

Abstract
Macrophages are phagocytes that play important roles in health and diseases. Acyl-CoA:cholesterol acyltransferase 1 (ACAT1) converts cellular cholesterol to cholesteryl esters and is expressed in many cell types. Unlike global Acat1 knockout (KO), myeloid-specific Acat1 KO (Acat1−) does not cause overt abnormalities in mice. Here, we performed analyses in age- and sex-matched Acat1−M/−M and wild-type mice on chow or Western diet and discovered that Acat1−M/−M mice exhibit resistance to Western diet-induced obesity. On both chow and Western diets, Acat1−M/−M mice display decreased adipocyte size and increased insulin sensitivity. When fed with Western diet, Acat1−M/−M mice contain fewer infiltrating macrophages in white adipose tissue (WAT), with significantly diminished inflammatory phenotype. Without Acat1, the Ly6Chi monocytes express reduced levels of integrin-β1, which plays a key role in the interaction between monocytes and the inflamed endothelium. Adoptive transfer experiment showed that the appearance of leukocytes from Acat1−M/−M mice to the inflamed WAT of wild-type mice is significantly diminished. Under Western diet, Acat1−M/−M causes suppression of multiple proinflammatory genes in WAT. Cell culture experiments show that in RAW 264.7 macrophages, inhibiting ACAT1 with a small-molecule ACAT1-specific inhibitor reduces inflammatory responses to lipopolysaccharide. We conclude that under Western diet, blocking ACAT1 in macrophages attenuates inflammation in WAT. Other results show that Acat1−M/−M does not compromise antiviral immune response. Our work reveals that blocking ACAT1 suppresses diet-induced obesity in part by slowing down monocyte infiltration to WAT as well as by reducing the inflammatory responses of adipose tissue macrophages.
Keywords: ACAT, cholesterol, diabetes, macrophage, obesity
INTRODUCTION
Macrophages are phagocytes that ingest foreign materials and cell debris and participate in various immune responses by producing and responding to cytokines. Macrophages are involved in various metabolic diseases, including atherosclerosis and obesity. In atherosclerosis, chronic inflammation causes macrophages to infiltrate from the blood vessels into the intimal region of the artery. Under dyslipidemia, the continuous accumulation of cholesterol in macrophages causes foam cell formation, which is recognized as one of the hallmarks of atherosclerosis. Different from atherosclerosis, which occurs primarily within the arterial wall, obesity primarily affects the adipose tissue (AT). Obesity can lead to insulin resistance and type 2 diabetes. In early stages of obesity, lipid overload and chronic, low-grade inflammation cause fat cells in AT to increase in size and in number. Studies in animal models show that, similar to early stages of atherosclerosis, in diet-induced obesity, the number of macrophages infiltrated to the adipose tissue (ATM) increases significantly (56, 58). Both the number and the phenotype of ATM affect the characteristics of fat cells (11, 39).
In macrophages and other cell types, massive buildup of free (unesterified) cholesterol within cells can be cytotoxic (52, 54). To prevent its buildup, the excess cellular cholesterol can be removed from the cells by the sterol efflux pathway (45); alternatively, it can be retained within cells by converting to cholesteryl esters. The cholesterol esterification process is carried out by the enzymes acyl-coenzyme A:cholesterol acyltransferases (ACATs), also called sterol O-acyltransferases (9). There are two ACAT genes, Acat1 and Acat2 (5, 8, 47). Most tissues and cells including macrophages express ACAT1 as the major isoenzyme and express ACAT2 as the minor isoenzyme (48). Pharmacological studies in mouse models showed that partial inhibition of both ACAT1 and ACAT2 (30) or of ACAT1 only (22) reduces foam cell content in plaques and benefits treatment against atherosclerosis. However, genetic studies show that global Acat1 knockout (KO) in atherosclerotic mouse models causes several undesirable phenotypes, including hair loss, dry eye, and cutaneous xanthomatosis (1, 59); whether the global Acat1 KO decreases the lesion size is controversial (1, 13, 59). Why global Acat1 KO may not decrease the lesion size remains unclear; however, we (20) have recently shown that global Acat1 KO increases proliferation of hematopoietic stem cells in bone marrow (BM) and leads to leukocytosis in mice. It is thus possible that in the global Acat1 KO mouse leukocytosis contributes to lesion size enlargement and acerbates atherosclerosis. To study the roles of ACAT1 in macrophages, we (21) generated a myeloid-specific Acat1 KO mouse model (Acat1−M/−M) and showed that in the apolipoprotein E (Apoe) KO mouse model for atherosclerosis, unlike global ACAT1 deficiency, myeloid ACAT1 deficiency reduces lesion macrophage content, decreases lesion size, and suppresses atherosclerosis progression; additional studies showed that myeloid ACAT1 deficiency causes paucity of the inflammatory monocytes/macrophages at the activated endothelium. Since migration of inflammatory monocytes/macrophages to the inflamed adipose tissue is also a key step that affects obesity and type 2 diabetes, we suspect that myeloid ACAT1 deficiency may also benefit treatment against diet-induced obesity. In the current study, we designed experiments to test this hypothesis and report our findings.
MATERIALS AND METHODS
Mice.
