

Abstract
Terminal or perisynaptic Schwann cells (TPSCs) are nonmyelinating, perisynaptic glial cells at the neuromuscular junction (NMJ) that respond to neural activity by increasing intracellular calcium (Ca2+) and regulate synaptic function. The onset of activity-induced TPSC Ca2+ responses, as well as whether axonal Schwann cells (ASCs) along the nerve respond to nerve stimulation during development, is unknown. Here, we show that phrenic nerve stimulation in developing male and female mice elicited Ca2+ responses in both ASCs and TPSCs at embryonic day 14. ASC responses were lost in a proximo-distal gradient over time, but could continue to be elicited by bath application of neurotransmitter, suggesting that a loss of release rather than a change in ASC competence accounted for this response gradient. Similar to those of early postnatal TPSCs, developing ASC/TPSC responses were mediated by purinergic P2Y1 receptors. The loss of ASC Ca2+ responses was correlated to the proximo-distal disappearance of synaptophysin immunoreactivity and synaptic vesicles in phrenic axons. Accordingly, developing ASC Ca2+ responses were blocked by botulinum toxin. Interestingly, the loss of ASC Ca2+ responses was also correlated to the proximo-distal development of myelination. Finally, compared with postnatal TPSCs, neonatal TPSCs and ASCs displayed Ca2+ signals in response to lower frequencies and shorter durations of nerve stimulation. Together, these results with GCaMP3-expressing Schwann cells provide ex vivo evidence that both axons and presynaptic terminals initially exhibit activity-induced vesicular release of neurotransmitter, but that the subsequent loss of axonal synaptic vesicles accounts for the postnatal restriction of vesicular release to the NMJ.
SIGNIFICANCE STATEMENT Neural activity regulates multiple aspects of development, including myelination. Whether the excitation of developing neurons in vivo results in the release of neurotransmitter from both axons and presynaptic terminals is unclear. Here, using mice expressing the genetically encoded calcium indicator GCaMP3 in Schwann cells, we show that both terminal/perisynaptic Schwann cells at the diaphragm neuromuscular junction and axonal Schwann cells along the phrenic nerve exhibit activity-induced calcium responses early in development, mediated by the vesicular release of ATP from the axons of motor neurons acting on P2Y1 receptors. These ex vivo findings corroborate classic in vitro studies demonstrating transmitter release by developing axons, and thus represent a tool to study the mechanisms and significance of this process during embryonic development.
Keywords: activity dependent, axo-glial, axonal, calcium imaging, myelination, neuromuscular
Introduction
In addition to providing structural and metabolic support to neurons, insulating axons, and mounting responses to injury, infection, or disease, glial cells dynamically regulate neuronal signaling, activity, and behavior (Araque et al., 2001). For example, optogenetic activation of astrocytes, a perisynaptic glial cell of the CNS, modulates sensory information processing, motor behavior, and sleep (Sasaki et al., 2012; Perea et al., 2014; Yamashita et al., 2014; Pelluru et al., 2016). Similarly, the pharmacogenetic activation of perisynaptic enteric glia is sufficient to excite smooth muscle of the ileum and colon (McClain et al., 2015). Recently, we showed that the activation of terminal/perisynaptic Schwann cells (TPSCs) of the postnatal neuromuscular junction (NMJ) regulates muscle fatigue (Heredia et al., 2018). These in vivo studies demonstrate that perisynaptic glia in multiple regions are important mediators of physiological function.
The activation of perisynaptic glia by neural activity represents an initial step in these examples of neuron–glia communication. The most well characterized component of this response is an increase of intracellular calcium (Ca2+) that occurs within these cells as a result of neurotransmitter release (Rousse and Robitaille, 2006; Scemes and Giaume, 2006). At the adult NMJ of the mouse and frog, activity-induced Ca2+ responses are mediated by multiple nerve-derived substances, including acetylcholine (ACh), purine adenine nucleotides, and adenosine (Darabid et al., 2014). In contrast to these adult responses, activity-induced Ca2+ responses of early postnatal TPSCs are mediated exclusively by purinergic signals (Darabid et al., 2013; Heredia et al., 2018). When TPSCs first become competent to respond to neural activity and whether developing axonal SCs (ASCs) along peripheral nerves respond to activity (Lev-Ram and Ellisman, 1995) are unknown. Because the absence of Schwann cells during embryogenesis results in the degeneration of spinal motor neurons (Woldeyesus et al., 1999; Lin et al., 2000; Wolpowitz et al., 2000), identifying the onset and consequence of Schwann cell responses to nerve stimulation is functionally significant. For example, the survival of embryonic motor neurons may be uniquely dependent on the pathways initiated within Schwann cells downstream of activity-induced Ca2+ signaling.
Using mice expressing the genetically encoded calcium indicator GCaMP3 in all Schwann cells (i.e., both ASCs and TPSCs), we examined activity-induced Ca2+ responses in SCs of the phrenic nerve and diaphragm at various stages of embryonic and postnatal development. We found that ASCs along the secondary phrenic intramuscular branches, which innervate the costal diaphragm and contain predominantly motor axons (Duron et al., 1978), as well as TPSCs at the NMJ, exhibited P2Y1R-sensitive Ca2+ responses to nerve stimulation at embryonic and neonatal stages. The response of ASCs to nerve stimulation was lost in a proximo-distal gradient over time, a phenomenon that corresponded to a reduction of synaptic vesicles (SVs) along developing axons. We also characterized the spatiotemporal and pharmacological features of activity-induced Ca2+ responses in neonatal ASCs and TPSCs. These results demonstrate the utility of genetic techniques to investigate the response features of Schwann cell populations and reveal that the presence of synaptic vesicles and accordingly the release of neurotransmitters is spatially regulated during development.
Materials and Methods
Ethical approval and use of mice.
P2ry1 mutant, GCaMP3 conditional knock-in, and Wnt1-Cre transgenic mice were all purchased from The Jackson Laboratory and maintained in the C57BL/6 strain. We refer to conditional GCaMP3-expressing mice produced from specific Cre-driver lines as Cre driver-GCaMP3, rather than Cre driver; conditional GCaMP3 (e.g., Wnt1-GCaMP3). We found no differences among male and female Wnt1-GCaMP3, P2ry1+/+, or P2ry1−/− mice, so we pooled these samples. For embryonic day 14 mice (E14), males and females were paired for several hours between 3:00 and 6:00 P.M. and pregnant dams were killed at 4:00 P.M. 2 weeks later. For E15.5 mice, males and females were paired for a night and separated the next morning; noon of that day was designated E0.5. Postnatal day 0 (P0) mice were killed within 8 h of their birth. Animal husbandry and experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the Institutional Animal Care and Use Committee at the University of Nevada.
Drugs.
The following reagents were used at the following concentrations: P2Y1R agonist ATP (10 μm; Sigma-Aldrich); P2Y1R antagonist MRS2500 (1 μm; Tocris Bioscience); P1R agonist adenosine (100 μm; Sigma-Aldrich); pan-cholinergic agonist; carbamoylcholine [carbachol (CCh); 10 μm; Sigma-Aldrich]; pan-muscarinic agonist muscarine (10 μm; Sigma-Aldrich); sarcoplasmic/endoplasmic reticulum Ca2+-ATPase inhibitor carbenoxolone (CBX; 100 μm; Cayman Chemical); meclofenamic acid (100 μm; Cayman Chemical); 18-α-glycyrrhetinic acid (10 μm; Sigma-Aldrich); 18-β-glycyrrhetinic acid (10 μm; Cayman Chemical); cyclopiazonic acid (CPA; 10 μm; Sigma-Aldrich); botulinum toxin E (BoNT/E; 10–100 nm; METAbiologics); potassium chloride (2–10 mm; Sigma-Aldrich); skeletal muscle myosin-blocker 3-(N-butylethanimidoyl)-4-hydroxy-2H-chromen-2-one (BHC; 100 μm; Hit2Lead); and 488-, 594-, and 633-conjugated α-bungarotoxin (α-BTX; 1 μg/ml; Biotium).
Calcium imaging.
Diaphragms of Wnt1-GCaMP3 mice were dissected and pinned on a 6 cm Sylgard-coated dish containing oxygenated Krebs'-Ringer's solution at room temperature according to standard procedure. The phrenic nerve was drawn into a suction electrode and stimulated with a suprathreshold square pulse (0.1 ms) once to ensure that the muscle contracted before treatment with 100 μm myosin inhibitor BHC (Heredia et al., 2016) to block movement (Heredia et al., 2016). The diaphragm was illuminated with a Spectra X light engine (Lumencor). To quantify maximal fluorescence exhibited by GCaMP3 in Schwann cells, 30 μm CPA was added to deplete sarcoplasmic reticular Ca2+ stores (Heredia et al., 2016). Image sequences were captured using an Andor Neo sCMOS camera (Oxford Instruments) and a Windows (Microsoft)-based PC using Nikon NIS Elements 4.1. Image sequences were recorded at 25 frames per second, and were exported as 8-bit TIFF files into custom-written software (Volumetry G8d; Hennig et al., 2015). Changes in background fluorescence were stabilized by subtracting the average intensity near the main phrenic nerve branch. An average intensity image was generated before stimulus application (prestim; ∼1 s) to quantify basal Ca2+ levels in Schwann cells. These images are presented using a blue → green color lookup table (CLUT). SD maps were calculated in similar fashion to the average intensity image, except the SD of 16-bit fluorescence intensity units (SD iu16) at every pixel before the application of the stimulus (0.5–1.0 s) extending to 60 s was calculated (Heredia et al., 2018). SD maps were color coded using a “fire” CLUT. Traces of fluorescence intensity were generated from movies and presented as changes in fluorescence with respect to initial fluorescence (ΔF/Favg(prestim) or ΔF/F0). Peak intensities of Ca2+ transients were calculated as a signal-to-noise ratio (SNR) in which peak SD values were divided by prestim SD value. The log10 of this ratio was generated and multiplied by 20 to standardize the SNR as decibels.
Electrophysiology.
Phrenic nerve-evoked muscle action potentials and endplate potentials (EPPs) were recorded from P1 diaphragm muscle after treatment with the striated muscle myosin inhibitor BHC (100 μm; Heredia et al., 2016) or the Nav1.4 antagonist μ-conotoxin GIIIb (2.3 μm), respectively. Signals were amplified, digitized, recorded, and analyzed as described previously (Heredia et al., 2016). Only muscle fibers with resting membrane potentials between −60 and −75 mV were included for analysis.
Fatigue.
Video recording of muscle shortening in hemidiaphragms (E14, P0) was performed as described previously (Heredia et al., 2016). The onset of fatigue was denoted as the onset of the decline of peak muscle shortening.
Immunohistochemistry.
Antibodies against GFP (Rockland) and synaptophysin (Santa Cruz Biotechnology) were used at 1/1000 in PBS containing 1% Triton X-100 and 10% fetal bovine serum to detect proteins in fixed, whole-mount diaphragms dissected from different aged wild-type or Wnt1-GCaMP3 mice. Fluorescently conjugated α-BTX was added with secondary antibodies. Tissues were confocally imaged with an Olympus Fluoview 1000.
Electron microscopy.
The phrenic nerve and costal diaphragm were dissected from P0, P1, and P7 mice, incubated in a fixative of 1.5% glutaraldehyde and 2% paraformaldehyde in 0.1 m sodium cacodylate at 4°C overnight, and then rinsed in 0.1 m sodium cacodylate for several hours at 4°C. Samples were postfixed in 2% osmium tetroxide, dehydrated, incubated in propylene oxide, embedded in Spurr's resin and polymerized at 60°C overnight. Ultrathin sections were cut at 90 μm and stained with uranyl acetate followed by lead citrate. Sections were photographed or digitized using a Philips CM10 Transmission Electron Microscope equipped with a Gatan BioScan Digital Imaging System. Axonal area and diameter, myelin thickness, and g-ratios were measured on images captured at 4600×, and lamellae were counted in images taken at 64,000×.
