

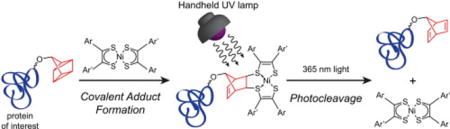
Abstract
The quadricyclane (QC) ligation is a bioorthogonal reaction between a quadricyclane moiety and a nickel bis(dithiolene) derivative. Here we show that a QC amino acid can be incorporated into a protein site-specifically using the pyrrolysine-based genetic code expansion platform, and subsequently used for ligation chemistry. Additionally, we exploited the photolability of the QC ligation product to render the adduct cleavable with a handheld UV lamp. We further developed a protein purification method that involves QC ligation of biotin to a protein of interest, capture on streptavidin resin, and finally release using only UV light. The QC ligation thus brings novel chemical manipulations to the realm of bioorthogonal chemistry.
Keywords: quadricyclane, bioorthogonal, pyrrolysine, unnatural amino acid, photocleavage, photolysis, protein purification
Graphical abstract
1. Introduction
The current scope of the bioorthogonal toolbox is much smaller than the repertoire of conventional synthetic chemical reactions. This is in part because the field is young, established less than two decades ago, but also because the constraints on a bioorthogonal reaction are formidable compared to those surrounding traditional organic transformations.1 The solvent is c 2018 Published by Elsevier Ltd. limited to water at near physiological pH, temperature is restricted to 37 °C or lower, and cross-reactivity with biological functionalities and other bioorthogonal reagents is not tolerated. In addition, the ligation has to be rapid enough to proceed on the timescale of the biological process of interest and one of the reagents must be capable of incorporation into a target biomolecule via the organism’s own biosynthetic machinery. Together, these requirements make the design of new bioorthogonal reactions a challenging pursuit.
Several bioorthogonal reactions involving phosphines,2–4 azides and other 1,3-dipoles, alkynes, alkenes and tetrazines are now in widespread use to generate covalent adducts under physiological conditions.5–7 However, there is a strong need for new bioorthogonal chemistries due to the demands of multiplexed imaging experiments and the lack of chemical manipulations accessible via the current bioorthogonal repertoire.6,8 Multiplexed imaging requires mutual orthogonality across bioorthogonal reactions, meaning the chemical functionalities unique to each ligation do not react with each other. Often, this requirement is a challenge due to inherent cross-reactivity between reagents.1,9–11
Lack of access to reactions other than ligations is the second driver of bioorthogonal reaction development. The majority of bioorthogonal reactions in the current toolbox produce a stable, covalent bond between two entities. Notable exceptions include uncaging reactions, bond cleavage reactions, and a recent report from Addy et al. in which the adduct of the bioorthogonal coupling of 5-hydroxytryptophan and an aromatic diazonium ion could be cleaved with the addition of the small molecule dithionite.12–14 The conventional synthetic toolbox contains additional diverse chemistries that are reversible, stereoselective, or triggered by environmental cues (i.e. photochemistries, pH sensitive reactions, etc.). The field of bioorthogonal chemistry has just begun to branch into these areas of new chemical functionality, and innovations in the design of unique ligations will drive this growth.
Rising to meet these needs, we proposed a new bioorthogonal reaction — the quadricyclane (QC) ligation.15 The reaction is unique because it exploits a chemical space previously unexplored through the lens of bioorthogonality. It is a formal [2σ + 2σ + 2π] cycloaddition proceeding between the strained hydrocarbon quadricyclane (QC, 1) and nickel bis(dithiolene) (2) to form adduct 3 (Scheme 1). We found that this reaction meets many of the requirements of bioorthogonality including rapid rate, selectivity, and compatibility with physiological conditions. Most importantly, the QC ligation was orthogonal to important protein bioconjugation chemistries such as oxime formation with aldehydes and aminooxy groups and copper-free click reaction.15
Scheme 1. The quadricyclane (QC) ligation.
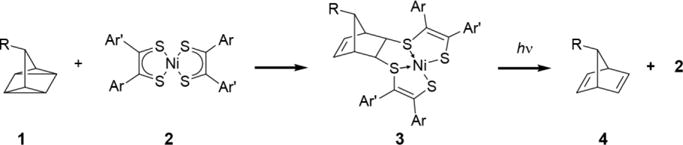
The QC ligation is a formal [2σ + 2σ + 2π] reaction between QC (1) and nickel bis(dithiolene) (2) to form adduct 3. This work describes the photocleavage of 3 with UV light to produce norbornadiene (4) and the original nickel bis(dithiolene) 2 applied in a biological context.
Here, we show that QC can be site-specifically incorporated into a protein of interest using the pyrrolysine-based genetic code expansion platform16,17 and employed for protein chemical modification. We also demonstrate that protein-bound QC adducts can be cleaved by mild photolysis to form norbornadiene 4 and the original nickel complex 2 (Scheme 1). Together, the QC ligation and photocleavage chemistries enabled a new avenue for protein purification.
2. Results
2.1 Site-specific incorporation of a QC amino acid into a protein
We first addressed the challenge of incorporating QC into a protein of interest, an important capability for bioorthogonal applications, and for this we chose the pyrrolysine method for unnatural amino acid incorporation. The PylRS/tRNACUA pair has proven a useful tool for non-natural amino acid incorporation in E. coli, mammalian cells, and even model organisms.18–24 We envisioned using PylRS/tRNACUA to site-specifically install a QC-containing amino acid in response to an in-frame amber stop codon in a protein of interest. Accordingly, we designed QC amino acid 6 (Figure 1A) based on its synthetic accessibility from known intermediate p-nitrophenyl QC carbonate (5, Figure 1A)15 and its structural similarity to norbornene amino acids that have been successfully incorporated into proteins using the PylRS/tRNACUA system.25–27 The previously reported N-ε-Boc-L-lysine (7, Figure 1B), a known substrate of wildtype PylRS, was used as a positive control in this study.25
Figure 1. Site-specific incorporation of QC amino acid into a protein.
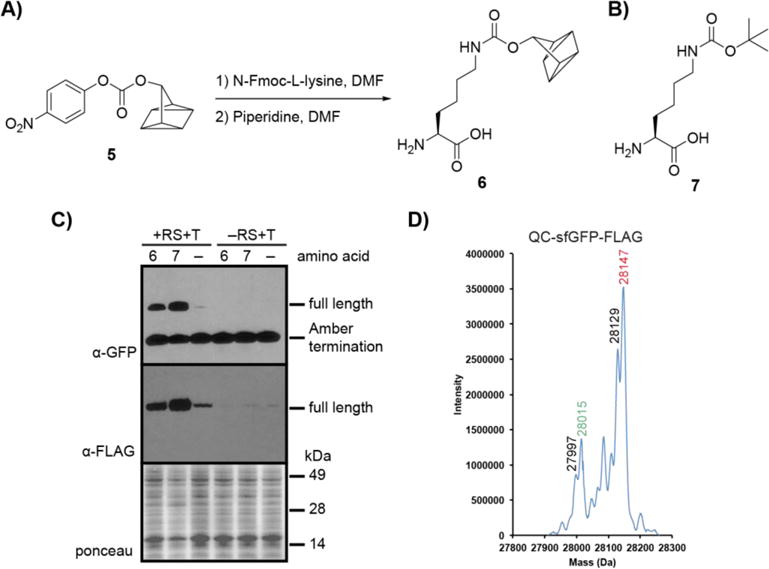
A) QC amino acid 6 was synthesized in a 2-step procedure from p-nitrophenyl carbonate 5: (1) N-Fmoc-L-lysine, DMF, rt, 16 h. (2) piperidine, DMF, rt, 2 h. B) N-ε-Boc-L-lysine (7), a known substrate of WT PylRS. C) Expression of full-length sfGFP-FLAG in the presence of 6 or 7. E. coli cells were transformed with plasmids pBKPylRS and psfGFP150TAGFLAGPylT (+RS+T) or with psfGFP150TAGFLAGPylT only (−RS+T) (see Supplementary data for plasmid maps and sequences) and cultures were supplemented with 1 mM 6, Boc-lysine (7), or vehicle (−). Protein was expressed for 3.5 h, after which lysates were analyzed by western blot probing with α-GFP and α-FLAG antibodies. D) High-resolution electrospray ionization (ESI) mass spectrometry (MS) analysis of purified QC-sfGFP-FLAG. Red represents the mass of the full-length protein and the green represents the mass of the full-length protein minus methionine. Black numbers represent neutral loss of H2O, which may occur during fragmentation using ESI-MS.28 QC-sfGFP-FLAG observed mass: 28147 Daltons, calculated mass: 28147.61 Daltons.
We next sought to determine if 6 is recognized by PylRS and incorporated into a recombinant protein. DH10β E. coli were transformed with pBKPylRS which encodes the gene for PylRS from Monosarchina barkeri, and psfGFP150TAGFLAGPylT which encodes tRNACUA and a FLAG-tagged superfolder GFP (sfGFP) with an in-frame amber stop codon at position 150. This site is known to be amenable to unnatural amino acid incorporation.29 Protein expression was induced in the presence of approximately 1 mM 6, 1 mM 7, or vehicle. For these experiments, vehicle was 0.1 M NaOH in ddH2O, the solvent in which the non-natural amino acid was dissolved for addition to E. coli culture. The solubility of 6 was not complete in 0.1 M NaOH, however, after a large screen of solvents it was found to be optimal. Following expression, cells were pelleted and subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were then transferred to nitrocellulose and analyzed via western blot for the presence of full-length QC- or Boc-sfGFP-FLAG by probing with an α-GFP antibody.
