

Abstract
Background
The essential branched-chain amino acids (BCAAs) leucine, isoleucine and valine have recently emerged as a potential novel treatment for medically-refractory epilepsy. Blood-derived BCAAs can readily enter the brain, where they contribute to glutamate biosynthesis and may either suppress or trigger acute seizures. However, the effects of BCAAs on chronic (i.e. spontaneous recurrent) seizures and epilepsy-associated neuron loss are incompletely understood.
Methods
Sixteen rats with mesial temporal lobe epilepsy (MTLE) were randomized into two groups that could drink, ad libitum, either a 4% solution of BCAAs in water (n = 8) or pure water (n = 8). The frequency and relative percent of convulsive and non-convulsive spontaneous seizures were monitored for a period of 21 days, and the brains were then harvested for immunohistochemical analysis.
Results
Although the frequency of convulsive and non-convulsive spontaneous recurrent seizures over a 3-week drinking/monitoring period were not different between the groups, there were differences in the relative percent of convulsive seizures in the first and third week of treatment. Moreover, the BCAA-treated rats had over 25% fewer neurons in the dentate hilus of the hippocampus compared with water-treated controls.
Conclusions
Acute BCAA supplementation reduces seizure propagation, while chronic oral supplementation with BCAAs worsens seizure propagation and causes neuron loss in rodents with MTLE. These findings raise the question of whether such supplementation has a similar effect in humans.
Keywords: branched-chain amino acids, glutamate, glutamine, glutamine synthetase, isoleucine, leucine, methionine sulfoximine, temporal lobe epilepsy, valine
Introduction
Epilepsy is a chronic and debilitating neurological disorder that affects approximately 1% of the population.1 Up to 40% of patients with epilepsy continue to experience seizures despite optimal medical management, or suffer intolerable side effects to currently available antiepileptic agents.2 Mesial temporal lobe epilepsy (MTLE), the most common form of focal epilepsy and most common intractable seizure disorder, is characterized by spontaneous recurrent seizures and an increased risk of comorbid conditions including anxiety, depression, cognitive impairment, and sudden death.3–5 Patients with MTLE are often referred for surgical resection of the seizure focus. Such surgery necessitates coordinated medical care with a multidisciplinary team that includes neurosurgeons, neurointensivists, neurologists, and neuroanesthesiologists. Although surgery is effective in reducing seizures in approximately 80% of patients for up to 2 years,6 up to 11% of these patients experience significant complications including neurocognitive and psychiatric sequelae, visual field deficits, infection, and delayed seizure recurrence.7 More effective treatments with favorable side effect profiles are therefore urgently needed to reduce the frequency of seizures in patients with MTLE.
In recent years, the essential branched-chain amino acids (BCAAs) leucine, isoleucine, and valine have emerged as a potential novel treatment for medically-refractory epilepsy. Although several laboratory studies have demonstrated anticonvulsant effects of BCAAs,8–13 nearly all investigations of BCAAs in epilepsy have been limited to acute seizures and seizure threshold after a single dose of the amino acids. Notably, no long-term studies have been carried out in MTLE. It is further unknown whether chronic, high-dose BCAA administration to patients with epilepsy can damage the brain. The latter is a concern because high levels of BCAAs and associated metabolites have been shown to cause neurodegeneration in vitro.14
Hence, the goal of the present study was to investigate the effects of chronic BCAA supplementation on recurrent seizures and neuron loss in a rodent model of MTLE. The model is created by inhibiting the enzyme glutamine synthetase (GS) in the right hippocampal formation in rats using a chronic intrahippocampal infusion of methionine sulfoximine (MSO).15 Because GS is necessary for efficient glutamate metabolism and clearance in the brain, we hypothesized that chronic, oral supplementation with all three BCAAs would increase the frequency and tissue propagation of recurrent seizures and aggravate hippocampal neuron loss in the MSO model of MTLE.
Materials and Methods
Chemicals and animals
All chemicals were purchased from Sigma Chemical Co. (St. Louis, Mo.) unless otherwise noted. A 4% BCAA solution was dissolved in water [1.38 g isoleucine, 1.38 g leucine, 1.24 g valine in 1000 mL water as described previously12] Male Sprague Dawley rats were obtained from Harlan (Indianapolis, IN.). Rats were individually housed and maintained in a temperature-controlled colony room (21°C–23°C) on a 12 h light–dark cycle. Rats were allowed free access to food (Teklad Global 18% Protein Rodent Diet, which contains 18 mcg/g leucine, 8 mcg/g isoleucine, and 9 mcg/g valine) and water, and were given at least 1 week of acclimation prior to the start of the experiment. All procedures were approved by the Institutional Animal Care and Use Committee at Yale University and were conducted in accordance with current guidelines.
Experimental design
The experimental protocol is summarized in Fig. 1. Sixteen rats (weight 455 – 615 g, ~16 weeks in age) were randomly separated into two groups: one group had unrestricted access to a 4% BCAA solution dissolved in water (n=8) and the other group to a plain water solution (n=8) ad libitum. Following 10 days of consumption, rats were implanted with the MSO pump, and the first set of venous blood samples were drawn. After surgery, rats were monitored for both convulsive and nonconvulsive spontaneous seizures over a period of 21 days. For the duration of the EEG recording period, MSO was continuously infused and rats were maintained on the BCAA or plain water solution. Water bottles were weighed daily to confirm consumption. After this monitoring period, rats were then perfused, during which time the second set of venous blood samples were drawn. The brains were then harvested for immunohistochemical analysis.
Figure 1. Experimental Protocol.
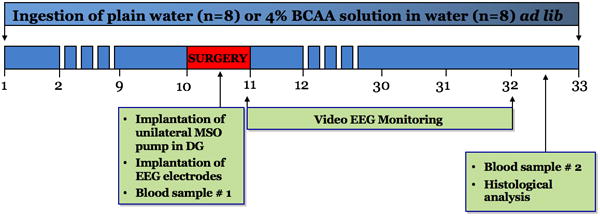
Sixteen rats were randomly separated to receive either unrestricted access to a 4% BCAA solution dissolved in water (n=8) or a plain water solution (n=8) ad libitum. Following 10 days of consumption, rats were implanted with the MSO pump, and seizure activity was monitored thereafter for a period of 21 days. For the duration of the EEG recording period, MSO was continuously infused and rats were maintained on the BCAA or plain water solution. After this monitoring period, rats were perfused and the brains were harvested for immunohistochemical analysis. BCAA concentrations were analyzed in venous blood samples that were drawn at two time points during the experiment. Three rats from the BCAA group were excluded from the final analysis after postmortem histological analysis confirmed that the MSO pump was outside of dentate gyrus.
