices impairs LTP, as shown by electrophysiological recordings [41]; however, the naturally occurring IL-1 receptor antagonist (IL-1RA) blocks the suppression of LTP by IL-18 [41], thus indicating that IL-18 impairs LTP indirectly, via an IL-1-dependent mechanism. Our data support this model by showing that IL-1β, but not IL-18, suppresses cLTP in isolated synaptosomes and neuronal cultures. We propose that, in contrast to the direct mechanism by which IL-1β suppresses LTP, IL-18 suppresses LTP via cytokine-cellular networks that may depend on multiple cell populations. A parsimonious possibility is that IL-18 induces production of IL-1β by microglia and astrocytes, as glial cells express IL-18 receptor (IL-18R) [104] and increase pro-IL-1β levels in response to IL-18 stimulation [104, 105]. Overall, based on a cytokine-cell network perspective, these evidences illustrate that IL-18 and IL-1β differ in their mechanisms to modulate synapses, despite of having a common activator.
A cytokine-cell network perspective may help to identify potential therapeutic targets for brain disorders characterized by neuroinflammation and cognitive decline. Dynamics of cytokine-cell maps could be defined by changes in steady-state levels of cytokine, in both periphery and brain in health and disease. During the course of AD, these steady-state levels may be reflecting adaptive cytokine network states, which could be supporting cellular mechanisms for healing and recovery such as tissue repair and protein clearance (e.g., Aβ clearance by microglia [106, 107]). However, above certain thresholds of either concentration or time of exposure, inflammatory cytokines including IL-1β, TNFα and IL-18 may gradually contribute to memory deficits [108], which could reflect impairments in synaptic plasticity mechanisms such as LTP. Our study identified IL-1β and TNFα as cytokines able to act as final effectors suppressing hippocampal LTP directly at the synapse. Notably, for synaptic dysfunction, IL-1β seems to be a common final point where several cytokine-cell networks converge to suppress LTP. Evidence suggests that IL-18 [41] and IFNγ [109, 110] activate glia-dependent molecular cascades that increase the levels of IL-1β, which impairs LTP directly in hippocampal neurons. In addition, as shown in a mouse model of postoperative cognitive decline, TNFα can act upstream to IL-1β and initiates a peripheral cytokine cascade that results in IL-1β-dependent memory impairment [14, 22]. In this model, a single dose of anti-TNFα antibody attenuates the increased IL-1β induction in hippocampus, and the IL-1β-dependent memory impairment [22]. Overall, these data provides experimental support to clinical and pre-clinical efforts to alleviate cognitive decline in AD by targeting IL-1β [111] and TNFα [112]. In particular, intrathecal [23] or perispinal [24] administration of biological antagonist of TNFα (infliximab, etanercept) induces a rapid cognitive improvement, beginning within minutes, thus suggesting a mechanism of action based on blocking direct effects of TNFα on synapses [24]. This proposed mechanism [24] is supported by our data demonstrating that TNFα can impair LTP at the synapse. Whether other cytokines impair LTP by directly acting on synapses remains an open question. However, the possibility that the signaling from multiple cytokines converges on synapses and contributes to LTP modulation might reflect a more realistic scenario.
In summary, our data support a model where brain inflammation can suppress LTP via multiple cytokine-dependent mechanisms. We show that TNFα and IL-1β, two highly expressed inflammatory cytokines in the diseased brain, suppress hippocampal LTP directly at the synapse. According to this finding, TNFα and IL-1β may act as final effectors of cytokine-cell networks and impair synaptic plasticity, thus supporting therapeutic approaches to target TNFα and IL-1β to improve cognition in brain disorders with an inflammatory component.
Acknowledgments
Work in the authors’ lab is supported by National Institutes of Health Grants R21-AG048506, P01-AG000538 and RO1-AG34667 (to C.W.C.), as well as by UC MEXUS-CONACYT Grant CN-16-170 (to G.A.P. and C.W.C.).
Footnotes
ORCID: 0000-0001-9517-6989
Conflict of interests The authors declare no competing financial interests.