We expected to see production of full-length sfGFP-FLAG only in samples treated with 6 or 7 and transformed with psfGFP150TAGFLAGPylT and pBKPylRS. For bacteria transformed with both plasmids (+RS+T), a dark band was visible at ~28 kDa in the +6 and +7 lanes whereas only a faint band was apparent without the amino acid supplements (−) lane (Figure 1C). This band represents full-length sfGFP-FLAG, and its presence in the +6 and +7 lanes suggested successful translational read-through at the amber site in both samples. These observations suggest that 6 is accepted by wildtype PylRS and incorporated site-specifically into proteins via the PylRS/tRNACUA system. The small amount of full-length sfGFP formed in the absence of amino acid supplement may be caused by misacylation of tRNACUA with native amino acids by PylRS. As expected, E. coli that were not transformed with the gene for PylRS (−RS+T) did not generate full-length sfGFP-FLAG, showing that PylRS is necessary for read-through of the amber stop codon. We further characterized full-length QC-sfGFP-FLAG by high-resolution ESI-MS and found that it matched the predicted mass of 28147 Daltons (Figure 1D).
The lower band in the blot shown in Figure 1C at ~14 kDa, present in all samples, represents the amber termination product, confirmed by mass spectrometry (Figure S1). This is a common byproduct of the unnatural amino acid incorporation process as the amber codon still acts as a stop codon and is recognized by release factors such as RF1 in E. coli.30
After confirming the presence of 6 in sfGFP-FLAG, we sought to label the QC handle with nickel bis(dithiolene) biotin 8 (Figure 2A). To start, we subjected purified QC-sfGFP-FLAG or Boc-sfGFP-FLAG to 200 μM 6 in PBS supplemented with 1mM K3Fe(CN)6 or with vehicle (1 mM K3Fe(CN)6 in PBS) for 1 h. The K3Fe(CN)6 is used as an additive to prevent reduction of Ni bis(dithiolene) adducts.15 After 1 h, the reaction was quenched with 100 equiv. of QC acetate (9, Figure 2B, hereafter referred to as QCOAc) relative to 8 followed by addition of sodium diethyldithiocarbamate (Na-DEDTC), which prevents photocleavage of the adduct.15 Ligation product was analyzed by western blot with an α-biotin-horseradish peroxidase (HRP) antibody (Figure 2C). We observed robust labeling of QC-sfGFP-FLAG in the presence of 8 (lane 1) with minimal signal from the Boc-sfGFP-FLAG negative control (lane 3). The smear observed in lane 1 may be due to the addition of a large PEG-containing group to QC-sfGFP, rather than off-target reactivity since no biotin signal is observed when Boc-sfGFP was incubated with 8, shown in lane 3. These data suggest selective reaction of 6 in sfGFP with 8 and minimal side reactivity of 8 with other amino acids in sfGFP-FLAG. Additional blot exposure times are shown in Figure S2.
Figure 2. Labeling QC-sfGFP-FLAG via the quadricyclane ligation.
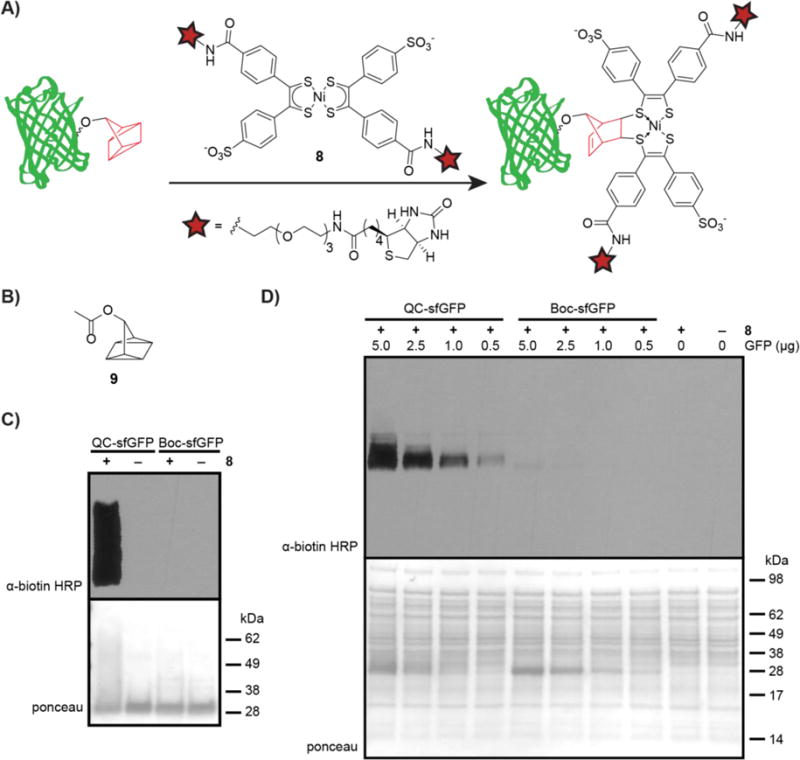
A) Schematic showing labeling QC-sfGFP-FLAG with 8. B) Structure of QC acetate (QCOAc, 9). C) Purified QC-sfGFP-FLAG or purified Boc-sfGFP-FLAG was incubated with 200 μM 8 in PBS with 1 mM K3Fe(CN)6 (+8) or with 1 mM K3Fe(CN)6 in PBS (−8) for 1 h at rt in the dark. The reaction was quenched with QCOAc (100 equiv. relative to 8) followed by treatment with sodium diethyldithiocarbamate (Na-DEDTC). Protein was analyzed by western blot with an α-biotin-HRP antibody. D) Various quantities of purified QC-sfGFP-FLAG or Boc-sfGFP-FLAG (5.0, 2.5, 1.0, 0.5, or 0 μg) were added to 10.0 μg bacterial lysate. The mixtures were incubated with 8 (200 μM) in K3Fe(CN)6 (1 mM) in PBS for 1 h at rt, quenched via addition of QCOAc (100 equiv. relative to 8), and treated with Na-DEDTC. Mixtures were analyzed via western blot with an α-biotin-HRP antibody.
We next sought to achieve selective labeling of QC-sfGFP-FLAG in the more complex environment of bacterial lysate. For this experiment, we doped varying amounts of purified QC- sfGFP-FLAG or Boc-sfGFP-FLAG into 10 μg samples of bacterial lysate (Figure 2D). Each sample was then incubated with Ni bis(dithiolene) biotin 8 (200 μM) and K3Fe(CN)6 (1 mM) in PBS for 1 h at 22 °C. The reaction was quenched with QCOAc (100 equiv. relative to 8), treated with Na-DEDTC, and samples were analyzed by western blot and the presence of biotin was probed with an α-biotin-HRP antibody. Results are shown in Figure 2D, where QC-sfGFP-FLAG can be seen to be selectively labeled by 8 in the presence of complex cell lysate at all QC-sfGFP-FLAG:lysate ratios, with more robust labeling at higher ratios (5.0 or 2.0 μg QC-sfGFP-FLAG added to 10 μg lysate). Samples with Boc-sfGFP-FLAG showed minor labeling at only the highest concentration (5.0 μg), suggesting occurrence of background labeling on sfGFP-FLAG. Thus, while it is clear that we have achieved labeling of QC-sfGFP-FLAG in the presence of bacterial lysate, background reactivity, both on Boc-GFP-FLAG as well as on lysate, remains a liability of the QC ligation.
In summary, we achieved site-specific incorporation of a QC amino acid into a protein of interest using wildtype PylRS and tRNACUA. Subsequently, this QC-labeled protein could be selectively modified in the presence of bacterial cell lysate. These developments demonstrate the potential applications of the QC ligation to native biological systems.
2.2 Photocleavage of the QC/nickel bis(dithiolene) adduct
Early reports of the photolability of the QC/metal bis(dithiolene) adduct piqued our interest for expanding the types of reactions accessible via the QC ligation. In 1965, Schrauzer and Mayweg were the first to describe the decomposition of adduct 10 (Figure 3A) when 31 exposed to daylight. Later studies by Wing and Sugimori describe the photolysis reaction to be that shown in Figure 3A, where adduct 10 is cleaved to form norbornadiene 11, an isomer of QC, and the starting nickel bis(dithiolene) 12.32–34 We envisioned taking advantage of the photolability of adducts such as 10 to render the product of the QC ligation cleavable. While there are numerous examples of cleavable bioconjugations that take advantage of native functionalities such as primary amines and thiols,35–42 or slowly reversible reactions such as aldehyde or ketone condensations,28,43–46 there is only one example of a fast and reversible bioorthogonal reaction that utilizes only non-native reactive groups.14 The ligation adduct of the chemoselective rapid azo-coupling reaction (CRACR) can be cleaved upon exposure to the small molecule dithionite.14 This requirement inherently limits the spatial control of this reaction in biological systems. A cleavable QC ligation reaction would enable selective tagging and photochemically controlled untagging of target biomolecules in a system, presenting opportunities for temporally and spatially controlled protein modification. Additionally, a cleavable bioorthogonal adduct could have applications in protein enrichment and purification. Thus, we sought to optimize the QC/nickel bis(dithiolene) photocleavage reaction for use on proteins.
Figure 3. Photocleavage of the QC/nickel bis(dithiolene) adduct.
