

Abstract
In this study, we report a unique dominantly inherited disorganized supernumerary cusp and single root phenotype presented by 11 affected individuals belonging to 5 north-eastern Thai families. Using whole exome sequencing (WES) we identified a common single missense mutation that segregates with the phenotype in exon 6 of CACNA1S (Cav1.1) (NM_000069.2: c.[865A > G];[=] p.[Ile289Val];[=]), the Calcium Channel, Voltage-Dependent, L Type, Alpha-1s Subunit, OMIM ∗ 114208), affecting a highly conserved amino-acid isoleucine residue within the pore forming subdomain of CACNA1S protein. This is a strong genetic evidence that a voltage-dependent calcium ion channel is likely to play a role in influencing tooth morphogenesis and patterning.
Keywords: rare disease, dental anomalies, patterning, mutations, NGS, human, calcium ion channel
Introduction
Advances in molecular biology have increased our in-depth understanding of major processes directing tooth morphogenesis (Jernvall and Thesleff, 2012). Odontogenesis occurs in sequential developmental stages initiated with dental epithelial placode induction (seen as localized thickening of the oral ectoderm), followed by the bud, cap, and bell morphogenetic stages. This is followed by the terminal differentiations of odontoblasts and ameloblasts. Subsequently, root formation and tooth eruption lead to the formation of sub-regional tooth types (incisors, canines, premolars and molars), which are distinguished by a unique shape and size of both the crown and root. This regionalized layout is defined with single cusps and roots present in incisors and canines and multiple cusps and roots in molars; premolars display an intermediate crown and root pattern (2–3 cusps and single to double roots). Studies of developing mouse teeth suggest that both cusp and root initiation and patterning repeatedly re-utilize conserved developmental pathways participating in interactions between the oral ectoderm and cephalic neural crest-derived ectomesenchymal cells (Thesleff and Mikkola, 2014; Balic and Thesleff, 2015).
During dental development, the number of cusps is controlled by enamel knots, which are morphogen-expressing signaling centers (Matalova et al., 2005; Ahtiainen et al., 2016). Indeed cusps form precisely at the location of secondary enamel knot signaling centers, which function as trophic regions inducing cusp growth and controlling the fine definition of cusp form (Jernvall et al., 2000). To date, molecular mechanisms regulating enamel knot development are understood in the overall context of dental morphogenesis. Cusp number regulation stems from primary patterning events relying on multiple known dental regulatory pathways such as sonic hedgehog, fibroblast growth factor, and ectodysplasin (Bei, 2009; Charles et al., 2009a,b Harjunmaa et al., 2012). Rare syndromic and non-syndromic alterations affecting human tooth development offer opportunities to understand unique mechanisms regulating tooth number or cusp morphology. To date, heritable genetic alterations increasing cusp number are relatively rare in human populations. In this study, we explore a dominantly inherited supernumerary cusp phenotype in 11 affected individuals belonging to 5 Thai families. Using next generation sequencing (NGS) we identified a single shared variant, a missense mutation in CACNA1S, the gene encoding the Calcium Channel, Voltage-Dependent, L Type, Alpha-1s Subunit, CaV.1 (1q32.1, OMIM × 114208), affecting a highly conserved amino-acid isoleucine residue within the pore forming subdomain of CACNA1S protein and segregating with the phenotype. This is a strong genetic evidence that a voltage-dependent calcium ion channel is likely to play a role in influencing tooth morphogenesis and patterning.
Materials and Methods
Patients
The patients and their families were examined at Khon Kaen University Faculty of Dentistry and Hospital in Thailand. Affected (11), non-affected (4), as well as unrelated control individuals (18) consented to participate in the study named “Identification of the gene involved in a unique dental anomaly phenotype: multiple tuberculated permanent teeth encountered in Thai families.” They were recruited between 2012 and 2017. This study ST 0514.13.7/7241, Reference No. HE542262, and accompanying documents (information, consent forms) were reviewed and approved in 2012 by the Khon Kaen University Ethics Committee for Human Research based on the Declaration of Helsinki and ICH Good Clinical Practice Guidelines.
The oral phenotypes (Figure 1) were documented using the D[4]/phenodent registry, a Diagnosing Dental Defects Database1, which is approved by CNIL (French National Commission for Informatics and Liberty, number 908416). This clinical study is registered at https://clinicaltrials.gov: NCT01746121 and NCT02397824, and with the MESR (French Ministry of Higher Education and Research) Bioethics Commission as a biological collection “Orodental Manifestations of Rare Diseases” DC-2012-1677 within DC-2012-1002 and was acknowledged by the CPP (person protection committee) Est IV December 11th 2012.
FIGURE 1.

A striking multiple cusps single root phenotype. Normal crown morphology (A,B) of the right upper second premolar (15) and first permanent molar (16) and of the right lower premolar (45) and first permanent molar (46) could be compared to the multicusp pattern phenotype clearly visible both in the upper (C 1_III.11, E 2_III.7) and lower (D 1_III.11, F 2_III.7) jaws. On the close up of the right premolar-molar area on panoramic radiographs (G control, H 1_III.11 affected) molars present a single root (1) pattern. Only the first lower permanent molar (46) shows a more apical root furcation leading to the taurodontic appearance of the tooth.
Affected and non-affected family members and enrolled subjects gave written informed consents in accordance with the Declaration of Helsinki, both for the D[4]/phenodent registry and for genetic analyses performed on the salivary samples included in the biological collection. A material transfer agreement (MTA) was signed between collaborating universities and laboratories.
Sample Collection
Genomic DNA was isolated from the saliva of patients, unaffected family members, as well as control unrelated individuals using the Oragene® DNA OG-510 or OG-250 commercial kits (DNA Genotek Inc., Kanata, ON, Canada 2) according to the manufacturer’s protocol. Whole exome sequencing (WES) was performed on eight individuals from families 1, 2, and 3, and Sanger sequencing was performed on exon 6 of the CACNA1S gene on all enrolled members of the five families (Figure 2), as well as on 18 unrelated and unaffected individuals from the same geographic region to serve, in combination with data provided by a Thaï3 and an Asian database (Ngamphiw et al., 2011), as single nucleotide polymorphism (SNP) controls.
FIGURE 2.

Pedigrees and sequences analysis of families 1–5. Black squares indicate the patients analyzed in this study. All available members from families 2 and 3 were analyzed by whole exome sequencing. In family 1 only the 2 individuals [indicated by a red arrow (1_II.11, 1_III.10) were analyzed by WES]. Families 4 and 5 were analyzed exclusively by Sanger sequencing. Electropherograms reveal a heterozygous missense mutation c.865A > G; p.Ile289Val in affected individuals (1_II.10, 1_III.10, 1_III.11, 2_II.5, 2_III.6, 2_III.7, 3_II.1, 3_II.2, 4_I.2, 4_II.3, 5_III.2).
Whole-Exome Sequencing
Whole exome sequencing (WES) was performed at the Centre National de Recherche en Génomique Humaine (CNRGH, Institut de Biologie François Jacob, CEA, Evry, France). For family 1 only the father and the son were sequenced, (1_II.11 and 1_III.10), for family 2 all the available members were sequenced (2_II.5, 2_II.6, 2_III.6, and 2_III.7) and for family 3 only the two daughters were sequenced (3_II.1 and 3_II.2).
After quality control, genomic DNA (3 μg) was captured using the in-solution enrichment methodology (Human All Exon v5 – 50 Mb, Agilent Technologies, CA, United States). Library preparation and exome enrichment protocol (∼20.000 targeted genes) have been performed using an automated platform, according to manufacturer’s instructions (SureSelect, Agilent Technologies, CA, United States). After normalization and quality control, exome enriched libraries were sequenced on a HiSeq 2000 from Illumina (Illumina Inc., CA, United States) as paired-end 100 bp reads. Image analysis and base calling were performed using the Illumina Real-Time Analysis (RTA) Pipeline. Sequence quality parameters were assessed throughout the sequencing run. The standard bioinformatics analysis of sequencing data was based on the Illumina pipeline to generate FASTQ files for each sample. Sequence data was then processed using the exome analysis platform developed at CNRGH, which follows GATK best practice. Coverage/depth statistics have been accessed as quality control criteria. The sequencing yield generated for each sample allowed a minimum of 10× coverage for 93% of the targets. An average sequencing depth of at least 60× was obtained for good quality sample DNA. Polymorphism detection for each sample was performed using reads mapping procedure onto the reference genome (hg19) followed by “SNP calling” algorithm implemented by GATK/SAMtools software. Results (FASTQ, BAM and non-annotated VCF files) were communicated through a dedicated secure web site at CNRGH.
Bioinformatics Analysis
Annotation, ranking and filtering of genetic variants were performed with the VaRank program (Geoffroy et al., 2015) in combination with the Alamut Batch software (Interactive Biosoftware, Rouen, France). Very stringent criteria were used for excluding non-pathogenic variants, in particular: (1) variants represented with an allele frequency of more than 1% in dbSNP 138 (Sherry et al., 2001), in the EXAC database (Karczewski et al., 2017), in the NHLBI Exome Sequencing Project Exome Variant Server (EVS) (NHLBI GO Exome Sequencing Project4) or in our internal exome database, (2) variants in the 5′ or 3′ UTR, (3) variants with intronic locations and no prediction of local splice effect, and (4) synonymous variants without prediction of local splice effect. Variant effect on the nearest splice site was predicted using MaxEntScan (Yeo and Burge, 2004), NNSplice (Reese et al., 1997) and Splice Site Finder (Shapiro and Senapathy, 1987). Our analysis focused on heterozygous variants (SNV/indel) shared by all affected individuals, consistent with a dominant mode of inheritance.