Surgery
Rats were anesthetized with 0.5 – 2% Isoflurane (Baxter, Deerfield, IL) in O2 and placed in a stereotaxic frame (David Kopf Instruments, Tujunga, CA). A 30-gauge stainless steel cannula, with a length of 8.1 mm, attached to a plastic pedestal (Plastics One, Roanoke, VA) was stereotaxically lowered into the right dentate gyrus using the following coordinates, with bregma marking zero for the mediolateral (ML) and anteroposterior (AP) directions, and the top of the skull marking zero for the dorsoventral (DV) direction: AP = −2.6 mm, ML = 4.6 mm, DV = −8.1 mm. The cannula was lowered into the brain till the pedestal touched the skull. The pedestal was then glued to the skull with medical grade cyanoacrylate (Vetbond Tissue Adhesive, Butler Animal Health Company, Chicago, IL). The cannula and pedestal were connected via plastic tubing to a subcutaneously implanted Alzet osmotic pump (Model 2004, Durect Corp., Cupertino, CA.) which delivers a continuous flow of 0.25 μL/h for ~28 days. The pumps were filled with MSO (2.5 mg/mL; dissolved in Dulbecco’s phosphate buffered saline (PBS) to achieve a delivery of 0.625 μg of MSO per h. This rate was used because previous studies demonstrated ipsilateral irreversible inhibition of GS without affecting glutathione levels.15 Following placement of the cannula and pedestal, four stainless steel epidural screw electrodes (Plastics One, Roanoke, VA) were implanted to record cortical electroencephalogram (EEG) activity. Two stainless-steel screw electrodes (one in each hemisphere) were positioned in the epidural space over the cortex. On the left side (contralateral to the injection), the electrode was placed in the skull above the dorsal anterior hippocampal formation (AP = −2.0 mm, ML = −2.5 mm). On the right side, the electrode was placed in the skull above the somatosensory cortex (AP = −6.25 mm, ML = 5.4 mm). One screw electrode was positioned in the epidural space (AP= 8.5 mm, ML= −2.2 mm) to serve as the reference. A fourth electrode, which was positioned in the skull above the cerebellum (AP = −10.0 mm, ML = 1.5 mm) served as the ground. Three additional stainless-steel screws were inserted into the skull to reduce the risk of head cap detachment. The female socket contacts on the ends of each electrode were inserted into a plastic pedestal (Plastics One), and the entire implant was secured by UV light cured acrylated urethane adhesive (Loctite 3106 Light Cure Adhesive, Henkel Corp., Rocky Hill, CT) to form a head cap.
Blood BCAA levels
Venous blood samples were collected in tubes containing lithium heparin anticoagulant at 2 time points during the experiment: during surgical implantation of the MSO pump at day 10 (drawn from the site of burr holes) and when the rats were sacrificed at 31 days (drawn from left ventricle of the heart). The blood was centrifuged at 12,000 g for 10 minutes at 4°C. The plasma was collected and stored at −80°C for later analysis. Concentrations of valine, leucine, and isoleucine were determined using the Ez:faast Free Amino Acid analysis kit (Phenomenex, Torrence, CA) followed by ultra-performance liquid chromatography-tandem mass spectrometry (UPLC/MS/MS) using a Waters Acquity, Xevo TQS mass spectrometer (Waters, Milford, MA). Briefly, 6 μL of sample was extracted according to the kit procedure. The samples were dried in N2 and reconstituted in 50 μL of 50% LC/MS grade methanol in water, and 5 μL of the sample was analyzed by UPLC/MS/MS. The sample was separated by UPLC using reverse-phase gradient elution at a flow rate of 0.5 mL/min. The separated sample was then subjected to MS/MS by multiple reaction monitoring. The area under the curve for each transition was determined, and the ratio of the area under the curve for each amino acid to its corresponding internal standard solution was used for relative concentration determinations. Absolute concentrations were calculated from a 5-point calibration curve, using standard solutions prepared from accurately weighed, analytical grade amino acids.
Video-intracranial EEG monitoring
After implantation of the MSO pump, rats were monitored for spontaneous seizures for 21 days. The experimental setup for recording video-EEG was adapted from Bertram et al.16 The rats were placed individually in custom-made Plexiglas cages. A spring-covered, 6-channel cable was connected to the electrode pedestal on one end and to a commutator (Plastics One) on the other. A second cable connected the commutator to the digital EEG recording unit (CEEGraph Vision LTM, Natus/Bio-Logic Systems Corp., San Carlos, CA). Digital cameras with infrared light detection capability were used to record animal behavior (two cages per camera). The digital video signal was encoded and synchronized to the digital EEG signals. Seizures were identified by visual inspection of the EEG record. As detailed by Avoli et al.,17 seizures were defined by EEG characteristics of the discharge. Specifically, seizures displayed distinct signal changes from background (interictal) activity. Such signal changes included sustained rhythmic or spiking EEG patterns and a clear evolution of signal characteristics from onset to termination (Fig. 2).
Figure 2. Two hundred-seventy seconds of continuous EEG recording from a screw electrode over the cortex in a rat chronically infused with MSO in the dentate gyrus.
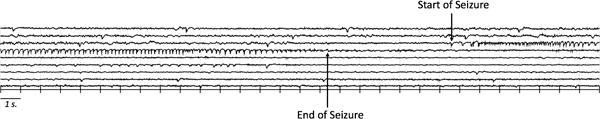
A seizure approximately 24 seconds in duration is identified by arrows indicating the start and end of the seizure. The seizures were like those previously described.20, 41
Subclinical seizures were distinguished from clinical seizures by examination of the video record. The start and stop points of seizures were identified by the following commonly used method. By visual inspection of the EEG, we determined a point that was unequivocally within the seizure. Next, we moved backward in time to determine the seizure start time as the first point where the EEG was different from background activity and forward in time to establish the seizure end time. The temporal distribution and total number of seizures in all animals was determined by reviewing the entire video-EEG record. Behavioral seizures were graded on the Racine seizure scale,18 and were subsequently classified as non-convulsive (subclinical to stage 2) or convulsive (stage 3–5) (Fig. 3). Rats were classified in this manner because we previously demonstrated a progressive change in regional involvement of the brain associated with behavioral severity, with the largest increase in signal power observed between Racine stage 2 and 3 seizures.19
Figure 3. Behavioral seizures were graded on the Racine seizure scale.
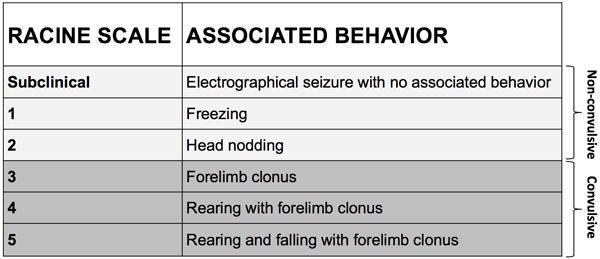
Seizure activity was monitored over 21 days with synchronized video and EEG. Seizures were identified by visual inspection of the EEG record, and behavioral seizures were graded from subclinical through stage 5 as previously described by Racine.18 Seizures were subsequently classified as non-convulsive (subclinical to stage 2) or convulsive (stage 3–5).
Histology
Rats were anesthetized with 0.5 – 2% Isoflurane in O2 and perfused transcardially with 0.9% NaCl followed by 4% formaldehyde in 0.1 M phosphate buffer, pH 7.4. The brains were removed and left in the same fixative at 4°C for 24 h and then transferred to phosphate buffered saline (PBS). The brains were stored at 4°C until being sectioned on a Vibratome at 50-μm thickness. Every fifth section through the hippocampus was mounted on gelatin-coated slides and stained with cresyl violet. Adjacent sections to the series above were stained with NeuN antibody (Millipore cat. # MAB377/lot # 2424507), diluted 1:10,000 in PB containing 0.3% Triton-X-100. The sections were processed using the avidin-biotin complex (ABC) kit (Vectastain, Burlingame, CA) with diaminobenzidine as the chromogen and mounted on a microscope slide for later analysis.
Our previous studies demonstrated a consistent and predictable pattern of seizure activity and neuronal loss when the MSO micro-infuser was localized to the hippocampal dentate gyrus.20 Therefore, only rats in which the location of the micro-infuser was confirmed in dentate gyrus were included in the final analysis.
Neurons were counted in the hilus of dentate gyrus, where the extent of neuron loss correlates with seizure activity in this model.20 Neuron counts were performed ipsilateral and contralateral to the infusion site using the optical fractionator method with StereoInvestigator, (Site Mark West, MBF Bioscience, Williston, VT). On the ipsilateral side, NeuN sections adjacent to the infusion site were counted (sections containing the infusion site were omitted). On the contralateral side, which did not have an infusion site, the series of sections stained with NeuN were counted. The sum of sections counted ranged from six to eight. After outlining the hilus, a 40 μm × 40 μm sector was randomly place by the software in the traced region and cells were counted within the sectors. The total area was calculated by multiplying the area of the sector by the number of sampling sites. The software randomly selected the number of sample sites to exclude bias, ranging from 60 – 110. Counts were calculated and expressed as neurons per 100 mm3.