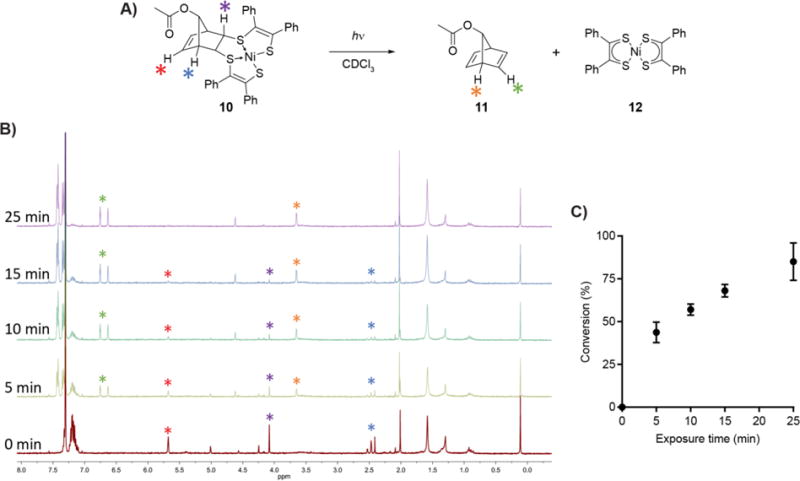
A) Reaction scheme for the model photocleavage. Adduct 10 was dissolved in CDCl3 to a concentration of (5 mM) and placed in a borosilicate glass tube. The solution was placed in a foil-lined container and irradiated with 365 nm light from a handheld lamp for 0-25 min. B) Representative 1H NMR spectra from Figure 3A at the indicated times. Diagnostic peaks are labeled with colors corresponding to the stars in Figure 3A. C) The progress of photocleavage was determined by comparing the integration of individual peaks of 10 to 11. Each measurement is the average of three separate experiments. Error bars represent one standard deviation.
We began by analyzing the efficiency of the photocleavage reaction in a flask, aiming to determine its half-life upon irradiation with a handheld UV lamp. Analyses were performed via 1H NMR, and we selected adduct 10 due to its relative stability to reduction in solution (the anionic form of the nickel bis(dithiolene) adduct is paramagnetic and challenging to analyze by NMR). Photocleavage was performed using a 365 nm light because of the high absorbance coefficient associated with this wavelength.33,34 We observed clean conversion of 10 to 11 and 12 within minutes (Figure 3B). Product formation was quantified by comparing the integration of 1H NMR peaks of product and starting material, and the half-life of this photocleavage was found to be ~8 min at 5 mM in CDCl3 (Figure 3C). A reaction occurring in minutes is useful on biological time-frames, and 8 min compares favorably to the half-life of the well-established bioorthogonal copper-free click reaction between the cyclooctyne BARAC and benzyl azide, with a maximum half-life of ~3.5 min when both reactants are present at 5 mM in CD3CN at room temperature.47 Given the rapid photolysis of adduct 10 at room temperature using a handheld lamp, we decided to test this approach on proteins labeled via the QC ligation.
To this end, we non-specifically labeled bovine serum albumin (BSA) with p-nitrophenyl quadricyclane carbonate (5, Figure 4A) as we previously reported15 to give QC-BSA, our model protein for optimization of the photocleavage reaction. QC-BSA was then labeled via QC ligation with nickel bis(dithiolene) biotin 8 to yield biotin-labeled BSA 13 (Figure 4A).15 As a negative control, BSA that was not QC-modified was exposed to the same reaction conditions with 8 (hereafter referred to as 8-treated BSA). We irradiated 13 and 8-treated BSA using a 365 nm handheld lamp and probed for release of 8 by western blot using an α-biotin-HRP antibody (Figure 4A and B). To obviate the possibility of the precedented reaction between the nickel bis(dithiolene) 8 and norbornadiene 14 (Figure 4A) released by the photocleavage,33 our samples were treated with scavenging QCOAc to sequester 8. When samples of 13 were irradiated with UV light, the amount of biotin signal was significantly reduced with increasing exposure times (Figure 4B). No biotin signal was observed with 8-treated BSA.
Figure 4. Photocleavage of the QC/nickel bis(dithiolene) adduct on a protein.
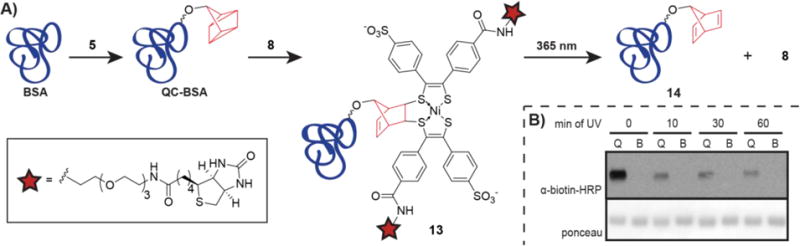
A) Lysines on bovine serum albumin (BSA) were capped via reaction with p-nitrophenyl quadricyclane carbonate 5. The resulting QC-BSA and untreated BSA (negative control) were treated with nickel bis(dithiolene) biotin 8 (50 μM in PBS) for 30 min to form adduct 13 or 8-treated BSA. The reactions were quenched with QCOAc and protein was purified from excess small molecule via centrifugal filtration with an Amicon Ultra 10 kDa molecular weight cutoff filter. 13 and 8-treated BSA in PBS were irradiated with a 365 nm handheld UV lamp for 0, 10, 30, or 60 min to cleave the adduct, providing 14 and 8. Protein products were separated from free 8 via centrifugal filtration as described above. C) Protein from Figure 4A was analyzed via western blot with an α-biotin-HRP antibody. Q = 13, B = 8-treated BSA.
With a readily cleavable biotin attached to a protein, we next sought to utilize the photocleavage capability of the QC ligation as a novel protein purification method. A schematic of purification using this photocleavage for QC-BSA or BSA is presented in Figure 5A. In the presence of just PBS or cell lysate, QC-BSA or BSA were incubated with 8 for 14 h in the dark. After removal of remaining small molecule by centrifugal filtration, the mixture was added to streptavidin resin for 30 min in the dark, and the beads were treated with PBS until no more protein washed from the resin (Washes). The beads were then irradiated with 365 nm light from a handheld lamp in 5-minute increments for a total of 20 minutes with elutions of PBS after every 5-minute mark (Elutions). Finally, the beads were washed with 8 M guanidine HCl (pH 1.5) to remove any remaining protein (not shown schematically). Following the collection of all washes and elutions, the proteins were visualized by SDS-PAGE and Coomassie staining. When either QC-BSA or BSA was used, protein was present in the initial washes (Figure 5B, Wash 1; Wash 2,3), but not in the final washes (Wash 6,7). After UV irradiation, a very faint band was observed when BSA was used (BSA, Elution 1 and 2). However, UV irradiation of the QC-BSA-containing mixture provided a significant amount of protein in the elution lanes (QC-BSA, Elution 1 and 2). This suggests that biotin attached to the protein specifically through the QC ligation was removed upon UV irradiation, thus releasing the protein from the streptavidin resin. Very little protein was left on the beads after UV irradiation (QC-BSA and BSA, Elution B).
Figure 5. QC photocleavage as a method for protein purification.
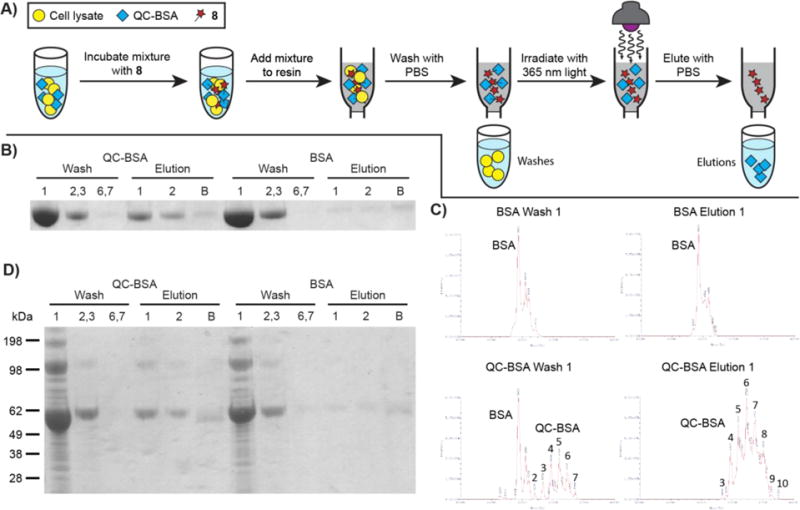
A) Schematic showing general protein purification procedure using QC-BSA and cell lysate. QC-BSA in a mixture with cell lysate was incubated with 8 for 14 h in the dark and subsequently quenched with QCOAc. The samples were transferred to Amicon Ultra centrifugal filters and washed with PBS three times. The concentrated samples were incubated with Pierce™ Streptavidin Agarose for 30 min. The resin was washed with PBS by spin filtration seven times (Washes 1-7). Following the washes, the resin was suspended in PBS and irradiated with 365 nm light by a handheld lamp for 5 min and the supernatant was eluted (Elution 1). The irradiation was repeated three more times with additional PBS each time (Elutions 2-4). The beads were then stripped of all remaining protein with 8 M guanidine HCl (pH = 1.5, 4 washes) and this was concentrated on an Amicon Ultra centrifugal filter (Elution B). Washes 1; 2,3; 6,7 and Elutions 1; 2; B were separated by gel electrophoresis and visualized with Coomassie staining. B) The general procedure from Figure 5A was followed with the following quantities of reagents: QC-BSA or BSA (100 μL in PBS, 1 mg/mL), 8 (100 μL, 50 μM in PBS), QCOAc (2.5 μL, 200 mM in DMSO), PBS rinse (200 μL, then twice with 350 μL), Pierce Streptavidin Agarose (100 μL), PBS washes (50 μL each, lanes Wash 1; 2,3; 6,7), elutions (50 μL PBS each, lanes Elution 1 and 2), guanidine HCl (50 μL washes, Elution B). C) Protein from Wash 1 and Elution 1 of BSA and QC-BSA samples from Figure 5B were subjected to ESI intact mass analysis. Unmodified BSA: BSA with no QC modifications. The numbers represent the number of QC moieties attached to BSA. D) The general procedure from Figure 5A and 5B was performed in the presence of bacterial cell lysate (10 μL, 4.94 g/mL) and K3Fe(CN)6 (10 μL, 22 mM). K3Fe(CN)6 and lysate were added sequentially to the solutions of QC-BSA or BSA before addition of 8. After QC ligation with 8, the capture, wash, and elutions sequence depicted in Figure 5A was performed. Lanes are as described in Figure 5B.