Structural variants were predicted using by default the CANOES program (Backenroth et al., 2014) and annotated thanks to our in-house script AnnotSV (Geoffroy et al., 2018) based on the classical annotations such as the Database of Genomic Variants (DGV).
For the evaluation of possible relationships between the eight sequenced individuals from families 1, 2, and 3 a multi-sample VCF was created. The generated multi-sample VCF was converted into a PLINK binary format and used by the KING program (v2.1.4) to estimate the pair-wise kinship coefficients (–kinship parameter) (Manichaikul et al., 2010). Close relatives were inferred based on the estimated kinship coefficients ranges >0.354, [0.177, 0.354], [0.0884, 0.177], and [0.0442, 0.0884] corresponding to duplicate/MZ twin, 1st-degree, 2nd-degree, and 3rd-degree relationships, respectively.
Sanger Sequencing and Segregation
Primer pair for exon 6 of the CACNA1S gene was designed using Primer 3 (sense: 5′-GACATAATTCCCGCTGCCTG; antisense: 5′- GTTTCCATTCTTCACCCGCC). The amplification of the specific region of interest was performed on 50 ng of genomic DNA template. The PCR product was then purified and bidirectional Sanger sequencing performed by GATC Sequencing Facilities (Konstanz, Germany). All enrolled members of the five families and control subjects were assessed by Sanger sequencing. For family 1 (1_II.10 and 1_III.11), family 4 (4 individuals including an affected mother and son, 4_I.2 and 4_II.3, respectively) and family 5 (male 5_III.2) only Sanger sequencing and not exome sequencing was performed.
Mutations in the Human CACNA1S Gene
The UCSC genome browser5 was used to model the human CACNA1S gene sequence. Previously described mutations extracted from the Human Genome Mutation Database (HGMD6) (Stenson et al., 2009) were represented on this gene sequence (Figure 3).
FIGURE 3.

CACNAS1 mutations and corresponding protein domain. (A) The CACNA1S human gene is located on chr1q32.1, extends over 73 kb, and contains 44 exons (each vertical black line denotes one exon). The position of the start codon (ATG) is indicated. The mutation detected in this study is highlighted by a red arrow on exon 6 (c.865A > G, p.Ile289Val). All other previously described mutations in the CACNA1S gene are symbolized by a single letter above the corresponding exon. (B) The corresponding protein domains are represented according to the PFAM database. (C) Schematic view of the CACNA1S encoded alpha 1 sub-unit of DHPR. The subunit has a total of four transmembrane domains (I, II, III, IV) composed by six segments (1–6) and three intracellular loop domains (loops I–II, loops II–III, and loops III–IV). The mutated isoleucine (red star) is located in the first pore-forming intramembrane domain close to one of the 3 amino-acids involved in calcium selectivity (green circles). The II_III loop interacts with RYR1 to allow excitation/contraction in muscle.
The Protein Families (PFAM) database (Punta et al., 2012) was used to search for protein domain organization and the Uniprot database7 to collect data for protein subdomains.
Multiple Protein Sequences Alignment
The CACNA1S Human pore-forming subdomain (280–301) protein sequence was aligned with the CACNA1S sequence of 25 species representative of mammalian lineages and 6 non-mammalian species using Uniprot website7. The data were then imported and visualized with Jalview8 and colored according to the ‘ClustalX’ coloring scheme (Figure 4). The percentage of amino acids identity compared to the Human protein was calculated using the Jalview pairwise alignment.
FIGURE 4.

Multiple protein sequence alignment. The sequence alignment of the pore forming subdomain (position 280–301) amongst mammalian 25 species. The alignment is displayed using Jalview and colored according to the ‘ClustalX’ coloring scheme. The isoleucine mutated in the affected individuals (red arrow) is unchanged in all the species.
Microarray Analysis
In 2013, we developed a transcriptomic atlas (Laugel-Haushalter et al., 2013) that has proven to be a successful tool to search for candidate genes, involved in rare diseases with dental abnormalities, based on their expression in mouse tooth at the E14.5 cap stage. For detailed protocols on microarrays see (Laugel-Haushalter et al., 2013).
Results
Clinical Phenotype
Eleven affected individuals from 5 not known to be related non-consanguineous families presented with a unique dental phenotype of disorganized supernumerary cusps pattern. Patients did not suffer from any other medical conditions as assessed by detailed medical history and examination. Patients’ medical records were scrutinized and reviewed a second time after the genotype results to search for specific phenotypes, such as myopathies, and no other existing disease was identified. They were examined clinically for in-depth analysis of their oral morphological changes. Strikingly, all of the affected subjects displayed supernumerary cusps throughout affected molars, [compare Figure 1 control molar (A, B) with 4/5 cusps versus affected patient with at least 10 extra-cusps (C–F)]. These extra-cusp alterations typically manifested in both maxillary and mandibular molars. The detailed oral phenotype can be seen in patient 1_III.11 (Supplementary Figures S1A–D), patient 2_III.7 (Supplementary Figures S1E–G), and patient 4_II.3 (Supplementary Figures S1I–L). These accessory structures created deep pits and grooves, which resembled “mulberry molars” (Nissanka-Jayasuriya et al., 2016). The location of these supernumerary molar cusps was clearly occlusal and appeared distinct from Carabelli cusps, the latter typically present on the external palatal side of the molar (Hunter et al., 2010) (Carabelli cusp is number 5 in Figure 1A). Even in the case of premolars, normally having 2–3 cusps, the unusual presence of multiple accessory cusps with one protruding cusp was clearly visible (see Figures 1D–F and Supplementary Figures S1D,L for lower premolars). Canines, which typically have a single cusp and a cingulum that could be considered as a small lingual or palatal cusp, also displayed an unusual multicusp pattern around one prominent vestibular cusp (Supplementary Figures S1K,L). Other dysmorphic changes in patients extended beyond simple cusp multiplications. About 80% of affected patients’ canine and incisors had exaggerated marginal ridges and unusually large cinguli. Incisor occlusal ridges showed groves or lobes termed “mamelons” (normally present in children’ permanent incisors at tooth eruption and disappearing with wear), that were larger than normal (Supplementary Figure S1I). Collectively, we observed widespread changes in the crown cusp pattern and morphology of all teeth in all affected patients.
Root Changes Accompanying Supernumerary Cusps
Tooth root branching shape anomalies were found in all affected patients and most commonly were root furcation branching deficits. Typically supernumerary cusped molars or premolars also had single tapered roots, instead of normal bifurcated or trifurcated ones found in unaffected individuals (Figures 1G,H). Taurodontism, a molar shape anomaly, visible on radiographs, consisting of elongated pulp chambers and apical displacement of the bifurcation or trifurcation of roots is illustrated in Figure 1H or in panoramic radiographs of patients from families 1, 2, 3, 4 (Supplementary Figures S1B,G,H,J). In all cases both molars and premolars with supernumerary cusps exhibited morphological alterations of root morphology. In the majority of cases (>97%) a single root was found (when normally 2–3 roots on specific premolars or molars). The only teeth that still demonstrated some multiple root formation were the first lower permanent molars (46 or 36; for the tooth numbering system refer to Supplementary Figure S3), with an apical displacement of the furcation leading to a taurodontic appearance (Figure 1H and Supplementary Figures S1B,G blue arrow).
Patient 2_III.7 also had abnormalities of tooth number with tooth agenesis and 2 missing premolars (second lower left 35, upper right 15 premolar). Anomalies of tooth number were also seen in family 3. Patient 3_II.2 had a supernumerary lower right permanent incisor (Supplementary Figure S1H).
Individual 4_II.3 (Supplementary Figures S1I–L) was the most severely affected, with numerous cusp duplications blocking normal dental occlusion (Supplementary Figure S1I).
Genetic Transmission
Fifteen individuals (11 affected and 4 unaffected) from 5 not known to be related non-consanguineous families were questioned concerning family genetics, then genotyped using salivary samples collected to obtain DNA. In all families, an autosomal dominant mode of inheritance was suspected. Figure 2 illustrates composite pedigrees/genetic lineages of all affected patients. The dominant inheritance was supported by results showing that no regions of homozygosity were found in affected patients (Supplementary Table S2). Moreover, a similar dominant phenotype has been described previously (Skrinjaric et al., 2016).
In the extended pedigree of family 1 with 28 individuals an autosomal dominant pattern of inheritance was clearly visible (Figure 2) with affected mother (1_II.10), unaffected father (1_II.11), affected son (1_III.10) and affected daughter (1_III.11). Family 2 comprised 15 individuals amongst which an affected mother (2_II.5), an unaffected father (2_II.6) and both affected children (2_III.6 and 2_III.7). Family 3 comprised two affected females (3_II.1 and 3_II.2). Families 1, 3, 4, 5 affected individuals and especially their parents or grand-parents originated from the same city-Maha Sarakham in the north-eastern region of Thailand. Family 2 was from Roi Et, a city 40 km away from Maha Sarakham.