Statistics
Parametric data were compared with Student’s t-test. Repeated measures ANOVA was used to compare the total frequency of non-convulsive (stage 1–2) and convulsive (stage 3–5) seizures over 21 days in 7-day bins (1–7, 8–14, 15–21). ANOVA was followed by a post hoc Fisher least significant difference (LSD) test. Chi-square was used to compare the relative percent of convulsive seizures over 21 days in 7-day bins. Statistical significance was defined as p ≤ 0.05. Data is presented as mean ± standard error (SE). Statistical analysis was performed with Statistica 8.0 (Statsoft; Tulsa, OK).
Results
Chronic BCAA ingestion increased blood concentrations of all three BCAAs
Three rats in the BCAA-treated group were excluded because the location of the micro-infuser was outside the dentate gyrus. To assess the palatability and effectiveness of the drinking approach, we first determined fluid consumption and plasma BCAA levels in all rats. There was no difference in fluid consumption between the BCAA- and plain water-treated rats (Fig. 4A). Rats in the BCAA-treated group consumed on average 1.06 ± 0.031g (1.75 g per kg body wt.) of BCAAs per day. After 10 days of drinking, the plasma concentrations of BCAAs were 29 to 75% higher in the BCAA-treated versus plain water-treated animals (Fig. 4B). After 31 days of drinking, the plasma concentrations of BCAAs continued to remain higher (35 to 52%) in BCAA-treated animals (Fig. 4C). There were no differences in BCAA concentrations between 7 and 31 days.
Figure 4. Volume consumption and blood BCAA concentrations.
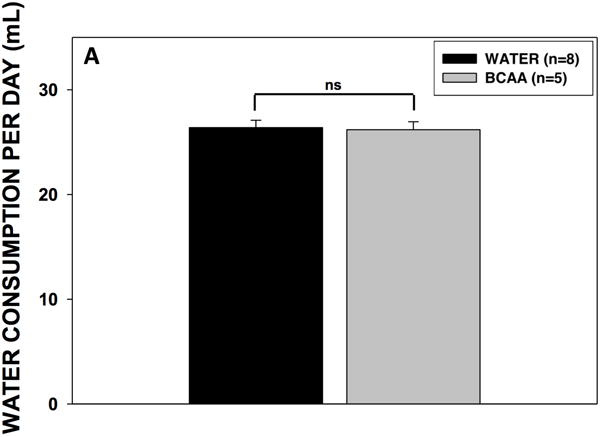
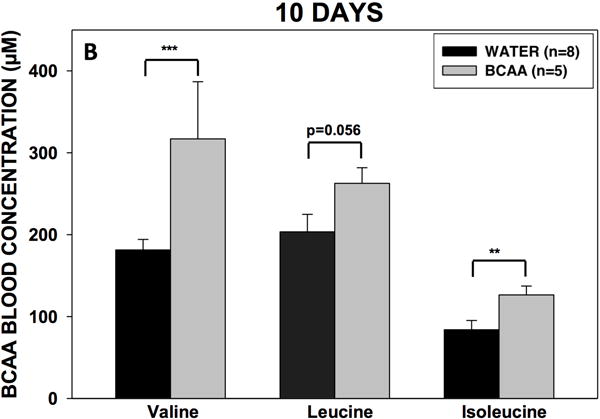
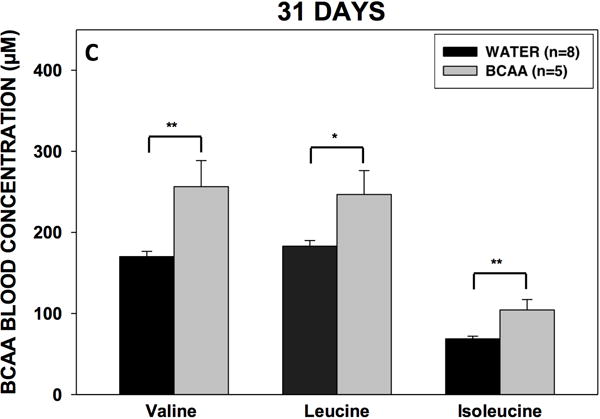
(A) Rats drank either plain water (n=8) or a solution of 4% BCAAs dissolved in water (n=5) ad libitum for 31 days. The water bottles were weighed daily for the duration of the experiment. There were no differences in water consumption between BCAA- and water-treated animals. Data is presented as mean water consumption (mL) per day ± SEM. Blood samples were drawn from the scalp during surgery for placement of methionine sulfoximine pump at 10 days (B) after drinking, and again from the heart during cardiac perfusion at 31 days (C) after drinking. Samples were analyzed for BCAAs using tandem mass spectrometry. At both time points, concentrations of BCAAs were higher in the rats that drank the BCAA solution compared with the rats that drank plain water. Data is presented as mean concentrations of branched-chain amino acids ± SEM. ***p < 0.001, **p < 0.01, *p < 0.05.
Chronic BCAA ingestion did not alter the frequency of convulsive or non-convulsive recurrent seizures
We next analyzed the video-EEG records from all rats for the entire 3-week collection period. There was no significant difference in the seizure frequency between the BCAA- and plain water-treated rats during weeks 1, 2, or 3 of the collection period (Fig. 5). However, during the second week of collection, there was a nonsignificant trend (p = 0.077) towards fewer seizures in the BCAA-treated rats. There was no difference in the average number of non-convulsive (i.e. subclinical to Racine stage 2) or convulsive (Racine stage 3 to 5) seizures during weeks 1, 2, of 3 of analysis.
Figure 5. Frequency and severity of recurrent seizures in BCAA- and water-treated animals.
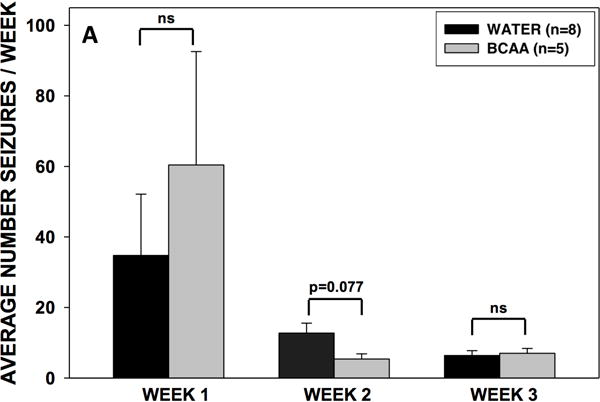
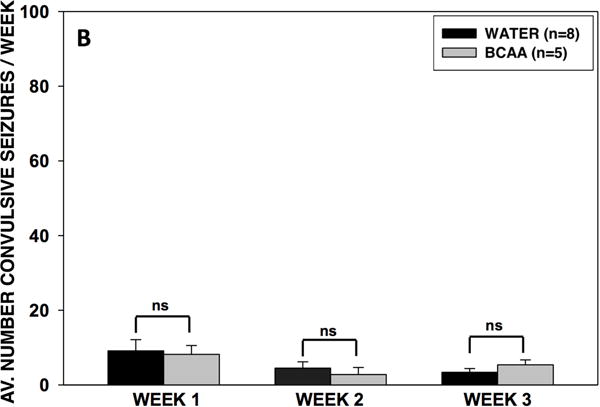
After 10 days of drinking either plain water or a solution of 4% BCAAs dissolved in water, rats were implanted with an osmotic pump through which MSO was chronically infused at a rate of 0.625 μg per hour in the right dentate gyrus. Rats were then subjected to 3 weeks of video-EEG analysis, whereby the frequency and severity (Racine scale 1–5) of seizures were recorded. (A) There were no differences in the total average number of seizures per day during weeks 1, 2, or 3 of EEG analysis. During week 2, there was a nonsignificant trend (p = 0.077) towards fewer seizures in rats who drank branched-chain amino acid solution. (B) There were no differences in the average number of non-convulsive (subclinical – Racine stage 2) or convulsive (Racine stage 3 – 5) seizures during weeks 1, 2, of 3 of EEG analysis. Data is presented as mean number of seizures per day ± SEM.