Curiously, when the QC-BSA mixture was incubated with the streptavidin beads, most of the total protein was present in the initial wash, rather than eluting only after irradiation. We hypothesized that since QC-BSA was non-specifically labeled with QC, the washes might contain BSA that was not labeled with QC, and thus could not undergo reaction with 8. We performed intact mass analysis of the protein present in Wash 1 and Elution 1 of both QC-BSA and BSA samples from Figure 5B. When the mixture with BSA was incubated with the streptavidin resin, only unmodified BSA was observed to wash off the resin (Figure 5C, BSA Wash 1). A small amount of unmodified BSA was also observed eluting from the column in the elution of BSA (Figure 5C, BSA Elution 1). Notably, the incubation of the QC-BSA mixture with streptavidin resin showed a large amount of unmodified BSA in addition to some QC-BSA in the initial wash (Figure 5C, QC-BSA Wash 1). This suggests that a significant portion of the QC-BSA sample was BSA that was not labeled by 5, and thus would not react specifically with 8. After irradiation of QC-BSA-containing resin, however, the elution contained only QC-BSA (Figure 5C, QC-BSA Elution 1). This suggests that BSA modified with QC could bind to the streptavidin after treatment with 8 and be subsequently released upon exposure to UV light. No protein was observed to be modified by 8 in the intact mass analysis. This could be due to quantitative photocleavage, or perhaps the QC ligation adduct does not ionize well or is not stable under the conditions present in ESI.
To further demonstrate the utility of this photocleavage, the isolation of a QC-modified protein from a complex mixture of biological material was attempted. To a solution of QC-BSA or BSA was added K3Fe(CN)6 and bacterial cell lysate. To this mixture, 8 was added and, following an incubation in the dark, the solutions were quenched with QCOAc and washed with PBS. The samples were then incubated on Pierce™ Streptavidin Agarose, followed by the washes and elutions described above. When BSA was used, most protein was found to come off the resin in the washes (BSA Washes, Figure 5D), while very little protein was observed in the elutions (BSA, Elutions). In contrast, when QC-BSA was used the bacterial lysate was only present in the washes (QC-BSA, Washes). Following irradiation, only QC-BSA was present in the elutions (QC-BSA, Elutions). In the complex mixture of cell lysate, QC-modified protein could be labeled and released selectively using the QC ligation and subsequent photocleavage. This presents a promising new protein purification method that is based on a photocleavable click chemistry adduct.
3. Conclusion
Over the past two decades, much attention has been given to bioorthogonal reactions that form adducts between two molecules. Here we have described the development of the QC ligation to be more biologically accessible and chemically useful beyond typical adduct-forming bioorthogonal reactions. As the number of chemical transformations that are adapted for use in biological systems increases, the impetus for discovery will shift to the creative application of these reactions. Using these new reactions, the ability to augment the complexity of life, rather than study it, will be a transformative force in the third decade of bioorthogonal chemistry.
4. Experimental
4.1 General Chemical Methods
Reagents and solvents were obtained commercially and used without further purification with the exception of anhydrous solvents which were prepared by the method developed by Pangborn et al.48 Anhydrous dimethylformamide was obtained from Sigma Aldrich in a Sure/Seal™ container. Flash column chromatography was performed according to the method described by Still et al. using Silicycle Silica Flash P60 silica (40-63 mm, 230-400 mesh).49 All high pressure liquid chromatography purifications were performed on a Varian ProStar HPLC equipped with UV/vis detector using a 100 Å C18 column (250 × 21.4 mm) at a flow rate of 20 mL/min.
Reaction progress was monitored by TLC with Silicycle glass backed extra hard layer TLC plates (60 Å, 250 μm) visualized with ultraviolet light or with potassium permanganate or p-anisaldehyde staining followed by brief heating on a hot plate. Unless otherwise noted, anhydrous magnesium sulfate was used to dry organic extracts and solutions were concentrated using a Buchi Rotovapor-114 equipped with a Welch 2026 self-cleaning dry vacuum system. Compounds were dried under reduced pressure on a Schlenk line equipped with an Edwards high vacuum. Lyophilization was performed using a Labconco FreeZone Plus 4.5 Liter benchtop lyophilizer equipped with a Labconco vacuum (Model 117).
All 1H and 13C spectra were obtained with a Bruker AV-600, AV-500, AVB-400, or AVQ-400 spectrometer. Peaks are reported in ppm and referenced to the solvent. Data are presented as follows: chemical shift in ppm, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, quin = quintet, m =multiplet), coupling constant in Hz, and integration value. High-resolution electrospray ionization (ESI) mass spectrometry data were obtained through the UC Berkeley Mass Spectrometry Facility on a Linear trap quadrupole fourier transform ion cyclotron resonance (LTQ-FT-ICR) mass spectrometer from Thermo Fisher. High-resolution electron ionization (EI) mass spectrometry data were obtained through the UC Berkeley Mass Spectrometry Facility on an AutoSpec mass spectrometer from Waters.
4.2 Synthesis of quadricyclane amino acid (6)
4.2.1 Fmoc-protected QC amino acid
To a flame dried round bottom flask was added 30 mL dry dimethylformamide followed by Nα-Fmoc-L-lysine (455 mg, 1.23 mmol, 1.00 equiv.). p-nitrophenyl carbonate 5 (508 mg, 1.86 mmol, 1.50 equiv.), prepared via the route published by Sletten et al.15 was added, and the reaction mixture immediately turned yellow. The solution was stirred at room temperature overnight, and the following day was concentrated to a yellow oil. Crude product was purified by flash column chromatography on silica gel that was pre-treated with 0.1% acetic acid in hexanes followed by washing with ~4 column volumes of hexanes. Product was eluted with hexanes:EtOAc:MeOH (10:1:0, 2:1:0, 0:1:0, 0:95:5, 0:9:1) to give the product in 85% yield (526 mg as a mixture with 140 mg DMF as determined by 1H NMR). Rf = 0.54 in 100% EtOAc; 1H NMR (600 MHz, MeOD) δ 7.78 (d, J = 7.5 Hz, 2H), 7.67 (dd, J = 11.3, 7.5 Hz, 2H), 7.38 (t, J = 7.5 Hz, 2H), 7.30 (t, J = 7.5 Hz, 2H), 5.53–5.46 (m, 1H), 4.34 (qd, J = 10.6, 7.1 Hz, 2H), 4.21 (t, J = 7.0 Hz, 1H), 4.14 (dd, J = 9.5, 4.6 Hz, 1H), 3.16–3.06 (m, 2H), 1.87–1.84 (m, 1H), 1.81–1.65 (m, 3H), 1.62–1.36 (m, 8H); 13C NMR (151 MHz, MeOD) δ 176.0, 159.6, 158.7, 145.3, 145.1, 142.6, 142.5, 128.8, 128.8, 128.2, 128.1, 126.3, 126.2, 120.9, 120.9, 83.7, 67.9, 55.3, 48.4, 41.4, 32.3, 30.5, 26.8, 26.8, 24.2, 16.8, 15.3; HRMS (ESI) calcd for C29H30O6N2Na [M+Na]+ : 525.1996; found: 525.1993.
4.2.2 QC amino acid (6)
Fmoc-protected QC amino acid (598 mg, 1.19 mmol, 1.00 equiv.) was dissolved in anhydrous dimethylformamide (10 mL) in a round bottom flask. Piperidine (400 μL, 4.05 mmol, 3.40 equiv.) was added, causing an immediate color change to yellow followed by cloudiness in the reaction mixture. The reaction mixture was stirred at room temperature for 2 hours at which point it was concentrated to an off white solid. The solid was triturated with cold EtOAc and filtered. The remaining white solid was washed with cold EtOAc to yield pure 6 (263 mg, 78% yield). Rf = 0.44 in 3:1 MeOH:EtOAC; 1H NMR (600 MHz, D2O) δ 5.50 (s, 1H), 3.70 (s, 1H), 3.14 (t, J = 6.9 Hz, 2H), 1.85–1.80 (m, 4H), 1.63 (s, 2H), 1.57–1.46 (m, 4H), 1.42–1.32 (m, 2H); 13C NMR (151 MHz, DMSO) δ 169.5, 156.5, 81.0, 54.1, 40.1, 30.8, 29.2, 25.5, 22.5, 15.6, 13.9; HRMS (ESI) calcd for C14H20O4N2Na [M+Na]+ : 303.1315; found: 303.131.