Mutation Analysis
To genetically investigate why this rare multicusp, single root pattern might occur, we analyzed the genotype of these patients by WES.
We also analyzed copy number variations using CANOES program (Backenroth et al., 2014) finding no region that was common to all the affected members in a single family.
Sequencing data processing and variant calling (SNV and InDels) revealed from 57,522 to 58,694 genetic variants per proband (Table 1). Variant filtering using stringent criteria reduced the number of genetics variants to, respectively, 770, 749, 771, 854, 926, 956, 934, and 798 variants per proband (1_II.11, 1_III.10, 2_II.5, 2_II.6, 2_III.6, 2_III.7, 3_ II.1, 3_ II.2, affected individuals in bold). As the pedigrees suggested an autosomal dominant mode of inheritance with a complete penetrance within a group of patients coming from the same region, we narrowed the selection to heterozygous variants, shared by affected and not present in non-affected individuals, and we were left with only one variant in the CACNA1S gene (NM_000069.2: c.865A > G; p.Ile289Val).
Table 1.
Summary of the WES analysis in families 1–3.
| Patient | 1_II.11 |
1_III.1 0 |
2_II.5 |
2_II.6 |
2_III.6 |
2_III.7 |
3_II.1 |
3_II.2 |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Type of variant | SNV | Indel | SNV | Indel | SNV | Indel | SNV | Indel | SNV | Indel | SNV | Indel | SNV | Indel | SNV | Indel |
| Total number of variants | 49953 | 7942 | 49610 | 7948 | 49531 | 7991 | 48192 | 7593 | 50611 | 8083 | 50284 | 7942 | 50070 | 7963 | 50544 | 8006 |
| Variants with an allele frequency <1% | 1782 | 590 | 1732 | 588 | 1761 | 615 | 1897 | 628 | 2065 | 671 | 2223 | 649 | 2100 | 626 | 1880 | 600 |
| Exclusion of 5′UTR, 3′UTR and intron locations without local splice effect prediction | 954 | 69 | 917 | 77 | 986 | 79 | 1076 | 79 | 1189 | 91 | 1240 | 75 | 1145 | 97 | 1011 | 87 |
| Exclusion of synonymous variants without local splice effect prediction | 706 | 64 | 679 | 70 | 696 | 75 | 781 | 73 | 839 | 87 | 887 | 69 | 840 | 94 | 716 | 82 |
| Variants consistent with dominant transmission | 1 heterozygous variant in CACNA1S | |||||||||||||||
One heterozygous variant common to all the affected patients (1_III.10, 2_II.5, 2_III.6, 2_III.7, 3_II.1, and 3_II.2) in the genes CACNA1S, was selected for further investigation after exclusion of mutations located in the 5′UTR, 3′UTR, intron, along with synonymous variants without predicted splicing effects.
To confirm this finding, we subsequently validated familial segregation by Sanger sequencing on all enrolled family members and confirmed that the mutation segregated properly with the disease within all the families (Figure 2).
To test for a possible increased presence of the identified variant in the Thai population independent of any pathological context, we sequenced exon 6 of the CACNA1S gene for 18 unaffected unrelated control subjects also originating from the north-eastern region of Thailand. Although limited in number, this data confirmed information found in a Thai database9 consisting of 32 genotyped individuals and an Asian database (Ngamphiw et al., 2011) of 1719 individuals in which the CACNA1S identified variant was never reported. In summary, control subjects never carried this mutation, excluding that this variant was a common polymorphism found in individuals originating from this region (Supplementary Figure S2).
The CACNA1S gene belongs to a family of genes encoding calcium channels-specifically the pore- forming alpha 1 subunit of the voltage-gated L-type Ca2+ channel (dihydropyridine receptor DHPR). It regulates calcium release from the muscle sarcoplasmic reticulum (Schartner et al., 2017). CACNA1S also known as DHPR1, Cav1.1 and CACN1 encodes the 176 kDa subunit of the DHPR channel. This unit is composed of four six-segments (S1–S6) transmembrane domains (forming ion transport domains I, II, III, and IV) and three intracellular loop domains (loops I and II, loops II and III, loops III and IV). The S4 segment of each repeated domain is thought to be the voltage sensor. Intramembrane domains are involved in the pore formation and play a role in the gating and the selectivity of the channel; the II–III loop interacts with Ryanodine receptor 1 (RYR1) to allow excitation/contraction of muscles. The mutation localized to the pore forming subdomain (280–301) of the first ion transport domain (50–345) of the CACNA1S protein (Figure 3).
Knowing that all the families shared the same variant and originated from the same region, we checked if the families could be closely related by estimating the kinship coefficient with KING (Supplementary Table S3), which suggested that the three families were unrelated. As the families were not closely related, we suspected a founder effect and we decided to look at the rare variants in the CACNA1S region. We realized that affected patient were sharing a region with exactly the same SNPs (Supplementary Table S4). This could suggest that a portion of chromosome with the mutation was transmitted along generations in a population, confirming a founder effect linked to the mutation and subsequent phenotype.
Mutations in the Human CACNA1S Gene
This mutation occurred at an isoleucine residue that is conserved in the 25 studied and aligned CACNA1S mammalian sequences. Moreover, this isoleucine residue is highly conserved in vertebrates but not in invertebrates (Figure 4). The mutation is also predicted to be disease causing by MutationTaster (Schwarz et al., 2014) and possibly damaging by PolyPhen-2 (Polymorphism Phenotyping v2) (PPH2) (Adzhubei et al., 2010). The list of all the bioinformatics algorithms used to determine the pathogenicity of variants and their scores are available for the 3 families in Supplementary Tables S5–S7). These findings suggest a putative important function for this amino acid.
Expression of CACNA1S Complex Partners and Interactors in Mouse Tooth Germs
CACNA1S or Cav1.1 is the pore forming subunit of a L-type calcium channel, composed of 4 subunits (CACNA1S, CACNAB1 or CACNAB2, CACNAG1, and CACNA2D1), named the dihydropyridine channel or DHP channel. In myocytes, it regulates calcium release from the sarcoplasmic reticulum by activating the ryanodine receptor (Schartner et al., 2017). To examine a potential role of calcium channels during odontogenesis, we checked for the presence and expression of Cacna1s complex partners and known interactors within a E14.5 mouse tooth transcriptomic atlas (Laugel-Haushalter et al., 2013) (Table 2).
Table 2.
Expression of CACNA1S complex partners (in bold) and interactors in E14.5 mouse tooth germ.
| Gene symbol | Lower molars ± STD | Lower incisors ± STD | Upper molars ± STD | FC lower molars versus lower incisors | p-value | FC lower versus upper molars | p-value2 |
|---|---|---|---|---|---|---|---|
| Cacna1s | 7.04 ± 0.06 | 6.39 ± 0.37 | 5.92 ± 0.1 | 1.7402 | 0.0000582054 | 1.58492 | 0.0000703422 |
| Cacnb1 | 8.12 ± 0.09 | 7.92 ± 0.17 | 7.78 ± 0.11 | 1.04509 | 0.46 | 1.0344 | 0.57 |
| Cacnb2 | 7.54 ± 0.11 | 7.73 ± 0.22 | 7.64 ± 0.1 | −1.16979 | 0.013 | 1.12838 | 0.04 |
| Cacna2d1 | 8.61 ± 0.01 | 8.91 ± 0.28 | 8.82 ± 0.08 | −1.25004 | 0.04 | −1.05355 | 0.52 |
| Cacng1 | 7.08 ± 0.07 | 6.51 ± 0.44 | 6.01 ± 0.07 | 1.43416 | 0.0000630813 | 1.5501 | 0.00000326276 |
| Calm1 | 11.81 ± 0.06 | 11.72 ± 0.14 | 11.67 ± 0.03 | −1.17861 | 0.0000945752 | 1.02461 | 0.28 |
| Calm2 | 10.92 ± 0.05 | 10.91 ± 0.12 | 10.92 ± 0.11 | −1.22554 | 0.00235493 | 1.04302 | 0.41 |
| Calm3 | 11.01 ± 0.04 | 10.91 ± 0.21 | 10.75 ± 0.05 | −1.12517 | 0.01 | 1.06782 | 0.09 |
| Dysf | 8.25 ± 0.21 | 8.57 ± 0.03 | 8.06 ± 0.04 | 1.08 | 0.11 | 1.21 | 0.00153247 |
| Jsrp1 | 7.54 ± 0.32 | 7.94 ± 0.02 | 7.28 ± 0.05 | 1.13 | 0.00534678 | 1.18 | 0.000235233 |
| Ryr1 | 8.06 ± 0.21 | 7.18 ± 0.57 | 6.43 ± 0.08 | 1.58111 | 0.00279044 | 1.88642 | 0.0000587608 |
| Stac | 7.97 ± 0.08 | 7.68 ± 0.12 | 7.92 ± 0.06 | −1.32 | 0.000197742 | 1.01 | 0.87 |
| Stac2 | 6.42 ± 0.17 | 6.4 ± 0.08 | 6.23 ± 0.07 | 1.02 | 0.77 | 1.01 | 0.96 |
| Stac3 | 6.63 ± 0.41 | 7.29 ± 0.12 | 6.17 ± 0.07 | 1.71 | 0.0000202013 | 1.38 | 0.000251342 |
All the CACNA1S partners and interactors are expressed in all tooth types (lower incisors, lower and upper molars) with the exception of Myh13. Most are more strongly expressed in lower molars.