Wild-type (WT), global Acat1−/− [total Acat1 knockout generated by Dr. Robert V. Farese, Jr., at University of California, San Francisco; received in C57BL/6J background from Dr. Sergio Fazio at Vanderbilt (13)], Acat1flox/flox, and Acat1−M/−M (myeloid-specific Acat1 knockout) mice (21) were all in C57BL/6J background, housed in a specific pathogen-free barrier facility under regular light-dark cycle, and fed with standard chow until they reached 8 wk old. Mice were continuously fed with regular chow diet (0.02% cholesterol; 13.5 kcal% from fat; 2018; Harlan) or with Western diet (rich in lard and in soybean oil; 60 kcal% from fat; D12492; Research Diets) for 8–19 wk as indicated. Unless indicated otherwise, only male mice were used. Animal care and procedures were performed in accordance with the guidelines of and were approved by the Dartmouth College Institutional Animal Care and Use Committee. Mouse genotype analysis was as described in Ref. 21.
Measurement of blood glucose and insulin concentration.
Blood glucose of mice fasted for 8 h was measured by using a blood glucose meter (Accu-Chek Aviva model via inserted 1-time-use strips) with tip insertion from the tail veins. Blood insulin levels were measured by using the ELISA kit (Crystal Chem) with a Luminex 200 machine (Millipore) as the readout.
Glucose tolerance tests.
Glucose levels in mice fasted for 8 h (fasting started at 10 AM) were measured and used as basal blood glucose levels. Starved mice were subjected to peritoneal glucose injection (1.5 g/kg; Sigma); blood glucose was monitored at 15, 30, 60, 90, and 120 min after injection. Glucose tolerance tests were evaluated quantitatively by measuring areas under the curve.
Stromal vascular cell isolation.
The stromal vascular cell fractions (SVF) of white fats were isolated as previously described (29). Briefly, gonadal adipose tissue was excised and minced in DMEM/F-12 (1:1) medium (GIBCO, Invitrogen) containing 1 mg/ml collagenase (type I; Worthington Biochemical) in a 37°C water bath for 30 min with gentle shaking. Nondigested tissues including cell debris were filtered using a 40-µm nylon sieve; the filtrates containing SVF were centrifuged at 1,000 rpm (5 min) at room temperature to separate floating adipocytes. The nonadipocyte layers including SVF were collected and further centrifuged at 3,000 rpm for 5 min at room temperature. The SVF was washed one time with PBS containing 3% FBS and treated (10 min) with 1-ml red blood cell lysis buffer (eBioscience) at room temperature for 10 min and washed again with PBS containing 3% FBS. After washing, cell suspension of the SVF was used for flow analysis.
Analysis of gonadal fat tissue sections with H&E staining and immunohistochemical staining.
Gonadal fat tissue was fixed in 10% formalin for 24 h, dehydrated, and embedded in paraffin. The tissue sections and hematoxylin-eosin (H&E) staining were done through Dartmouth core facility (http://cancer.dartmouth.edu/researchers/pathology-translational-services.html). Quantification of area of fat cells was calculated by using ImageJ from 20 individual adipocytes randomly selected from each slide for chow-fed mice and 50 individual adipocytes randomly selected from each slide for Western diet-fed mice; 5 slides were randomly chosen from each male mouse, with 4 mice per group. For the detection of crownlike structures in adipose tissue, immunohistochemistry was performed using deparaffinized tissue sections (5 µm) that were incubated in blocking buffer for 1 h (1% BSA, 10% normal goat serum in PBS) and permeabilized with 0.1% Triton X-100. The sections were incubated in blocking buffer containing primary antibodies of F4/80 biotin (Invitrogen) for 30 min. Endogenous horseradish peroxidase activity was quenched by incubation with 3% hydrogen peroxide for 10 min. The primary antibodies were detected by incubation with streptavidin-horseradish peroxidase. Sections were counterstained with H&E before dehydration and mounting to measure fat cell size. Quantification of the crownlike structure number is from five slides randomly chosen from each male mouse for a total of four mice per group.
Analysis of mRNAs of various genes associated with inflammation in gonadal and brown fat tissues.
Gonadal and brown adipose tissues were harvested and stored in liquid nitrogen. Total RNA was isolated using the RNeasy Plus Universal Kit (QIAGEN) according to the manufacturer’s protocol. To prevent DNA contamination, an on-column DNase treatment using the 5 Prime RNase-Free DNase kit was performed. The reverse-transcription step involved utilizing the Bio-Rad iScript cDNA Synthesis Kit to synthesize cDNA from 1 µg of RNA. Quantitative PCR analysis of SYBR Green reactions was performed on an Applied Biosystems StepOne Real-Time PCR System. The ΔΔCT comparative threshold cycle method was used to calculate target gene expression relative to hypoxanthine-guanine phosphoribosyltransferase. Quantitative PCR results for white adipose tissue (WAT) and brown adipose tissue (BAT) were normalized to mRNA levels from chow-fed WT mice. The primer sequences for various genes associated with WAT inflammatory process were according to Ref. 61. The primer sets used for analyzing M2-like genes were according to Refs. 29 and 35.
Measurement of leukocyte migration into gonadal fat tissue in vivo.