Statistics.
Power analyses were performed using G*power of 3.010 to determine the numbers of samples required. Differences between vehicle- and drug-treated diaphragms were assessed by unpaired two-tailed Student's t tests between two independent means, assuming equal variance. A p value of <0.05 was considered significant.
Results
We first assessed the onset of activity-induced Ca2+ responses in Schwann cells of the diaphragm of Wnt1-GCaMP3 mice. E14 is the first age at which we could reliably isolate and stimulate the phrenic nerve with a suction electrode and obtain a contraction of the diaphragm. At this age, 30 s of nerve stimulation at frequencies of ≥10 Hz induce muscle contraction, indicated by muscle fiber shortening, followed by fatigue, indicated by failure to maintain peak shortening (average fatigue onset, 19.3 ± 1.5 vs 6.2 ± 2.3 vs 2.3 ± 1.3 s, for 10 vs 20 vs 40 Hz; n = 3). Because 30 s of 40 Hz stimulation induced a quick and robust fatigue, we used these parameters to investigate Ca2+ responses in Schwann cells at E14. ASCs along secondary phrenic intramuscular branches, as well as those along smaller tertiary branches and those at nascent NMJs, exhibited a transient increase of cytosolic Ca2+ in response to nerve stimulation. The intensity of these transients was not different between ASCs and TPSCs (peak intensity, 17.5 ± 3.1 vs 17.2 ± 2.7 dB, for E14 ASC vs TPSC; p = 0.83, n = 3, c = 9; Fig. 1A; Video 1), and was also not significantly different from Ca2+ transient intensity in P7 TPSCs (peak intensity, 17.1 + 1.3 dB; n = 3, c = 9). The onset after nerve stimulation and duration of Ca2+ transients were also not different between ASCs and TPSCs (onset, or time from stimulation to 10% of peak Ca2+ response, 3.1 ± 1.7 vs 3.8 ± 1.4 s, for E14 ASC vs TPSC; p = 0.37, n = 3, c = 9; duration, 11.7 ± 1.3 vs 10.5 ± 2.0 s, for E14 ASC vs TPSC; p = 0.37, n = 3, c = 9). In contrast to older ages (e.g., P7; Heredia et al., 2018), however, when all TPSCs and no phrenic nerve branch-associated ASCs displayed activity-induced Ca2+ responses, some but not all Schwann cells in each of these populations exhibited Ca2+ responses at E14 (Video 1). This did not result from a general inability of all of each population to mount Ca2+ responses, as bath application of the ACh mimetic carbachol induced a response in all ASCs and TPSCs (Fig. 1A; Video 2). Nor was it caused by the failure of all phrenic nerve branch-associated Schwann cells to express GCaMP3 (Fig. 1B).
Figure 1.

ASCs and TPSCs both exhibit robust Ca2+ responses to phrenic nerve stimulation (Stim) at E14. A, Left, Spatial map of the SD iu16 values of Ca2+ responses in a population of ASCs and TPSCs in the diaphragm of an E14 Wnt1-GCaMP3 mouse imaged in response to 30 s of 40 Hz nerve stimulation; fire CLUT heatmap in SD iu16 and scale bar in micrometers. Note the responses by some but not all ASCs along the secondary phrenic intramuscular branch (arrow) as well as by some but not all TPSCs (arrowhead). Middle, Spatial fluorescence intensity map (in SD iu16) of the same sample in the left panel after treatment with the pan-cholinergic agonist CCh. In contrast to nerve stimulation, CCh induces Ca2+ responses in all ASCs and TPSCs. Right, An average fluorescence intensity image generated before application of a stimulus (prestim, in black and white) shows the overall structure of GCaMP3-expressing Schwann cell elements, including the distal end of the primary phrenic trunk (1), the secondary phrenic intramuscular branch (2), as well as several color-coded boxes enclosing ASC or TPSC Ca2+ responses that were collected for analysis in the graph to the right. Transient averages of ASC or TPSCs were plotted over time as changes in fluorescence divided by initial fluorescence (ΔF/F0). B, Left, A prestim image in green of the diaphragm of an E14 Wnt1-GCaMP3 mouse. Middle left, Spatial fluorescence intensity map after 30 s of 40 Hz phrenic nerve stimulation. Note the response of ASCs and TPSCs again. Middle, Enlarged area of inset box in first panel imaged after the diaphragm was immunostained with antibodies against green fluorescent protein (GFP; green; to label GCaMP3-positive Schwann cells) and synaptophysin (SYP; red; to label synaptic vesicle-containing nerves and nerve terminals). Middle right, right, Enlarged regions of inset box in second panel showing α-BTX-labeled nicotinic ACh receptor-enriched postsynaptic apparati in blue together with GFP and synaptophysin staining. Note the presence of GFP-immunoreactive Schwann cells near every synaptophysin-labeled presynaptic nerve ending (right, arrow). At this age, not every α-BTX-labeled postsynaptic ACh receptor (AChR) cluster is innervated (arrowhead).
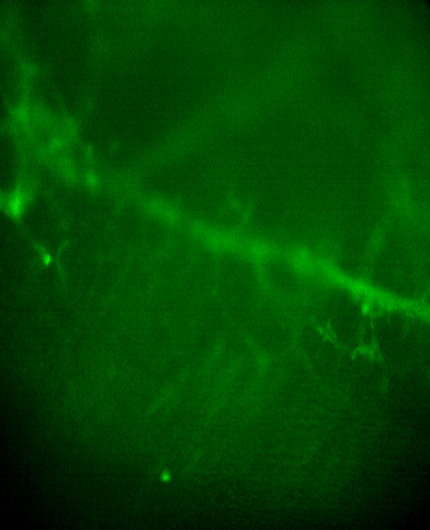
Some but not all Schwann cells along axons (ASCs) and some but not all TPSCs at NMJs exhibit a robust increase in intracellular Ca2+ in response to nerve stimulation at E14.
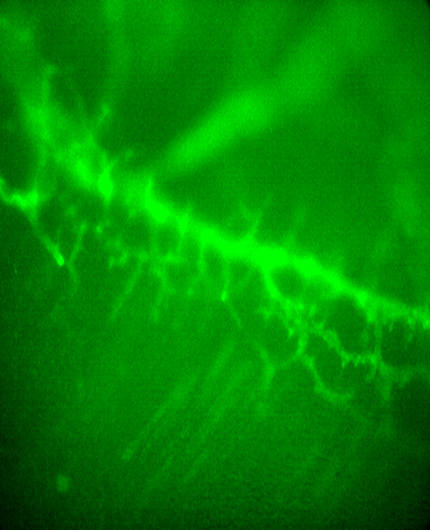
All ASCs and all TPSCs exhibit a Ca2+ response (respond) to the ACh mimetic CCh at E14.
To determine when ASCs stop displaying activity-induced Ca2+ responses, we examined them at several ages between E14 and P15. We found that in contrast to E14, ASCs along the secondary phrenic intramuscular branches no longer mounted significant responses to nerve stimulation at P1 (Fig. 2; Videos 3, 4; Fig. 3-1). However, ASCs along tertiary axon branches and preterminal axons still responded at P1. At P5–P7, TPSCs and preterminal ASCs responded to nerve stimulation (Videos 5, 6), and by P15 only TPSCs displayed activity-induced Ca2+ responses, (Fig. 2; Video 7).
Figure 2.
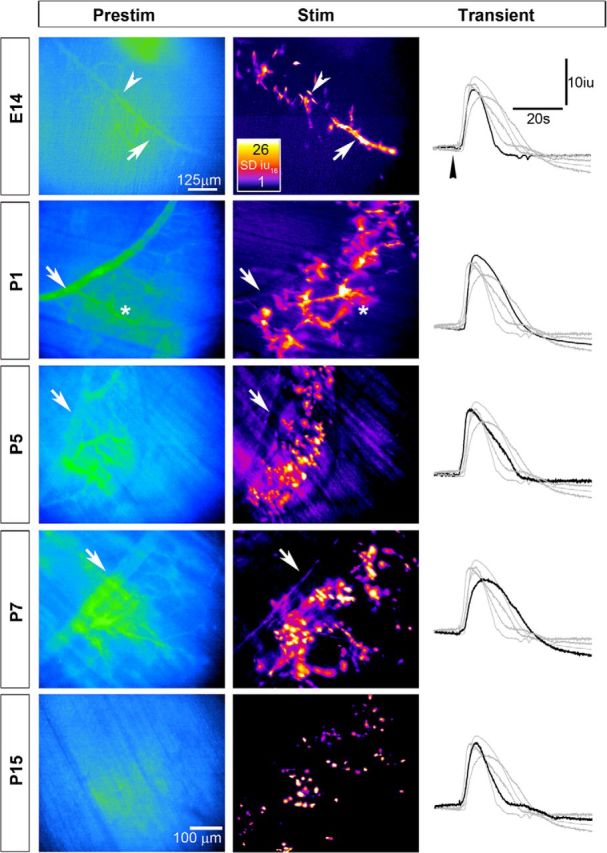
The population of developing Schwann cells exhibiting activity-induced Ca2+ responses becomes restricted over time along a proximo-distal gradient. Average fluorescence intensity images (Prestim, blue and green; left panels), spatial fluorescence intensity maps (in SD iu16; middle panels), and transients (right panels) of Ca2+ responses in a population of Schwann cells in the diaphragm of Wnt1-GCaMP3 mice at different ages in response to 30 s of 20 Hz phrenic nerve stimulation; fire CLUT heatmap in SD iu16 and scalebar in micrometers. Note that in contrast to E14, when both ASCs (arrow) and TPSCs (arrowhead) exhibit responses, similar to Figure 1, at older ages, the populations of responding Schwann cells become restricted to increasingly peripheral populations. For example, at P1, ASCs along secondary phrenic intramuscular branches (arrow) no longer respond to nerve stimulation, whereas ASCs along tertiary branches (asterisk) do. At older ages, these ASCs lose their response to activity, leading to an increasingly circular, punctate pattern of spatial fluorescence maps. Representative transients of stimulation-induced Ca2+ responses in Schwann cells over the course of development are similar in intensity and duration (right panels; arrowhead denotes onset of nerve stimulation, and black trace shows a representative response at the age denoted to the left, while gray traces show representative responses of each of the other ages).
ASCs along secondary phrenic intramuscular branches and tertiary branches, as well as TPSCs at NMJs, respond to nerve stimulation at E14.

ASCs along secondary phrenic intramuscular branches no longer respond to nerve stimulation, whereas ASCs along tertiary branches, as well as TPSCs at NMJs, respond to nerve stimulation at P1.
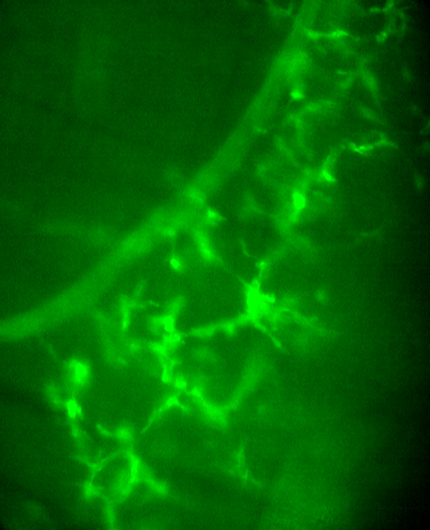
ASCs along secondary phrenic intramuscular and most tertiary branches no longer respond to nerve stimulation, whereas ASCs along preterminal axons, as well as TPSCs at NMJs, respond to nerve stimulation at P5.