Chronic BCAA ingestion altered the relative percent of convulsive seizures during weeks 1 and 3 of treatment
Next, we analyzed the relative percent of convulsive (Racine stage 3 to 5) seizures during weeks 1, 2, and 3 of the EEG recording period (Fig. 6). In week 1, the BCAA-treated rats had a lower relative percent of convulsive seizures compared with water-treated rats (13.6% vs. 26.3%, respectively, p < 0.001). In week 2, there were no significant differences in the relative percent of convulsive seizures between the BCAA- and water-treated rats. In week 3, the BCAA-treated rats had a higher relative percent of convulsive seizures compared with water-treated rats (77.1% vs. 53.0%, p < 0.05).
Figure 6. Relative percent of convulsive in BCAA- and water-treated animals.
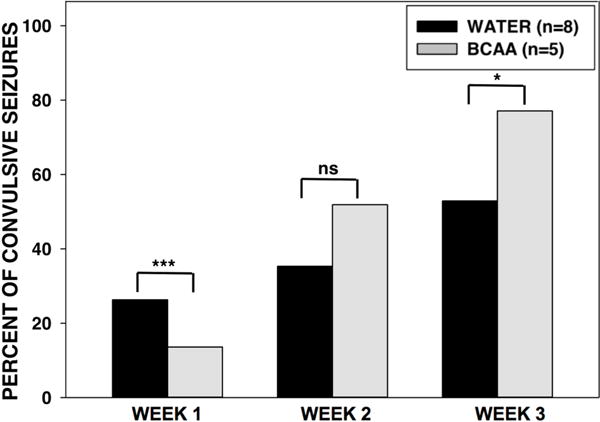
The relative percent of convulsive (Racine stage 3 to 5) seizures during weeks 1, 2, and 3 of the EEG recording period was analyzed. In the first week of the recording period, the BCAA-treated rats had a lower relative percent of convulsive seizures compared with water-treated rats (p < 0.001). In week 2, there were no significant differences in the relative percent of convulsive seizures between the BCAA- and water-treated rats, whereas in week 3 the BCAA-treated rats had a higher relative percent of convulsive seizures (p < 0.05). Data is presented as percent of convulsive seizures. ***p < 0.001, *p < 0.05.
Chronic BCAA ingestion aggravated the neuronal loss in the dentate hilus
Finally, we determined the density of NeuN-stained neurons in the hilus of the left and right dentate gyrus of BCAA- and plain water-treated rats (Fig. 7). BCAA-treated rats had 36% fewer neurons in the ipsilateral (right) dentate hilus compared with the plain water-treated group (575.1 ± 67.9 vs. 894.3 ± 59.5 cells per 100 mm3, respectively, p<0.0001), and 29% fewer neurons in the contralateral (left) dentate hilus compared with the plain water-treated group (499.5 ± 58.3 vs. 707.7 ± 33.6 cells per 100 mm3, respectively, p<0.001).
Figure 7. Hippocampal neuron counts.
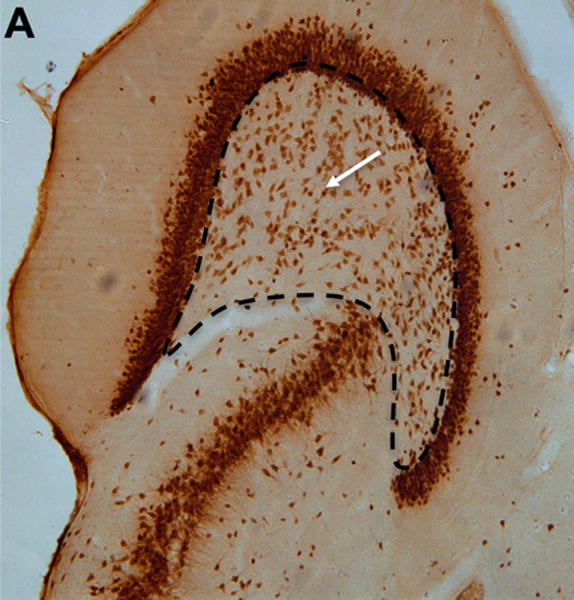
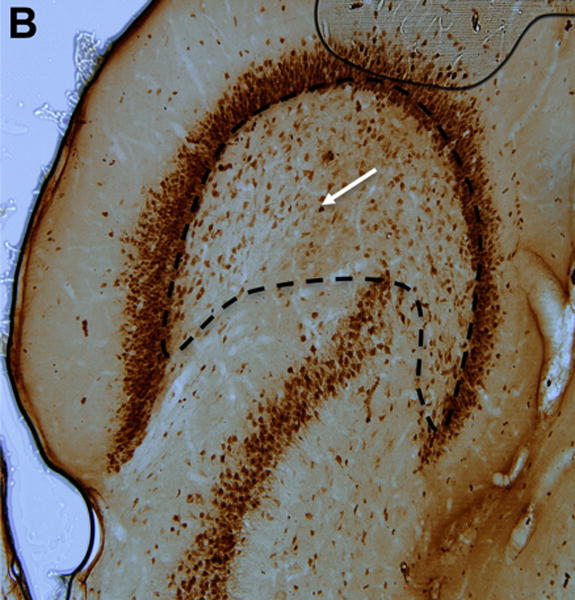
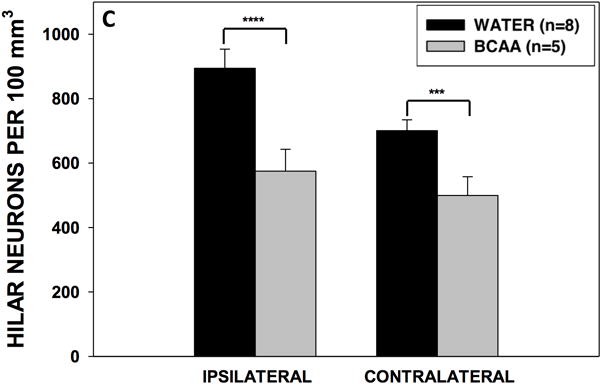
(A) Section stained with NeuN demonstrating neurons within the dentate hilus (borders outlined with dashed lines) in a representative control and (B) branched-chain amino acid (BCAA) rat. The white arrows point to a stained neuron. (C) Rats in the branched-chain amino acid vs. control group had 36% fewer neurons in the dentate hilus of the hemisphere ipsilateral (p<0.0001) to the methionine sulfoximine infusion and 29% fewer neurons contralateral (p<0.001) to the infusion. Data is presented as mean number of cells per 100 mm3 ± SEM. ****p < 0.0001, ***p < 0.001
Discussion
To our knowledge, this is the first study of the effect of high-dose, oral BCAA supplementation on recurrent seizures and brain pathology. We demonstrated that chronic BCAA ingestion was ineffective in reducing the frequency of convulsive and non-convulsive recurrent spontaneous seizures. However, there were differences between groups with respect to the relative percent of convulsive seizures, which reflects the propagation of seizure activity from the hippocampus to neocortical areas, such as the motor cortex. While BCAAs initially reduced the propagation of seizures during the first week of treatment, the seizure propagation subsequently worsened with a longer duration of BCAA treatment. Lastly, chronic BCAA treatment resulted in increased loss of hippocampal hilar neurons.