This procedure was based on the method described in Ref. 38 with modification. WT and Acat1−M/−M leukocytes (CD11b+ cells) were isolated from BM by using a cell sorter and labeled with two different fluorescent dyes by using the cell-surface cross-linking reagents that carry two different fluorescent colors (PKH67GL and PKH26GL from Sigma). After labeling, equal numbers of monocytes from WT and Acat1−M/−M BMs were mixed and served as the donor. The monocytes were transferred at 4 × 106 cells per mouse into recipient WT mice fed with either chow diet or Western diet as indicated. After 16 h, gonadal fats were isolated, and the percentages of infiltrated fluorescent-labeled WT or Acat1−M/−M cells present in SVF were examined by using flow cytometry.
Flow cytometry.
Methods for cytometry analysis relating to blood and bone marrow studies were as described in Ref. 20. Cells isolated from SVF were used to examine macrophages present in the fat tissue. In these experiments, we stained macrophages with antibodies against cell surface antigens, including F4/80-APC, CD206-PerCP, CD11c-FITC, CD301-PE, CD11b-APC/Cy7, and CD86-PE/Cy7, or matching isotype control antibodies, all from BioLegend; Siglec-F PE was from BD Biosciences. Cells were also stained with a primary antibody to NIMP-R14 (Santa Cruz Biotechnology) and detected using FITC-conjugated anti-rat IgG (Santa Cruz Biotechnology). Cells isolated from blood were used to examine the expression levels of integrins expressed on Ly6Chi monocyte subsets by using the cell-surface antigens including CD29-PE, CD11c-FITC, and CD11b-PE, all from BioLegend. For flow analysis, cells were stained with these antibodies at room temperature for 15 min and washed once with PBS containing 3% FBS. Flow cytometry analysis was performed by using a BD FACSCanto flow cytometry (BD Biosciences). Data were analyzed with FlowJo software (Tree Star).
LPS treatment of RAW 264.7 macrophages.
RAW 264.7 murine macrophages maintained in DMEM + 10% heat-inactivated calf serum were plated at 5 × 105 cells/ml in 12-well dishes (used for RT-PCR analysis) and 6-well dishes (used for Western blot analysis). Cells were treated with 1 ng/ml LPS (cat. no. 437627; Calbiochem) with or without the addition of the ACAT1-specific inhibitor K604 (0.5 µM in DMSO; a research gift from Kowa Pharmaceuticals) for 8 h (collected for RT-PCR analysis) or 24 h (collected for Western blot analysis). DMSO treatment served as the control. To analyze mRNA levels of inflammatory genes, the cells were directly lysed with TRIzol for RNA extraction. The RNA was extracted according to the manufacturer’s protocol (Thermo Fisher Scientific). β-Actin was used as the housekeeping gene when calculating the relative gene expression by the ΔΔCT method. The data for K604-, LPS-, and LPS + K604-treated samples were normalized to the mRNA levels from control (DMSO treated) cells. For Western blot analysis, the cells treated without or with 1 ng/ml LPS, without or with K604 for 24 h, were washed two times with PBS and then lysed in 1× radioimmunoprecipitation assay buffer supplemented with protease inhibitor. The cell lysates were sonicated on ice for 5 s (duty cycle 3, pulsed on 50% output) and then centrifuged for 5 min at 1,500 rpm. The protein concentrations of the supernatants were determined by the Lowry method. Lysates were run on 10% gels and transferred to a nitrocellulose membrane for 4 h at 400 mA. After blocking with 5% milk, the membranes were blotted for proteins using various primary antibodies, including inducible nitric oxide synthase (iNOS; Abcam), cyclooxygenase 2 (COX2; Santa Cruz Biotechnology), and β-tubulin (Santa Cruz Biotechnology).
Isolation and culture of bone marrow-derived macrophages.
Bone marrow-derived macrophage (BMDM) isolation protocol was essentially according to the method of Ref. 40. In brief, bone marrow was flushed from femurs and tibias of WT and Acat1−M/−M mice with DMEM using a 25-gauge needle. The cells were then passed through a 100-µm cell strainer (Fisher Scientific), treated with red blood cell lysis buffer (BioLegend), and centrifuged. Bone marrow from 3 mice of the same genotype were pooled together and plated in a 6-well dish. The cells were maintained in DMEM +10% calf serum + 20% L929-conditioned media containing macrophage colony-stimulating factor in a 37°C incubator for 7 days (with 1 media change on day 3). The L929 cells (a gift from Dr. Brent Berwin at Dartmouth) were cultured in DMEM + 10% calf serum.
AcLDL treatment and free cholesterol analysis.
To prepare the acetylated low-density lipoprotein (AcLDL), we employed methods described in Ref. 2. In short, acetyl modified LDL was prepared by slowly adding acetic anhydride to LDL at 5 µl/mg LDL for 1 h with continuous stirring on ice. After mixing for an additional 30 min, the acetyl-LDL was then dialyzed in 12 liters of PBS at 4°C for 24 h. For experiments employing the RAW 264.7, 100 µg/ml AcLDL (±0.5 µM K604) was added 24 h after seeding. The cells were harvested after a 48-h incubation. Similarly, differentiated BMDM from WT and Acat1−M/−M mice were incubated in DMEM +10% calf serum with L929-supplemented media with 100 µg/ml AcLDL (±0.5 µM K604) and then collected after a 48-h incubation. Following the AcLDL treatment, the cells were washed two times with PBS and pelleted. The total cellular lipids were extracted from pellets by using the Bligh and Dyer method and then dried down under N2. The lipids were resuspended in ethanol, spotted onto a 96-well dish, and analyzed for free cholesterol content according to the manufacturer’s protocol (Wako Free Cholesterol Kit E).