References
- 1.Prieto-Moreno GA, Rosenstein Y. The links between the neuroendocrine and the immune systems: Views of an immunologist. In: Joseph-Bravo P, editor. Molecular Endocrinology. 1. Research Signpost; Kerala, India: 2006. pp. 171–192. [Google Scholar]
- 2.Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun. 2011;25:181–213. doi: 10.1016/j.bbi.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 3.Griffin WS. Neuroinflammatory cytokine signaling and Alzheimer’s disease. N Engl J Med. 2013;368:770–771. doi: 10.1056/NEJMcibr1214546. [DOI] [PubMed] [Google Scholar]
- 4.Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci. 2015;16:358–372. doi: 10.1038/nrn3880. [DOI] [PubMed] [Google Scholar]
- 5.Becher B, Spath S, Goverman J. Cytokine networks in neuroinflammation. Nat Rev Immunol. 2017;17:49–59. doi: 10.1038/nri.2016.123. [DOI] [PubMed] [Google Scholar]
- 6.Prieto GA, Cotman CW. Cytokines and cytokine networks target neurons to modulate long-term potentiation. Cytokine Growth Factor Rev. 2017;34:27–33. doi: 10.1016/j.cytogfr.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andreasson KI, Bachstetter AD, Colonna M, Ginhoux F, Holmes C, Lamb B, Landreth G, Lee DC, Low D, Lynch MA, Monsonego A, O’Banion MK, Pekny M, Puschmann T, Russek-Blum N, Sandusky LA, Selenica ML, Takata K, Teeling J, Town T, Van Eldik LJ. Targeting innate immunity for neurodegenerative disorders of the central nervous system. J Neurochem. 2016;138:653–693. doi: 10.1111/jnc.13667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM, Herrup K, Frautschy SA, Finsen B, Brown GC, Verkhratsky A, Yamanaka K, Koistinaho J, Latz E, Halle A, Petzold GC, Town T, Morgan D, Shinohara ML, Perry VH, Holmes C, Bazan NG, Brooks DJ, Hunot S, Joseph B, Deigendesch N, Garaschuk O, Boddeke E, Dinarello CA, Breitner JC, Cole GM, Golenbock DT, Kummer MP. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14:388–405. doi: 10.1016/S1474-4422(15)70016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vitkovic L, Konsman JP, Bockaert J, Dantzer R, Homburger V, Jacque C. Cytokine signals propagate through the brain. Mol Psychiatry. 2000;5:604–615. doi: 10.1038/sj.mp.4000813. [DOI] [PubMed] [Google Scholar]
- 10.Brosseron F, Krauthausen M, Kummer M, Heneka MT. Body fluid cytokine levels in mild cognitive impairment and Alzheimer’s disease: a comparative overview. Molecular neurobiology. 2014;50:534–544. doi: 10.1007/s12035-014-8657-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lai KSP, Liu CS, Rau A, Lanctot KL, Kohler CA, Pakosh M, Carvalho AF, Herrmann N. Peripheral inflammatory markers in Alzheimer’s disease: a systematic review and meta-analysis of 175 studies. J Neurol Neurosurg Psychiatry. 2017 doi: 10.1136/jnnp-2017-316201. [DOI] [PubMed] [Google Scholar]
- 12.Swardfager W, Lanctot K, Rothenburg L, Wong A, Cappell J, Herrmann N. A meta-analysis of cytokines in Alzheimer’s disease. Biol Psychiatry. 2010;68:930–941. doi: 10.1016/j.biopsych.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 13.Barrientos RM, Hein AM, Frank MG, Watkins LR, Maier SF. Intracisternal interleukin-1 receptor antagonist prevents postoperative cognitive decline and neuroinflammatory response in aged rats. J Neurosci. 2012;32:14641–14648. doi: 10.1523/JNEUROSCI.2173-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cibelli M, Fidalgo AR, Terrando N, Ma D, Monaco C, Feldmann M, Takata M, Lever IJ, Nanchahal J, Fanselow MS, Maze M. Role of interleukin-1beta in postoperative cognitive dysfunction. Ann Neurol. 2010;68:360–368. doi: 10.1002/ana.22082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frank MG, Barrientos RM, Hein AM, Biedenkapp JC, Watkins LR, Maier SF. IL-1RA blocks E. coli-induced suppression of Arc and long-term memory in aged F344xBN F1 rats. Brain Behav Immun. 2010;24:254–262. doi: 10.1016/j.bbi.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hein AM, Stasko MR, Matousek SB, Scott-McKean JJ, Maier SF, Olschowka JA, Costa AC, O’Banion MK. Sustained hippocampal IL-1beta overexpression impairs contextual and spatial memory in transgenic mice. Brain Behav Immun. 2010;24:243–253. doi: 10.1016/j.bbi.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trompet S, de Craen AJ, Slagboom P, Shepherd J, Blauw GJ, Murphy MB, Bollen EL, Buckley BM, Ford I, Gaw A, Macfarlane PW, Packard CJ, Stott DJ, Jukema JW, Westendorp RG. Genetic variation in the interleukin-1 beta-converting enzyme associates with cognitive function. The PROSPER study. Brain. 2008;131:1069–1077. doi: 10.1093/brain/awn023. [DOI] [PubMed] [Google Scholar]