4.3 Synthesis of nickel bis(dithiolene) biotin (8)
4.3.1 Dithiocarbonate ligand (S2)
Dithiocarbonate S1 (Scheme S1) was synthesized according to the route developed by Sletten et al.15 EZ-link amine-PEG3-biotin from Thermo Scientific (53 mg) was transferred to a round bottom flask as a solution in water, concentrated to a solid on a rotary evaporator, and azeotroped from toluene (2 × 2 mL). The amine-PEG3-biotin was then dissolved in 1 mL dry dimethylformamide under an atmosphere of nitrogen. In a separate flask S1 (24.2 mg, 0.06 mmol, 1.00 equiv.) was dissolved in 1 mL dry dimethylformamide. 500 μL of the amine-PEG3-biotin solution (26.5 mg, 0.07 mmol, 1.17 equiv.) was added to the solution of S1 followed by addition of EDC•HCl (22.6 mg, 0.12 mmol, 2.00 equiv.), HOBt hydrate (13.0 mg) and triethylamine (20 μL, 0.14 mmol, 2.33 equiv.). The reaction mixture was stirred at room temperature for 3 days, then concentrated to a yellow oil. Crude product was dissolved in 1:2 MeOH:H2O, filtered through a 0.2 micron filter, and purified by HPLC on a C18 prep column, 30 to 95% MeOH in H2O over 30 min, 20 mL/min flow rate. The product eluted as a broad peak between 8 and 12 min, yielding S2 as a yellow oil (28 mg, 0.035 mmol, 58% yield). 1H NMR (600 MHz, MeOD) δ 7.76 (t, J = 8.5 Hz, 4H), 7.34 (dd, J = 11.2, 8.1 Hz, 4H), 4.49–4.46 (m, 1H), 4.30–4.28 (m, 1H), 3.65–3.61 (m, 10H), 3.57–3.53 (m, 5H), 3.50–3.48(m, 2H), 3.21–3.17 (m, 2H), 3.15–3.11 (m, 1H), 2.92–2.87 (m, 2H), 2.70–2.68 (m, 1H), 2.49–2.48 (m, 1H), 2.20 (t, J = 7.5 Hz, 2H), 1.75–1.69 (m, 1H), 1.66–1.54 (m, 3H), 1.44–1.39 (m, 2H); 13C NMR (151 MHz, MeOD) δ 190.7, 176.1, 169.1, 166.0, 147.2, 136.2, 135.8, 134.4, 130.9, 130.7, 130.2, 129.8, 129.0, 127.7, 71.5, 71.5, 71.3, 71.2, 70.6, 70.5, 63.4, 61.6, 57.0, 49.9, 43.4, 41.1, 41.0, 40.4, 40.3, 40.3, 37.2, 36.7, 35.9, 32.3, 29.7, 29.5, 27.0, 26.8, 19.3, 15.7. HRMS (ESI) calcd for C34H41O10N4S4 [M–H]− : 793.1711; found: 793.1698.
4.3.2 Nickel bis(dithiolene) biotin (8)
Dithiocarbonate S2 (17.3 mg, 0.02 mmol, 1.00 equiv.) was dissolved in 400 μL THF and 400 μL H2O. Tetramethylammonium hydroxide pentahydrate (8.4 mg, 0.05 mmol, 2.5 equiv.) was added to the reaction mixture as a solution in 60 μL MeOH and the mixture quickly became a dark yellow/orange color. After stirring the resulting mixture at room temperature for 30 minutes, nickel(II) chloride hexahydrate (2.8 mg, 0.01 mmol, 0.5 equiv.) was added. The solution immediately became dark red, and was stirred at room temperature overnight. The following morning, iodine (3.2 mg, 0.01 mmol, 0.5 equiv.) was added and the reaction mixture immediately became dark green. After stirring at room temperature for 3 h, the reaction mixture was concentrated to a green solid on a rotary evaporator. The crude product was purified by flash column chromatography and eluted with acetonitrile:H2O (10:1, 5:1, 3:1, 2:1). Fractions containing product were pooled, acetonitrile was evaporated on a rotary evaporator, and the remaining aqueous solution was lyophilized overnight to yield 8 as a dark green, fluffy solid (7.2 mg, 0.005 mmol, 21% yield). Rf = 0.64 in 5:1 acetonitrile:H2O; UV/vis/NIR 225–900 nm (water): 859 nm (1.3 au), 311 nm (3.9 au), 282 nm (4.1 au), 232 nm (44.3 au); HRMS (ESI) calcd for C66H82O18N8NiS8 [M–2H]2− : 794.1439; found: 794.1417.
4.4 Non-natural amino acid incorporation and QC ligation labeling experiments
4.4.1 General methods
Nε-Boc-L-lysine (Boc-lysine) was purchased from Chem-Impex and used without further purification. 6 was synthesized and purified as described above. Phosphate-buffered saline 1X, – calcium –magnesium (PBS) was obtained from Corning Cellgro. All protein expression experiments were performed using DH10β cells from Invitrogen (MAX efficiency, chemically competent). A New Brunswick Scientific C25 incubator shaker was used to incubate E. coli liquid cultures at 37 °C with shaking at 225 rpm. A Lab-line Imperial II incubator was used to incubate LB agar plates at 37 °C. LB agar plates were prepared using Luria Broth Base (Miller’s LB Broth Base, Invitrogen) and agar (granulated, Fisher Scientific). LB liquid media and LB agar plates were supplemented with kanamycin to 50 μg/mL and/or tetracycline to 25 μg/mL depending on the combination of plasmids used. Sample centrifugation was performed in a Thermo Scientific Sorvall RC 6+ centrifuge, Sorvall RC 5C Plus centrifuge, a Sorvall Legent RT centrifuge, or an Eppendorf centrifuge 5417C.
Gel electrophoresis was performed using BioRad Criterion gel electrophoresis chambers equipped with a BioRad Power Pac 200. All gels (Biorad Criterion XT pre-cast) were run in MES buffer. Western blot transfers were performed using BioRad Criterion blotter transfer boxes equipped with a BioRad Power Pac 200. Gels were transferred to nitrocellulose (BioRad 0.45 μm) in tris-glycine buffer (20% MeOH). See Blue Plus 2 pre-stained protein ladder from Life Technologies was used for all gels and western blots. α-GFP (4B10) Mouse mAb (#2955), α-Mouse IgG horseradish-peroxidase-linked (HRP) antibody (#7076), and α-FLAG®-HRP rabbit polyclonal antibody (DYKDDDDK Tag antibody HRP conjugate, #2044) were obtained from Cell Signaling Technologies. Mouse α-biotin-HRP was obtained from Southern Biotech. Protein loading was assessed via staining with Ponceau (5% acetic acid, 0.1% (w/v) ponceau). Blots were visualized on Premium X-ray film (blue) from Phenix Research Products. Densitometry analyses were performed using ImageJ 1.48v (National Institutes of Health). Fast protein liquid chromatography (FPLC) was performed on a Pharmacia Biotech Akta explorer 10. High resolution mass spectrometry data were obtained on a Linear Trap Quadrupole LTQ-Orbitrap-XL mass spectrometer (Thermo Scientific) equipped with an electrospray ionization (ESI) source.
4.4.2 General Procedure for Transformation of E. coli with plasmids psfGFP150TAGFLAGPylT and/or pBKPylRS
DH10β E. coli were transformed with psfGFP150TAGFLAGPylT and pBKPylRS (+RS+T) or with psfGFP150TAGFLAGPylT and sterile ddH2O (−RS+T) by heat shock (30 sec, 42 °C, 2 min, wet ice) and recovered in 250 μL super optimal broth with 20 mM glucose (S.O.C. medium) for 35 min at 37 °C and 225 rpm. Cells were then plated on kanamycin/tetracycline (+RS+T E. coli) or tetracycline (−RS+T E. coli) LB agar plates and incubated at 37 °C overnight. The following day, 5 mL liquid cultures of LB media supplemented with kanamycin/tetracycline (+RS+T E. coli) or tetracycline (−RS+T E. coli) were inoculated with one colony from +RS+T and one colony from −RS+T respectively. Cultures were incubated at 37 °C and 225 rpm overnight. The following morning, glycerol stocks were prepared (375 μL culture and 125 μL sterile 80% glycerol in ddH2O) and frozen to −80 °C for future use.
4.4.3 Expression of QC- and Boc-sfGFP-FLAG
DH10β cells were transformed with the relevant plasmids (+RS+T or −RS+T) as described above and the resulting 5 mL overnight cultures were used immediately. Alternatively, glycerol stabs of transformed DH10β cells stored at −80 °C were used to streak fresh LB agar plates (supplemented with the requisite antibiotics). The plates were grown overnight at 37 °C, and 5 mL liquid media cultures (supplemented with the requisite antibiotics) were inoculated with single colonies from each plate. These 5 mL cultures were then incubated overnight at 37 °C and 225 rpm and used the next day to inoculate larger cultures for protein expression. All protein expression protocols begin with a 5 mL overnight culture.
4.4.3.1 Expression of QC-sfGFP-FLAG and Boc-sfGFP-FLAG for use in the experiment shown in Figure 1C
DH10β E. coli were transformed with pBKPylRS and/or psfGFP150TAGFLAGPylT and 5 mL overnight cultures were prepared as described in previous experiments. The following morning, 2 mL from each culture was used to inoculate 50 mL LB media cultures (supplemented with the requisite antibiotics). Each culture was incubated at 37 °C and 225 rpm to OD600 ~0.4 at which point the cultures were divided into three separate 15 mL polystyrene tubes (5 mL/tube) and supplemented with 6 or BocLys (from 22 mM stock solution of amino acid in sterile 0.1 M NaOH ddH2O, actual concentration may be lower due to limited solubility) to ~1 mM or supplemented with the same volume of vehicle (0.1 M NaOH) and incubated at 37 °C and 225 rpm for 30 min. Protein expression was induced via addition of 20% L-arabinose in sterile ddH2O to 0.2% and expressed for 3.5 hours. Following expression, cultures were transferred to 15 mL falcon tubes and pelleted at 3500 rom and 4 °C for 10 min. Pellets were washed 3× via resuspension in LB media followed by pelleting at 9,700 rpm for 5 min and aspiration of the supernatant. Washed pellets were stored at −80 °C until further use.
Samples were thawed on ice, then warmed to rt. 1/6 of each pellet was isolated and resuspended in 40 μL 1X SDS loading buffer supplemented with 2.5% β-mercaptoethanol. The suspension was centrifuged at 14,000 rpm for 15 min, and samples were loaded onto two, 18-well 4–12% bis-tris Criterion XT precast polyacrylamide gels (15 μl sample/per well). Gels were treated as described in the General Western Blotting Procedure section with α-GFP Mouse mAb and α-Mouse-HRP or with α-FLAG-HRP rabbit polyclonal antibody.