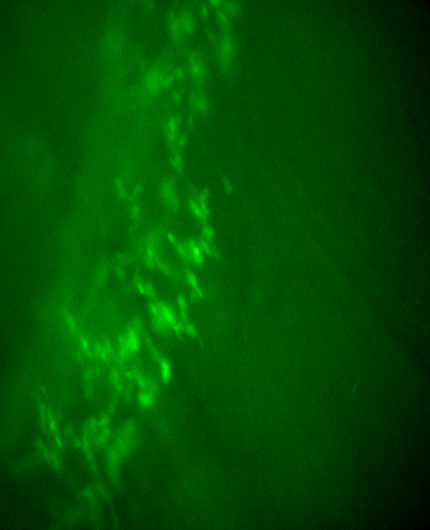
TPSCs at NMJs, but not ASCs, respond to nerve stimulation at P7.
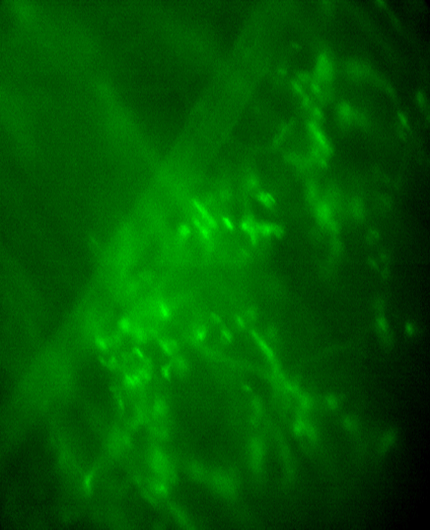
TPSCs at NMJs, but not ASCs, respond to nerve stimulation at P15.
To test whether ASCs fail to exhibit activity-induced Ca2+ responses because they lose competence to respond to nerve stimulation, we evaluated the effects of the bath application of neurotransmitter. In addition to ACh, presynaptic terminals of motor neurons corelease ATP (Silinsky, 1975). Released ATP can activate purinergic receptors (P2Rs) on TPSCs, motor nerve terminals, or muscle cells, or can be converted to other P2R-stimulating derivatives (ADP, AMP) or to adenosine, which can activate adenosinergic receptors (P1Rs) on Schwann cells or presynaptic nerve terminals. We focused on P1, an age at which ASCs of the secondary phrenic intramuscular branches no longer displayed these responses. For these studies, we induced responses with 30 s of 20 Hz nerve stimulation, which is sufficient to trigger Ca2+ responses many times in succession, as long as each episode is separated by a 15 min recovery episode (intensity of eighth vs first nerve stimulation-induced Ca2+ response, 97.3 ± 3%; n = 3, c = 8). In contrast to nerve stimulation, treatment with the long-acting ACh mimetic carbachol or the P2R agonist ATP each induced robust Ca2+ responses in ASCs of these main intramuscular branches supplying the costal diaphragm (Fig. 3). However, the Ca2+ response of ASCs to each of these agents was delayed, compared with that of TPSCs (Fig. 3-1). This was not a result of a delay in diffusion, because it occurred when the agonist was bath applied more closely to the ASCs of these main branches (Fig. 3A, left) or to the TPSCs of the NMJs (Fig. 3A, right; n = 3).
Figure 3.
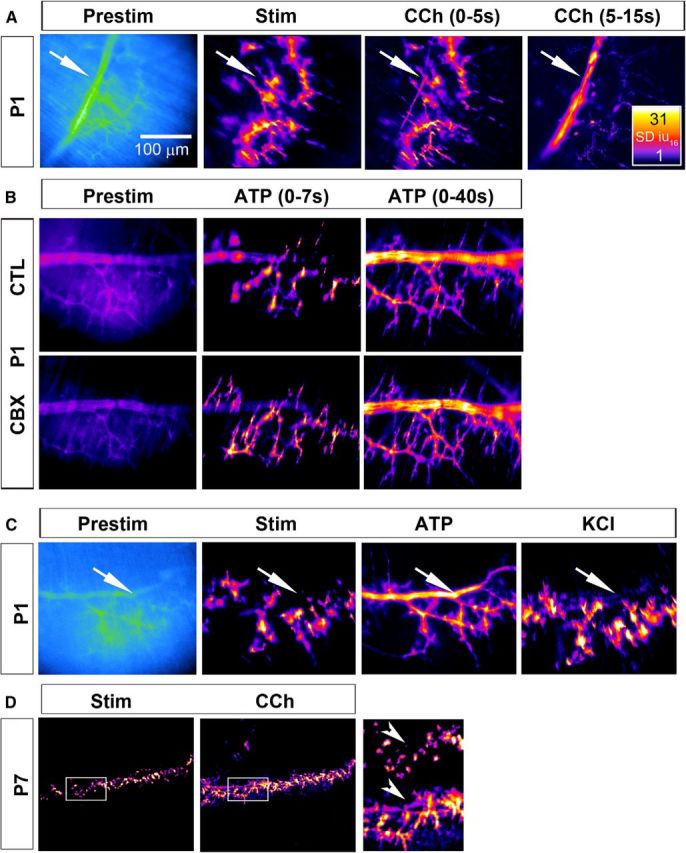
The loss of activity-induced Ca2+ responses in ASCs does not result from a loss of competence to respond to nerve stimulation. A, Left, Average fluorescence intensity image (Prestim, blue and green) of a diaphragm of a P1 Wnt1-GCaMP3 mouse. Arrow marks the secondary phrenic intramuscular branch. Middle left, Spatial intensity map of Ca2+ responses to 30 s of 20 Hz phrenic nerve stimulation; note the presence of TPSC and tertiary ASC responses, but the absence of ASC responses along the secondary branch (arrow). Middle right, right, In contrast, treatment with CCh produces a response in both TPSCs and secondary/tertiary ASCs. The Ca2+ response in ASCs occurs after that of TPSCs (compare signal along secondary branch in right panel, 5–15 s after nerve stimulation onset, with that in middle right panel, 0–5 s after onset) (see Figure 3-1). B, Left panels, Prestimulation images (in black and purple) of the same P1 Wnt1-GCaMP3 diaphragm. Middle, right panels, Spatial intensity map of Ca2+ responses in response to ATP (0–7 and 0–40 s after ATP treatment; note the delayed response, similar to the response to CCh, of secondary branch-associated ASCs, relative to tertiary ASCs and TPSCs). This distal-to-proximal activation pattern of SCs (top row, control; CTL) was not affected by treatment with 100 μm carbenoxolone, an inhibitor of gap junctions (bottom row; CBX). C, Prestim images (in blue and green) of the same P1 Wnt1-GCaMP3 diaphragm after nerve stimulation, ATP or KCl. The robust activation of Ca2+ responses in ASCs after TPSCs (arrow) was not observed after treatment with KCl, in contrast to ATP. D, ASCs still respond to neurotransmitter at P7, despite not being activated by nerve stimulation. In the enlarged images to the right, note the response of ASCs to CCh but not to nerve stimulation (arrowheads) in the same diaphragm.
Spatial identification of Schwann cell Ca2+ responses at P1. Ca2+ transients of spatially identified ASCs and TPSCs in response to nerve stimulation (Stim;left) or carbachol (CCh; right) at P1 as identified by color coding and numbering. Note the reduced heterogeneity in amplitude and shape of Ca2+ transients in response to nerve stimulation at P0 vs. E14. Note also the small response to nerve stimulation in ASCs of the phrenic intramuscular branch (Cells 1-8) vs. that within TPSCs at the NMJ (Cells 9-15). In contrast, ASCs along the phrenic intramuscular branch all exhibit prolonged responses to CCh, although this response is delayed compared to that within TPSCs (bar running through transients is set to peak amplitude of CCh-induced Ca2+ response in TPSCs). Download Figure 1-1, TIF file (3.1MB, tif)
The delayed activation of ASCs by bath-applied agonists suggests the possibility that activated TPSCs release a molecule that in turn activates ASCs. Such a molecule could be secreted, released exocytotically, or passed through gap junctions. Alternatively, the delayed activation could reflect a lower relative sensitivity of ASCs than TPSCs to these agonists. To test whether delayed neurotransmitter-induced Ca2+ responses of ASCs were mediated indirectly by prior activation of TPSCs, we evaluated this response after pretreatment with the gap junction inhibitor CBX. In response to ATP, the activation of TPSCs preceded that of ASCs in a temporally similar manner in the presence or absence of 100 μm CBX, arguing against this idea (Fig. 3B). This disto-proximal pattern of activation of Schwann cell Ca2+ signaling also persisted in the presence of the gap junction inhibitors 18α- or 18β-glycyrrhetinic acid or meclofenamic acid (onset of ATP-induced ASC Ca2+ response after onset of ATP-induced TPSC Ca2+ response, 13.2 ± 5.2 vs 11.8 ± 3.4 vs 14.4 ± 4.3 s, for control vs 18α-glycyrrhetinic acid vs meclofenamic acid, n = 3). These data suggest that the delayed activation of ASCs relative to TPSCs is not mediated by gap junctions between them. Moreover, TPSC Ca2+ responses themselves are not a prerequisite for delayed, robust ASC responses induced by CCh or ATP, because bath application of K+ produced intense TPSC Ca2+ responses but only minor ASC Ca2+ responses in secondary phrenic intramuscular branches (Fig. 3C). Together, these data suggest that the delayed activation of ASCs is not indirectly caused by the prior activation of TPSCs, but cannot rule out the possibility that it results from the indirect activation of other cells such as muscle.
Similar to those at E14 and P1, ASCs at P7 also responded to bath application of neurotransmitter, but not to nerve stimulation (Fig. 3D). Therefore, these studies demonstrate that ASCs initially exhibit robust Ca2+ responses to nerve stimulation, but gradually lose this response in a proximo-distal gradient from E14 to P15. Because bath application of neurotransmitter continues to induce these responses in ASCs, these results suggest that the activity-induced release of neurotransmitter by phrenic axons may be spatially regulated during development.
First, we examined the mechanism by which neurotransmitter-induced Ca2+ responses were transduced in developing ASCs. The robustness of these responses (ΔF/F0 = approximately sixfold to sevenfold; Fig. 1) was similar to those of postnatal TPSCs (Heredia et al., 2018). We found that treatment with MRS2500, the highly specific antagonist to P2Y1R, abolished these robust, activity-induced ASC (and TPSC) Ca2+ responses along secondary phrenic intramuscular branches at E15.5 and smaller tertiary branches at P1; these ASC responses in smaller tertiary branches were also eliminated in P2ry1 mutant mice at P1 (Fig. 4A,B). The absence of these responses was not caused by gross differences in P2ry1 mutant ASCs, which appeared normal ultrastructurally at P0 (Fig. 4C) and at P7 [axonal/fiber diameter ratio (g-ratio), 0.71 ± 0.071 vs 0.73 ± 0.061, for P2ry1 WT vs mutant secondary branch; p = 0.0005, n = 3, c = 9). Together, these results suggest that the developmentally transient, robust Ca2+ response of ASCs to nerve stimulation is completely dependent on P2Y1R expression by ASCs, similar to early postnatal TPSC responses.
Figure 4.