We showed that Cacna1s and all complex partners were expressed in E14.5 mouse incisors and molars. Indeed, the signal value from genes expressed in the tissue are between 4 and 13, so the genes with a ratio above 4 will be considered as expressed in the tissue. For Cacna1s and Cacnag1, expression was stronger in lower molars compared to lower incisors and upper molars.
Calmodulin is a calcium sensor known to bind the isoleucine glutamine (IQ) sequence motifs of CACNA1S (Halling et al., 2009). We showed that Calm1, Calm2, and Calm3 were strongly expressed in all tooth types suggesting that the CACNA1S channel may play a role in calcium sensing and transport in teeth. Ryanodine receptor 1 (RYR1) and STAC proteins have also been described to interact with CACNA1S during muscle contraction (Wong King Yuen et al., 2017). Ryr1 and Stac were strongly expressed in teeth, with a higher expression than Ryr1, Stac2, and Stac3 in lower molars. Dysf (Dysferlin) and Jsrp1 (Junctional Sarcoplasmic Reticulum Protein 1) are other known CACNA1S interactors found in E14.5 fetal teeth.
These expression data suggest a possible role of calcium regulated channels directing tissue morphogenic events. The Cacna1s gene is indeed expressed during mouse E14.5 cap stage tooth development, consistent with a possible role of this gene in tooth shape patterning and modulation of cusp signaling center activity.
Discussion
Previous Reports of CACNA1S-Like Dental Defects
In this paper, we identified mutations in the CACNA1S gene as the probable cause of a dental rare disease consisting of major tooth morphogenetic modifications enhancing the number of cusps, altering tooth crown cusp pattern, and inhibiting root branching. (Skrinjaric et al., 2016) described a similar phenotype in three unrelated Croatian families with 17 affected members presenting with cone-shaped premolars, multitubercular molar crowns, pyramidal molar roots with single root canals, shovel-shaped incisors with palatal invaginations, and hypodontia-a condition estimated to be present in less than 1:1,000,000 with an autosomal dominant mode of inheritance. In Ather et al. (2013) microdontia and multicusp patterns were reported, along with tooth discoloration, hypercementosis, and multiple pulp stones suggesting a different disease. Another similar phenotype is lobodontia (Brook and Winder, 1979; Gorlin, 1998; Gorlin et al., 2001; Hennekam et al., 2010; Ather et al., 2013; Kiyan et al., 2013; Skrinjaric et al., 2016), tooth morphology changes somewhat like Canis lupus familiaris (dog) or wolf teeth with multiple abnormalities -fanged-like teeth (accentuated cusps), molar multituberculisms, shovel-shaped incisors, hypodontia, and microdontia. In all described cases, potential genetic effectors are to date unknown. Moreover, children born from mothers with syphilis have multicusp mulberry-like molars (Hillson et al., 1998).
Possible Role of Calcium Signaling in Dental Development
Mutations in the CACNA1S gene encoding a calcium ion channel have been a suspected cause of a variety of different diseases. Indeed according to HGMD, 43 mutations involved in seven different diseases have been described in the CACNA1S gene (listed in Supplementary Table S1 and Figure 3). These include malignant hyperthermia susceptibility 5, MIM #601887 (Eltit et al., 2012), thyrotoxic periodic paralysis, susceptibility to, 1, MIM #188580 (Kung, 2006), hypokalemic periodic paralysis, type 1, MIM #170400 (Fialho et al., 2018), normokalaemic periodontic paralysis (Fan et al., 2013), myopathy (Schartner et al., 2017), rhabdomyolysis (Vivante et al., 2017), exertional heat illness (Fiszer et al., 2015), and schizophrenia (Fromer et al., 2014; Purcell et al., 2014). Six mutations, localized to the first ion transport domain, were previously reported to result in myopathies, malignant hyperthermia, and schizophrenia. Uniquely, our families had a mutation in the pore-forming subdomain of this first domain (Figure 3). Affected individuals did not suffer from any of these conditions. Collectively, our data strongly implicate the observed c.865A > G missense mutation as the most likely cause of the dental phenotype observed in our families.
In all of our patients, the mutation segregates with the phenotype and likely produces defects such as extra cusps, deficient roots, along with other dental alterations. CACNA1S is the pore-forming subunit of a voltage-activated channel, acting as a sensor of depolarization, allowing calcium release and diffusion. In general, channelopathies are diseases resulting from the dysfunction of voltage-gated ion channels, and can induce a number of neurological, muscular, and cardiac defects. Calcium influx and consequent regulation through regulated entry are increasingly recognized as key regulators of various developmental processes (Bates, 2015). To date, no dental malformations have been described in patients with CACNA1S mutations. Disruption of other voltage-regulated calcium channels, though, has been shown to alter dentition, produce microdontia or tooth agenesis, dentin formation, tooth eruption defects, or amelogenesis imperfecta [reviewed in Duan (2013)]. For instance, CACNA1C disruption in Timothy Syndrome produces small, misplaced teeth due to enamel hypoplasia (Splawski et al., 2004). Roles of ion transporters affecting enamel and dentin biomineralization could stem from calcium roles regulating mineralization, ion homeostasis, pH, or endocytosis. Recently the TRPM4 channel knockdown in dental follicle progenitors was found to cause reduced calcium influx, which actually increases mineralization by reducing dental stem proliferation (Nelson et al., 2013).
Fatemifar et al. (2013) identified through genome wide association study CACNB2 associated with primary tooth eruption. The voltage-gated calcium channel CACNA1C and calcium influx was shown to be involved in regulating mandible development, providing further evidence of a role of these molecules in non-excitable cells during development (Ramachandran et al., 2013).
Role of the Identified Mutation in CACNA1S
We identified a missense mutation in exon 6 of CACNA1S (NM_000069.2: c.865A > G; p.Ile289Val). However, we could not exclude that this variant could co-segregate with another non-coding variant. It is likely that this variant produces the dental phenotype we here describe, based on the fact that some association of the CACNA1S region with tooth development/anomalies such as with permanent tooth eruption (Geller et al., 2011) and with tooth agenesis (Jonsson et al., 2018) were already reported. The variant was found once among 30974 genomes based on the gnomAD database (Lek et al., 2016) and is present in dbSNP (rs139920212) as supported by several submissions but it could be that the individual dental phenotype was never assessed.
The amino-acid change (p.Ile289Val) is in the intramembranous domain of the protein portion constituting the pore-forming subunit of the voltage-gated calcium channel. Loss of function mutations observed in the CACNA1S first domain (p.F275L) and the nearby pore forming subdomain resulted in myopathy (Schartner et al., 2017), a disease that is clearly not present in the affected individuals of our cohort. The mutation that we identified is 3 amino-acids before the amino-acid 292 involved in calcium ion selectivity and permeability. As the mutation is very close to this amino-acid involved in calcium selectivity (Figure 3), we propose that changing an isoleucine into a valine could result in a higher accessibility to this amino acid leading to potential alteration of the channel gating or specificity. The mutation that we identified is most probably a gain of function or a modification of the channel permeability possibly allowing other ions to travel through the lumen or increasing the calcium influx. A second review targeting medical assessment found no CACNA1S mutation-specific phenotypes (myopathy, for example) in our patients. However, we cannot exclude a possible risk of malignant hyperthermia caused by general anesthesia.
How Alterations in Ion Gradients Could Alter Cusp Pattern by Placode Signaling Centers
Tooth crown morphology as defined by the number of cusps, their spatial distribution, shape and size is clearly dependent of signaling centers called primary and then secondary enamel knots (Jernvall et al., 1994; Vaahtokari et al., 1996; Thesleff and Jernvall, 1997; Thesleff et al., 2001; Matalova et al., 2005; Harjunmaa et al., 2012). The increase in cusp number does not result from an increase in tooth size, but from an altered primary patterning phase of development (Harjunmaa et al., 2012). Multiple signaling pathways (NF-kappaB, SHH, BMP…) are involved in setting up this complexity which can be increased substantially by adjusting these pathways simultaneously (Harjunmaa et al., 2012).
Teeth are highly evolvable because only small developmental changes are needed to produce large changes in size and number of small cusps (Jernvall, 2000). BMP4 is clearly involved in the induction of the tooth signaling centers (Jernvall et al., 1998).
In mammals, the design that is considered one of the most important characteristics is a three-cusp shape (protocone, paracone, metacone) called a tribosphenic molar. Quadrate (also called quadritubercular) molars have an additional fourth cusp on the lingual side called the hypocone that is present in primates and humans. The modified multiple cusps molar phenotype reported here nevertheless allows the recognition of 3–4 prominent cusps, reminiscent of this primary cusp organization that allowed the evolution and diversity of cusps formation in mammalian species. The complexity and number of cusps may arise by budding of the crown due to the multiplication of signaling centers.