- 18.Zhu W, Cao FS, Feng J, Chen HW, Wan JR, Lu Q, Wang J. NLRP3 inflammasome activation contributes to long-term behavioral alterations in mice injected with lipopolysaccharide. Neuroscience. 2017;343:77–84. doi: 10.1016/j.neuroscience.2016.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bossu P, Ciaramella A, Salani F, Bizzoni F, Varsi E, Di Iulio F, Giubilei F, Gianni W, Trequattrini A, Moro ML, Bernardini S, Caltagirone C, Spalletta G. Interleukin-18 produced by peripheral blood cells is increased in Alzheimer’s disease and correlates with cognitive impairment. Brain Behav Immun. 2008;22:487–492. doi: 10.1016/j.bbi.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 20.Ohgidani M, Kato TA, Sagata N, Hayakawa K, Shimokawa N, Sato-Kasai M, Kanba S. TNF-alpha from hippocampal microglia induces working memory deficits by acute stress in mice. Brain Behav Immun. 2016;55:17–24. doi: 10.1016/j.bbi.2015.08.022. [DOI] [PubMed] [Google Scholar]
- 21.Sahin TD, Karson A, Balci F, Yazir Y, Bayramgurler D, Utkan T. TNF-alpha inhibition prevents cognitive decline and maintains hippocampal BDNF levels in the unpredictable chronic mild stress rat model of depression. Behav Brain Res. 2015;292:233–240. doi: 10.1016/j.bbr.2015.05.062. [DOI] [PubMed] [Google Scholar]
- 22.Terrando N, Monaco C, Ma D, Foxwell BM, Feldmann M, Maze M. Tumor necrosis factor-alpha triggers a cytokine cascade yielding postoperative cognitive decline. Proc Natl Acad Sci U S A. 2010;107:20518–20522. doi: 10.1073/pnas.1014557107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi JQ, Wang BR, Jiang WW, Chen J, Zhu YW, Zhong LL, Zhang YD, Xu J. Cognitive improvement with intrathecal administration of infliximab in a woman with Alzheimer’s disease. J Am Geriatr Soc. 2011;59:1142–1144. doi: 10.1111/j.1532-5415.2011.03445.x. [DOI] [PubMed] [Google Scholar]
- 24.Tobinick EL, Gross H. Rapid cognitive improvement in Alzheimer’s disease following perispinal etanercept administration. J Neuroinflammation. 2008;5:2. doi: 10.1186/1742-2094-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McKim DB, Niraula A, Tarr AJ, Wohleb ES, Sheridan JF, Godbout JP. Neuroinflammatory Dynamics Underlie Memory Impairments after Repeated Social Defeat. J Neurosci. 2016;36:2590–2604. doi: 10.1523/JNEUROSCI.2394-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alvarez-Arellano L, Pedraza-Escalona M, Blanco-Ayala T, Camacho-Concha N, Cortes-Mendoza J, Perez-Martinez L, Pedraza-Alva G. Autophagy impairment by caspase-1-dependent inflammation mediates memory loss in response to beta-Amyloid peptide accumulation. J Neurosci Res. 2018;96:234–236. doi: 10.1002/jnr.24130. [DOI] [PubMed] [Google Scholar]
- 27.Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 28.Paula-Lima AC, Brito-Moreira J, Ferreira ST. Deregulation of excitatory neurotransmission underlying synapse failure in Alzheimer’s disease. Journal of neurochemistry. 2013;126:191–202. doi: 10.1111/jnc.12304. [DOI] [PubMed] [Google Scholar]
- 29.Nicholson DA, Yoshida R, Berry RW, Gallagher M, Geinisman Y. Reduction in size of perforated postsynaptic densities in hippocampal axospinous synapses and age-related spatial learning impairments. J Neurosci. 2004;24:7648–7653. doi: 10.1523/JNEUROSCI.1725-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morrison JH, Baxter MG. The ageing cortical synapse: hallmarks and implications for cognitive decline. Nat Rev Neurosci. 2012;13:240–250. doi: 10.1038/nrn3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 32.Murray CA, Lynch MA. Evidence that increased hippocampal expression of the cytokine interleukin-1 beta is a common trigger for age- and stress-induced impairments in long-term potentiation. J Neurosci. 1998;18:2974–2981. doi: 10.1523/JNEUROSCI.18-08-02974.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vereker E, O’Donnell E, Lynch MA. The inhibitory effect of interleukin-1beta on long-term potentiation is coupled with increased activity of stress-activated protein kinases. J Neurosci. 