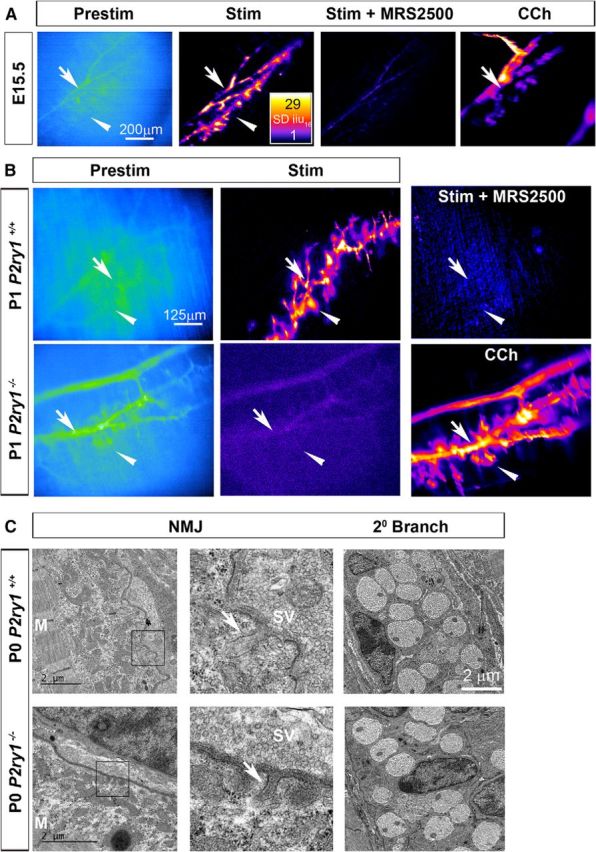
The developmentally transient Ca2+ response of ASCs to nerve stimulation is mediated by purinergic P2Y1 receptors. A, Prestimulation and poststimulation intensity maps, before and after treatment with the P2Y1R antagonist MRS2500, of a diaphragm of an E15.5 Wnt1-GCaMP3 mouse. Note the loss of both ASC (arrow) and TPSC (arrowhead) Ca2+ responses after MRS2500 treatment. Right, Carbachol-induced responses in ASCs and TPSCs are maintained after MRS2500 treatment. B, Left, middle panels, Prestimulation and poststimulation intensity maps of the diaphragm of P1 P2ry1 wild-type (top row) or mutant (bottom row) Wnt1-GCaMP3 mice. Note that activity-induced Ca2+ responses in tertiary ASCs (arrow, top middle) as well as TPSCs (arrowhead) of P2ry1 wild-type mice are absent in P2ry1 mutant mice (bottom middle). Right top, These Ca2+ responses are also eliminated in the diaphragm of P2ry1 wild-type mice after treatment with MRS2500. Bottom right, Carbachol-induced responses in ASCs and TPSCs are maintained in P2ry1 mutant mice. C, Electron micrographs of NMJs (left panels, inset boxes) from P2ry1 wild-type and mutant mice each show similarly nascent junctional folds (arrows). M, Muscle filaments. Right panels show axons and Schwann cells in a secondary phrenic intramuscular branch. Each Schwann cell at P0 is associated with five to eight axons at this promyelinating stage.
The axons of Xenopus spinal neurons cultured together with muscle cells in vitro release neurotransmitter “en passant” as they grow (Hume et al., 1983; Young and Poo, 1983), but whether this occurs in vivo is unknown. Our observations above suggest that ASCs may function as an in situ sensor of neurotransmitter release, similar to the ACh receptor-enriched patch of muscle used in these in vitro studies. To test whether the release of neurotransmitter along developing phrenic axons occurs through vesicular or nonvesicular pathways such as pannexins (Horton et al., 2017) or volume-activated anion channels (Fields and Ni, 2010), we imaged activity-dependent Ca2+ responses in ASCs of embryonic Wnt1-GCaMP3 mice in the presence of BoNT/E, a potent and fast-acting inhibitor of calcium-triggered exocytotic release of synaptic vesicles (Lawrence et al., 2007; Rasetti-Escargueil et al., 2009) through its proteolytic cleavage of the target-soluble N-ethylmaleimide-sensitive factor attachment protein (SNAP) receptor (t-SNARE) protein SNAP of 25 kDa (SNAP-25; Schiavo et al., 2000). As a control, we treated P1 diaphragm muscle with 100 nm BoNT/E for 30 min at room temperature, and observed that nerve stimulation-induced muscle fiber shortening was completely blocked (peak muscle fiber shortening, 0 vs 435 ± 65 μm; n = 3), similar to previous studies (Lawrence et al., 2007). Next, we imaged TPSC Ca2+ responses in P7 diaphragm after intoxication with BoNT/E. Similar to its effects on muscle contraction (mediated by a block of vesicular release of ACh from presynaptic terminals), BoNT/E inhibited TPSC Ca2+ responses (mediated by a block of vesicular corelease of ATP from presynaptic terminals; Silinsky, 1975; Heredia et al., 2018; 0.4 ± 0.8 vs 15.4 ± 3.2 dB). Finally, we imaged ASCs in response to nerve stimulation at E14 in the presence of BoNT/E. We observed a dramatic loss of activity-induced Ca2+ responses in ASCs, as well as in TPSCs, at this age (Fig. 5A). These results provide functional evidence that developing activity-induced Ca2+ responses in ASCs along secondary phrenic intramuscular phrenic branches are mediated by the vesicular release of ATP.
Figure 5.

The loss of synaptic vesicles from phrenic axons occurs in a proximo-distal gradient in the phrenic nerve. A, Left, Average fluorescence intensity image (Prestim, blue and green) of a diaphragm of an E14 Wnt1-GCaMP3 mouse. Arrow marks a secondary phrenic intramuscular branch. Middle, right, Spatial intensity maps of Ca2+ responses to 30 s of 40 Hz phrenic nerve stimulation before and after intoxication with BoNT/E. Note the absence of responses in both ASCs (arrow) and TPSCs (arrowhead) after BoNT/E treatment. B, Synaptophysin immunoreactivity in diaphragms during development. Note the progressive loss over time of axonal immunoreactivity (arrow) and the maintenance of synaptic immunoreactivity (arrowhead). C, Electron micrographs of a secondary phrenic intramuscular branch [nerve (N)] at E14 imaged at 2600× (left) or at 25,000× (middle) display abundant vesicles in axons, including some clustered toward the edge of one axon next to a Schwann cell [right, arrow (enlarged region of inset box in middle, which is enlarged region of inset box in left)]. Note the electron-dense region of axonal membrane near this cluster of vesicles (right, asterisk). Arrowhead in middle panel also shows vesicles in the rounded bouton-like structure at the edge of the nerve, also next to the Schwann cell (S) in the left panel.
To determine whether the proximo-distal pattern of ASC Ca2+ responses to nerve activity was caused by a similar gradient in the distribution of vesicles, we examined the expression of synaptophysin, a protein associated with synaptic vesicles that is reportedly expressed in a proximo-distal gradient during embryonic and early postnatal development (Lupa and Hall, 1989; Dahm and Landmesser, 1991). We found that at E14, when ASCs along secondary phrenic intramuscular branches respond to activity-induced vesicular release of neurotransmitter, synaptophysin was robustly expressed by these branches (Fig. 5B). At P1, when ASC Ca2+ responses along these branches are no longer observed, synaptophysin immunoreactivity along these branches was dramatically reduced. At P7, when activity-induced vesicular responses were largely restricted to TPSCs, synaptophysin expression was concordantly expressed only at the NMJ (ratio of axonal/presynaptic synaptophysin immunoreactivity intensity, 1.1 ± 0.2 vs 1.22 ± 0.3 vs 0.04 ± 0.006 vs 0 ± 0, for E14 vs E15.5 vs P1 vs P7, n = 3; Fig. 5B). When we examined secondary phrenic intramuscular branches at E14 by electron microscopy, we found many vesicles near 50 nm in diameter along the edges of axons, adjacent to electron-dense regions of axonal membrane that may represent active zones, opposed by the plasma membrane of a Schwann cell (n = 3; Fig. 5C). These results provide morphological support to the idea that neurotransmitter-containing vesicles containing synaptic proteins are released from developing phrenic axons in response to stimulation during development.
We noted that the time during which ASCs along secondary phrenic intramuscular branches no longer exhibited activity-dependent Ca2+ responses, P1, was closely related to the onset of myelination (Song et al., 1999). In the CNS, proximal pathways exhibit myelination before distal ones, but whether this extends to the proximal and distal parts of individual axons is unclear (Kinney et al., 1988). In the one example where this has been examined, myelination of the optic nerve occurs earlier in distal than proximal axons (Friede and Hu, 1967). In contrast, whether myelination of peripheral axons exhibits either of these patterns is unknown. To address the possibility that myelination influences the proximo-distal gradient of activity-induced ASC Ca2+ responses, we examined the myelination of the phrenic nerve ultrastructurally at P7 in the primary trunk and in the more distal, secondary intramuscular branch. First, we examined axonal caliber, because a recent report of the adult mouse phrenic nerve trunk showed that proximal axons are larger than distal axons (Shen et al., 2010). We first measured the diameter and observed no significant difference (axonal diameter, 0.61 ± 0.13 vs 0.81 ± 0.32 μm, for primary vs secondary; p = 0.09, n = 3, c = 9). Because the axons of the intramuscular branch were not all circular in our sections, we also measured the area, but this too was not significantly different (axonal area, 1.2 ± 0.33 vs 1.18 ± 0.51 μm2, for primary vs secondary; p = 0.9, n = 3, c = 9). To compare myelination, we measured the g-ratio and found that it was statistically larger in the secondary branch compared with the proximal nerve (g-ratio, 0.58 ± 0.06 vs 0.71 ± 0.071, for primary vs secondary; p = 0.0005, n = 3, c = 9), indicating that the myelin was less thick in these more distal branches. This was also supported by the direct measurement of myelin thickness (thickness, 0.40 ± 0.07 vs 0.289 ± 0.07 μm, for primary vs secondary; p = 0.002, n = 3, c = 9) as well as the number of lamellae measured at high magnification (lamellae, 32.4 ± 5.8 vs 21 ± 4.3, for primary vs secondary; p = 0.0002, n = 3, c = 9). These data suggest that the myelination of proximal phrenic axons is more advanced than that of secondary intramuscular axons at P7, an age when only TPSCs but not ASCs exhibit activity-induced Ca2+ responses. However, nearly all phrenic axons at this age already exhibited compact myelin (Fig. 6). Therefore, we examined myelination in proximal and distal locations at P1. Although every axon had established a 1:1 relationship with a Schwann cell in the proximal trunk (i.e., radial sorting), many Schwann cells were still in contact with multiple axons in the more distal secondary phrenic intramuscular branches (Fig. 6). Similarly, compact myelin could be clearly detected around some axons in the proximal trunk but not around any in the distal branches (lamellae, 7.3 ± 3.8 vs 0.9 ± 0.8, for primary vs secondary; p = 0.0001, n = 3, c = 9). Together, these data are consistent with the notion that myelination proceeds proximo-distally during early postnatal development, which is similar to the pattern of ASC Ca2+ response by axons.
Figure 6.