Several mammalian species naturally possess multicuspid incisors, suggesting that mammals have the capacity to form multicuspid teeth regardless of location in the oral jaw. Misregulation of Shh and Bmp signaling in Lrp4 null mice led to the formation of incisors with molar like cusps (Ohazama et al., 2010). Clearly the multiple cusps pattern encountered in case of CACNA1S mutation is not restricted to the molar area.
Ion channels influence developmental processes involving BMP signaling (Bates, 2015). Every cell has a membrane potential set up by the concentration of ions in and out the cell. Ion channels can influence this membrane potential. Voltage gated calcium channels are depolarizing channels making the membrane potential more positive. Ions can travel through the ion channels or gap junctions. BMP signaling pathway and secretion may rely on ion channel function, as demonstrated in hair follicle stem cells and by craniofacial or digit phenotypes observed in Kir2.1 or Cav1.2 dysfunction.
Ion current alterations could change osmotic balance, affecting cell volume, increasing physical stress. Patients with K+ channel Kir2.1 mutations can have cleft palate, reduced jaw size, and dental alterations (Yoon et al., 2006), similar to the mouse knockout model implicating its role [(Dahal et al., 2012) and references therein]. Compromised expression of the Drosophila Kir2.1 homolog reduces bone morphogenetic protein BMP signaling, a change that can dramatically alter tooth patterning. Reducing zebrafish BMP levels induce supernumerary cusps and teeth (Jackman et al., 2013). How any ion-induced alterations in dental placode alter tooth development is relatively unexplored. Alterations in dental placode activators (ectodysplasin, activin, and FGF), or inhibitors (BMP or SHH) seem to collectively act to regulate tooth cusp number complexity. Reduced BMP levels acting within the established network of tooth morphogens might increase cusp number or cause other malformations (Harjunmaa et al., 2012). In addition, regulation of intracellular calcium content, through channel-regulated osmotic control might have influenced dental evolution, especially concerning the transition from ocean to land-based species.
It is intriguing, however, why Cacna1s transgenic mice were not reported to display a dental phenotype. Knockout-mice with a total loss of function of Cacna1s suffered from myopathy mimicking the phenotype observed in Human with an equivalent CACNA1S loss of function mutation (Filipova et al., 2018). Mice were severely affected and died by asphyxia at birth but were a good model to study myopathy during embryonic stages. Mice with a mutation in one of the voltage sensor segment R528H (see Figure 3C) reproduced the phenotype observed in the Human hypokalemic periodic paralysis and suffered from hindlimb weakness (Wu et al., 2012). Those mouse models confirm that the effects of the mutations (gain or loss of function) are linked to their nature and position in Cacna1s. Another mouse model with a mutation in the third pore-forming domain (Figure 3C, third intramembrane domain) was also engineered and suffered from fatty acid metabolism alteration (Georgiou et al., 2015). This mutation impacted the channel selectivity and abolished Ca2+ binding within Cacna1s. Those mice were not investigated for tooth abnormalities so we could not exclude that they could be affected. However, we suggest that the mutation in our patients, localized in the first pore-forming domain, changes the selectivity or the gating of the channel rather than totally abolishing the calcium binding and that this different mechanism results in a different pathophysiology and phenotype.
Root Patterning
CACNA1S mutation also altered tooth root patterning. Tooth crown and root may have different control mechanisms. The formation of the roots and their branching relies on Hertwig’s epithelial root sheath (HERS), which promotes root initiation and elongation through many signaling pathways including BMP (Wang and Feng, 2017). Bmp2-cKOSp7-Cre-EGFP mouse have short roots (Rakian et al., 2013). However, the mechanism leading to the formation of a single root, or 2 to 3 roots, is unknown. Taurodontism may appear as a failure of early tooth root division leading to an extended pulp chamber along the tooth main axis. This phenotype was recently associated with Wnt10a downregulation (Yang et al., 2015). Elevated extracellular calcium was shown to increase BMP2 via L-type Ca2+ channel and ERK pathway in human dental pulp cells (Tada et al., 2010). This mechanism may account for the root patterning defect encountered in patient presenting with CACNA1S mutation.
This is a first report of the possible involvement of a voltage L-type Ca2+ channel, CACNA1S, in tooth crown and root patterning, as discovered by next generation sequencing analysis of families presenting striking multicusp and single root defects. This suggests CACNA1S alterations may produce major dysfunctions in signaling centers shaping odontogenesis.
Author Contributions
PP and SM collected the salivary samples and detailed the patients’ phenotype. VL-H, CS, VG, JM, AB, and J-FD identified the molecular basis of the disease through NGS assays. VL-H, SM, PP, KN, VG, WP, KC, HD, and AB-Z analyzed the data and wrote the manuscript. PP and AB-Z designed the study and were involved from conception, funding seeking to drafting, and critical review of the manuscript. All authors therefore contributed to conception, design, data acquisition, analysis and interpretation, drafted, and critically revised the manuscript. All authors gave final approval and agreed to be accountable for all aspects of the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to the families for their participation and invaluable contribution. We are grateful to IGBMC GenomEast platform (Christelle Thibault-Carpentier, Bernard Jost, Stéphanie Le Gras) for their support. The computing resources for this work were provided by the BICS and BISTRO bioinformatics platforms in Strasbourg. We would like to thank Khon Kaen University Faculty of Dentistry, who inspired this project and supported this long-lasting collaboration (PHC Hubert Curien Siam 20634-RG 2009–2010). We would like to thank also Dr. M. K. Prasad for her help during the project as well as Professor Ophir Klein for critical reading of the manuscript.
Funding. This work was financed by grants from: La Fondation Maladies Rares (http://fondation-maladiesrares.org) for the project “Morphodent: Identification of a gene involved in the enamel knot signaling center and dental cusps morphogenesis and anomalies” 2013; the University of Strasbourg; the Hôpitaux Universitaires de Strasbourg (API, 2009–2012, “Development of the oral cavity: from gene to clinical phenotype in Human”); the EU-funded project (ERDF) A27 “Oro-dental manifestations of rare diseases”, supported by the RMT-TMO Offensive Sciences initiative, INTERREG IV Upper Rhine program, and the INTERREG V RARENET program. This study was also supported by the grant ANR-10-LABX-0030-INRT, a French State fund managed by the Agence Nationale de la Recherche under the frame programme Investissements d’Avenir labelled ANR-10-IDEX-0002-02. AB-Z is a fellow of University of Strasbourg Institute for Advanced Study (USIAS, 2015). SM received a Junior Research Fellowship (Ambassade de France en Thaïlande, Service de Coopération et d’Action Culturelle, 2018), a Franco-Thai fellowship (Bourse doctorale Franco-Thaï, Ministère des Affaires Etrangères, France) in co-partnership with Khon Kaen University (2014, 2015–2017) for Master and Ph.D.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2018.01329/full#supplementary-material
Tooth cusp patterning defects found in affected families. The intraoral photographs and panoramic radiograph of affected daughter from family 1 (1_III.11) showed tooth morphology alteration especially multiple accessory cusps in both premolars (A–D) or upper (C) and lower (D) molars. Panoramic radiograph (B) showed root branching anomalies, with single roots present for all teeth except the first lower permanent molars presenting with a taurodontism-like phenotype (B). Lateral upper incisors (A,C) and multicusp premolars were microdont (C). In the lower arch a similar cusp pattern alteration was visible but each lower premolar showed a prominent single protruding cusp (D). The affected son from family 2 (2_III.7) showed the same tooth dysmorphology phenotype (E,F). The panoramic radiograph from this person showed the similar root alterations (G), the blue arrow pointed toward taurodontic lower right first permanent molar (46). The contralateral tooth (36) had the same taurodontic dysmorphic appearance. In addition, two premolars were missing in both the maxilla and the mandible (red ∗). Tooth morphology defects and single root pattern were also observed on the panoramic radiograph from the female (3_II.2) from family 3 (H). A supernumerary lower incisor was present (H). Family 4 affected son (4_II.3) showed the most severe phenotype (I–L). The occlusal ridge of the lower incisors exhibited “mamelons” or indentations. Both upper and lower canines showed multiple cusps instead of their tapered shape (K,L). Multiple cusps interfered with the occlusion (I).