2000;20:6811–6819. doi: 10.1523/JNEUROSCI.20-18-06811.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tong L, Prieto GA, Kramar EA, Smith ED, Cribbs DH, Lynch G, Cotman CW. Brain-derived neurotrophic factor-dependent synaptic plasticity is suppressed by interleukin-1beta via p38 mitogen-activated protein kinase. J Neurosci. 2012;32:17714–17724. doi: 10.1523/JNEUROSCI.1253-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chapman TR, Barrientos RM, Ahrendsen JT, Maier SF, Patterson SL. Synaptic correlates of increased cognitive vulnerability with aging: peripheral immune challenge and aging interact to disrupt theta-burst late-phase long-term potentiation in hippocampal area CA1. J Neurosci. 2010;30:7598–7603. doi: 10.1523/JNEUROSCI.5172-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cunningham AJ, Murray CA, O’Neill LA, Lynch MA, O’Connor JJ. Interleukin-1 beta (IL-1 beta) and tumour necrosis factor (TNF) inhibit long-term potentiation in the rat dentate gyrus in vitro. Neurosci Lett. 1996;203:17–20. doi: 10.1016/0304-3940(95)12252-4. [DOI] [PubMed] [Google Scholar]
- 37.Bellinger FP, Madamba S, Siggins GR. Interleukin 1 beta inhibits synaptic strength and long-term potentiation in the rat CA1 hippocampus. Brain Res. 1993;628:227–234. doi: 10.1016/0006-8993(93)90959-q. [DOI] [PubMed] [Google Scholar]
- 38.Hoshino K, Hasegawa K, Kamiya H, Morimoto Y. Synapse-specific effects of IL-1beta on long-term potentiation in the mouse hippocampus. Biomed Res. 2017;38:183–188. doi: 10.2220/biomedres.38.183. [DOI] [PubMed] [Google Scholar]
- 39.Cumiskey D, Curran BP, Herron CE, O’Connor JJ. A role for inflammatory mediators in the IL-18 mediated attenuation of LTP in the rat dentate gyrus. Neuropharmacology. 2007;52:1616–1623. doi: 10.1016/j.neuropharm.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 40.Cumiskey D, Pickering M, O’Connor JJ. Interleukin-18 mediated inhibition of LTP in the rat dentate gyrus is attenuated in the presence of mGluR antagonists. Neurosci Lett. 2007;412:206–210. doi: 10.1016/j.neulet.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 41.Curran B, O’Connor JJ. The pro-inflammatory cytokine interleukin-18 impairs long-term potentiation and NMDA receptor-mediated transmission in the rat hippocampus in vitro. Neuroscience. 2001;108:83–90. doi: 10.1016/s0306-4522(01)00405-5. [DOI] [PubMed] [Google Scholar]
- 42.Butler MP, O’Connor JJ, Moynagh PN. Dissection of tumor-necrosis factor-alpha inhibition of long-term potentiation (LTP) reveals a p38 mitogen-activated protein kinase-dependent mechanism which maps to early-but not late-phase LTP. Neuroscience. 2004;124:319–326. doi: 10.1016/j.neuroscience.2003.11.040. [DOI] [PubMed] [Google Scholar]
- 43.Tancredi V, D’Arcangelo G, Grassi F, Tarroni P, Palmieri G, Santoni A, Eusebi F. Tumor necrosis factor alters synaptic transmission in rat hippocampal slices. Neurosci Lett. 1992;146:176–178. doi: 10.1016/0304-3940(92)90071-e. [DOI] [PubMed] [Google Scholar]
- 44.Prieto GA, Snighda S, Baglietto-Vargas D, Smith ED, Berchtold N, Tong L, Ajami D, LaFerla FM, Rebek J, Cotman CW. Synapse-specific IL-1 receptor subunit reconfiguration augments vulnerability to IL-1β in the aged hippocampus. Proc Natl Acad Sci U S A. 2015;112:E5078–5087. doi: 10.1073/pnas.1514486112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Whittaker VP. Thirty years of synaptosome research. J Neurocytol. 1993;22:735–742. doi: 10.1007/BF01181319. [DOI] [PubMed] [Google Scholar]
- 46.Wilhelm BG, Mandad S, Truckenbrodt S, Krohnert K, Schafer C, Rammner B, Koo SJ, Classen GA, Krauss M, Haucke V, Urlaub H, Rizzoli SO. Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science. 2014;344:1023–1028. doi: 10.1126/science.1252884. [DOI] [PubMed] [Google Scholar]
- 47.Sandoval ME, Horch P, Cotman CW. Evaluation of glutamate as a hippocampal neurotransmitter: glutamate uptake and release from synaptosomes. Brain Res. 1978;142:285–299. doi: 10.1016/0006-8993(78)90636-4. [DOI] [PubMed] [Google Scholar]
- 48.Daniel JA, Malladi CS, Kettle E, McCluskey A, Robinson PJ. Analysis of synaptic vesicle endocytosis in synaptosomes by high-content screening. Nat Protoc. 2012;7:1439–1455. doi: 10.1038/nprot.2012.070. [DOI] [PubMed] [Google Scholar]