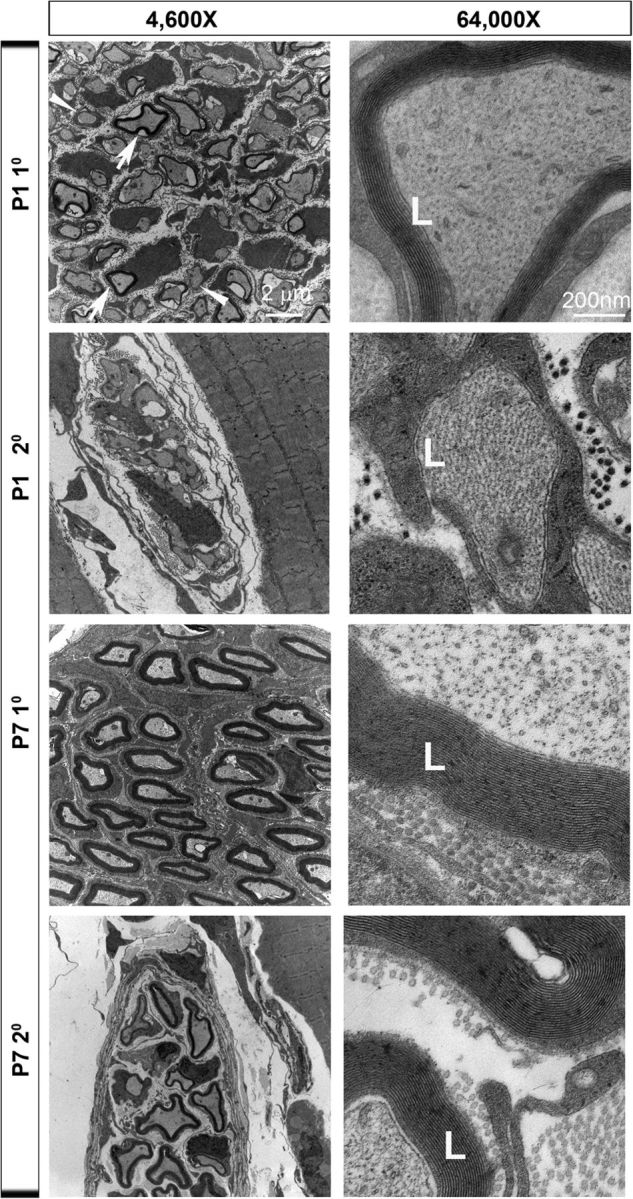
Myelination in phrenic axons develops in a proximo-distal gradient. Electron micrographs of cross sections of the primary phrenic nerve trunk at P1 or P7 (top row, third row) and the secondary phrenic intramuscular branch at P1 or P7 (second row, fourth row) at low or high magnification (left, right columns). Note the heterogeneity of myelination among proximal phrenic trunk axons at P1 (arrows indicate axons surrounded by compact myelin; arrowheads indicate axons that are not surrounded by compact myelin). An example of a P1 proximal axon surrounded by several lamellae (L) of compact myelin is shown. Note also the increased myelin thickness of the primary vs the secondary phrenic nerve at low magnification at P7, as well as the increased number of lamellae at high magnification.
Finally, to test whether the basic response features of developing ASCs and TPSCs to nerve stimulation were similar to those of postnatal TPSCs, we stimulated the phrenic nerve at a variety of frequencies and durations and compared these responses. In each case, the responses of developing ASCs along secondary phrenic intramuscular branches and TPSCs at NMJs were similar in magnitude, onset, and duration, so we pooled them. For these studies, we focused on P1. We observed that relative to P7 TPSCs, P1 ASCs/TPSCs exhibited Ca2+ responses to lower frequency stimulation. For example, whereas the lowest nerve stimulation frequency capable of inducing Ca2+ responses in P7 TPSCs was 5 Hz, P1 ASCs/TPSCs responded to 1 Hz stimulation (Fig. 7A,B). However, in both cases, the number of Schwann cells exhibiting Ca2+ responses to low-frequency stimulation was lower than that responding to higher-frequency stimulation (compare 1 to 2 Hz stimulation at P1, or 5 to 20 Hz stimulation at P7; Fig. 7A,B). These low-frequency Ca2+ responses were slower and less intense, and exhibited onsets that were more temporally heterogeneous after nerve stimulation (Figs. 7C, 8). Interestingly, ASC/TPSC Ca2+ transients at P1 achieved peak intensity after 20 Hz stimulation but exhibited a more dramatic decline over time compared with low-frequency stimulations (Fig. 7A,C). This decline reflected a corresponding larger reduction in the amount of neurotransmitter released over time in response to 20 Hz than to 10 Hz nerve stimulation, as indicated by the amplitude of ACh-mediated endplate potentials, resulting in reduced functional neurotransmission (Fig. 7D,E). In addition to responding to lower frequencies, ASCs/TPSCs at P1 also responded to shorter durations of stimuli relative to P7 (Fig. 9). For example, although 0.1 s of 20 Hz nerve stimulation was sufficient to induce a brief Ca2+ response within ASCs and TPSCs at P1, the smallest duration that resulted in TPSC Ca2+ responses at this frequency at P7 was 5 s (Fig. 9). Together, these data suggest that the sensitivity of Schwann cells to small amounts of neurotransmitter release is enhanced in P1 compared with P7 Schwann cells.
Figure 7.
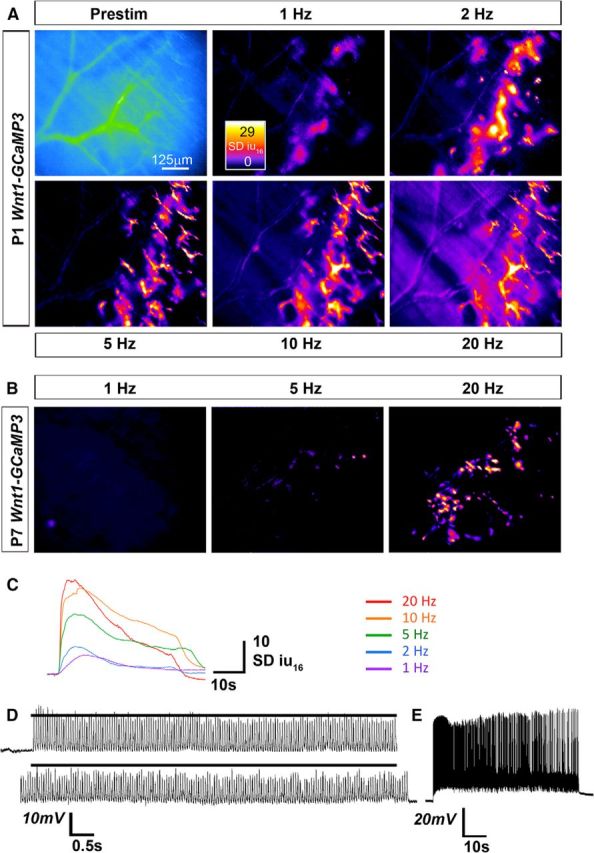
Neonatal ASCs/TPSCs exhibit Ca2+ responses to lower frequencies of nerve stimulation than postnatal TPSCs. Average fluorescence intensity image (Prestim, blue, and green) and spatial intensity maps of Ca2+ responses to 30 s of different frequencies of phrenic nerve stimulation from the same diaphragm of a P1 Wnt1-GCaMP3 mouse. Note the response to 1 Hz stimulation. B, Spatial intensity maps of Ca2+ responses to 30 s of different frequencies of phrenic nerve stimulation from the same diaphragm of a P7 Wnt1-GCaMP3 mouse. In contrast to ASCs/TPSCs at P1, TPSCs at P7 fail to exhibit a substantial number of Ca2+ responses to frequencies <5 Hz. Note too the fewer TPSCs responding to 5 Hz, as opposed to 20 Hz, nerve stimulation. C, Individual P1 ASC/TPSC Ca2+ transients sampled from the same cell in response to different phrenic nerve stimulation frequencies. Note that as the frequency is increased, the rate of rise, and the amplitude and duration of transient rise commensurately, with the exception that transient duration is relatively shorter in response to 20 than to 10 Hz stimulation. D, Intracellular recording of muscle endplate potentials (EPPs) in P1 diaphragm in the presence of μ-conotoxin in response to 30 s of 20 Hz stimulation. Top trace, first 7 s; bottom, last 7 s. Black line in both traces indicates the average EPP amplitude of the first 7 s; note the lower amplitude of EPPs in the final vs the first 7 s, indicating reduced transmitter release. E, Intracellular recording of muscle action potentials (APs) in P1 diaphragm in the presence of BHC in response to 30 s of 20 Hz stimulation. In last third of the train, note the increasing prevalence of failed muscle APs (white in between spikes).
Figure 8.
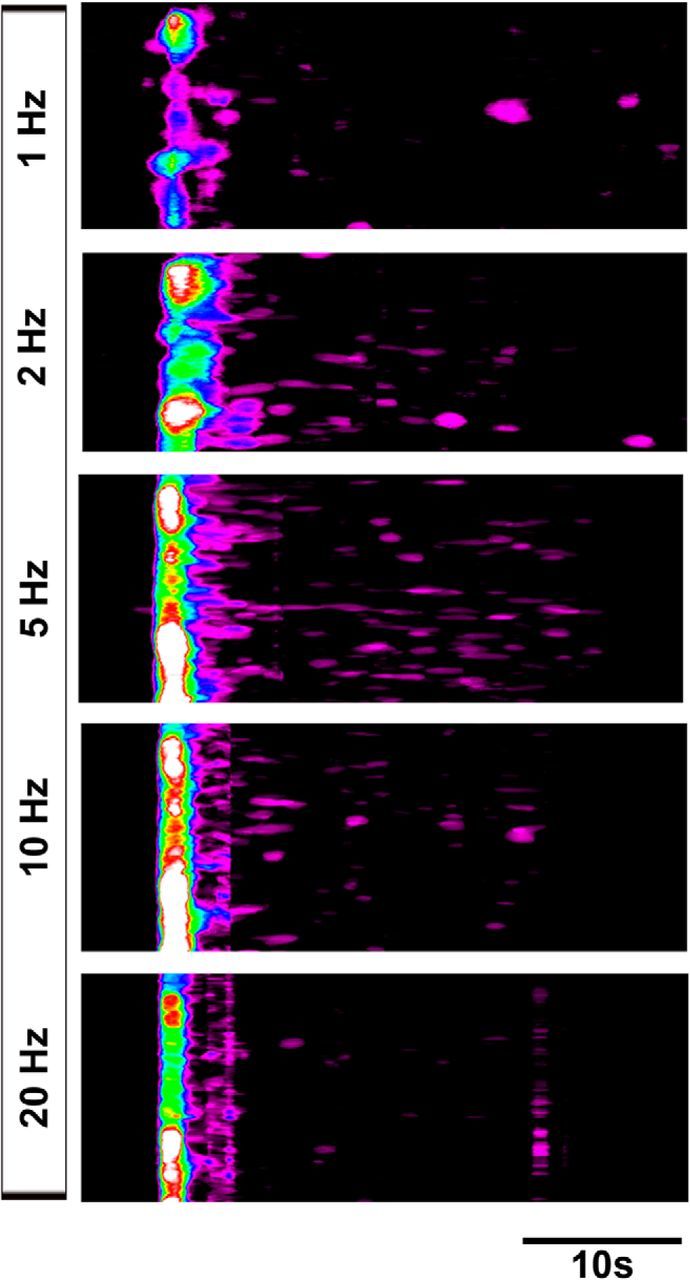
Low-frequency nerve stimulation induces temporally heterogeneous Ca2+ responses in neonatal ASCs/TPSCs. Spatiotemporal maps of ASC/TPSC Ca2+ responses to different frequencies of phrenic nerve stimulation from the same diaphragm of a P1 Wnt1-GCaMP3 mouse. The onset of response after nerve stimulation becomes shorter with increasing frequency, as indicated by the shift to the left of the high-intensity band of cells. Note the increase of responses at times significantly after the onset of nerve stimulation in response to 2 and 5 Hz stimulation (purple signal to the right of the high-intensity band.
Figure 9.

Neonatal ASCs/TPSCs exhibit Ca2+ responses to shorter durations of nerve stimulation than postnatal TPSCs. A, Spatial intensity maps of ASC/TPSC Ca2+ responses to different durations of 20 Hz phrenic nerve stimulation from the same diaphragm of a P1 Wnt1-GCaMP3 mouse. Note that peak responses are observed in response to 1 and 5 s, relative to shorter and longer durations. B, Spatial intensity maps of TPSC Ca2+ responses to different durations of 20 Hz phrenic nerve stimulation from the same diaphragm of a P7 Wnt1-GCaMP3 mouse. In contrast to ASC/TPSCs at P1, TPSCs at P7 fail to exhibit Ca2+ responses in response to 0.1 s of 20 Hz stimulation.