4.4.3.2 Expression of QC-sfGFP-FLAG and Boc-sfGFP-FLAG for use in the experiment shown in Figures 1D and 2C-D
+RS+T DH10β and −RS+T DH10β E. coli 5 mL overnight cultures were prepared as described above using pBKPylRS and psfGFP15TLAGFLAGPylT plasmids. The following morning, 2 mL from each culture was used to inoculate 100 mL LB media (supplemented with the requisite antibiotics). Cultures were grown to OD600 = 0.3–0.5 and supplemented with Boc-Lys or 6 (from 50 mM amino acid stock solution in 0.1 M NaOH ddH2O, actual concentration may be lower due to limited solubility) to ~1 mM. Flasks were incubated for an additional 30 min at 37 °C and 225 rpm after which protein expression was induced via addition of 20% L-arabinose in sterile ddH2O to 0.2%. Protein was expressed for 3.5 hours and then each culture was separated into two 50 mL falcon tubes and pelleted at 3,500 rpm for 10 min at 4 °C. The supernatant was aspirated and pellets from each culture were combined and washed 3× by resuspension in 20 mL LB media, pelleting at 3,500 rpm for 10 min at 4 °C, and aspiration of supernatant. Pellets were stored at −20 °C until lysis, at which point they were resuspended in 3 mL lysis buffer (tris-HCl (10 mM), NaCl (200 mM), imidazole (20 mM), pH = 8, Roche protease inhibitor) and, lysed via sonication (Misonix Sonicator 3000, power level 2, 4 min on total: 10 sec on, 10 sec off). The resulting lysate was clarified by centrifugation (20 min, 21,000 × g, 4 °C).
FLAG-tagged protein was then purified via affinity column at room temperature using ANTI-FLAG® M2 Affinity Gel from Sigma Aldrich. The gel was equilibrated with 5 column volumes of TBS buffer (150 mM NaCl, 50 mM tris-HCl, pH = 7.4), lysate was loaded via three passes through the column, and unbound protein was washed off the column with 20 column volumes of TBS buffer. Bound protein was eluted with five column volumes 100 μg/mL 3XFLAG peptide (Sigma Aldrich) in TBS. Elutions were pooled and concentrated to ~500 μL using an Amicon Ultra 4 mL, 10 kDa molecular weight cutoff centrifugal filter. Protein was further purified by fast protein liquid chromatography (FPLC) at 4 °C on a Superdex 75 10/300 GL (GE Healthcare Life Science) column using FPLC buffer (tris-HCl (20 mM), NaCl (100 mM), pH = 7.4) at a flow rate of 0.5–0.6 mL/min Fractions (0.5 mL each) were analyzed by polyacrylamide gel electrophoresis followed by protein visualization with Coomassie stain (0.25% (w/v) Coomassie, 40% methanol, 10% acetic acid). Following FPLC purification, protein was transferred to PBS via solvent exchange using Amicon Ultra 10 kDa molecular weight cutoff filters.
4.4.4 General QC ligation procedure for sfGFP
Purified QC- or Boc-sfGFP (~0.2 mg/mL) in the presence or absence of E. coli lysate (0.33 mg/mL) was combined with 6 (200 μM) and K3Fe(CN)6 (1 mM) in PBS and incubated in the dark at room temperature for 1-2 h. Reactions were quenched via addition of 100 equiv. QCOAc (200 mM solution in DMSO) relative to sfGFP. Samples were then combined with Na-DEDTC (100 mM solution in PBS).
4.4.5 General Western Blotting Procedure
All samples were combined with 4X sodium dodecyl sulfate (SDS) loading buffer (supplemented with 10% β-mercaptoethanol) to a final loading buffer concentration of 1X for separation via sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). All SDS-PAGE was performed using 4–12% bis-tris Criterion XT precast polyacrylamide gels. Gels were run at 175 V for 40 min and then were transferred to nitrocellulose at 75 V for 90 min. The blots were treated with Ponceau stain to visualize proteins.
Blots to be analyzed with α-biotin-HRP were blocked with 5% bovine serum albumin (BSA) in PBST at 4 °C in the dark overnight. All other blots were blocked with 5% milk in PBST at 4 °C in the dark overnight. After blocking, α-biotin-HRP blots were incubated in a 1:200,000 dilution of α-biotin-HRP in 5% BSA, PBST for 15 min at room temperature in the dark and then washed 3 × 10 min with PBST. α-FLAG blots were incubated in a 1:3,000 dilution of α-FLAG-HRP in 5% milk PBST followed by washing 3 × 10 min PBST. α-GFP blots were incubated in a 1:3,000 dilution of α-GFP in 5% milk, PBST for 1 h at room temperature followed by washing 3 × 10 min with PBST. After washing, blots were incubated in a 1:10,000 dilution of α-mouse-HRP in 5% milk, PBST for 1 h at room temperature followed by washing 3 × 10 min with PBST.
For all blots, detection was performed by chemiluminescence using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific). Blots were developed on a Kodak X-OMAT 500RA processor.
4.4.6 Analysis of Termination Product
We expressed His6-tagged QC-sfGFP and Boc-sfGFP according to the procedure described in section 4.4.3.2 using plasmids psfGFP150TAGHis6PylT and pBKPylRS. Upon lysis, His6-tagged proteins were purified on a Ni2+-NTA column. Qiagen Ni2+-NTA beads (500 μl) were washed 3× via suspension in binding buffer (imidazole (5 mM), tris-HCl (10 mM), NaCl (200 mM), pH = 8) followed by centrifugation and aspiration of supernatant. Beads were then added to clarified lysate as a suspension in 500 μL binding buffer, and the mixture was incubated with agitation for 1–1.25 h at 4 °C, after which beads were transferred to a fritted column. At 4 °C, the beads were washed 3 × 3 mL wash buffer (tris-HCl (10 mM), imidazole (20 mM), NaCl (200 mM), pH = 8) followed by elution with 6 × 1 mL wash buffer supplemented with imidazole to 200 mM. SDS-PAGE analysis of the fractions indicated co-elution of termination product along with full length QC- and Boc-sfGFP-His6. Elutions were pooled and concentrated to ~500 μL using an Amicon Ultra 4 mL, 10 kDa molecular weight cutoff centrifugal filter. Full length QC- and Boc-sfGFP-His6 was separated from amber termination product via FPLC purification according to the protocol described in section 4.4.3.2. Purified proteins were further characterized by high resolution mass spectrometry (Figure S1).
4.5 Cloning to exchange His6 tag for FLAG tag
The sfGFP gene from psfGFP150TAGHis6PylT (sequence below, plasmid obtained from Jason Chin Lab, Medical Research Council, Cambridge, UK) was amplified using forward primer and reverse primers purchased from Elim Biopharma with PCR. Forward primer: 5′- CAGGAGGAATTAACCATGGTTAGCAAAG-3′, reverse primer: 5′-ATCGTCGTCCTTGTAGTCGCTGCCTTTATACAGTTCATCCATACC-3′. The PCR reaction was purified by agarose gel electrophoresis, and the band corresponding to the PCR product was excised from the gel and purified using a Zymoclean Gel DNA Recovery Kit (Zymo Research) to give gene sfGFP150TAGFLAG1. Gene sfGFP150TAGFLAG1 was further amplified with forward and reverse primers to form full-length insert sfGFP150TAGFLAG2. Forward primer: as above, reverse primer: 5′-TTCGCTCGAGCTTTACTTGTCATCGTCGTCCTTGTAGTCGCTG-3′. The insert was isolated by agarose gel electrophoresis as described above, and the gene ends were digested for 1 h 45 min at 37 °C with XhoI and NcoI-HF (both from New England Biolabs) and purified using a Zymo DNA Clean & Concentrator Kit (Zymo Research). Vector psfGFP150TAGHis6PylT was linearized with XhoI and NcoI-HF (both from New England Biolabs) at 37 °C for 1 h 40 min, and linearized vector was isolated by agarose gel electrophoresis and treated with Antarctic phosphatase (New England Biolabs) at 37 °C for 1 h followed by purification with a Zymo DNA Clean & Concentrator Kit. The full-length sfGFP150TAGFLAG insert was ligated to linearized vector with T4 DNA ligase (New England Biolabs) at rt for 50 min to form psfGFP150TAGFLAGPylT.
One Shot, chemically competent DH5α E. coli (Life Technologies) were transformed with 2 μL ligation reaction containing the full plasmid psfGFP150TAGFLAGPylT. E. coli were transformed by heat shock (30 sec, 42 °C, 2 min, wet ice) and recovered in 250 μL super optimal broth with 20 mM glucose (S.O.C. medium) for 35 min at 37 °C and 225 rpm. Cells were then plated on tetracycline LB agar plates (~100–200 μL S.O.C. culture per plate) and incubated at 37 °C overnight. Single colonies were selected for inoculation of 5 mL liquid LB media cultures (supplemented with tetracycline) and cultures were incubated overnight at 37 °C and 225 rpm. The resulting cultures were used to prepare psfGFP150TAGFLAGPylT plasmid DNA using a Qiagen Miniprep kit. The sfGFP150TAGFLAG portion of the plasmid was sequenced using forward and reverse primers by Elim Bioharma. Forward primer: 5′-CAGGAGGAATTAACCATGGTTAGCAAAG-3′, reverse primer: 5′-TTCGCTCGAGCTTTACTTGTCATCGTCGTCCTTGTAGTCGCTG-3′.
4.6 1H NMR Studies of Photocleavage of 10
QC-Ni-bis(dithiolene) adduct 10 was prepared as reported by Sletten et al.15 A 5 mM solution of 10 in CDCl3 was added to 5 NMR tubes (600 μL each) wrapped in foil. An NMR was taken of one sample (0 min), and the others were placed in a dark container and irradiated with 365 nm light from a handheld UV lamp (Spectroline model ENF-260c, 254/365 nm). At 5, 10, 15, and 25 minutes a sample was removed from the box and a 1H NMR spectrum was taken. The amount of product was determined by measuring the integration of a product peak and comparing it to a starting material peak. This procedure was replicated 3 times, and the amount of product formation at each time point was averaged.