- 49.Michaelis EK, Michaelis ML, Chang HH, Kitos TE. High affinity Ca2+-stimulated Mg2+-dependent ATPase in rat brain synaptosomes, synaptic membranes, and microsomes. J Biol Chem. 1983;258:6101–6108. [PubMed] [Google Scholar]
- 50.Michaelis ML, Michaelis EK, Myers SL. Adenosine modulation of synaptosomal dopamine release. Life Sci. 1979;24:2083–2092. doi: 10.1016/0024-3205(79)90082-1. [DOI] [PubMed] [Google Scholar]
- 51.Prieto GA, Trieu BH, Dang CT, Bilousova T, Gylys KH, Berchtold NC, Lynch G, Cotman CW. Pharmacological Rescue of Long-Term Potentiation in Alzheimer Diseased Synapses. J Neurosci. 2017;37:1197–1212. doi: 10.1523/JNEUROSCI.2774-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron. 2001;29:243–254. doi: 10.1016/s0896-6273(01)00194-5. [DOI] [PubMed] [Google Scholar]
- 53.Musleh W, Bi X, Tocco G, Yaghoubi S, Baudry M. Glycine-induced long-term potentiation is associated with structural and functional modifications of alpha-amino-3-hydroxyl-5-methyl-4-isoxazolepropionic acid receptors. Proc Natl Acad Sci U S A. 1997;94:9451–9456. doi: 10.1073/pnas.94.17.9451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kramar EA, Lin B, Lin CY, Arai AC, Gall CM, Lynch G. A novel mechanism for the facilitation of theta-induced long-term potentiation by brain-derived neurotrophic factor. J Neurosci. 2004;24:5151–5161. doi: 10.1523/JNEUROSCI.0800-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen TJ, Wang DC, Chen SS. Amyloid-beta interrupts the PI3K-Akt-mTOR signaling pathway that could be involved in brain-derived neurotrophic factor-induced Arc expression in rat cortical neurons. J Neurosci Res. 2009;87:2297–2307. doi: 10.1002/jnr.22057. [DOI] [PubMed] [Google Scholar]
- 56.Rex CS, Lin CY, Kramar EA, Chen LY, Gall CM, Lynch G. Brain-derived neurotrophic factor promotes long-term potentiation-related cytoskeletal changes in adult hippocampus. J Neurosci. 2007;27:3017–3029. doi: 10.1523/JNEUROSCI.4037-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smith ED, Prieto GA, Tong L, Sears-Kraxberger I, Rice JD, Steward O, Cotman CW. Rapamycin and Interleukin-1beta Impair Brain-derived Neurotrophic Factor-dependent Neuron Survival by Modulating Autophagy. J Biol Chem. 2014;289:20615–20629. doi: 10.1074/jbc.M114.568659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fortin DA, Davare MA, Srivastava T, Brady JD, Nygaard S, Derkach VA, Soderling TR. Long-term potentiation-dependent spine enlargement requires synaptic Ca2+-permeable AMPA receptors recruited by CaM-kinase I. J Neurosci. 2010;30:11565–11575. doi: 10.1523/JNEUROSCI.1746-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Park M, Salgado JM, Ostroff L, Helton TD, Robinson CG, Harris KM, Ehlers MD. Plasticity-induced growth of dendritic spines by exocytic trafficking from recycling endosomes. Neuron. 2006;52:817–830. doi: 10.1016/j.neuron.2006.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen RQ, Wang SH, Yao W, Wang JJ, Ji F, Yan JZ, Ren SQ, Chen Z, Liu SY, Lu W. Role of glycine receptors in glycine-induced LTD in hippocampal CA1 pyramidal neurons. Neuropsychopharmacology. 2011;36:1948–1958. doi: 10.1038/npp.2011.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Baumgarth N, Roederer M. A practical approach to multicolor flow cytometry for immunophenotyping. J Immunol Methods. 2000;243:77–97. doi: 10.1016/s0022-1759(00)00229-5. [DOI] [PubMed] [Google Scholar]
- 62.Hulspas R, O’Gorman MR, Wood BL, Gratama JW, Sutherland DR. Considerations for the control of background fluorescence in clinical flow cytometry. Cytometry B Clin Cytom. 2009;76:355–364. doi: 10.1002/cyto.b.20485. [DOI] [PubMed] [Google Scholar]
- 63.Menon V, Thomas R, Ghale AR, Reinhard C, Pruszak J. Flow cytometry protocols for surface and intracellular antigen analyses of neural cell types. J Vis Exp. 2014 doi: 10.3791/52241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cotman CW, Haycock JW, White WF. Stimulus-secretion coupling processes in brain: analysis of noradrenaline and gamma-aminobutyric acid release. J Physiol. 1976;254:475–505. doi: 10.1113/jphysiol.1976.sp011241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Michaelis ML, Jiang L, Michaelis EK. Isolation of Synaptosomes, Synaptic Plasma Membranes, and Synaptic Junctional Complexes. Methods Mol Biol. 2017;1538:107–119. doi: 10.1007/978-1-4939-6688-2_9. [DOI] [PubMed] [Google Scholar]