Body composition and food intake measurements.
The effects of chow- or Western diet-fed mice on body composition were monitored by noninvasively measuring whole body fat and lean mass using 1H-magnetic resonance spectroscopy (Echo Medical Systems, Houston, TX). The food intake was measured using metabolic cages (TSE Systems, Chesterfield, MO).
Hyperinsulinemic-euglycemic clamp to assess insulin sensitivity in conscious mice.
Hyperinsulinemic-euglycemic clamp was conducted in conscious mice as previously described (31), with a primed and continuous infusion of human insulin (150 mU/kg body wt priming followed by 2.5 mU·kg−1·min−1; Novolin; Novo Nordisk). To maintain euglycemia, 20% glucose was infused at variable rates during clamps. Whole body glucose turnover was assessed with a continuous infusion of [3-3H]glucose (PerkinElmer), and 2-deoxy-d-[1-14C]glucose (2-[14C]DG) was administered as a bolus (10 µCi) at 75 min after the start of clamps to measure insulin-stimulated glucose uptake in individual organs. At the end of the clamps, mice were anesthetized, and tissues were taken for biochemical analysis.
Biochemical assays.
Glucose concentrations during clamps were analyzed by using 10 µl of plasma by a glucose oxidase method on an Analox GM9 Glucose Analyser (Analox Instruments, Hammersmith, London, United Kingdom). Plasma concentrations of [3-3H]glucose, 2-[14C]DG, and 3H2O were determined following deproteinization of plasma samples as previously described. For the determination of tissue 2-[14C]DG-6-phosphate (2-[14C]DG-6-P) content, tissue samples were homogenized, and the supernatants were subjected to an ion exchange column to separate 2-[14C]DG-6-P from 2-[14C]DG. Insulin-stimulated glucose uptake in individual tissues was assessed by determining the tissue content of 2-[14C]DG-6-P and plasma 2-[14C]DG profile.
Calculation.
Rates of basal hepatic glucose production (HGP) and insulin-stimulated whole body glucose turnover were determined as described previously (28). Insulin-stimulated rate of HGP during the clamp was determined by subtracting the glucose infusion rate from whole body glucose uptake. Whole body glycolysis and glycogen and lipid synthesis from glucose were calculated as described previously. Insulin-stimulated glucose uptake in individual tissues was assessed by determining the tissue (e.g., skeletal muscle, heart) content of 2-[14C]DG-6-P and plasma 2-[14C]DG profile.
Vaccinia primary infection, plaque assay for measuring viral load, and vaccinia virus neutralization assay.
Primary infection (1,000 plaque-forming units) with vaccinia virus, Western Reserve strain (VV-WR), was administered via the intranasal route under anesthesia with isoflurane, according to procedure described in Ref. 19. To assess virus load in lung after primary vaccinia infection, lung tissues taken at day 10 postinfection were homogenized, diluted, and added onto 143B cells in 12-well plates. Cells and virus were incubated for 1 h at 37°C to allow absorption before addition of 2 ml of prewarmed medium containing carboxymethylcellulose. Cells were incubated at 37°C for 2 days and then fixed with methanol-acetone and stained with Giemsa stain. Plaque number was counted, and the viral titer was calculated by multiplying the relevant dilution. Serum was taken at day 10 postinfection and diluted to different ratio with cell medium. Vaccinia virus was added into serum samples and incubated for 1 h. Virus-serum mixture was added to 143B cells in 12-well plates. Viral plaque assay was performed as described above.
Statistics.
Results are reported as means ± SE. Two-tailed parametric Student’s t-test and two-way ANOVA with Bonferroni multiple-comparison posttest were used to evaluate the statistical significance among various study groups. Analysis was performed and plotted by using GraphPad Prism 5.0.
RESULTS
Global Acat1−/−, but not Acat1−M/−M mice, affects hematopoietic recovery.
We had previously shown that unlike global Acat1−/−, Acat1−M/−M does not increase cell proliferation in BM. To document this finding further, here we compared the rate of hematopoietic recovery in WT, global Acat1−/−, and Acat1−M/−M mice after a single injection of the myelosuppressive agent 5-fluorouracil (5-FU), which rapidly eliminated myeloid cells within BM (32). The result showed that, as expected, on day 0 before injection, global Acat1−/− mice, but not Acat1−M/−M mice, display higher levels of myeloid cells than WT mice. On day 6 postinjection of 5-FU, almost all of the myeloid cells in three genotypes were depleted. During the 6–21 days postinjection period, 5-FU was cleared from the body, and global Acat1−/− mice, but not Acat1−M/−M mice, consistently displayed increased recovery in myeloid cells compared with the WT mice (Fig. 1). This result indicates that global Acat1−/−, but not Acat1−M/−M, boosts myeloid cell proliferation de novo. These data strengthen our previous findings and show that, unlike global Acat1−/−, Acat1−M/−M does not affect hematopoiesis in BM.
Fig. 1.