Discussion
The widespread expression of GCaMP3 in both ASCs and TPSCs early in embryogenesis in Wnt1-GCaMP3 mice led us to investigate the factors that activate Ca2+ signaling in each of these populations. Similar to previous studies of individual or small groups of TPSCs loaded with chemical Ca2+ indicators at the normal, developing, or diseased NMJ (Todd et al., 2010; Darabid et al., 2013; Arbour et al., 2015), the current study of populations of both ASCs and TPSCs using this genetic method demonstrates that Ca2+ signaling with Schwann cells is sensitive to distinct levels of neuromuscular activity. Therefore, we used Wnt1-GCaMP3 mice to determine the temporal and spatial features of neuromuscular activity in an ex vivo preparation of the developing phrenic nerve and diaphragm.
The main finding is that phrenic nerve stimulation during embryonic development induces robust Ca2+ responses in ASCs along proximal and distal phrenic axons as well as in TPSCs at distal NMJs, whereas stimulation at early postnatal stages results in the activation of only TPSCs. This proximo-distal loss of ASC Ca2+ responses is not caused by a change in the competence of these cells to respond to nerve stimulation, because bath application of either ACh or the purine adenine nucleotide ATP, which are coreleased by the synaptic vesicles of motor neurons in response to nerve stimulation (Silinsky, 1975), each induce ASC responses even when nerve stimulation does not. Similar to those of early postnatal TPSCs (Darabid et al., 2013; Heredia et al., 2018), activity-induced Ca2+ responses in developing ASCs and TPSCs are mediated by P2Y1R, suggesting that they are induced by ATP released from developing phrenic axons and presynaptic terminals. These responses appear to reflect the release of ATP from exocytosing synaptic vesicles, rather than through nonvesicular mechanisms such as pannexins, because they are abrogated in the presence of the vesicular release inhibitor BoNT/E. This interpretation is strengthened by the observation that immunoreactivity of the synaptic vesicle protein synaptophysin and synaptic vesicles themselves are both found transiently in developing phrenic axons but then become restricted to presynaptic terminals along the same spatiotemporal gradient as activity-induced Schwann cell Ca2+ responses. Together, these data suggest that the transient response of ASCs along phrenic axons to nerve stimulation derives from the transient presence of synaptic vesicles within these axons. These ex vivo studies corroborate earlier in vitro findings of axonal transmitter release from Xenopus spinal neurons (Hume et al., 1983; Young and Poo, 1983; Zakharenko et al., 1999), rat hippocampal neurons (Matteoli et al., 1992), and mouse hypothalamic neurons (Gao and van den Pol, 2000).
The presence of synaptic vesicles in axons is not by itself sufficient to result in the exocytotic release of vesicular neurotransmitter in response to activity. In order for these vesicles to be docked to the plasma membrane, primed and eventually fused to the plasma membrane in response to activity-induced Ca2+ influx into the axon, the constituents of the cytomatrix at the active zone (CAZ) are also required (Südhof, 2000). Additionally, the presence of a dedicated molecular machinery underlying endocytosis and SV reformation is required during periods of sustained neurotransmission (Saheki and De Camilli, 2012). Live imaging studies of developing hippocampal and cortical neurons in vitro have shown that the presynaptic components of the synaptic vesicle and the CAZ are packaged in the cell body into two distinct types of vesicle, the piccolo-bassoon transport vesicle (PTV) for CAZ proteins and the synaptic vesicle protein transport vesicles for proteins associated with synaptic vesicles (Zhai et al., 2001; Sabo and McAllister, 2003). Synaptic vesicles are transported from the cell body through the axon and to presynaptic terminals along microtubules by the kinesin superfamily proteins 1A/B (Yonekawa et al., 1998; Nakamura et al., 2002), whereas PTVs are transported along microtubules by KIF5B (kinesin family member 5B) and the adaptor protein syntabulin (Cai et al., 2007). Aggregates of both kinds of these vesicles are detected ultrastructurally in developing axons (Tao-Cheng, 2007), consistent with the coordination of their transport along axons even before synapse formation (Bury and Sabo, 2011). The presence of each of these presynaptic protein packets in the axons of developing neurons underlies the observation that neurotransmitter release from them occurs through the exocytosis of synaptic vesicles (Matteoli et al., 1992).
Although recent in vitro studies demonstrate that mature hippocampal neurons continue to exhibit exocytotic release of neurotransmitter from axons (Ratnayaka et al., 2011), the current ex vivo studies show that phrenic axons only transiently display this form of release, likely resulting from the loss of synaptic vesicles in older axons. Alternatively, the loss of axonal neurotransmitter release in postnatal nerves may reflect a change in the competence of SVs to undergo targeting to or release or reformation from axons (Rizzoli, 2014). On the one hand, ultrastructural studies of adult mouse brain demonstrate the continued presence of CAZ molecules such as the t-SNARE SNAP-25 in the axonal plasma membrane (Tao-Cheng et al., 2000). On the other hand, biochemical and functional studies of several molecules mediating exo-endocytosis have shown a temporally dynamic pattern of expression that may contribute to the loss of axonal neurotransmitter release. For example, mice lacking neural cell adhesion molecule (NCAM) continue to exhibit neurotransmitter release from preterminal axons adjacent to the NMJ at P30 (Polo-Parada et al., 2001; Hata et al., 2007). This aberrant maintenance of axonal release ex vivo is blocked by treatment with the fungal toxin brefeldin A (BFA), which also inhibits axonal transmitter release in vitro (Zakharenko et al., 1999). In addition to blocking all forms of axonal transport by interfering with Golgi vesicle formation (Helms and Rothman, 1992; Donaldson et al., 1992; Campenot et al., 2003), BFA blocks the exo-endocytosis of SVs by inhibiting the ADP-ribosylation factor-1-dependent recruitment of the cytoplasmic coat protein adaptor protein-3 (AP-3) to SV membranes (Faúndez et al., 1998; Ooi et al., 1998; Scheuber et al., 2006). Therefore, NCAM may normally prevent axonal neurotransmitter release by regulating SV exo-endocytosis. Supporting this idea, NCAM changes its biochemical affinity during development from the adaptor protein AP-3, which targets protein complexes to the axon, to the AP-2, which mediates SV exocytosis in mature presynaptic terminals (Kim and Ryan, 2009; Shetty et al., 2013; Li et al., 2016). Collectively, these data suggest that the loss of phrenic axonal neurotransmitter release may be caused by a loss of this exo-endocytic machinery in addition to the loss of SVs themselves.
What is the role of developmental axonal release of the neurotransmitters ACh and ATP by developing phrenic axons? Recent studies support the idea that neuronal activity during development regulates myelination (Hines et al., 2015; Mensch et al., 2015), but whether activity in this context is transduced through the synaptic vesicular release of neurotransmitter onto myelinating glial progenitor cells or through nonsynaptic vesicular or nonvesicular release (i.e., axo-glial) is unclear (Micu et al., 2018). For example, a recent in vitro study demonstrated that the preferential myelination of electrically active versus silent neurons by oligodendrocyte progenitor cells occurred by axonal nonsynaptic vesicular release rather than synaptic vesicular release of the excitatory transmitter glutamate (Wake et al., 2015). Our current results show that the robust Schwann cell ASC Ca2+ responses reflecting neurotransmitter release are lost by P1, an age at which myelination of proximal phrenic axons is just beginning. Therefore, if the axonal release of neurotransmitter facilitates or is required for myelination, it would have to be mediated by a signaling pathway that regulates the onset but not the duration of this process, which continues significantly after P1.
Although ASC Ca2+ responses reflect the release of ACh and ATP, the analysis of P2Y1R mice lacking them reflects only the loss of the P2Y1R-stimulating purines ATP/ADP, and not that of ACh or adenosine, which is rapidly produced in the synaptic cleft from ATP by ecto-nucleotidases (Cunha and Sebastião, 1991), nor that of ATP on Schwann cells, axons, or muscle through purinergic receptors other than P2Y1R. In other words, the absence of delays of or defects in the myelination of phrenic axons in P2ry1 wild-type mice in the current study cannot be taken as evidence that activity-induced axo-glial signaling fails to regulate myelination. Moreover, even if there were differences, it would be hard to ascribe them to an alteration of axonal vesicular ATP release, because P2ry1 mutants also lack TPSC Ca2+ responses mediated by synaptic vesicular ATP release. On the other hand, for precisely this reason, these results offer insight into the effects of axo-glial vesicular communication during development, insofar as they demonstrate that one component of this pathway, the activity-induced vesicular ATP/P2Y1R-mediated axonal Schwann cell Ca2+ response, is apparently dispensable for myelination.
Although myelination was not overtly disrupted in mice lacking P2Y1Rs, a relationship between activity and myelination was observed during peripheral nerve development. Specifically, the spatiotemporal gradient of myelination is correlated with that of axonal release. For example, at both P1 and P7, myelination appeared more advanced in proximal compared with distal phrenic axons. To our knowledge, this is the first examination of the development of myelination along different proximo-distal regions of a peripheral nerve (for a similar analysis of an adult sensory nerve, see Yagihashi et al., 1990). These data are therefore consistent with those from previous studies showing that axonal neurotransmitter release regulates myelination (Wake et al., 2015). To test this hypothesis in vivo, it would be necessary to block ACh signaling in a small cohort of phrenic axons using genetic methods (Buffelli et al., 2003), because the elimination of ACh synthesis, release, or transduction in muscle in all motor neurons leads to paralysis and death upon birth (Misgeld et al., 2002; Washbourne et al., 2002; An et al., 2010), before the development of significant levels of myelination. Conversely, these results could suggest the opposite relationship, namely that myelination regulates the fast anterograde transport of synaptic vesicles and thus the axonal release of neurotransmitter, similar to the effects of myelinating Schwann cells on slow anterograde transport (de Waegh et al., 1992). One way to test this idea would be to treat phrenic secondary intramuscular branches at P1–P7 with the acute demyelinating agent LPC (Hall and Gregson, 1971), an age at which ASCs no longer exhibit activity-induced Ca2+ responses, and test whether this treatment restores these responses.
We used activity-induced ASC Ca2+ responses in Wnt1-GCaMP3 mice as a monitor of axonal transmitter release and observed their disappearance early in the developing phrenic nerve. These observations are consistent with early investigations of Schwann cell Ca2+ responses in the neuromuscular system (Jahromi et al., 1992; Reist and Smith, 1992), but at odds with those of other studies (Lev-Ram and Ellisman, 1995). One potential difference is the duration of nerve stimulation, as noted by Fields and Stevens (2000). A second possibility is that the phrenic nerve, composed largely of myelinated motor axons, responds differently than the sciatic nerve used in the study by Lev-Ram and Ellisman (1995), which contains many unmyelinated sensory as well as myelinated sensory and motor axons. Alternatively, because the activity-induced Ca2+ response of these ASCs was noted to be of significantly less intensity than that of TPSCs, its ability to be optically detected in secondary phrenic intramuscular branches, which course through the muscle, may be reduced compared with that of the sciatic nerve. Future studies of the sciatic nerve of Wnt1-GCaMP3 mice will help to determine whether this response is detectable in response to similar stimuli and, if so, whether it depends on vesicular transmitter release and P2Y1Rs or distinct molecular mechanisms.