BCAAs are known to play important roles in normal physiology, and are widely used by athletes to promote muscle growth and recovery.21 BCAAs account for 35% of the essential amino acids in muscle22 and stimulate muscle growth by activating the mechanistic target of rapamycin (mTOR) signaling pathway.23 Moreover, diet-derived BCAAs contribute to approximately one-third of de novo synthesized glutamate in the rodent central nervous system (CNS).24
Ingested BCAAs undergo minimal first-pass metabolism, enter the systemic circulation and readily distribute to the CNS via several types of amino acid transporters, particularly the large neutral amino acid transporter 1 (LAT1)25 and the solute carrier family 6 member 15 (SLC6A15).26 Once in the CNS, each BCAA can donate its amine group to alpha-ketoglutarate, forming glutamate and the corresponding branched-chain keto acid, via the branched-chain aminotransferase (BCAT) reaction.27 Diet-derived BCAAs in the blood can therefore modulate brain glutamate levels, which are normally under tight homeostatic control to facilitate efficient neurotransmission and prevent glutamate-mediated excitotoxicity.28 It is well known that sustained high levels of extracellular glutamate in the brain lead to excessive stimulation of neuronal glutamate receptors and subsequent abnormal neurotransmission, increased excitability, seizures and neuronal loss.29 Furthermore, the actions of glutamate and gamma-aminobutyric acid (GABA; a metabolite of glutamate) have been implicated in both the toxic and therapeutic effects of many general anesthetics.30–32 Therefore, understanding the mechanisms of glutamate and GABA metabolism is important because it may lead to the development of targeted neuroprotective agents, antiepileptic therapies, and general anesthetics.
While some studies report neurotoxic and disease-promoting properties of BCAAs and their associated metabolites, others demonstrate protective effects. For example, perturbations in the homeostasis of brain BCAAs have been implicated in the pathophysiology of several neurological disorders such as maple syrup urine disease-associated encephalopathy,33 malignant gliomas,34 autism spectrum disorders,35 amyotrophic lateral sclerosis36 and epilepsy.8, 9, 13, 37 With respect to epilepsy, a small number of studies have shown that administration of BCAAs alone, or in combination with a ketogenic diet, reduce the frequency of seizures and increase the seizure threshold in children with epilepsy8 as well as in the kainic acid9 and pentylenetetrazole13 models of acute convulsive seizures. However, in the Genetic Absence Epilepsy Rats from Strasbourg (GAERS) model of absence seizures, a single intraperitoneal dose of BCAAs increases the frequency of electrographic seizures.37
In the present study, BCAAs were ineffective in reducing the frequency of convulsive and non-convulsive recurrent seizures in the epileptic rats. This observation contrasts with previous studies in other animal models that show clear anticonvulsant effects of BCAAs.8–13 Several animal models have been used to study the mechanisms of epileptogenesis and seizure generation in MTLE, in which acute seizures are induced by chemoconvulsants, electrical or sound stimuli, or traumatic brain injury. Although each model has unique neuropathological and behavioral characteristics38, the MSO model of MTLE we developed is advantageous in that it recapitulates several important features of the human MTLE condition, making the model highly translatable. Some of these features include inhibition in GS in hippocampal astrocytes,29, 39 an initial insult followed by recurrent spontaneous seizures that originate from the mesial temporal lobe,15 loss of hippocampal neurons,40, 41 worsening of the seizures as the disease progresses over time,20 comorbid depressive features42 and perturbations in the brain glutamate homeostasis.43, 44
Although the mechanisms by which BCAAs prevent seizures in other epilepsy models are poorly understood, an efficient metabolism and clearance of glutamate might be necessary. For example, administration of BCAAs has been shown to favor the synthesis of GABA over glutamate, by inhibiting the activities of glutamate dehydrogenase (GDH)45, 46 and glutamate oxaloacetate transaminase (GOT).47 Moreover, administration of BCAAs could potentially reduce neuronal glutamate concentrations via the glial-neuronal BCAA shuttle,37 as follows. Blood-derived BCAAs taken up astrocytes may transaminate α-ketoglutarate, thereby producing glutamate and branched-chain ketoacids (BCKAs). The BCKAs are poorly oxidized in astrocytes and accumulate in the extracellular fluid.48 Neurons take up the BCKAs, which are transaminated back to BCAAs in a reaction that consumes glutamate.27, 37, 48 The glutamate produced in astrocytes from the BCAA transamination reaction are likely converted to glutamine via the GS reaction. The excess astroglial glutamine are then exported from the brain to the blood.49
However, when GS is deficient, such as in patients with MTLE and in the rodent model used here, the astrocyte glutamate excess caused by the BCAAs cannot be effectively eliminated from the brain.29, 43, 50 It is well known that glutamate is a potent excitotoxin, and its accumulation in the extracellular compartment of the brain has deleterious effects on neuronal function and survival in laboratory animals.51 As such, the accumulation of extracellular glutamate that results from the GS deficiency is thought to play a critical role in the pathophysiology of MTLE.52 Moreover, slowed glutamate-glutamine cycling may further contribute to seizures by reducing neuronal glutamate available for GABA synthesis.53 While we did not directly measure glutamate levels in the brain in this experiment, it is well-known that BCAAs readily the blood brain barrier in both the healthy and injured brain.54 Moreover, our unpublished microdialysis experiments with MSO-treated rats have demonstrated that BCAAs readily cross the blood brain barrier, where they are subsequently converted to glutamate.
Although generalized tonic-clonic seizures are not the typical primary manifestation of MTLE, these types of seizures can certainly be observed, and patients often experience secondarily generalized seizures.55 Unlike the human condition, there is a rapid spread of seizure activity in the rodent MSO model, which might result in a larger fraction of generalized seizures in rats compared with humans. While the present study failed to demonstrate differences in seizure frequency in BCAA- vs. water-treated rats, the relative percent of convulsive seizures changed as the duration of treatment increased. This observation is important because convulsive seizures (compared with non-convulsive seizures) are known to reflect a spread of seizure activity from the hippocampal seizure focus to neocortical areas such as the motor cortex.19 Here we demonstrated that during the early treatment period, BCAAs inhibited the propagation of seizures to the motor cortex, whereas with a longer duration of exposure, chronic BCAA treatment facilitated motor cortical propagation. This suggests that the effect of BCAA treatment on the propagation of seizure activity changes as the duration of exposure to BCAAs increases. Although the underlying mechanism for time-related difference in effect is unclear, its implications might significantly preclude the use of BCAAs as a chronic treatment for refractory epilepsy.
Hippocampal neurons, particularly cells in the dentate hilus, are often lost in patients with MTLE and animal models of the disease, including the model used here.20, 56, 57 We have previously shown that rats treated with MSO exhibit various degrees of hippocampal neuron loss. Most animals demonstrate minimal or modest neuron loss throughout the hippocampus, with a minority (< 25%) displaying classic hippocampal sclerosis, defined as loss of CA1, CA3 and dentate neurons along with shrinkage of the hippocampal formation.15, 41 There is considerable variability in neuron loss among patients with MTLE as well; however, up to one-third of all patients with surgically-treated MTLE exhibit classic hippocampal sclerosis,58 which is higher than in the MSO model. The reason for the difference in prevalence of hippocampal sclerosis between the MSO and human MTLE is no understood, but might reflect a longer duration of the disease in the surgically resected patients with more time for the sclerosis to develop. Despite these differences, the overall patterns of neuropathology are very similar between our MSO model and humans with MTLE.
Our study is the first to show that high-dose, oral supplementation with BCAAs worsens the loss of neurons associated with MTLE. Although dentate granule cells are generally relatively resistant to seizures, hilar interneurons are known to be particularly vulnerable to degeneration in patients with epilepsy and in several animal models.59, 60 Because it would be a formidable task to count all neuron populations, we chose a sentinel and particularly vulnerable neuronal population as a first screening to determine whether treatment with BCAAs enhances the neuron loss in epilepsy. Importantly, the BCAA-induced hilar neuron loss demonstrated in the present study highlights the need for follow up studies that systematically investigate other neuronal populations, as well as the underlying mechanisms of neurodegeneration with chronic BCAA administration.