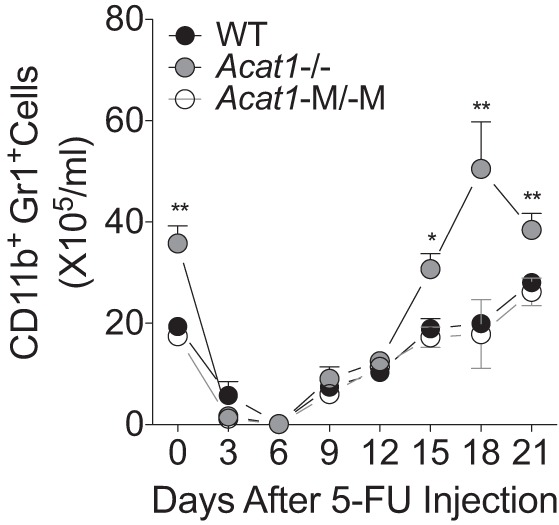
Global total Acat1 knockout mouse model (Acat1−/−), but not myeloid-specific Acat1 knockout mouse model (Acat1−M/−M), increases hematopoietic recovery. On day 0, wild-type (WT), Acat1−/−, and Acat1−M/−M mice were treated with 5-fluorouracil (5-FU; 200 mg/kg ip injection); the number of blood myeloid cells (CD11b+Gr1+) was measured at various time points as indicated. Eight-week-old mice with comparable numbers in male and female populations were used (n = 18–22 per group). Results reported are means ± SE. *P < 0.05; **P < 0.01.
Acat1−M/−M mice are resistant to diet-induced obesity.
We first performed paired studies to monitor food/water intake in age-matched male WT and Acat1−M/−M mice fed with 10-wk chow or 16-wk Western diet. The results of the metabolic cage study (Table 1) showed that on either diet, daily food/water intake between the two genotypes was not significantly different. We next monitored the weight gain in male WT and Acat1−M/−M mice fed with chow or Western diet starting at 8 wk of age. The results showed that, under chow diet for ≤20 wk, there was no significant difference in body weight or in fasting glucose levels between two genotypes (Fig. 2A, left and right). In contrast, under Western diet for ≤16 wk, Acat1−M/−M mice gained less weight (by ≤15%) compared with WT mice (Fig. 2B, left). In WT mice, Western diet feeding for 8–10 wk increased the fasting blood glucose level significantly; such increase was much smaller in the Acat1−M/−M mice (Fig. 2B, right). In the experiment that involved Western diet, we also included global Acat1−/− mice; the results show that unlike the Acat1−M/−M mice, Western diet caused the global Acat1−/− mice to gain more weight (by ≤35%) and to have more elevated fasting blood glucose levels than the WT mice (by ≤68%; Fig. 2B). This result further supports the notion that it is important to use Acat1−M/−M mice to gain a better understanding of ACAT1 function in monocytes/macrophages. Next, to determine the whole body lean and fat mass, WT and Acat1−M/−M mice fed with chow diet for 10 wk or fed Western diet for 10 or 16 wk were analyzed by using 1H-magnetic resonance spectroscopy. The results show that on chow diet, no detectable difference existed between WT and Acat1−M/−M mice in their body composition (Fig. 2C). Following Western diet for 10 or 16 wk, the Acat1−M/−M and WT mice showed comparable amounts of lean mass (Fig. 2C, left), but Acat1−M/−M mice showed significantly less fat mass than the WT mice (Fig. 2C, right; by 37% in 10 wk and 15% in 16 wk), demonstrating that the lower body weight of Acat1−M/−M mice is largely due to reduced adiposity in these mice.
Table 1.
Food and water intake
| Chow |
Western for 16 wk |
||||
|---|---|---|---|---|---|
| Parameter | Cycle | WT | Acat1−M/−M | WT | Acat1−M/−M |
| Food intake, g | Total | 4.6 ± 0.2 | 4.9 ± 0.3 | 2.0 ± 0.1 | 2.0 ± 0.1 |
| Light | 1.3 ± 0.1 | 1.5 ± 0.1* | 0.5 ± 0.1 | 0.8 ± 0.1† | |
| Dark | 3.3 ± 0.2 | 3.4 ± 0.2 | 1.5 ± 0.1 | 1.3 ± 0.1* | |
| Daily water intake, ml | Total | 3.6 ± 0.2 | 3.1 ± 0.1* | 1.5 ± 0.1 | 0.9 ± 0.1* |
| Light | 0.8 ± 0.1 | 0.7 ± 0.1 | 0.4 ± 0.1 | 0.3 ± 0.0 | |
| Dark | 2.8 ± 0.1 | 2.5 ± 0.1* | 1.1 ± 0.1 | 0.6 ± 0.1* | |
Results are presented as means ± SE for a 3-day measurement with 10 mice per group. Measurement of food/water intake, using TSE Systems metabolic cages with male mice fed with 10-wk chow or 16-wk Western diet. Acat1−M/−M, myeloid-specific Acat1 knockout mouse model; WT, wild-type.
P < 0.05;
P < 0.01.
Fig. 2.