4.7 Preparation of QC-BSA
BSA (10 mg) was dissolved in PBS (0.5 mL) and to this mixture was added p-nitrophenyl carbonate 5 (1.4 mg, 0.056 mmol) dissolved in DMSO (83.3 μL) with a small amount of DMF (16.7 μL). An addition 200 uL of DMSO was added and the resulting yellow mixture was incubated at 22 °C for 3 h with gentle agitation. The protein was purified in illustra NAP-5 columns (GE Healthcare Life Sciences, Pittsburgh) as per manufacturer’s protocol, eluting in PBS. Protein concentration was measured by Pierce BCA Protein Assay (Thermo Fisher Scientific, Waltham). Resulting protein solutions were stored at 4 °C until use.
4.8 Photocleavage of the QC/nickel bis(dithiolene) adduct on a protein
A solution of QC-BSA or BSA (80 μL, 1 mg/mL in PBS) was combined with a solution of nickel bis(dithiolene) biotin 8 (80 μL of 100 μM in PBS, 50 μM final concentration 8). Reactions were incubated in the dark at room temperature for 30 min, then quenched via addition of 4 μL of a QCOAc in DMSO (200 mM, 100 equiv QCOAc relative to 8). Each reaction was diluted with 300 μl PBS, then transferred to an Amicon Ultra centrifugal filter (10k MW cutoff) and centrifuged at 14,000 × g for 5 min. Concentrated reactions were diluted with 350 μl PBS followed by centrifugation in a centrifugal filter at 14,000 × g for 5 min. This wash procedure was repeated one additional time to give the resulting proteins as solutions in ~50 μl PBS.
In fresh 1.5 mL eppendorf tubes, 50 μL of each sample was diluted with 380 μL PBS. The resulting solutions were aliquoted into borosilicate glass tubes (50 μL protein solution/tube). The protein in each tube was then diluted with an additional 550 μl PBS and 15 μl 200 mM QCOAc in DMSO to a final QCOAc concentration of 5 mM (solution is cloudy due to minimal solubility of QCOAc in PBS at this concentration). Tubes were irradiated at 365 nm with a handheld UV lamp or were irradiated with ambient light for 0, 10, 30, or 60 min (0 min samples were kept in the dark for the duration of the experiment).
From each sample, 400 μL was transferred to a 0.5 ml Amicon Ultra centrifugal filter and centrifuged at 14,000 × g for 5 min. The remaining sample (~200 μL) was then added to each filter and centrifuged at 14,000 × g for 5 min. PBS (350 μL) was added to each sample and they were centrifuged at 14,000 × g for 5 min. This process was repeated one additional time. The samples were subjected to western blot using an α-biotin HRP antibody were performed according to the general procedures.
4.9 Purification of biotin-labeled protein from streptavidin resin by photocleavage
QC-BSA or BSA (100 μL, 1 mg/mL in PBS) was mixed with a solution of 8 (100 μL, 50 μM in PBS) and gently agitated for 14 h in the dark. In purification experiments where cell lysate was used, K3Fe(CN)6 (10 μL of 22 mM in ddH2O) and bacterial cell lysate (10 μL, 4.94 mg/mL) were added sequentially prior to the addition of 8. QCOAc (2.5 μL, 200 mM in DMSO) was added to quench the reaction. The mixtures were transferred to Amicon Ultra centrifugal filters, 10k MW cutoff, containing 200 μL PBS, then centrifuged at 14000 × g, 5 min. PBS (350 μL) was added and the samples were spun at 14000 × g, 5 min. This was repeated once more before the samples were collected, spun 1000 × g, 2 min.
For each sample, Pierce™ Streptavidin Agarose (100 μL, ThermoFisher Scientific) was added to a Micro Bio-Spin™ Chromatography Column (Bio-Rad, Hercules). The storage buffer was removed via centrifugation 500 × g, 1 min. The gel was washed three times with PBS (100 μL), and centrifuged 500 × g, 1 min each wash. The samples were then loaded onto the beads and allowed to incubate in the dark for 30 min. The supernatant was removed with centrifugation, 500 × g, 1 min (Wash 1). The beads were washed 6 more times with PBS (50 μL) and collected by centrifugation, 500 × g, 1 min, (Washes 2-7). Washes 2 and 3, 4 and 5, and 6 and 7 were combined as pairs. Each sample was resuspended in PBS (50 μL) and irradiated with 365 nm light from a handheld UV lamp (Spectroline model ENF-260c, 254/365 nm) in a foil-lined container for 5 min. The supernatant was collected by centrifugation (500 × g, 1 min, Elution 1). This irradiation procedure was repeated three more times (Elutions 2-4). Finally, the beads were stripped of any remaining bound protein with the procedure above with five washes of 8 M guanidine HCl (pH 1.5, 50 μL each). These were combined and transferred to an Amicon Ultra spin concentrator (10k MW cutoff) and centrifuged at 14000 × g, 5 min. The concentrated solution was washed twice with PBS (250 μL) before being collected (Beads, lane B). The samples were then separated by gel electrophoresis and visualized with Coomassie staining.
Supplementary Material
Acknowledgments
We thank Jason Chin for providing plasmids encoding the pyrrolysine synthetase and transferase. C.G.G was supported by a predoctoral fellowship from the National Science Foundation. E.M.S. was supported by a predoctoral fellowship from the American Chemical Society. This work was supported by a grant from the National Institutes of Health (GM058867) to C.R.B.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sletten EM, Bertozzi CR. From Mechanism to Mouse: A Tale of Two Bioorthogonal Reactions. Acc Chem Res. 2011;44(9):666–676. doi: 10.1021/ar200148z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shih HW, Prescher JA. A Bioorthogonal Ligation of Cyclopropenones Mediated by Triarylphosphines. J Am Chem Soc. 2015 doi: 10.1021/jacs.5b06969. [DOI] [PubMed] [Google Scholar]
- 3.Vallée MRJ, Artner LM, Dernedde J, Hackenberger CPR. Alkyne Phosphonites for Sequential Azide-Azide Couplings. Angew Chemie Int Ed. 2013;52(36):9504–9508. doi: 10.1002/anie.201302462. [DOI] [PubMed] [Google Scholar]
- 4.Vallée MRJ, Majkut P, Krause D, Gerrits M, Hackenberger CPR. Chemoselective Bioconjugation of Triazole Phosphonites in Aqueous Media. Chem - A Eur J. 2015;21(3):970–974. doi: 10.1002/chem.201404690. [DOI] [PubMed] [Google Scholar]
- 5.Chen X, Wu Y-W. Selective Chemical Labeling of Proteins. Org Biomol Chem. 2016;14(24):5417–5439. doi: 10.1039/c6ob00126b. [DOI] [PubMed] [Google Scholar]
- 6.Patterson DM, Nazarova LA, Prescher JA. Finding the Right (Bioorthogonal) Chemistry. ACS Chem Biol. 2014;9(3):592–605. doi: 10.1021/cb400828a. [DOI] [PubMed] [Google Scholar]
- 7.McKay CS, Finn MG. Click Chemistry in Complex Mixtures: Bioorthogonal Bioconjugation. Chem Biol. 2014;21(9):1075–1101. doi: 10.1016/j.chembiol.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shih H-W, Kamber DN, Prescher JA. Building Better Bioorthogonal Reactions. Curr Opin Chem Biol. 2014;21:103–111. doi: 10.1016/j.cbpa.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 9.Sachdeva A, Wang K, Elliott T, Chin JW. Concerted, Rapid, Quantitative, and Site-Specific Dual Labeling of Proteins. J Am Chem Soc. 2014;136(22):7785–7788. doi: 10.1021/ja4129789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karver MR, Weissleder R, Hilderbrand SA. Bioorthogonal Reaction Pairs Enable Simultaneous, Selective, Multi-Target Imaging. Angew Chemie - Int Ed. 2012;51(4):920–922. doi: 10.1002/anie.201104389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang K, Sachdeva A, Cox DJ, Wilf NW, Lang K, Wallace S, Mehl RA, Chin JW. Optimized Orthogonal Translation of Unnatural Amino Acids Enables Spontaneous Protein Double-Labelling and FRET. Nat Chem. 2014;6(5):393–403. doi: 10.1038/nchem.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim J, Bertozzi CR. A Bioorthogonal Reaction of N-Oxide and Boron Reagents. Angew Chemie Int Ed. 2015;54(52):15777–15781. doi: 10.1002/anie.201508861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li J, Chen PR. Development and Application of Bond Cleavage Reactions in Bioorthogonal Chemistry. Nat Chem Biol. 2016;12(3):129–137. doi: 10.1038/nchembio.2024. [DOI] [PubMed] [Google Scholar]