- 66.Snigdha S, Prieto GA, Petrosyan A, Loertscher BM, Dieskau AP, Overman LE, Cotman CW. H3K9me3 Inhibition Improves Memory, Promotes Spine Formation, and Increases BDNF Levels in the Aged Hippocampus. J Neurosci. 2016;36:3611–3622. doi: 10.1523/JNEUROSCI.2693-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Carlos AJ, Tong L, Prieto GA, Cotman CW. IL-1beta impairs retrograde flow of BDNF signaling by attenuating endosome trafficking. J Neuroinflammation. 2017;14:29. doi: 10.1186/s12974-017-0803-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fu Y, Huang ZJ. Differential dynamics and activity-dependent regulation of alpha- and beta-neurexins at developing GABAergic synapses. Proc Natl Acad Sci U S A. 2010;107:22699–22704. doi: 10.1073/pnas.1011233108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mondin M, Labrousse V, Hosy E, Heine M, Tessier B, Levet F, Poujol C, Blanchet C, Choquet D, Thoumine O. Neurexin-neuroligin adhesions capture surface-diffusing AMPA receptors through PSD-95 scaffolds. J Neurosci. 2011;31:13500–13515. doi: 10.1523/JNEUROSCI.6439-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Prieto GA, Cotman CW. On the road towards the global analysis of human synapses. Neural Regen Res. 2017;12:1586–1589. doi: 10.4103/1673-5374.217321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- 72.Manabe T, Renner P, Nicoll RA. Postsynaptic contribution to long-term potentiation revealed by the analysis of miniature synaptic currents. Nature. 1992;355:50–55. doi: 10.1038/355050a0. [DOI] [PubMed] [Google Scholar]
- 73.Park M, Penick EC, Edwards JG, Kauer JA, Ehlers MD. Recycling endosomes supply AMPA receptors for LTP. Science. 2004;305:1972–1975. doi: 10.1126/science.1102026. [DOI] [PubMed] [Google Scholar]
- 74.Man HY, Wang Q, Lu WY, Ju W, Ahmadian G, Liu L, D’Souza S, Wong TP, Taghibiglou C, Lu J, Becker LE, Pei L, Liu F, Wymann MP, MacDonald JF, Wang YT. Activation of PI3-kinase is required for AMPA receptor insertion during LTP of mEPSCs in cultured hippocampal neurons. Neuron. 2003;38:611–624. doi: 10.1016/s0896-6273(03)00228-9. [DOI] [PubMed] [Google Scholar]
- 75.Sui L, Wang J, Li BM. Role of the phosphoinositide 3-kinase-Akt-mammalian target of the rapamycin signaling pathway in long-term potentiation and trace fear conditioning memory in rat medial prefrontal cortex. Learn Mem. 2008;15:762–776. doi: 10.1101/lm.1067808. [DOI] [PubMed] [Google Scholar]
- 76.Hu H, Qin Y, Bochorishvili G, Zhu Y, van Aelst L, Zhu JJ. Ras signaling mechanisms underlying impaired GluR1-dependent plasticity associated with fragile X syndrome. J Neurosci. 2008;28:7847–7862. doi: 10.1523/JNEUROSCI.1496-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pen Y, Borovok N, Reichenstein M, Sheinin A, Michaelevski I. Membrane-tethered AKT kinase regulates basal synaptic transmission and early phase LTP expression by modulation of post-synaptic AMPA receptor level. Hippocampus. 2016;26:1149–1167. doi: 10.1002/hipo.22597. [DOI] [PubMed] [Google Scholar]
- 78.Tong L, Balazs R, Soiampornkul R, Thangnipon W, Cotman CW. Interleukin-1 beta impairs brain derived neurotrophic factor-induced signal transduction. Neurobiol Aging. 2008;29:1380–1393. doi: 10.1016/j.neurobiolaging.2007.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Alboni S, Montanari C, Benatti C, Sanchez-Alavez M, Rigillo G, Blom JM, Brunello N, Conti B, Pariante MC, Tascedda F. Interleukin 18 activates MAPKs and STAT3 but not NF-kappaB in hippocampal HT-22 cells. Brain Behav Immun. 2014;40:85–94. doi: 10.1016/j.bbi.2014.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhou J, Ping FF, Lv WT, Feng JY, Shang J. Interleukin-18 directly protects cortical neurons by activating PI3K/AKT/NF-kappaB/CREB pathways. Cytokine. 2014;69:29–38. doi: 10.1016/j.cyto.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 81.Pfau ML, Menard C, Russo SJ. Inflammatory Mediators in Mood Disorders: Therapeutic Opportunities. Annu Rev Pharmacol Toxicol. 2017 doi: 10.1146/annurev-pharmtox-010617-052823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Erion JR, Wosiski-Kuhn M, Dey A, Hao S, Davis CL, Pollock NK, Stranahan AM. Obesity elicits interleukin 1-mediated deficits in hippocampal synaptic plasticity. J Neurosci. 2014;34:2618–2631. doi: 10.1523/JNEUROSCI.4200-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Blum-Degen D, Muller T, Kuhn W, Gerlach M, Przuntek H, Riederer P. Interleukin-1 beta and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci Lett. 1995;202:17–20. doi: 10.1016/0304-3940(95)12192-7. [DOI] [PubMed] [Google Scholar]