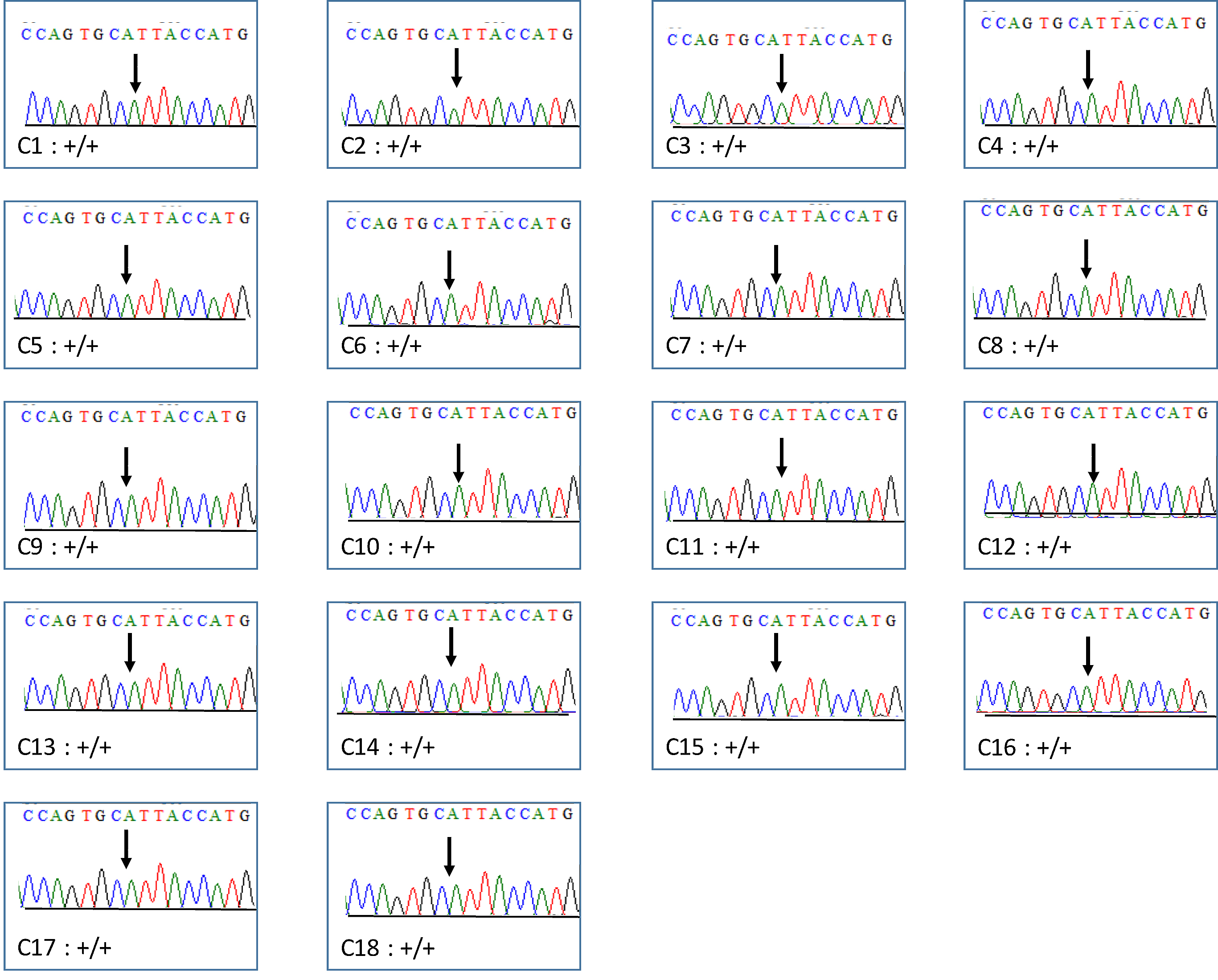
Sanger sequencing of CACNA1S exon 6 from 18 unaffected and unrelated Thai-patient controls originating from the same region as our index cases. None of the control samples presented the mutation, excluding a SNPs carried by individuals from this region.
{kind=link}
Human permanent teeth nomenclature. Numerotation designing human permanent teeth according to their type and location following the FDI two digits (No Authors, 1988) and ISO 3950:2016 (Dentistry — Designation system for teeth and areas of the oral cavity) recommendations. Ca, Canines; PreM, Premolars. The image is extracted from the D[4]/phenodent Diagnosing Dental Defect Database (www.phenodent.org).
{kind=link}
CACNA1S mutations and associated pathologies reported in the literature. The 43 CACNA1S known mutations according to HGMD are shown in this table.
Homozygosity regions detected in families 1–3. Homozygosity regions for the nine individuals analyzed by whole exome sequencing (1_II.11, 1_III.10, 2_II.5, 2_II.6, 2_III.6, 2_III.7, 3_II.1, 3_II.2) were reported in this table. No homozygosity region was common to all affected individuals (1_III.10, 2_II.5, 2_III.6, 2_III.7, 3_II.1, 3_II.2) suggesting that the phenotype did not arise because of shared homozygous mutation.
Estimation of family relationship inference for family 1, 2, and 3 by KING program. N_SNP: The number of SNPs that do not have missing genotypes in either of the individual, HetHet: Proportion of SNPs with double heterozygotes (e.g., AG and AG), IBS0: Proportion of SNPs with zero IBS (identical-by-state) (e.g., AA and GG), Kinship: Estimated kinship coefficient from the SNP data.
Common rare variants shared by affected individuals in the CACNA1S region. Both alleles of each individual are represented, the blue region on one allele of each affected individual corresponds to the region with common rare variants shared by all affected individuals. Affected individuals are in bold. The variant corresponding to the CACNA1S mutation is in red.
Variant pathogenicity predictions in family 1. This table summarizes the scores of the bioinformatics algorithms used to predict the pathogenicity of variants. The columns were annotated using the Varank software (Geoffroy et al., 2015).
Variant pathogenicity predictions in family 2. This table summarizes the scores of the bioinformatics algorithms used to predict the pathogenicity of variants. The columns were annotated using the Varank software (Geoffroy et al., 2015).
Variant pathogenicity predictions in family 3. This table summarizes the scores of the bioinformatics algorithms used to predict the pathogenicity of variants. The columns were annotated using the Varank software (Geoffroy et al., 2015).
References
- Adzhubei I. A., Schmidt S., Peshkin L., Ramensky V. E., Gerasimova A., Bork P., et al. (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7 248–249. 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahtiainen L., Uski I., Thesleff I., Mikkola M. L. (2016). Early epithelial signaling center governs tooth budding morphogenesis. J. Cell Biol. 214 753–767. 10.1083/jcb.201512074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ather A., Ather H., Acharya S. R., Radhakrishnan R. A. (2013). Lobodontia: the unravelling of the wolf teeth. Rom. J. Morphol. Embryol. 54 215–217. [PubMed] [Google Scholar]
- Backenroth D., Homsy J., Murillo L. R., Glessner J., Lin E., Brueckner M., et al. (2014). CANOES: detecting rare copy number variants from whole exome sequencing data. Nucleic Acids Res. 42:e97. 10.1093/nar/gku345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balic A., Thesleff I. (2015). Tissue interactions regulating tooth development and renewal. Curr. Top. Dev. Biol. 115 157–186. 10.1016/bs.ctdb.2015.07.006 [DOI] [PubMed] [Google Scholar]
- Bates E. (2015). Ion channels in development and cancer. Annu. Rev. Cell Dev. Biol. 31 231–247. 10.1146/annurev-cellbio-100814-125338 [DOI] [PubMed] [Google Scholar]
- Bei M. (2009). Molecular genetics of tooth development. Curr. Opin. Genet. Dev. 19 504–510. 10.1016/j.gde.2009.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brook A. H., Winder M. (1979). Lobodontia–a rare inherited dental anomaly. Report of an affected family. Br. Dent. J. 147 213–215. 10.1038/sj.bdj.4804326 [DOI] [PubMed] [Google Scholar]
- Charles C., Lazzari V., Tafforeau P., Schimmang T., Tekin M., Klein O., et al. (2009a). Modulation of Fgf3 dosage in mouse and men mirrors evolution of mammalian dentition. Proc. Natl. Acad. Sci. U.S.A. 106 22364–22368. 10.1073/pnas.0910086106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles C., Pantalacci S., Tafforeau P., Headon D., Laudet V., Viriot L. (2009b). Distinct impacts of Eda and Edar loss of function on the mouse dentition. PLoS One 4:e4985. 10.1371/journal.pone.0004985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahal G. R., Rawson J., Gassaway B., Kwok B., Tong Y., Ptacek L. J., et al. (2012). An inwardly rectifying K+ channel is required for patterning. Development 139 3653–3664. 10.1242/dev.078592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan X. (2013). ion channels, channelopathies, and tooth formation. J. Dent. Res. 93 117–125. 10.1177/0022034513507066 [DOI] [PubMed] [Google Scholar]
- Eltit J. M., Bannister R. A., Moua O., Altamirano F., Hopkins P. M., Pessah I. N., et al. (2012). Malignant hyperthermia susceptibility arising from altered resting coupling between the skeletal muscle L-type Ca2+ channel and the type 1 ryanodine receptor. Proc. Natl. Acad. Sci. U.S.A. 109 7923–7928. 10.1073/pnas.1119207109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan C., Lehmann-Horn F., Weber M. A., Bednarz M., Groome J. R., Jonsson M. K., et al. (2013). Transient compartment-like syndrome and normokalaemic periodic paralysis due to a Ca(v)1.1 mutation. Brain 136 3775–3786. 10.1093/brain/awt300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemifar G., Hoggart C. J., Paternoster L., Kemp J. P., Prokopenko I., Horikoshi M., et al. (2013). Genome-wide association study of primary tooth eruption identifies pleiotropic loci associated with height and craniofacial distances. Hum. Mol. Genet. 22 3807–3817. 10.1093/hmg/ddt231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fialho D., Griggs R. C., Matthews E. (2018). Periodic paralysis. Handb. Clin. Neurol. 148 505–520. 10.1016/B978-0-444-64076-5.00032-6 [DOI] [PubMed] [Google Scholar]
- Filipova D., Henry M., Rotshteyn T., Brunn A., Carstov M., Deckert M., et al. (2018). Distinct transcriptomic changes in E14.5 mouse skeletal muscle lacking RYR1 or Cav1.1 converge at E18.5. PLoS One 13:e0194428. 10.1371/journal.pone.0194428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiszer D., Shaw M. A., Fisher N. A., Carr I. M., Gupta P. K., Watkins E. J., et al. (2015). Next-generation sequencing of RYR1 and CACNA1S in malignant hyperthermia and exertional heat illness. Anesthesiology 122 1033–1046. 10.1097/ALN.0000000000000610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromer M., Pocklington A. J., Kavanagh D. H., Williams H. J., Dwyer S., Gormley P., et al. (2014). De novo mutations in schizophrenia implicate synaptic networks. Nature 506 179–184. 10.1038/nature12929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geller F., Feenstra B., Zhang H., Shaffer J. R., Hansen T., Esserlind A. L., et al. (2011). Genome-wide association study identifies four loci associated with eruption of permanent teeth. PLoS Genet. 7:e1002275. 10.1371/journal.pgen.1002275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoffroy V., Herenger Y., Kress A., Stoetzel C., Piton A., Dollfus H., et al. (2018). AnnotSV: an integrated tool for structural variations annotation. Bioinformatics 10.1093/bioinformatics/bty304 [Epub ahead ofprint]. [DOI] [PubMed] [Google Scholar]