- 14.Addy PS, Erickson SB, Italia JS, Chatterjee A. A Chemoselective Rapid Azo-Coupling Reaction (CRACR) for Unclickable Bioconjugation. J Am Chem Soc. 2017;139(34):11670–11673. doi: 10.1021/jacs.7b05125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sletten EM, Bertozzi CR. A Bioorthogonal Quadricyclane Ligation. J Am Chem Soc. 2011;133(44):17570–17573. doi: 10.1021/ja2072934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neumann H, Peak-Chew SY, Chin JW. Genetically Encoding Nε-Acetyllysine in Recombinant Proteins. Nat Chem Biol. 2008;4(4):232–234. doi: 10.1038/nchembio.73. [DOI] [PubMed] [Google Scholar]
- 17.Hancock SM, Uprety R, Deiters A, Chin JW. Expanding the Genetic Code of Yeast for Incorporation of Diverse Unnatural Amino Acids via a Pyrrolysyl-tRNA Synthetase/tRNA Pair. J Am Chem Soc. 2010;132(42):14819–14824. doi: 10.1021/ja104609m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nozawa K, Ishitani R, Nureki O. Pyrrolysyl-tRNA Synthetase-tRNAPyl Structure Reveals the Molecular Basis of Orthogonality. Seikagaku. 2010;82(7):617–623. [PubMed] [Google Scholar]
- 19.Chen PR, Groff D, Guo J, Ou W, Cellitti S, Geierstanger BH, Schultz PG. A Facile System for Encoding Unnatural Amino Acids in Mammalian Cells. Angew Chemie - Int Ed. 2009;48(22):4052–4055. doi: 10.1002/anie.200900683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mukai T, Kobayashi T, Hino N, Yanagisawa T, Sakamoto K, Yokoyama S. Adding L-Lysine Derivatives to the Genetic Code of Mammalian Cells with Engineered Pyrrolysyl-tRNA Synthetases. Biochem Biophys Res Commun. 2008;371(4):818–822. doi: 10.1016/j.bbrc.2008.04.164. [DOI] [PubMed] [Google Scholar]
- 21.Lang K, Chin JW. Cellular Incorporation of Unnatural Amino Acids and Bioorthogonal Labeling of Proteins. Chem Rev. 2014;114(9):4764–4806. doi: 10.1021/cr400355w. [DOI] [PubMed] [Google Scholar]
- 22.Blight SK, Larue RC, Mahapatra A, Longstaff DG, Chang E, Zhao G, Kang PT, Green-Church KB, Chan MK, Krzycki JA. Direct Charging of tRNACUA with Pyrrolysine in Vitro and in Vivo. Nature. 2004;431(7006):333–335. doi: 10.1038/nature02895. [DOI] [PubMed] [Google Scholar]
- 23.Greiss S, Chin JW. Expanding the Genetic Code of an Animal. J Am Chem Soc. 2011;133(36):14196–14199. doi: 10.1021/ja2054034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bianco A, Townsley FM, Greiss S, Lang K, Chin JW. Expanding the Genetic Code of Drosophila Melanogaster. Nat Chem Biol. 2012;8(9):748–750. doi: 10.1038/nchembio.1043. [DOI] [PubMed] [Google Scholar]
- 25.Lang K, Davis L, Torres-Kolbus J, Chou C, Deiters A, Chin JW. Genetically Encoded Norbornene Directs Site-Specific Cellular Protein Labelling via a Rapid Bioorthogonal Reaction. Nat Chem. 2012;4(4):298–304. doi: 10.1038/nchem.1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaya E, Vrabel M, Deiml C, Prill S, Fluxa VS, Carell T. A Genetically Encoded Norbornene Amino Acid for the Mild and Selective Modification of Proteins in a Copper-Free Click Reaction. Angew Chemie - Int Ed. 2012;51(18):4466–4469. doi: 10.1002/anie.201109252. [DOI] [PubMed] [Google Scholar]
- 27.Plass T, Milles S, Koehler C, Szymański J, Mueller R, Wießler M, Schultz C, Lemke EA. Amino Acids for Diels-Alder Reactions in Living Cells. Angew Chemie - Int Ed. 2012;51(17):4166–4170. doi: 10.1002/anie.201108231. [DOI] [PubMed] [Google Scholar]
- 28.Carrico IS, Carlson BL, Bertozzi CR. Introducing Genetically Encoded Aldehydes into Proteins. Nat Chem Biol. 2007;3(6):321–322. doi: 10.1038/nchembio878. [DOI] [PubMed] [Google Scholar]
- 29.Seitchik JL, Peeler JC, Taylor MT, Blackman ML, Rhoads TW, Cooley RB, Refakis C, Fox JM, Mehl RA. Genetically Encoded Tetrazine Amino Acid Directs Rapid Site-Specific in Vivo Bioorthogonal Ligation with Trans-Cyclooctenes. J Am Chem Soc. 2012;134(6):2898–2901. doi: 10.1021/ja2109745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang K, Neumann H, Peak-Chew SY, Chin JW. Evolved Orthogonal Ribosomes Enhance the Efficiency of Synthetic Genetic Code Expansion. Nat Biotechnol. 2007;25(7):770–777. doi: 10.1038/nbt1314. [DOI] [PubMed] [Google Scholar]
- 31.Schrauzer GN, Mayweg VP. Preparation, Reactions, and Structure of Bisdithio-α-Diketone Complexes of Nickel, Palladium, and Platinum 1,2. J Am Chem Soc. 1965;87(7):1483–1489. [Google Scholar]
- 32.Baker JR, Hermann A, Wing RM. Mechanism of Oxidative Cycloaddition of Olefins to Metal Dithiolenes. J Am Chem Soc. 1971;93(24):6486–6489. [Google Scholar]
- 33.Kajitani M, Kohara M, Kitayama T, Akiyama T, Sugimori A. Formation of 1:1 Adducts between bis(1,2-Diaryl-1,2-ethylenedithiolato)metal(0) (Metal = NI, PD, and PT) and Quadricyclane and Their Photodissociation. J Phys Org Chem. 1989;2(2):131–145. [Google Scholar]
- 34.Kajitani M, Kohara M, Kitayama T, Asano Y, Sugimori A. Photosensitive 1:1 Adduct between Bis(1,2-Diphenyl-1,2-ethylenedithiolato)nickel(0) and Quadricyclane. Chem Lett. 1986;15(12):2109–2112. [Google Scholar]
- 35.Lin YA, Boutureira O, Lercher L, Bhushan B, Paton RS, Davis BG. Rapid Cross-Metathesis for Reversible Protein Modifications via Chemical Access to Se-Allyl-Selenocysteine in Proteins. J Am Chem Soc. 2013;135(33):12156–12159. doi: 10.1021/ja403191g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Siegel D. Applications of Reversible Covalent Chemistry in Analytical Sample Preparation. Analyst. 2012;137(23):5457. doi: 10.1039/c2an35697j. [DOI] [PubMed] [Google Scholar]
- 37.Kramer JR, Deming TJ. Reversible Chemoselective Tagging and Functionalization of Methionine Containing Peptides. Chem Commun. 2013;49(45):5144. doi: 10.1039/c3cc42214c. [DOI] [PubMed] [Google Scholar]
- 38.Lin YA, Chalker JM, Floyd N, Bernardes GJL, Davis BG. Allyl Sulfides Are Privileged Substrates in Aqueous Cross-Metathesis: Application to Site-Selective Protein Modification. J Am Chem Soc. 2008;130(30):9642–9643. doi: 10.1021/ja8026168. [DOI] [PubMed] [Google Scholar]
- 39.Foettinger A, Leitner A, Lindner W. Solid-Phase Capture and Release of Arginine Peptides by Selective Tagging and Boronate Affinity Chromatography. J Chromatogr A. 2005;1079:187–196. doi: 10.1016/j.chroma.2005.03.038. (1–2 SPEC. ISS.) [DOI] [PubMed] [Google Scholar]
- 40.Egawa Y, Seki T, Takahashi S, Anzai J. Electrochemical and Optical Sugar Sensors Based on Phenylboronic Acid and Its Derivatives. Mater Sci Eng C. 2011;31(7):1257–1264. [Google Scholar]
- 41.Foettinger A, Melmer M, Leitner A, Lindner W. Reaction of the Indole Group with Malondialdehyde: Application for the Derivatization of Tryptophan Residues in Peptides. Bioconjug Chem. 2007;18(5):1678–1683. doi: 10.1021/bc070001h. [DOI] [PubMed] [Google Scholar]
- 42.Foettinger A, Leitner A, Lindner W. Selective Enrichment of Tryptophan-Containing Peptides from Protein Digests Employing a Reversible Derivatization with Malondialdehyde and Solid-Phase Capture on Hydrazide Beads. J Proteome Res. 2007;6(9):3827–3834. doi: 10.1021/pr0702767. [DOI] [PubMed] [Google Scholar]
- 43.Wu P, Shui W, Carlson BL, Hu N, Rabuka D, Lee J, Bertozzi CR. Site-Specific Chemical Modification of Recombinant Proteins Produced in Mammalian Cells by Using the Genetically Encoded Aldehyde Tag. Proc Natl Acad Sci U S A. 2009;106(9):3000–3005. doi: 10.1073/pnas.0807820106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen I, Howarth M, Lin W, Ting AY. Site-Specific Labeling of Cell Surface Proteins with Biophysical Probes Using Biotin Ligase. Nat Methods. 2005;2(2):99–104. doi: 10.1038/nmeth735. [DOI] [PubMed] [Google Scholar]
- 45.Zeng Y, Ramya TNC, Dirksen A, Dawson PE, Paulson JC. High-Efficiency Labeling of Sialylated Glycoproteins on Living Cells. Nat Methods. 2009;6(3):207–209. doi: 10.1038/nmeth.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Z, Smith BAC, Wang L, Brock A, Cho C, Schultz PG. A New Strategy for the Site-Specific Modification of Proteins in Vivo. Biochemistry. 2003;42(22):6735–6746. doi: 10.1021/bi0300231. [DOI] [PubMed] [Google Scholar]
- 47.Jewett JC, Sletten EM, Bertozzi CR. Rapid Cu-Free Click Chemistry with Readily Synthesized Biarylazacyclooctynones. J Am Chem Soc. 2010;132(11):3688–3690. doi: 10.1021/ja100014q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Safe and Convenient Procedure for Solvent Purification. Organometallics. 1996;15(5):1518–1520. [Google Scholar]
- 49.Still WC, Kahn M, Mitra A. Rapid Chromatographic Technique for Preparative Separations with Moderate Resolution. J Org Chem. 1978;43(14):2923–2925. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.