Several mechanisms may explain the neurotoxic effects of BCAAs. First, as described above, an excess of brain BCAAs in the setting of astroglial GS deficiency may lead to increased astroglial and extracellular glutamate, resulting in excitotoxic neuronal loss. Second, BCAAs, especially leucine, stimulate ammonia production in the brain by activating GDH, which results in glutamate oxidation in synaptosomes.37, 61 Ammonia is neurotoxic, possibly via mechanisms that involve astrocyte swelling or shrinking, increased astrocyte calcium signaling, and reduced potassium clearance by saturating potassium uptake mechanisms.62–64 There are several metabolic pathways through which ammonia is normally cleared from the brain, including the GS pathway in astrocytes.65 Therefore, the combination of increased ammonia production via BCAAs and reduced ammonia clearance via GS inhibition may account for the enhanced neuron loss observed in BCAA-treated rats. Third, BCAAs compete with entry of other large amino acids, resulting in amino acid deficiency and altered protein synthesis in the brain.66 Some of these amino acids, including phenylalanine and tyrosine, are precursors for neurotransmitter synthesis, and competition for transport into the brain might therefore interfere with neurotransmitter synthesis.66 Fourth, accumulation of BCAAs can stimulate lipid peroxidation and increase the production and accumulation of free radicals, resulting in oxidant cortical injury.67
The BCAA-potentiated neuronal loss is of significant interest not only with respect to epilepsy, but also in the developing brain. Several in vivo animal studies have demonstrated that general anesthetics are toxic to the developing brain,68 especially the hippocampus,69 and alterations in glutamate metabolism are thought to play an important mechanistic role.70, 71 To better understand the underlying metabolic pathways implicated in anesthesia-induced neurotoxicity, recent studies have used cerebral metabolomics profiling to characterize chemical changes in the rodent72 and human73 brain during exposure to volatile and intravenous anesthetics. Interestingly, prolonged exposure to volatile anesthetics has been shown to significantly alter amino acid metabolism in the developing brain,74 suggesting a possible mechanistic link between amino acid metabolism and anesthesia-induced neurotoxicity. In this way, the developing brain might be particularly susceptible to the neurotoxic effects of a BCAA-rich diet, and potentiated by prolonged or repeated exposure to inhaled general anesthetics. This possibility is intriguing and should be further evaluated in future studies.
Moreover, although not specifically investigated in the present study, BCAA-induced neuron loss might have important implications for healthy individuals who use BCAAs as nutritional supplements. In our study, BCAA-treated rats consumed on average approximately 1.75 g/kg body wt. of BCAAs per day. While it is unclear precisely how this dose translates in humans, BCAA supplementation in high-performance athletes is highly variable and often well exceeds 1.0 g/kg per body wt. per day.75 It is currently unknown whether high-dose BCAA supplementation affects brain physiology, cognition or behavior in healthy individuals. Similarly, it is unknown whether BCAAs can lead to neuron loss in the absence of epilepsy. Further studies are needed to assess these possibilities.
In summary, we demonstrated for the first time the effects of chronic BCAA ingestion on spontaneous seizures in a translationally-relevant model of MTLE. Chronic BCAA ingestion aggravated hilar neuron loss, but had no significant effect on the frequency of convulsive or non-convulsive spontaneous seizures in GS-inhibited, epileptic rats. Although the cortical spread of seizure activity was inhibited in the first week of BCAA supplementation, a longer duration of treatment facilitated the spread of seizures and aggravated neuron loss. These findings suggest that GS function may be necessary for the previously demonstrated anticonvulsant effects of BCAAs, and that chronic BCAA supplementation may facilitate seizure spread and aggravate neuron loss in a setting of MTLE.
Acknowledgments
TE and RD are supported by grants from the National Institutes of Health (NIH): NINDS R01 NS070824. SG is supported by grants from the Foundation for Anesthesia Education and Research (FAER) and the NIH: T32 GM086287. This work was also made possible by a grant from the National Center for Advancing Translational Sciences (NCATS; UL1 TR000142), a component of the NIH and the NIH Roadmap for Medical Research.
References
- 1.Jennum P, Gyllenborg J, Kjellberg J. The social and economic consequences of epilepsy: a controlled national study. Epilepsia. 2011;52:949–56. doi: 10.1111/j.1528-1167.2010.02946.x. [DOI] [PubMed] [Google Scholar]
- 2.French JA. Refractory epilepsy: clinical overview. Epilepsia. 2007;48(Suppl 1):3–7. doi: 10.1111/j.1528-1167.2007.00992.x. [DOI] [PubMed] [Google Scholar]
- 3.Steiger BK, Jokeit H. Why epilepsy challenges social life. Seizure. 2017;44:194–198. doi: 10.1016/j.seizure.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 4.Engel J., Jr Mesial temporal lobe epilepsy: what have we learned? Neuroscientist. 2001;7:340–52. doi: 10.1177/107385840100700410. [DOI] [PubMed] [Google Scholar]
- 5.Wandschneider B, Koepp M, Scott C, et al. Structural imaging biomarkers of sudden unexpected death in epilepsy. Brain. 2015;138:2907–19. doi: 10.1093/brain/awv233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tellez-Zenteno JF, Hernandez Ronquillo L, Moien-Afshari F, et al. Surgical outcomes in lesional and non-lesional epilepsy: a systematic review and meta-analysis. Epilepsy Res. 2010;89:310–8. doi: 10.1016/j.eplepsyres.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 7.McClelland S, 3rd, Guo H, Okuyemi KS. Population-based analysis of morbidity and mortality following surgery for intractable temporal lobe epilepsy in the United States. Arch Neurol. 2011;68:725–9. doi: 10.1001/archneurol.2011.7. [DOI] [PubMed] [Google Scholar]
- 8.Evangeliou A, Spilioti M, Doulioglou V, et al. Branched chain amino acids as adjunctive therapy to ketogenic diet in epilepsy: pilot study and hypothesis. J Child Neurol. 2009;24:1268–72. doi: 10.1177/0883073809336295. [DOI] [PubMed] [Google Scholar]
- 9.Hartman AL, Santos P, O’Riordan KJ, et al. Potent anti-seizure effects of D-leucine. Neurobiol Dis. 2015;82:46–53. doi: 10.1016/j.nbd.2015.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeske J, Misterek K, Dorociak A. Some pharmacological effects of amino acid 4 aminoantipyrine derivatives. Bull Pol Med Sci Hist. 1974;15 II/- [Google Scholar]
- 11.Pinto JMB, Maher TJ. Administration of aspartame potentiates pentylenetetrazole- and fluorothyl-induced seizures in mice. Neuropharmacology. 1988;27:51–55. doi: 10.1016/0028-3908(88)90200-6. [DOI] [PubMed] [Google Scholar]
- 12.Skeie B, Petersen AJ, Manner T, et al. Branched-chain amino acids increase the seizure threshold to picrotoxin in rats. Pharmacology, Biochemistry and Behavior. 1992;43:669–671. doi: 10.1016/0091-3057(92)90393-t. [DOI] [PubMed] [Google Scholar]