- Geoffroy V., Pizot C., Redin C., Piton A., Vasli N., Stoetzel C., et al. (2015). VaRank: a simple and powerful tool for ranking genetic variants. PeerJ 3:e796. 10.7717/peerj.796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiou D. K., Dagnino-Acosta A., Lee C. S., Griffin D. M., Wang H., Lagor W. R., et al. (2015). Ca2+ binding/permeation via calcium channel, CaV1.1, regulates the intracellular distribution of the fatty acid transport protein, CD36, and fatty acid metabolism. J. Biol. Chem. 290 23751–23765. 10.1074/jbc.M115.643544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlin R. J. (1998). Otodental syndrome, oculo-facio-cardio-dental (OFCD) syndrome, and lobodontia: dental disorders of interest to the pediatric radiologist. Pediatr. Radiol. 28 802–804. 10.1007/s002470050469 [DOI] [PubMed] [Google Scholar]
- Gorlin R. J., Cohen M. M., Hennekam R. C. M. (2001). Syndromes of the Head and Neck. Oxford: Oxford: University Press. [Google Scholar]
- Halling D. B., Georgiou D. K., Black D. J., Yang G., Fallon J. L., Quiocho F. A., et al. (2009). Determinants in CaV1 channels that regulate the Ca2+ sensitivity of bound calmodulin. J. Biol. Chem. 284 20041–20051. 10.1074/jbc.M109.013326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harjunmaa E., Kallonen A., Voutilainen M., Hamalainen K., Mikkola M. L., Jernvall J. (2012). On the difficulty of increasing dental complexity. Nature 483 324–327. 10.1038/nature10876 [DOI] [PubMed] [Google Scholar]
- Hennekam R., Krantz I., Allanson J. (2010). Gorlin’s Syndromes of the Head and Neck. Oxford: Oxford University Press. [Google Scholar]
- Hillson S., Grigson C., Bond S. (1998). Dental defects of congenital syphilis. Am. J. Phys. Anthropol. 107 25–40. 10.1002/(SICI)1096-8644(199809)107:1<25::AID-AJPA3>3.0.CO;2-C [DOI] [PubMed] [Google Scholar]
- Hunter J. P., Guatelli-Steinberg D., Weston T. C., Durner R., Betsinger T. K. (2010). Model of tooth morphogenesis predicts carabelli cusp expression, size, and symmetry in humans. PLoS One 5:e11844. 10.1371/journal.pone.0011844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman W. R., Davies S. H., Lyons D. B., Stauder C. K., Denton-Schneider B. R., Jowdry A., et al. (2013). Manipulation of Fgf and Bmp signaling in teleost fishes suggests potential pathways for the evolutionary origin of multicuspid teeth. Evol. Dev. 15 107–118. 10.1111/ede.12021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jernvall J. (2000). Linking development with generation of novelty in mammalian teeth. Proc. Natl. Acad. Sci. U.S.A. 97 2641–2645. 10.1073/pnas.050586297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jernvall J., Aberg T., Kettunen P., Keranen S., Thesleff I. (1998). The life history of an embryonic signaling center: BMP-4 induces p21 and is associated with apoptosis in the mouse tooth enamel knot. Development 125 161–169. [DOI] [PubMed] [Google Scholar]
- Jernvall J., Keranen S. V., Thesleff I. (2000). From the cover: evolutionary modification of development in mammalian teeth: quantifying gene expression patterns and topography. Proc. Natl. Acad. Sci. U.S.A. 97 14444–14448. 10.1073/pnas.97.26.14444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jernvall J., Kettunen P., Karavanova I., Martin L. B., Thesleff I. (1994). Evidence for the role of the enamel knot as a control center in mammalian tooth cusp formation: non-dividing cells express growth stimulating Fgf-4 gene. Int. J. Dev. Biol. 38 463–469. [PubMed] [Google Scholar]
- Jernvall J., Thesleff I. (2012). Tooth shape formation and tooth renewal: evolving with the same signals. Development 139 3487–3497. 10.1242/dev.085084 [DOI] [PubMed] [Google Scholar]
- Jonsson L., Magnusson T. E., Thordarson A., Jonsson T., Geller F., Feenstra B., et al. (2018). Rare and common variants conferring risk of tooth agenesis. J. Dent. Res. 97 515–522. 10.1177/0022034517750109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski K. J., Weisburd B., Thomas B., Solomonson M., Ruderfer D. M., Kavanagh D., et al. (2017). The ExAC browser: displaying reference data information from over 60 000 exomes. Nucleic Acids Res. 45 D840–D845. 10.1093/nar/gkw971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyan A., Allen C., Damm D., Trotter L. (2013). Lobodontia: report of a family with a rare inherited dental anomaly. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 116 e508–e509. 10.1016/j.oooo.2013.09.075 290365 [DOI] [Google Scholar]
- Kung A. W. (2006). Clinical review: thyrotoxic periodic paralysis: a diagnostic challenge. J. Clin. Endocrinol. Metab. 91 2490–2495. 10.1210/jc.2006-0356 [DOI] [PubMed] [Google Scholar]
- Laugel-Haushalter V., Paschaki M., Thibault-Carpentier C., Dembele D., Dolle P., Bloch-Zupan A. (2013). Molars and incisors: show your MICROARRAY IDs. BMC Res. Notes 6:113. 10.1186/1756-0500-6-113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M., Karczewski K. J., Minikel E. V., Samocha K. E., Banks E., Fennell T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manichaikul A., Mychaleckyj J. C., Rich S. S., Daly K., Sale M., Chen W. M. (2010). Robust relationship inference in genome-wide association studies. Bioinformatics 26 2867–2873. 10.1093/bioinformatics/btq559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matalova E., Antonarakis G. S., Sharpe P. T., Tucker A. S. (2005). Cell lineage of primary and secondary enamel knots. Dev. Dyn. 233 754–759. 10.1002/dvdy.20396 [DOI] [PubMed] [Google Scholar]
- Nelson P., Ngoc Tran T. D., Zhang H., Zolochevska O., Figueiredo M., Feng J. M., et al. (2013). Transient receptor potential melastatin 4 channel controls calcium signals and dental follicle stem cell differentiation. Stem Cells 31 167–177. 10.1002/stem.1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngamphiw C., Assawamakin A., Xu S., Shaw P. J., Yang J. O., Ghang H., et al. (2011). PanSNPdb: the Pan-Asian SNP genotyping database. PLoS One 6:e21451. 10.1371/journal.pone.0021451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissanka-Jayasuriya E. H., Odell E. W., Phillips C. (2016). Dental Stigmata of congenital syphilis: a historic review with present day relevance. Head Neck Pathol. 10 327–331. 10.1007/s12105-016-0703-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- No Authors (1988). FDI Director calls on more countries to adopt the FDI two-digit tooth-numbering system. J. Ir. Dent. Assoc. 34 49. [PubMed] [Google Scholar]
- Ohazama A., Blackburn J., Porntaveetus T., Ota M. S., Choi H. Y., Johnson E. B., et al. (2010). A role for suppressed incisor cuspal morphogenesis in the evolution of mammalian heterodont dentition. Proc. Natl. Acad. Sci. U.S.A. 107 92–97. 10.1073/pnas.0907236107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punta M., Coggill P. C., Eberhardt R. Y., Mistry J., Tate J., Boursnell C., et al. (2012). The Pfam protein families database. Nucleic Acids Res. 40 D290–D301. 10.1093/nar/gkr1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S. M., Moran J. L., Fromer M., Ruderfer D., Solovieff N., Roussos P., et al. (2014). A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506 185–190. 10.1038/nature12975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakian A., Yang W. C., Gluhak-Heinrich J., Cui Y., Harris M. A., Villarreal D., et al. (2013). Bone morphogenetic protein-2 gene controls tooth root development in coordination with formation of the periodontium. Int. J. Oral Sci. 5 75–84. 10.1038/ijos.2013.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran K. V., Hennessey J. A., Barnett A. S., Yin X., Stadt H. A., Foster E., et al. (2013). Calcium influx through L-type CaV1.2 Ca2+ channels regulates mandibular development. J. Clin. Invest. 123 1638–1646. 10.1172/JCI66903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese M. G., Eeckman F. H., Kulp D., Haussler D. (1997). Improved splice site detection in Genie. J. Comput. Biol. 4 311–323. 10.1089/cmb.1997.4.311 [DOI] [PubMed] [Google Scholar]
- Schartner V., Romero N. B., Donkervoort S., Treves S., Munot P., Pierson T. M., et al. (2017). Dihydropyridine receptor (DHPR, CACNA1S) congenital myopathy. Acta Neuropathol. 133 517–533. 10.1007/s00401-016-1656-8 [DOI] [PubMed] [Google Scholar]
- Schwarz J. M., Cooper D. N., Schuelke M., Seelow D. (2014). MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods 11 361–362. 10.1038/nmeth.2890 [DOI] [PubMed] [Google Scholar]