Finally, we observed that in contrast to postnatal TPSCs, neonatal ASCs and TPSCs responded to low frequencies and the duration of nerve stimulation. Such low frequencies are not sufficient to induce muscle fatigue, suggesting that these Schwann cell Ca2+ responses did not contribute to potassium buffering or other mechanisms to regulate fatigue (Heredia et al., 2018). The ability of low-frequency stimulation to activate neonatal but not postnatal ASCs/TPSCs is not likely due to differences in the release of ATP, but rather to the sensitivity of the Schwann cell Ca2+ response, because the level of release, measured by the amplitude of ACh-induced EPPs obtained in response to 1 Hz stimulation, is not sufficiently different at P1 versus P7 (EPP amplitude, 22.7 ± 3.6 vs 23.8 ± 2.8 mV, for P1 vs P7; p = 0.55, n = 3, c = 7). Therefore, these results imply that differences in ATP catabolism in the synaptic cleft, the affinity of ATP to P2Y1Rs or the robustness of P2Y1R-induced signaling, exist between neonatal and postnatal Schwann cells.
In conclusion, we have used Schwann cell population Ca2+ responses in Wnt1-GCaMP3 mice as a tool to measure the spatial and temporal patterns of neurotransmitter release during embryonic development. Similar to earlier in vitro reports, the current ex vivo study supports the notion that axons transiently release neurotransmitter early in development, most likely due to the transient presence of axonal synaptic vesicles, which are trafficked to terminals from the cell body at these stages, before local recycling from presynaptic endosomes is established. Because developing Schwann cell Ca2+ responses were dependent on ATP release and P2Y1R, we were able to assess, using genetic methods, the effect of eliminating this signal on myelination. Future genetic studies manipulating the level of ACh release at these ages will help to determine whether early axonal release of ACh, rather than ATP, modifies myelination or other aspects of Schwann cell development. Last, because the regrowth and reinnervation of peripheral motor axons after injury depends on a sequence of changes to Schwann cells, including the restoration of axo-glial relationships and remyelination (Fex Svennigsen and Dahlin, 2013), the data presented in the current and proposed studies may be useful for enhancing repair after such an insult.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants GM-103554 and GM-110767 (T.W.G); National Center for Research Resources Grant 5P20-RR-018751-09; and National Institute of General Medical Sciences Grant 8P20-GM-103513-09 (G.W.H). Work performed in a core laboratory was supported by NIH Grant GM-103554. We thank Chris von Bartheld for assistance with the use of the electron microscope.
The authors declare no competing financial interests.
References
- An MC, Lin W, Yang J, Dominguez B, Padgett D, Sugiura Y, Aryal P, Gould TW, Oppenheim RW, Hester ME, Kaspar BK, Ko CP, Lee KF (2010) Acetylcholine negatively regulates development of the neuromuscular junction through distinct cellular mechanisms. Proc Natl Acad Sci U S A 107:10702–10707. 10.1073/pnas.1004956107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Carmignoto G, Haydon PG (2001) Dynamic signaling between astrocytes and neurons. Annu Rev Physiol 63:795–813. 10.1146/annurev.physiol.63.1.795 [DOI] [PubMed] [Google Scholar]
- Arbour D, Tremblay E, Martineau É, Julien JP, Robitaille R (2015) Early and persistent abnormal decoding by glial cells at the neuromuscular junction in an ALS model. J Neurosci 35:688–706. 10.1523/JNEUROSCI.1379-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffelli M, Burgess RW, Feng G, Lobe CG, Lichtman JW, Sanes JR (2003) Genetic evidence that relative synaptic efficacy biases the outcome of synaptic competition. Nature 424:430–434. 10.1038/nature01844 [DOI] [PubMed] [Google Scholar]
- Bury LA, Sabo SL (2011) Coordinated trafficking of synaptic vesicle and active zone proteins prior to synapse formation. Neural Dev 6:24. 10.1186/1749-8104-6-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q, Pan PY, Sheng ZH (2007) Syntabulin-kinesin-1 family member 5B-mediated axonal transport contributes to activity-dependent presynaptic assembly. J Neurosci 27:7284–7296. 10.1523/JNEUROSCI.0731-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campenot RB, Soin J, Blacker M, Lund K, Eng H, MacInnis BL (2003) Block of slow axonal transport and axonal growth by brefeldin A in compartmented cultures of rat sympathetic neurons. Neuropharmacology 44:1107–1117. 10.1016/S0028-3908(03)00042-X [DOI] [PubMed] [Google Scholar]
- Cunha RA, Sebastião AM (1991) Extracellular metabolism of adenine nucleotides and adenosine in the innervated skeletal muscle of the frog. Eur J Pharmacol 197:83–92. 10.1016/0014-2999(91)90368-Z [DOI] [PubMed] [Google Scholar]
- Dahm LM, Landmesser LT (1991) The regulation of synaptogenesis during normal development and following activity blockade. J Neurosci 11:238–255. 10.1523/JNEUROSCI.11-01-00238.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darabid H, Arbour D, Robitaille R (2013) Glial cells decipher synaptic competition at the mammalian neuromuscular junction. J Neurosci 33:1297–1313. 10.1523/JNEUROSCI.2935-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darabid H, Perez-Gonzalez AP, Robitaille R (2014) Neuromuscular synaptogenesis: coordinating partners with multiple functions. Nat Rev Neurosci 15:703–718. [PubMed] [Google Scholar]
- de Waegh SM, Lee VM, Brady ST (1992) Local modulation of neurofilament phosphorylation, axonal caliber, and slow axonal transport by myelinating schwann cells. Cell 68:451–463. 10.1016/0092-8674(92)90183-D [DOI] [PubMed] [Google Scholar]
- Donaldson JG, Finazzi D, Klausner RD (1992) Brefeldin A inhibits Golgi membrane-catalysed exchange of guanine nucleotide onto ARF protein. Nature 360:350–352. 10.1038/360350a0 [DOI] [PubMed] [Google Scholar]
- Duron B, Jung-Caillol MC, Marlot D (1978) Myelinated nerve fiber supply and muscle spindles in the respiratory muscles of cat: quantitative study. Anat Embryol (Berl) 152:171–192. 10.1007/BF00315923 [DOI] [PubMed] [Google Scholar]
- Faúndez V, Horng JT, Kelly RB (1998) A function for the AP3 coat complex in synaptic vesicle formation from endosomes. Cell 93:423–432. 10.1016/S0092-8674(00)81170-8 [DOI] [PubMed] [Google Scholar]
- Fex Svennigsen A, Dahlin LB (2013) Repair of the peripheral nerve-remyelination that works. Brain Sci 3:1182–1197. 10.3390/brainsci3031182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields RD, Ni Y (2010) Nonsynaptic communication through ATP release from volume-activated anion channels in axons. Sci Signal 3:ra73. 10.1126/scisignal.2001128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields RD, Stevens B (2000) ATP: an extracellular signaling molecule between neurons and glia. Trends Neurosci 23:625–633. 10.1016/S0166-2236(00)01674-X [DOI] [PubMed] [Google Scholar]
- Friede RL, Hu KH (1967) Proximo-distal differences in myelin development in human optic fibers. Z Zellforsch Mikrosk Anat 79:259–264. 10.1007/BF00369289 [DOI] [PubMed] [Google Scholar]
- Gao XB, van den Pol AN (2000) GABA release from mouse axonal growth cones. J Physiol 523:629–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall SM, Gregson NA (1971) The in vivo and ultrastructural effects of injection of lysophosphatidyl choline into myelinated peripheral nerve fibres of the adult mouse. J Cell Sci 9:769–789. [DOI] [PubMed] [Google Scholar]
- Hata K, Polo-Parada L, Landmesser LT (2007) Selective targeting of different neural cell adhesion molecule isoforms during motoneuron myotube synapse formation in culture and the switch from an immature to mature form of synaptic vesicle cycling. J Neurosci 27:14481–14493. 10.1523/JNEUROSCI.3847-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helms JB, Rothman JE (1992) Inhibition by brefeldin A of a golgi membrane enzyme that catalyses exchange of guanine nucleotide bound to ARF. Nature 360:352–354. 10.1038/360352a0 [DOI] [PubMed] [Google Scholar]
- Hennig GW, Gould TW, Koh SD, Corrigan RD, Heredia DJ, Shonnard MC, Smith TK (2015) Use of genetically encoded calcium indicators (GECIs) combined with advanced motion tracking techniques to examine the behavior of neurons and glia in the enteric nervous system of the intact murine colon. Front Cell Neurosci 9:436. 10.3389/fncel.2015.00436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heredia DJ, Schubert D, Maligireddy S, Hennig GW, Gould TW (2016) A novel striated muscle-specific myosin-blocking drug for the study of neuromuscular physiology. Front Cell Neurosci 10:276. 10.3389/fncel.2016.00276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heredia DJ, Feng CY, Hennig GW, Renden RB, Gould TW (2018) Activity-induced Ca2+ signaling in perisynaptic Schwann cells of the early postnatal mouse is mediated by P2Y1 receptors and regulates muscle fatigue. eLife 7:e30839. 10.7554/eLife.30839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines JH, Ravanelli AM, Schwindt R, Scott EK, Appel B (2015) Neuronal activity biases axon selection for myelination in vivo. Nat Neurosci 18:683–689. 10.1038/nn.3992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton SM, Luna Lopez C, Blevins E, Howarth H, Weisberg J, Shestopalov VI, Makarenkova HP, Shah SB (2017) Pannexin 1 modulates axonal growth in mouse peripheral nerves. Front Cell Neurosci 11:365. 10.3389/fncel.2017.00365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume RI, Role LW, Fischbach GD (1983) Acetylcholine release from growth cones detected with patches of acetylcholine receptor-rich membranes. Nature 305:632–634. 10.1038/305632a0 [DOI] [PubMed] [Google Scholar]
- Jahromi BS, Robitaille R, Charlton MP (1992) Transmitter release increases intracellular calcium in perisynaptic Schwann cells in situ. Neuron 8:1069–1077. 10.1016/0896-6273(92)90128-z [DOI] [PubMed] [Google Scholar]
- Kim SH, Ryan TA (2009) Synaptic vesicle recycling at CNS snapses without AP-2. J Neurosci 29:3865–3874. 10.1523/JNEUROSCI.5639-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney HC, Brody BA, Kloman AS, Gilles FH (1988) Sequence of central nervous system myelination in human infancy. II. patterns of myelination in autopsied infants. J Neuropathol Exp Neurol 47:217–234. 10.1097/00005072-198805000-00003 [DOI] [PubMed] [Google Scholar]
- Lawrence G, Wang J, Chion CK, Aoki KR, Dolly JO (2007) Two protein trafficking processes at motor nerve endings unveiled by botulinum neurotoxin E. J Pharmacol Exp Ther 320:410–418. 10.1124/jpet.106.108829 [DOI] [PubMed] [Google Scholar]