Myeloid-specific Acat1 knockout (Acat1−M/−M) mice are resistant to Western diet-induced obesity and exhibit improved glucose tolerance. A: male wild-type (WT) and Acat1−M/−M mice on chow diet have comparable body weights (left) and fasting glucose levels (right; n = 8 per group). B: male Acat1−M/−M mice on Western diet are resistant to weight gain and have lower fasting glucose levels than WT and global total Acat1 knockout mouse model (Acat1−/−) mice. Body weight (left) and blood glucose levels (right) of mice fed with Western diet were measured at the indicated times (n = 6–14 per group). For A and B, glucose fasting time was 8 h. C: male Acat1−M/−M mice on Western diet have less fat mass but have the same lean mass. Lean and fat mass in WT and Acat1−M/−M mice on chow diet for 10 wk or on Western diet for 10 or 16 wk as indicated were measured (n = 10 per group). D: glucose tolerance test in male (♂; left) or female (♀; right) WT and Acat1−M/−M mice on Western diet for 10 wk (n = 5–9 per group). AUC, area under the curve. E: the fasting blood insulin levels of male WT and Acat1−M/−M mice on chow or 8 wk on Western diet before and after refeeding were measured by ELISA (n = 7 per group). Results are means ± SE. *P < 0.05; **P < 0.01; ***P < 0.001.
Acat1−M/−M mice under Western diet display improved glucose tolerance and lower blood insulin levels.
We next performed glucose tolerance tests using a procedure described previously (12, 25) in male WT and Acat1−M/−M mice fed with Western diet for 10 wk. Our data showed that following intraperitoneal injection of glucose, blood glucose levels in Acat1−M/−M mice were significantly lower compared with those in the WT mice (Fig. 2D, 1st panel). Similar results were found in female mice, but the difference between the two genotypes was smaller (Fig. 2D, 3rd panel). Consistent with these results, in both male and female Acat1−M/−M mice, area under the curve of blood glucose levels was significantly smaller than those of the WT mice (Fig. 2D, 2nd and 4th panels), suggesting that under Western diet Acat1−M/−M mice have improved glucose tolerance. We also noted that the chow-fed Acat1−M/−M mice had a trend toward decreased fasting blood insulin levels (Fig. 2E, 1st 2 bars). In addition, following 8 wk of Western diet, the fasting blood insulin levels in the Acat1−M/−M mice were significantly lower (by >50%; Fig. 2E, 3rd and 4th bars) than WT mice. After fasting and refeeding, both genotypes had large increases in blood insulin levels to a very similar level (Fig. 2E, 5th and 6th bars), suggesting that the insulin secretion capacity in pancreatic β-cells was not affected by Acat1−M/−M. Together, these results show that on Western diet, Acat1−M/−M improves blood glucose tolerance and insulin sensitivity.
Acat1−M/−M mice display increased insulin sensitivity in hyperinsulinemic-euglycemic clamp study.
We next performed hyperinsulinemic-euglycemic clamp study in male WT and Acat1−M/−M mice fed with chow or Western diet for 10 wk. The results show that on both diets, the Acat1−M/−M mice exhibit higher glucose infusion rates during clamps than the WT mice (Fig. 3A), indicating that the Acat1−M/−M mice are more insulin-sensitive than WT mice. Additional analyses showed that insulin-stimulated whole body glucose turnover rates are also significantly higher in the Acat1−M−M mice (Fig. 3D). Whole body glycolysis rates tend to be higher in Acat1−M/−M mice (Fig. 3E), but whole body glycogen synthesis rates do not differ between these 2 genotypes (Fig. 3F). Results of basal hepatic glucose production (HGP) rates show no significant difference between the 2 genotypes on chow (Fig. 3B, 1st 2 bars); however, following Western diet, basal HGP rates are lower in Acat1−M/−M mice (Fig. 3B, 3rd and 4th bars), which is consistent with lower fasting glucose levels in these mice shown earlier (Fig. 2B, right). Hepatic insulin action (insulin-mediated percentage suppression of basal HGP) between these 2 genotypes is comparable (Fig. 3C), indicating that Acat1−M/−M may not affect diet-induced insulin resistance in the liver. Insulin-stimulated glucose uptake in individual organs was measured by using [14C]2-deoxyglucose injection during clamps. The results show that on chow diet, insulin-stimulated glucose uptake in skeletal muscle was significantly elevated in the Acat1−M/−M mice (Fig. 3G). Following Western diet for 10 wk, insulin-stimulated glucose uptake in brown adipose tissue was significantly elevated in the Acat1−M/−M mice (Fig. 3H, right). At present, we are unable to provide a simple rationale to explain the interesting but puzzling observations described in Fig. 3, G and H. Nevertheless, overall, the studies in Fig. 3 support the notion that Acat1−M/−M mice display increased insulin sensitivity and glucose metabolism mainly in peripheral organs in vivo. These results cannot rule out the possibility that Acat1−M/−M may also have secondary effects in the liver.
Fig. 3.

Myeloid-specific Acat1 knockout (Acat1−M/−M) mice improve glucose homeostasis through increased peripheral insulin sensitivity. Hyperinsulinemic-euglycemic clamp was performed to measure insulin sensitivity and glucose metabolism in live male wild-type (WT) and Acat1−M/−M mice on chow or Western diet for 10 wk. A–F: various parameters as indicated to measure whole body glucose metabolism. G: insulin-stimulated glucose uptake in different regions of skeletal muscles as indicated. H: insulin-stimulated glucose uptake in white adipose tissue (WAT) or in brown adipose tissue (BAT) as indicated. Results are means ± SE. n = 7–11 Per group. *P < 0.05.
Acat1−M/−M mice display decreased adipocyte size.