- Shapiro M. B., Senapathy P. (1987). RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res. 15 7155–7174. 10.1093/nar/15.17.7155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry S. T., Ward M. H., Kholodov M., Baker J., Phan L., Smigielski E. M., et al. (2001). dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29 308–311. 10.1093/nar/29.1.308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skrinjaric T., Gorseta K., Skrinjaric I. (2016). Lobodontia: genetic entity with specific pattern of dental dysmorphology. Ann. Anat. 203 100–107. 10.1016/j.aanat.2015.04.007 [DOI] [PubMed] [Google Scholar]
- Splawski I., Timothy K. W., Sharpe L. M., Decher N., Kumar P., Bloise R., et al. (2004). Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119 19–31. 10.1016/j.cell.2004.09.011 [DOI] [PubMed] [Google Scholar]
- Stenson P. D., Mort M., Ball E. V., Howells K., Phillips A. D., Thomas N. S., et al. (2009). The human gene mutation database: 2008 update. Genome Med. 1:13. 10.1186/gm13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada H., Nemoto E., Kanaya S., Hamaji N., Sato H., Shimauchi H. (2010). Elevated extracellular calcium increases expression of bone morphogenetic protein-2 gene via a calcium channel and ERK pathway in human dental pulp cells. Biochem. Biophys. Res. Commun. 394 1093–1097. 10.1016/j.bbrc.2010.03.135 [DOI] [PubMed] [Google Scholar]
- Thesleff I., Jernvall J. (1997). The enamel knot: a putative signaling center regulating tooth development. Cold Spring Harb. Symp. Quant. Biol. 62 257–267. 10.1101/SQB.1997.062.01.032 [DOI] [PubMed] [Google Scholar]
- Thesleff I., Keranen S., Jernvall J. (2001). Enamel knots as signaling centers linking tooth morphogenesis and odontoblast differentiation. Adv. Dent. Res. 15 14–18. 10.1177/08959374010150010401 [DOI] [PubMed] [Google Scholar]
- Thesleff I., Mikkola M. L. (2014). Development of ectodermal organs. Semin. Cell Dev. Biol. 2 1–2. 10.1016/j.semcdb.2014.02.002 [DOI] [PubMed] [Google Scholar]
- Vaahtokari A., Aberg T., Jernvall J., Keranen S., Thesleff I. (1996). The enamel knot as a signaling center in the developing mouse tooth. Mech. Dev. 54 39–43. 10.1016/0925-4773(95)00459-9 [DOI] [PubMed] [Google Scholar]
- Vivante A., Ityel H., Pode-Shakked B., Chen J., Shril S., Van Der Ven A. T., et al. (2017). Exome sequencing in Jewish and Arab patients with rhabdomyolysis reveals single-gene etiology in 43% of cases. Pediatr. Nephrol. 32 2273–2282. 10.1007/s00467-017-3755-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Feng J. Q. (2017). Signaling pathways critical for tooth root formation. J. Dent. Res. 96 1221–1228. 10.1177/0022034517717478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong King Yuen S. M., Campiglio M., Tung C. C., Flucher B. E., Van Petegem F. (2017). Structural insights into binding of STAC proteins to voltage-gated calcium channels. Proc. Natl. Acad. Sci. U.S.A. 114 E9520–E9528. 10.1073/pnas.1708852114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F., Mi W., Hernandez-Ochoa E. O., Burns D. K., Fu Y., Gray H. F., et al. (2012). A calcium channel mutant mouse model of hypokalemic periodic paralysis. J. Clin. Invest. 122 4580–4591. 10.1172/JCI66091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Wang S. K., Choi M., Reid B. M., Hu Y., Lee Y. L., et al. (2015). Taurodontism, variations in tooth number, and misshapened crowns in Wnt10a null mice and human kindreds. Mol. Genet. Genomic Med. 3 40–58. 10.1002/mgg3.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G., Burge C. B. (2004). Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 11 377–394. 10.1089/1066527041410418 [DOI] [PubMed] [Google Scholar]
- Yoon G., Oberoi S., Tristani-Firouzi M., Etheridge S. P., Quitania L., Kramer J. H., et al. (2006). Andersen-Tawil syndrome: prospective cohort analysis and expansion of the phenotype. Am. J. Med. Genet. A 140 312–321. 10.1002/ajmg.a.31092 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tooth cusp patterning defects found in affected families. The intraoral photographs and panoramic radiograph of affected daughter from family 1 (1_III.11) showed tooth morphology alteration especially multiple accessory cusps in both premolars (A–D) or upper (C) and lower (D) molars. Panoramic radiograph (B) showed root branching anomalies, with single roots present for all teeth except the first lower permanent molars presenting with a taurodontism-like phenotype (B). Lateral upper incisors (A,C) and multicusp premolars were microdont (C). In the lower arch a similar cusp pattern alteration was visible but each lower premolar showed a prominent single protruding cusp (D). The affected son from family 2 (2_III.7) showed the same tooth dysmorphology phenotype (E,F). The panoramic radiograph from this person showed the similar root alterations (G), the blue arrow pointed toward taurodontic lower right first permanent molar (46). The contralateral tooth (36) had the same taurodontic dysmorphic appearance. In addition, two premolars were missing in both the maxilla and the mandible (red ∗). Tooth morphology defects and single root pattern were also observed on the panoramic radiograph from the female (3_II.2) from family 3 (H). A supernumerary lower incisor was present (H). Family 4 affected son (4_II.3) showed the most severe phenotype (I–L). The occlusal ridge of the lower incisors exhibited “mamelons” or indentations. Both upper and lower canines showed multiple cusps instead of their tapered shape (K,L). Multiple cusps interfered with the occlusion (I).
Sanger sequencing of CACNA1S exon 6 from 18 unaffected and unrelated Thai-patient controls originating from the same region as our index cases. None of the control samples presented the mutation, excluding a SNPs carried by individuals from this region.
Human permanent teeth nomenclature. Numerotation designing human permanent teeth according to their type and location following the FDI two digits (No Authors, 1988) and ISO 3950:2016 (Dentistry — Designation system for teeth and areas of the oral cavity) recommendations. Ca, Canines; PreM, Premolars. The image is extracted from the D[4]/phenodent Diagnosing Dental Defect Database (www.phenodent.org).
CACNA1S mutations and associated pathologies reported in the literature. The 43 CACNA1S known mutations according to HGMD are shown in this table.
Homozygosity regions detected in families 1–3. Homozygosity regions for the nine individuals analyzed by whole exome sequencing (1_II.11, 1_III.10, 2_II.5, 2_II.6, 2_III.6, 2_III.7, 3_II.1, 3_II.2) were reported in this table. No homozygosity region was common to all affected individuals (1_III.10, 2_II.5, 2_III.6, 2_III.7, 3_II.1, 3_II.2) suggesting that the phenotype did not arise because of shared homozygous mutation.
Estimation of family relationship inference for family 1, 2, and 3 by KING program. N_SNP: The number of SNPs that do not have missing genotypes in either of the individual, HetHet: Proportion of SNPs with double heterozygotes (e.g., AG and AG), IBS0: Proportion of SNPs with zero IBS (identical-by-state) (e.g., AA and GG), Kinship: Estimated kinship coefficient from the SNP data.
Common rare variants shared by affected individuals in the CACNA1S region. Both alleles of each individual are represented, the blue region on one allele of each affected individual corresponds to the region with common rare variants shared by all affected individuals. Affected individuals are in bold. The variant corresponding to the CACNA1S mutation is in red.
Variant pathogenicity predictions in family 1. This table summarizes the scores of the bioinformatics algorithms used to predict the pathogenicity of variants. The columns were annotated using the Varank software (Geoffroy et al., 2015).
Variant pathogenicity predictions in family 2. This table summarizes the scores of the bioinformatics algorithms used to predict the pathogenicity of variants. The columns were annotated using the Varank software (Geoffroy et al., 2015).
Variant pathogenicity predictions in family 3. This table summarizes the scores of the bioinformatics algorithms used to predict the pathogenicity of variants. The columns were annotated using the Varank software (Geoffroy et al., 2015).