- 84.Tarkowski E, Blennow K, Wallin A, Tarkowski A. Intracerebral production of tumor necrosis factor-alpha, a local neuroprotective agent, in Alzheimer disease and vascular dementia. J Clin Immunol. 1999;19:223–230. doi: 10.1023/a:1020568013953. [DOI] [PubMed] [Google Scholar]
- 85.Gale SC, Gao L, Mikacenic C, Coyle SM, Rafaels N, Murray Dudenkov T, Madenspacher JH, Draper DW, Ge W, Aloor JJ, Azzam KM, Lai L, Blackshear PJ, Calvano SE, Barnes KC, Lowry SF, Corbett S, Wurfel MM, Fessler MB. APOepsilon4 is associated with enhanced in vivo innate immune responses in human subjects. J Allergy Clin Immunol. 2014;134:127–134. doi: 10.1016/j.jaci.2014.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vitek MP, Brown CM, Colton CA. APOE genotype-specific differences in the innate immune response. Neurobiol Aging. 2009;30:1350–1360. doi: 10.1016/j.neurobiolaging.2007.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, Tsai RM, Spina S, Grinberg LT, Rojas JC, Gallardo G, Wang K, Roh J, Robinson G, Finn MB, Jiang H, Sullivan PM, Baufeld C, Wood MW, Sutphen C, McCue L, Xiong C, Del-Aguila JL, Morris JC, Cruchaga C, Alzheimer’s Disease Neuroimaging I. Fagan AM, Miller BL, Boxer AL, Seeley WW, Butovsky O, Barres BA, Paul SM, Holtzman DM. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature. 2017;549:523–527. doi: 10.1038/nature24016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. 2011;117:3720–3732. doi: 10.1182/blood-2010-07-273417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Friedman WJ. Cytokines regulate expression of the type 1 interleukin-1 receptor in rat hippocampal neurons and glia. Exp Neurol. 2001;168:23–31. doi: 10.1006/exnr.2000.7595. [DOI] [PubMed] [Google Scholar]
- 90.Farrar WL, Kilian PL, Ruff MR, Hill JM, Pert CB. Visualization and characterization of interleukin 1 receptors in brain. J Immunol. 1987;139:459–463. [PubMed] [Google Scholar]
- 91.Smith DE, Lipsky BP, Russell C, Ketchem RR, Kirchner J, Hensley K, Huang Y, Friedman WJ, Boissonneault V, Plante MM, Rivest S, Sims JE. A central nervous system-restricted isoform of the interleukin-1 receptor accessory protein modulates neuronal responses to interleukin-1. Immunity. 2009;30:817–831. doi: 10.1016/j.immuni.2009.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Neumann H, Schweigreiter R, Yamashita T, Rosenkranz K, Wekerle H, Barde YA. Tumor necrosis factor inhibits neurite outgrowth and branching of hippocampal neurons by a rho-dependent mechanism. J Neurosci. 2002;22:854–862. doi: 10.1523/JNEUROSCI.22-03-00854.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhu CB, Lindler KM, Owens AW, Daws LC, Blakely RD, Hewlett WA. Interleukin-1 receptor activation by systemic lipopolysaccharide induces behavioral despair linked to MAPK regulation of CNS serotonin transporters. Neuropsychopharmacology. 2010;35:2510–2520. doi: 10.1038/npp.2010.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Murray CA, McGahon B, McBennett S, Lynch MA. Interleukin-1 beta inhibits glutamate release in hippocampus of young, but not aged, rats. Neurobiol Aging. 1997;18:343–348. doi: 10.1016/s0197-4580(97)80317-x. [DOI] [PubMed] [Google Scholar]
- 95.Zhu CB, Blakely RD, Hewlett WA. The proinflammatory cytokines interleukin-1beta and tumor necrosis factor-alpha activate serotonin transporters. Neuropsychopharmacology. 2006;31:2121–2131. doi: 10.1038/sj.npp.1301029. [DOI] [PubMed] [Google Scholar]