We next examined whether Acat1−M/−M also affects adipocyte cell size. Gonadal fat pads from the WT and Acat1−M/−M mice on chow or Western diet for 8 wk were isolated, and adipocyte size was measured after H&E staining. The results showed that, on both chow and Western diet (Fig. 4A), the adipocyte size in Acat1−M/−M mice was significantly decreased. We next performed quantitative analyses of fat cell size (as adipocyte cross-sectional area in square micrometers). The result (reported as means ± SE) showed that on chow diet (Fig. 4B), the value was 2,971 ± 802 for WT and 1,712 ± 566 for Acat1−M/−M; on Western diet (Fig. 4C), the value was 5,449 ± 2,398 for WT and 3,577 ± 1,180 for Acat1−M/−M.
Fig. 4.

Myeloid-specific Acat1 knockout (Acat1−M/−M) decreases adipocyte size in chow- and Western-fed mice and reduces the adipose tissue crownlike structure (CLS) in Western-fed mice. Wild-type (WT) and Acat1−M/−M male mice fed with chow or Western diet for 8 wk were used. A: histogram of gonadal fat pad adipocytes and CLS, identified in dark red color. Scale bar = 50 μm. B and C: quantification of fat cell size based on adipocyte-size histograms from mice on chow diet (B) or on Western diet (C). For adipocyte area, the box on the top right represents the upper and lower quartiles, the whiskers or bars indicate the 5th or 95th percentiles, and the line in the box represents the median. D: quantification of CLS number per slide from mice on Western diet. HPF, high-power field. Results are means ± SE. n = 4 Per group. ***P < 0.001.
Acat1−M/−M mice fed with Western diet contain less adipose tissue macrophage number with altered polarization.
During the onset of high-fat-diet-induced obesity, macrophages infiltrating the AT are arranged around dead adipocytes and form crownlike structures (Ref. 33). Through the course of fat cell size analyses described above, we noted that when fed with Western diet, gonadal fat pads from Acat1−M/−M mice contained significantly less crownlike structures than those in WT mice (by ~50%; Fig. 4A, right, and Fig. 4D), suggesting that on Western diet, Acat1−M/−M decreases the ATM number. To validate this finding further, we utilized immunohistochemical (IHC) analysis to compare the accumulation of F4/80-positive macrophages within WAT, BAT, and muscle tissues isolated from 8-wk Western diet-fed WT and Acat1−M/−M mice. The results (Fig. 5, A and B) showed that in WAT, Acat1−M/−M mice exhibited a marked reduction in macrophage number; in BAT, the macrophage numbers were comparable between the two genotypes. The result present in BAT needs to be qualified because the IHC antibody stain (brown color) in tissue might be partially masked by the brown color of BAT. In muscles, Acat1−M/−M mice might contain more macrophages than the WT mice, but the difference did not reach statistical significance. We next performed flow cytometry analysis, which demonstrated that, when fed with Western diet, the number of F4/80-positive cells present in the stromal vascular fraction (SVF) of WAT from Acat1−M/−M mice was significantly reduced compared with those of WT mice (Fig. 5C, 3rd and 4th panels). When fed with chow diet, Acat1−M/−M mice showed a trend toward decrease in ATM, but the difference between the 2 genotypes did not reach statistical difference (Fig. 5C, 1st and 2nd panels).
Fig. 5.

Myeloid-specific Acat1 knockout (Acat1−M/−M) mice fed with Western diet contain less adipose tissue macrophages with M2-like polarization. A: immunohistochemical analysis of F4/80-positive cells in in white adipose tissue (WAT), brown adipose tissue (BAT), and muscle tissues from Western-fed wild-type (WT) and Acat1−M/−M mice [brown color for F4/80 antibody staining, blue color for hematoxylin-eosin (H&E) staining]. B: quantification of F4/80-positive macrophages per slide. N.S., not significant. C: flow cytometry quantification of macrophages infiltrated to the adipose tissue (ATM) in the stromal vascular fraction (SVF) per gram gonadal fat pad. D and E: quantitation of M1-like (F4/80+CD11c+CD206−; D) or M2-like (F4/80+CD11c−CD206+; E) percentage of total F4/80+ macrophage within SVF of gonadal fat pad. F and G: quantitation of M2-like percentage of total F4/80+ macrophage within SVF of gonadal fat pad from Western-fed mice by using 2 additional markers (CD301 in F and CD86 in G). H: M2-like-to-M1-like ratio based on results shown in D and E. Results are means ± SE. For A–C, n = 4 per group; for D–F, n = 7 per group. *P < 0.05; **P < 0.01; ***P < 0.001. MFI, mean fluorescence intensity. I: representative flow cytometry image analysis with gating and with isotype controls. I1: after excluding cell debris, stromal vascular macrophages (F4/80+) were analyzed for expressions of CD206 vs. CD11c. FSC-A, forward scatter area; SSC-A, side scatter area. I2 and I3: analyses of stromal vascular macrophages from WT (I2) and Acat1−M−M mice (I3) on Western diet for 8 wk. I4: isotype controls for CD206 (left) and CD11c (right). Stromal vascular cells stained with F4/80, CD206, and CD11c isotype control (left) or F4/80, CD11c, and CD206 isotype control (right) were analyzed. Values obtained by using the isotype controls, representing the nonspecific fluorescence signal (due to Fc receptor-mediated binding and other nonspecific interactions), were subtracted as the background values in calculations.