- Lev-Ram V, Ellisman MH (1995) Axonal activation-induced calcium transients in myelinating Schwann cells, sources, and mechanisms. J Neurosci 15:2628–2637. 10.1523/JNEUROSCI.15-04-02628.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Merrill SA, Jorgensen EM, Shen K (2016) Two clathrin adaptor protein complexes instruct axon-dendrite polarity. Neuron 90:564–580. 10.1016/j.neuron.2016.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Sanchez HB, Deerinck T, Morris JK, Ellisman M, Lee KF (2000) Aberrant development of motor axons and neuromuscular synapses in erbB2-deficient mice. Proc Natl Acad Sci U S A 97:1299–1304. 10.1073/pnas.97.3.1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupa MT, Hall ZW (1989) Progressive restriction of synaptic vesicle protein to the nerve terminal during development of the neuromuscular junction. J Neurosci 9:3937–3945. 10.1523/JNEUROSCI.09-11-03937.1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matteoli M, Takei K, Perin MS, Südhof TC, De Camilli P (1992) Exo-endocytotic recycling of synaptic vesicles in developing processes of cultured hippocampal neurons. J Cell Biol 117:849–861. 10.1083/jcb.117.4.849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClain JL, Fried DE, Gulbransen BD (2015) Agonist-evoked Ca2+ signaling in enteric glia drives neural programs that regulate intestinal motility in mice. Cell Mol Gastroenterol Hepatol 1:631–645. 10.1016/j.jcmgh.2015.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mensch S, Baraban M, Almeida R, Czopka T, Ausborn J, El Manira A, Lyons DA (2015) Synaptic vesicle release regulates myelin sheath number of individual oligodendrocytes in vivo. Nat Neurosci 18:628–630. 10.1038/nn.3991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micu I, Plemel JR, Caprariello AV, Nave KA, Stys PK (2018) Axo-myelinic neurotransmission: a novel mode of cell signalling in the central nervous system. Nat Rev Neurosci 19:49–58. 10.1038/nrn.2017.128 [DOI] [PubMed] [Google Scholar]
- Misgeld T, Burgess RW, Lewis RM, Cunningham JM, Lichtman JW, Sanes JR (2002) Roles of neurotransmitter in synapse formation: development of neuromuscular junctions lacking choline acetyltransferase. Neuron 36:635–648. 10.1016/S0896-6273(02)01020-6 [DOI] [PubMed] [Google Scholar]
- Nakamura N, Miyake Y, Matsushita M, Tanaka S, Inoue H, Kanazawa H (2002) KIF1Bbeta2, capable of interacting with CHP, is localized to synaptic vesicles. J Biochem 132:483–491. 10.1093/oxfordjournals.jbchem.a003246 [DOI] [PubMed] [Google Scholar]
- Ooi CE, Dell'Angelica EC, Bonifacino JS (1998) ADP-ribosylation factor 1 (ARF1) regulates recruitment of the AP-3 adaptor complex to membranes. J Cell Biol 142:391–402. 10.1083/jcb.142.2.391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelluru D, Konadhode RR, Bhat NR, Shiromani PJ (2016) Optogenetic stimulation of astrocytes in the posterior hypothalamus increases sleep at night in C57BL/6J mice. Eur J Neurosci 43:1298–1306. 10.1111/ejn.13074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G, Yang A, Boyden ES, Sur M (2014) Optogenetic astrocyte activation modulates response selectivity of visual cortex neurons in vivo. Nat Commun 5:3262. 10.1038/ncomms4262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo-Parada L, Bose CM, Landmesser LT (2001) Alterations in transmission, vesicle dynamics, and transmitter release machinery at NCAM-deficient neuromuscular junctions. Neuron 32:815–828. 10.1016/S0896-6273(01)00521-9 [DOI] [PubMed] [Google Scholar]
- Rasetti-Escargueil C, Jones RG, Liu Y, Sesardic D (2009) Measurement of botulinum types A, B and E neurotoxicity using the phrenic nerve-hemidiaphragm: improved precision with in-bred mice. Toxicon 53:503–511. 10.1016/j.toxicon.2009.01.019 [DOI] [PubMed] [Google Scholar]
- Ratnayaka A, Marra V, Branco T, Staras K (2011) Extrasynaptic vesicle recycling in mature hippocampal neurons. Nat Commun 2:531. 10.1038/ncomms1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reist NE, Smith SJ (1992) Neurally evoked calcium transients in terminal Schwann cells at the neuromuscular junction. Proc Natl Acad Sci U S A 89:7625–7629. 10.1073/pnas.89.16.7625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzoli SO. (2014) Synaptic vesicle recycling: steps and principles. EMBO J 33:788–822. 10.1002/embj.201386357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousse I, Robitaille R (2006) Calcium signaling in schwann cells at synaptic and extra-synaptic sites: active glial modulation of neuronal activity. Glia 54:691–699. 10.1002/glia.20388 [DOI] [PubMed] [Google Scholar]
- Sabo SL, McAllister AK (2003) Mobility and cycling of synaptic protein-containing vesicles in axonal growth cone filopodia. Nat Neurosci 6:1264–1269. 10.1038/nn1149 [DOI] [PubMed] [Google Scholar]
- Saheki Y, De Camilli P (2012) Synaptic vesicle endocytosis. Cold Spring Harb Perspect Biol 4:a005645. 10.1101/cshperspect.a005645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Beppu K, Tanaka KF, Fukazawa Y, Shigemoto R, Matsui K (2012) Application of an optogenetic byway for perturbing neuronal activity via glial photostimulation. Proc Natl Acad Sci U S A 109:20720–20725. 10.1073/pnas.1213458109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scemes E, Giaume C (2006) Astrocyte calcium waves: what they are and what they do. Glia 54:716–725. 10.1002/glia.20374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuber A, Rudge R, Danglot L, Raposo G, Binz T, Poncer JC, Galli T (2006) Loss of AP-3 function affects spontaneous and evoked release at hippocampal mossy fiber synapses. Proc Natl Acad Sci U S A 103:16562–16567. 10.1073/pnas.0603511103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavo G, Matteoli M, Montecucco C (2000) Neurotoxins affecting neuroexocytosis. Physiol Rev 80:717–766. 10.1152/physrev.2000.80.2.717 [DOI] [PubMed] [Google Scholar]
- Shen H, Barry DM, Garcia ML (2010) Distal to proximal development of peripheral nerves requires the expression of neurofilament heavy. Neuroscience 170:16–21. 10.1016/j.neuroscience.2010.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty A, Sytnyk V, Leshchyns'ka I, Puchkov D, Haucke V, Schachner M (2013) The neural cell adhesion molecule promotes maturation of the presynaptic endocytotic machinery by switching synaptic vesicle recycling from adaptor protein 3 (AP-3)- to AP-2-dependent mechanisms. J Neurosci 33:16828–16845. 10.1523/JNEUROSCI.2192-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silinsky EM. (1975) On the association between transmitter secretion and the release of adenine nucleotides from mammalian motor nerve terminals. J Physiol 247:145–162. 10.1113/jphysiol.1975.sp010925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song A, Tracey DJ, Ashwell KW (1999) Development of the rat phrenic nerve and the terminal distribution of phrenic afferents in the cervical cord. Anat Embryol (Berl) 200:625–643. 10.1007/s004290050310 [DOI] [PubMed] [Google Scholar]
- Südhof TC. (2000) The synaptic vesicle cycle revisited. Neuron 28:317–320. 10.1016/S0896-6273(00)00109-4 [DOI] [PubMed] [Google Scholar]
- Tao-Cheng JH. (2007) Ultrastructural localization of active zone and synaptic vesicle proteins in a preassembled multi-vesicle transport aggregate. Neuroscience 150:575–584. 10.1016/j.neuroscience.2007.09.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao-Cheng JH, Du J, McBain CJ (2000) Snap-25 is polarized to axons and abundant along the axolemma: an immunogold study of intact neurons. J Neurocytol 29:67–77. 10.1023/A:1007168231323 [DOI] [PubMed] [Google Scholar]
- Todd KJ, Darabid H, Robitaille R (2010) Perisynaptic glia discriminate patterns of motor nerve activity and influence plasticity at the neuromuscular junction. J Neurosci 30:11870–11882. 10.1523/JNEUROSCI.3165-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake H, Ortiz FC, Woo DH, Lee PR, Angulo MC, Fields RD (2015) Nonsynaptic junctions on myelinating glia promote preferential myelination of electrically active axons. Nat Commun 6:7844. 10.1038/ncomms8844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washbourne P, Thompson PM, Carta M, Costa ET, Mathews JR, Lopez-Benditó G, Molnár Z, Becher MW, Valenzuela CF, Partridge LD, Wilson MC (2002) Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat Neurosci 5:19–26. 10.1038/nn783 [DOI] [PubMed] [Google Scholar]
- Woldeyesus MT, Britsch S, Riethmacher D, Xu L, Sonnenberg-Riethmacher E, Abou-Rebyeh F, Harvey R, Caroni P, Birchmeier C (1999) Peripheral nervous system defects in erbB2 mutants following genetic rescue of heart development. Genes Dev 13:2538–2548. 10.1101/gad.13.19.2538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolpowitz D, Mason TB, Dietrich P, Mendelsohn M, Talmage DA, Role LW (2000) Cysteine-rich domain isoforms of the neuregulin-1 gene are required for maintenance of peripheral synapses. Neuron 25:79–91. 10.1016/S0896-6273(00)80873-9 [DOI] [PubMed] [Google Scholar]
- Yagihashi S, Kamijo M, Watanabe K (1990) Reduced myelinated fiber size correlates with loss of axonal neurofilaments in peripheral nerve of chronically streptozotocin diabetic rats. Am J Pathol 136:1365–1373. [PMC free article] [PubMed] [Google Scholar]
- Yamashita A, Hamada A, Suhara Y, Kawabe R, Yanase M, Kuzumaki N, Narita M, Matsui R, Okano H, Narita M (2014) Astrocytic activation in the anterior cingulate cortex is critical for sleep disorder under neuropathic pain. Synapse 68:235–247. 10.1002/syn.21733 [DOI] [PubMed] [Google Scholar]
- Yonekawa Y, Harada A, Okada Y, Funakoshi T, Kanai Y, Takei Y, Terada S, Noda T, Hirokawa N (1998) Defect in synaptic vesicle precursor transport and neuronal cell death in KIF1A motor protein-deficient mice. J Cell Biol 141:431–441. 10.1083/jcb.141.2.431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young SH, Poo MM (1983) Spontaneous release of transmitter from growth cones of embryonic neurones. Nature 305:634–637. 10.1038/305634a0 [DOI] [PubMed] [Google Scholar]
- Zakharenko S, Chang S, O'Donoghue M, Popov SV (1999) Neurotransmitter secretion along growing nerve processes: comparison with synaptic vesicle exocytosis. J Cell Biol 144:507–518. 10.1083/jcb.144.3.507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai RG, Vardinon-Friedman H, Cases-Langhoff C, Becker B, Eckart D, Gundelfinger ED, Ziv NE, Garner CC (2001) Assembling the presynaptic active zone: characterization of an AZ precursor vesicle. Neuron 29:131–143. 10.1016/S0896-6273(01)00185-4 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Some but not all Schwann cells along axons (ASCs) and some but not all TPSCs at NMJs exhibit a robust increase in intracellular Ca2+ in response to nerve stimulation at E14.
All ASCs and all TPSCs exhibit a Ca2+ response (respond) to the ACh mimetic CCh at E14.
ASCs along secondary phrenic intramuscular branches and tertiary branches, as well as TPSCs at NMJs, respond to nerve stimulation at E14.
ASCs along secondary phrenic intramuscular branches no longer respond to nerve stimulation, whereas ASCs along tertiary branches, as well as TPSCs at NMJs, respond to nerve stimulation at P1.
ASCs along secondary phrenic intramuscular and most tertiary branches no longer respond to nerve stimulation, whereas ASCs along preterminal axons, as well as TPSCs at NMJs, respond to nerve stimulation at P5.
TPSCs at NMJs, but not ASCs, respond to nerve stimulation at P7.
TPSCs at NMJs, but not ASCs, respond to nerve stimulation at P15.
Spatial identification of Schwann cell Ca2+ responses at P1. Ca2+ transients of spatially identified ASCs and TPSCs in response to nerve stimulation (Stim;left) or carbachol (CCh; right) at P1 as identified by color coding and numbering. Note the reduced heterogeneity in amplitude and shape of Ca2+ transients in response to nerve stimulation at P0 vs. E14. Note also the small response to nerve stimulation in ASCs of the phrenic intramuscular branch (Cells 1-8) vs. that within TPSCs at the NMJ (Cells 9-15). In contrast, ASCs along the phrenic intramuscular branch all exhibit prolonged responses to CCh, although this response is delayed compared to that within TPSCs (bar running through transients is set to peak amplitude of CCh-induced Ca2+ response in TPSCs). Download Figure 1-1, TIF file (3.1MB, tif)