- 13.Skeie B, Petersen AJ, Manner T, et al. Effects of valine, leucine, isoleucine, and a balanced amino acid solution on the seizure threshold to picrotoxin in rats. Pharmacol Biochem Behav. 1994;48:101–3. doi: 10.1016/0091-3057(94)90504-5. [DOI] [PubMed] [Google Scholar]
- 14.Contrusciere V, Paradisi S, Matteucci A, et al. Branched-chain amino acids induce neurotoxicity in rat cortical cultures. Neurotox Res. 2010;17:392–8. doi: 10.1007/s12640-009-9115-0. [DOI] [PubMed] [Google Scholar]
- 15.Eid T, Ghosh A, Wang Y, et al. Recurrent seizures and brain pathology after inhibition of glutamine synthetase in the hippocampus in rats. Brain. 2008;131:2061–70. doi: 10.1093/brain/awn133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bertram EH, Williamson JM, Cornett JF, et al. Design and construction of a long-term continuous video-EEG monitoring unit for simultaneous recording of multiple small animals. Brain Res Brain Res Protoc. 1997;2:85–97. doi: 10.1016/s1385-299x(97)00033-0. [DOI] [PubMed] [Google Scholar]
- 17.Avoli M, Gloor P. Pathophysiology of focal and generalized convulsive seizures versus that of generalized non-convulsive seizures. In: Wolf P, editor. Epileptic Seizures and Syndromes. Montrouge, France: John Libby Eurotext Ltd; 1994. pp. 547–561. [Google Scholar]
- 18.Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–94. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- 19.Blumenfeld H, Rivera M, Vasquez JG, et al. Neocortical and thalamic spread of amygdala kindled seizures. Epilepsia. 2007;48:254–62. doi: 10.1111/j.1528-1167.2006.00934.x. [DOI] [PubMed] [Google Scholar]
- 20.Dhaher R, Wang H, Gruenbaum SE, et al. Effects of site-specific infusions of methionine sulfoximine on the temporal progression of seizures in a rat model of mesial temporal lobe epilepsy. Epilepsy Res. 2015;115:45–54. doi: 10.1016/j.eplepsyres.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dudgeon WD, Kelley EP, Scheett TP. In a single-blind, matched group design: branched-chain amino acid supplementation and resistance training maintains lean body mass during a caloric restricted diet. J Int Soc Sports Nutr. 2016;13:1. doi: 10.1186/s12970-015-0112-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu G. Functional amino acids in nutrition and health. Amino Acids. 2013;45:407–11. doi: 10.1007/s00726-013-1500-6. [DOI] [PubMed] [Google Scholar]
- 23.Anthony JC, Yoshizawa F, Anthony TG, et al. Leucine stimulates translation initiation in skeletal muscle of postabsorptive rats via a rapamycin-sensitive pathway. J Nutr. 2000;130:2413–9. doi: 10.1093/jn/130.10.2413. [DOI] [PubMed] [Google Scholar]
- 24.Yudkoff M. Glia. United States: 1997. Brain metabolism of branched-chain amino acids; pp. 92–8. [DOI] [PubMed] [Google Scholar]
- 25.Matsuo H, Tsukada S, Nakata T, et al. Expression of a system L neutral amino acid transporter at the blood-brain barrier. Neuroreport. 2000;11:3507–11. doi: 10.1097/00001756-200011090-00021. [DOI] [PubMed] [Google Scholar]
- 26.Takanaga H, Mackenzie B, Peng JB, et al. Characterization of a branched-chain amino-acid transporter SBAT1 (SLC6A15) that is expressed in human brain. Biochem Biophys Res Commun. 2005;337:892–900. doi: 10.1016/j.bbrc.2005.09.128. [DOI] [PubMed] [Google Scholar]
- 27.Hutson SM, Berkich D, Drown P, et al. Role of branched-chain aminotransferase isoenzymes and gabapentin in neurotransmitter metabolism. J Neurochem. 1998;71:863–74. doi: 10.1046/j.1471-4159.1998.71020863.x. [DOI] [PubMed] [Google Scholar]
- 28.Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- 29.Eid T, Thomas MJ, Spencer DD, et al. Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet. 2004;363:28–37. doi: 10.1016/s0140-6736(03)15166-5. [DOI] [PubMed] [Google Scholar]
- 30.Solt K, Forman SA. Correlating the clinical actions and molecular mechanisms of general anesthetics. Curr Opin Anaesthesiol. 2007;20:300–6. doi: 10.1097/ACO.0b013e32816678a5. [DOI] [PubMed] [Google Scholar]
- 31.Han B, McCarren HS, O’Neill D, et al. Distinctive recruitment of endogenous sleep-promoting neurons by volatile anesthetics and a nonimmobilizer. Anesthesiology. 2014;121:999–1009. doi: 10.1097/ALN.0000000000000383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tao F, Chen Q, Sato Y, et al. Inhalational anesthetics disrupt postsynaptic density protein-95, Drosophila disc large tumor suppressor, and zonula occludens-1 domain protein interactions critical to action of several excitatory receptor channels related to anesthesia. Anesthesiology. 2015;122:776–86. doi: 10.1097/ALN.0000000000000609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zinnanti WJ, Lazovic J, Griffin K, et al. Dual mechanism of brain injury and novel treatment strategy in maple syrup urine disease. Brain. 2009;132:903–18. doi: 10.1093/brain/awp024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tonjes M, Barbus S, Park YJ, et al. BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type IDH1. Nat Med. 2013 doi: 10.1038/nm.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Novarino G, El-Fishawy P, Kayserili H, et al. Mutations in BCKD-kinase lead to a potentially treatable form of autism with epilepsy. Science. 2012;338:394–7. doi: 10.1126/science.1224631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Belli S, Vanacore N. Proportionate mortality of Italian soccer players: is amyotrophic lateral sclerosis an occupational disease? Eur J Epidemiol. 2005;20:237–42. doi: 10.1007/s10654-004-6879-7. [DOI] [PubMed] [Google Scholar]
- 37.Dufour F, Nalecz KA, Nalecz MJ, et al. Modulation of absence seizures by branched-chain amino acids: correlation with brain amino acid concentrations. Neurosci Res. 2001;40:255–63. doi: 10.1016/s0168-0102(01)00232-2. [DOI] [PubMed] [Google Scholar]
- 38.Kandratavicius L, Balista PA, Lopes-Aguiar C, et al. Animal models of epilepsy: use and limitations. Neuropsychiatr Dis Treat. 2014;10:1693–705. doi: 10.2147/NDT.S50371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van der Hel WS, Notenboom RG, Bos IW, et al. Reduced glutamine synthetase in hippocampal areas with neuron loss in temporal lobe epilepsy. Neurology. 2005;64:326–33. doi: 10.1212/01.WNL.0000149636.44660.99. [DOI] [PubMed] [Google Scholar]
- 40.Sommer W. Erkrankung des Ammonshorns als aetiologisches Moment der Epilepsie. Arch Psychiatr Nervenkr. 1880;10:631–675. [Google Scholar]
- 41.Wang Y, Zaveri HP, Lee TS, et al. The development of recurrent seizures after continuous intrahippocampal infusion of methionine sulfoximine in rats: a video-intracranial electroencephalographic study. Exp Neurol. 2009;220:293–302. doi: 10.1016/j.expneurol.2009.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gruenbaum SE, Wang H, Zaveri HP, et al. Inhibition of glutamine synthetase in the central nucleus of the amygdala induces anhedonic behavior and recurrent seizures in a rat model of mesial temporal lobe epilepsy. Epilepsy Behav. 2015;51:96–103. doi: 10.1016/j.yebeh.2015.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cavus I, Kasoff WS, Cassaday MP, et al. Extracellular metabolites in the cortex and hippocampus of epileptic patients. Ann Neurol. 2005;57:226–35. doi: 10.1002/ana.20380. [DOI] [PubMed] [Google Scholar]