- 96.Gardoni F, Boraso M, Zianni E, Corsini E, Galli CL, Cattabeni F, Marinovich M, Di Luca M, Viviani B. Distribution of interleukin-1 receptor complex at the synaptic membrane driven by interleukin-1beta and NMDA stimulation. J Neuroinflammation. 2011;8:14. doi: 10.1186/1742-2094-8-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cheng X, Yang L, He P, Li R, Shen Y. Differential activation of tumor necrosis factor receptors distinguishes between brains from Alzheimer’s disease and non-demented patients. J Alzheimers Dis. 2010;19:621–630. doi: 10.3233/JAD-2010-1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Iosif RE, Ekdahl CT, Ahlenius H, Pronk CJ, Bonde S, Kokaia Z, Jacobsen SE, Lindvall O. Tumor necrosis factor receptor 1 is a negative regulator of progenitor proliferation in adult hippocampal neurogenesis. J Neurosci. 2006;26:9703–9712. doi: 10.1523/JNEUROSCI.2723-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Naude PJ, Dobos N, van der Meer D, Mulder C, Pawironadi KG, den Boer JA, van der Zee EA, Luiten PG, Eisel UL. Analysis of cognition, motor performance and anxiety in young and aged tumor necrosis factor alpha receptor 1 and 2 deficient mice. Behav Brain Res. 2014;258:43–51. doi: 10.1016/j.bbr.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 100.Saresella M, La Rosa F, Piancone F, Zoppis M, Marventano I, Calabrese E, Rainone V, Nemni R, Mancuso R, Clerici M. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol Neurodegener. 2016;11:23. doi: 10.1186/s13024-016-0088-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Singhal G, Jaehne EJ, Corrigan F, Toben C, Baune BT. Inflammasomes in neuroinflammation and changes in brain function: a focused review. Front Neurosci. 2014;8:315. doi: 10.3389/fnins.2014.00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Youm YH, Grant RW, McCabe LR, Albarado DC, Nguyen KY, Ravussin A, Pistell P, Newman S, Carter R, Laque A, Munzberg H, Rosen CJ, Ingram DK, Salbaum JM, Dixit VD. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 2013;18:519–532. doi: 10.1016/j.cmet.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, Gelpi E, Halle A, Korte M, Latz E, Golenbock DT. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493:674–678. doi: 10.1038/nature11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Felderhoff-Mueser U, Schmidt OI, Oberholzer A, Buhrer C, Stahel PF. IL-18: a key player in neuroinflammation and neurodegeneration? Trends Neurosci. 2005;28:487–493. doi: 10.1016/j.tins.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 105.Wheeler RD, Brough D, Le Feuvre RA, Takeda K, Iwakura Y, Luheshi GN, Rothwell NJ. Interleukin-18 induces expression and release of cytokines from murine glial cells: interactions with interleukin-1 beta. J Neurochem. 2003;85:1412–1420. doi: 10.1046/j.1471-4159.2003.01787.x. [DOI] [PubMed] [Google Scholar]
- 106.Guillot-Sestier MV, Town T. Innate immunity in Alzheimer’s disease: a complex affair. CNS Neurol Disord Drug Targets. 2013;12:593–607. doi: 10.2174/1871527311312050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Montgomery SL, Mastrangelo MA, Habib D, Narrow WC, Knowlden SA, Wright TW, Bowers WJ. Ablation of TNF-RI/RII expression in Alzheimer’s disease mice leads to an unexpected enhancement of pathology: implications for chronic pan-TNF-alpha suppressive therapeutic strategies in the brain. Am J Pathol. 2011;179:2053–2070. doi: 10.1016/j.ajpath.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Donzis EJ, Tronson NC. Modulation of learning and memory by cytokines: signaling mechanisms and long term consequences. Neurobiology of learning and memory. 2014;115:68–77. doi: 10.1016/j.nlm.2014.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Maher FO, Clarke RM, Kelly A, Nally RE, Lynch MA. Interaction between interferon gamma and insulin-like growth factor-1 in hippocampus impacts on the ability of rats to sustain long-term potentiation. J Neurochem. 2006;96:1560–1571. doi: 10.1111/j.1471-4159.2006.03664.x. [DOI] [PubMed] [Google Scholar]
- 110.Kelly RJ, Minogue AM, Lyons A, Jones RS, Browne TC, Costello DA, Denieffe S, O’Sullivan C, Connor TJ, Lynch MA. Glial Activation in AbetaPP/PS1 Mice is Associated with Infiltration of IFNgamma-Producing Cells. J Alzheimers Dis. 2013;37:63–75. doi: 10.3233/JAD-130539. [DOI] [PubMed] [Google Scholar]
- 111.Kitazawa M, Cheng D, Tsukamoto MR, Koike MA, Wes PD, Vasilevko V, Cribbs DH, LaFerla FM. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal beta-catenin pathway function in an Alzheimer’s disease model. J Immunol. 2011;187:6539–6549. doi: 10.4049/jimmunol.1100620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chang R, Yee KL, Sumbria RK. Tumor necrosis factor alpha Inhibition for Alzheimer’s Disease. J Cent Nerv Syst Dis. 2017;9 doi: 10.1177/1179573517709278. 1179573517709278. [DOI] [PMC free article] [PubMed] [Google Scholar]