- 44.Perez EL, Lauritzen F, Wang Y, et al. Evidence for astrocytes as a potential source of the glutamate excess in temporal lobe epilepsy. Neurobiol Dis. 2012;47:331–7. doi: 10.1016/j.nbd.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Plaitakis A, Constantakakis E, Smith J. The neuroexcitotoxic amino acids glutamate and aspartate are altered in the spinal cord and brain in amyotrophic lateral sclerosis. Ann Neurol. 1988;24:446–9. doi: 10.1002/ana.410240314. [DOI] [PubMed] [Google Scholar]
- 46.Plaitakis A. Abnormal glutamatergic mechanisms and branched-chain aminoacids in amyotrophic lateral sclerosis. Lancet. 1989;1:157. doi: 10.1016/s0140-6736(89)91170-7. [DOI] [PubMed] [Google Scholar]
- 47.Yudkoff M, Daikhin Y, Nissim I, et al. Ketogenic diet, brain glutamate metabolism and seizure control. Prostaglandins Leukot Essent Fatty Acids. 2004;70:277–85. doi: 10.1016/j.plefa.2003.07.005. [DOI] [PubMed] [Google Scholar]
- 48.Hutson SM, Lieth E, LaNoue KF. Function of leucine in excitatory neurotransmitter metabolism in the central nervous system. J Nutr. 2001;131:846S–850S. doi: 10.1093/jn/131.3.846S. [DOI] [PubMed] [Google Scholar]
- 49.Broer S, Brookes N. Transfer of glutamine between astrocytes and neurons. J Neurochem. 2001;77:705–19. doi: 10.1046/j.1471-4159.2001.00322.x. [DOI] [PubMed] [Google Scholar]
- 50.Laake JH, Slyngstad TA, Haug FM, et al. Glutamine from glial cells is essential for the maintenance of the nerve terminal pool of glutamate: immunogold evidence from hippocampal slice cultures. J Neurochem. 1995;65:871–81. doi: 10.1046/j.1471-4159.1995.65020871.x. [DOI] [PubMed] [Google Scholar]
- 51.Olney JW, Sharpe LG, Feigin RD. Glutamate-induced brain damage in infant primates. J Neuropathol Exp Neurol. 1972;31:464–88. doi: 10.1097/00005072-197207000-00006. [DOI] [PubMed] [Google Scholar]
- 52.Coulter DA, Eid T. Astrocytic regulation of glutamate homeostasis in epilepsy. Glia. 2012;60:1215–26. doi: 10.1002/glia.22341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liang SL, Carlson GC, Coulter DA. Dynamic regulation of synaptic GABA release by the glutamate-glutamine cycle in hippocampal area CA1. J Neurosci. 2006;26:8537–48. doi: 10.1523/JNEUROSCI.0329-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cole JT, Mitala CM, Kundu S, et al. Dietary branched chain amino acids ameliorate injury-induced cognitive impairment. Proc Natl Acad Sci U S A. 2010;107:366–71. doi: 10.1073/pnas.0910280107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bone B, Fogarasi A, Schulz R, et al. Secondarily generalized seizures in temporal lobe epilepsy. Epilepsia. 2012;53:817–24. doi: 10.1111/j.1528-1167.2012.03435.x. [DOI] [PubMed] [Google Scholar]
- 56.Kobayashi M, Buckmaster PS. Reduced inhibition of dentate granule cells in a model of temporal lobe epilepsy. J Neurosci. 2003;23:2440–52. doi: 10.1523/JNEUROSCI.23-06-02440.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bouilleret V, Ridoux V, Depaulis A, et al. Recurrent seizures and hippocampal sclerosis following intrahippocampal kainate injection in adult mice: electroencephalography, histopathology and synaptic reorganization similar to mesial temporal lobe epilepsy. Neuroscience. 1999;89:717–29. doi: 10.1016/s0306-4522(98)00401-1. [DOI] [PubMed] [Google Scholar]
- 58.de Lanerolle NC, Kim JH, Williamson A, et al. A retrospective analysis of hippocampal pathology in human temporal lobe epilepsy: evidence for distinctive patient subcategories. Epilepsia. 2003;44:677–87. doi: 10.1046/j.1528-1157.2003.32701.x. [DOI] [PubMed] [Google Scholar]
- 59.Sun C, Mtchedlishvili Z, Bertram EH, et al. Selective loss of dentate hilar interneurons contributes to reduced synaptic inhibition of granule cells in an electrical stimulation-based animal model of temporal lobe epilepsy. J Comp Neurol. 2007;500:876–93. doi: 10.1002/cne.21207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marx M, Haas CA, Haussler U. Differential vulnerability of interneurons in the epileptic hippocampus. Front Cell Neurosci. 2013;7:167. doi: 10.3389/fncel.2013.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Erecinska M, Nelson D. Activation of glutamate dehydrogenase by leucine and its nonmetabolizable analogue in rat brain synaptosomes. J Neurochem. 1990;54:1335–43. doi: 10.1111/j.1471-4159.1990.tb01967.x. [DOI] [PubMed] [Google Scholar]
- 62.Butterworth RF. Pathophysiology of hepatic encephalopathy: a new look at ammonia. Metab Brain Dis. 2002;17:221–7. doi: 10.1023/a:1021989230535. [DOI] [PubMed] [Google Scholar]
- 63.Norenberg MD, Jayakumar AR, Rama Rao KV, et al. New concepts in the mechanism of ammonia-induced astrocyte swelling. Metab Brain Dis. 2007;22:219–34. doi: 10.1007/s11011-007-9062-5. [DOI] [PubMed] [Google Scholar]
- 64.Rangroo Thrane V, Thrane AS, Wang F, et al. Ammonia triggers neuronal disinhibition and seizures by impairing astrocyte potassium buffering. Nat Med. 2013;19:1643–8. doi: 10.1038/nm.3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Eid T, Lee TS. Reassessing the role of astrocytes in ammonia neurotoxicity. Nat Med. 2013;19:1572–4. doi: 10.1038/nm.3420. [DOI] [PubMed] [Google Scholar]
- 66.Burrage LC, Nagamani SC, Campeau PM, et al. Branched-chain amino acid metabolism: from rare Mendelian diseases to more common disorders. Human Molecular Genetics. 2014;23:R1–8. doi: 10.1093/hmg/ddu123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bridi R, Araldi J, Sgarbi MB, et al. Induction of oxidative stress in rat brain by the metabolites accumulating in maple syrup urine disease. Int J Dev Neurosci. 2003;21:327–32. doi: 10.1016/s0736-5748(03)00074-1. [DOI] [PubMed] [Google Scholar]
- 68.Bilotta F, Evered LA, Gruenbaum SE. Neurotoxicity of anesthetic drugs: an update. Curr Opin Anaesthesiol. 2017;30:452–457. doi: 10.1097/ACO.0000000000000482. [DOI] [PubMed] [Google Scholar]
- 69.Amrock LG, Starner ML, Murphy KL, et al. Long-term effects of single or multiple neonatal sevoflurane exposures on rat hippocampal ultrastructure. Anesthesiology. 2015;122:87–95. doi: 10.1097/ALN.0000000000000477. [DOI] [PubMed] [Google Scholar]
- 70.Rappaport BA, Suresh S, Hertz S, et al. Anesthetic neurotoxicity–clinical implications of animal models. N Engl J Med. 2015;372:796–7. doi: 10.1056/NEJMp1414786. [DOI] [PubMed] [Google Scholar]
- 71.Han XD, Li M, Zhang XG, et al. Single sevoflurane exposure increases methyl-CpG island binding protein 2 phosphorylation in the hippocampus of developing mice. Mol Med Rep. 2015;11:226–30. doi: 10.3892/mmr.2014.2751. [DOI] [PubMed] [Google Scholar]
- 72.Makaryus R, Lee H, Yu M, et al. The metabolomic profile during isoflurane anesthesia differs from propofol anesthesia in the live rodent brain. J Cereb Blood Flow Metab. 2011;31:1432–42. doi: 10.1038/jcbfm.2011.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jacob Z, Li H, Makaryus R, et al. Metabolomic profiling of children’s brains undergoing general anesthesia with sevoflurane and propofol. Anesthesiology. 2012;117:1062–71. doi: 10.1097/ALN.0b013e31826be417. [DOI] [PubMed] [Google Scholar]
- 74.Liu B, Gu Y, Xiao H, et al. Altered metabolomic profiles may be associated with sevoflurane-induced neurotoxicity in neonatal rats. Neurochem Res. 2015;40:788–99. doi: 10.1007/s11064-015-1529-x. [DOI] [PubMed] [Google Scholar]
- 75.Aminiaghdam S, Baturak K, Panahi PM, et al. The effects of BCAA supplementation on muscle damage following a lower-body resistance exercise bout in soccer players. Football Science. 2012;9:62–69. [Google Scholar]