Blood monocytes can be classified into ≥2 distinct phenotypic and functional subsets, Ly6Clo and Ly6Chi (16). The Ly6Chi circulating monocyte subset is thought to be the precursor of inflammatory macrophages recruited to inflamed tissues. Depending on the environment where they reside in, mature macrophages can adopt ≥2 different activation states, broadly classified as M1-like (i.e., proinflammatory) and M2-like [i.e., anti-inflammatory; the M1-like and M2-like classifications of macrophages are undergoing continuous revision (34)]. In obese adipose tissue, most macrophages are M1-like, secreting cytokines such as tumor necrosis factor-α and IL-6 that promote inflammation within the WAT and lead to decreased insulin sensitivity. On the other hand, the M2-like macrophages present in WAT in mice can play a beneficial role (35, 36). We asked whether Acat1−M/−M alters ATM polarization and examined this issue by using flow cytometry by employing an M1-like marker (F4/80+CD11c+CD206−) as well as an M2-like marker (F4/80+CD11c−CD206+; Ref. 17). Based on the markers used, the results showed that, on chow diet, ATM polarization between these two genotypes is similar (Fig. 5D). On Western diet, in WT mice, 42% ATM is M1-like, and 24% is M2-like phenotype, whereas in Acat1−M−M mice, 22% ATM is M1-like, and 46% is M2-like phenotype (Fig. 5E). The M2-like-to-M1-like ratio of ATMs is greatly increased in Acat1−M/−M mice on Western diet (Fig. 5H). The results shown in Fig. 5E were confirmed by using two other M2 markers [Fig. 5, F and G; M2a (CD301) and M2b (CD86); Ref. 46]. A representative flow cytometry image analysis with gating and isotype control is shown in Fig. 5I; this result demonstrated that the antibodies employed for CD206 and CD11c were specific. To summarize, on Western diet, Acat1−M/−M decreases the content of ATMs. Based on limited numbers of markers employed here, these results suggest that Acat1−M/−M alters ATM polarization to exhibit a less inflammatory phenotype.
Depletion of myeloid ACAT1 slows down leukocyte migration and reduces WAT inflammation.
Western diet causes accumulation of macrophages and elevated expressions of proinflammatory genes in WAT. The results described in Fig. 5 suggest that Acat1−M/−M may cause monocytes/macrophages to migrate to inflamed adipose tissue less efficiently in vivo; in addition, those macrophages residing in WAT tend to polarize to M2-like cells. To test the possibility that Acat1−M/−M reduces monocyte infiltration into ATMs, we performed adoptive transfer experiment to monitor leukocyte migration in vivo by using the method described by Oh and colleagues (38). Monocytes isolated from chow-fed WT or Acat1−M/−M mice were used as donors; WT mice on chow diet or on Western diet for 2 or 8 wk as indicated were used as the recipients. The results showed that, with chow-fed WT mice as recipients, ~0.3% of total cells in SVF can be attributed to donor monocytes, and no difference was detectable between the 2 donors (Fig. 6A, 1st 2 bars). When 2-wk Western diet-fed WT mice were used as recipients, percentage of donor monocytes present in SVF increased in both donor cells; percentage of Acat1−M/−M donor monocytes had a trend to be less than percentage of WT donor monocytes, but the difference was not statistically significant (Fig. 6A, 3rd and 4th bars). When 8-wk Western diet-fed WT mice were used as recipients, percentage of donor monocytes present in SVF increased even higher for both donors; under this condition, percentage of Acat1−M/−M donor was significantly less than percentage of WT donor (Fig. 6A, 5th and 6th bars). These results are consistent with the interpretation that Acat1−M/−M may slow down the migration of monocytes/macrophages to inflamed adipose tissue. Integrin-β1 (CD29) present in monocytes/macrophages plays a key role in facilitating the interaction between monocytes/macrophages and the activated endothelium, as reviewed in Ref. 23. Previously, we (21) reported that in an apolipoprotein E (Apoe) KO model for atherosclerosis, inhibiting ACAT1 enzyme activity directly decreased the CD29 protein content in activated macrophages. To test whether a similar situation occurs in a different disease model, here we monitored the cell-surface expressions of CD29 in Ly6Chi monocytes isolated from WT and Acat1−M/−M mice by flow cytometry (Fig. 6E) and showed that on Western diet but not on chow diet the monocyte expression of CD29 from Acat1−M/−M mice was decreased compared with WT mice (Fig. 6, C and F). In the same experiment, we also compared the percentage of Ly6Chi monocytes between these two genotypes and found that the values were the same on chow but were decreased in Acat1−M/−M mice on Western diet (Fig. 6B). Results of a control experiment showed that on both diets, the expression of the pan-macrophage marker CD11b remained the same between these two genotypes (Fig. 6, D and F). The gating strategy employed for flow cytometry analysis reported in Fig. 6B is shown in Fig. 6E. The result presented in Fig. 6F demonstrated that the antibodies employed for CD11b and for CD29 were specific, and the background values were minimal.
Fig. 6.

Myeloid-specific Acat1 knockout (Acat1−M/−M) retards leukocyte migration into inflamed wild-type (WT) adipose tissue. A: comparison of WT and Acat1−M/−M myeloid cell migration to the adipose tissue of WT mouse in vivo. Bone-marrow CD11b+ myeloid cells isolated from 8-wk-old WT and Acat1−M/−M mice fed with chow diet were labeled with the fluorescent probe PKH67 (green) or PKH26 (red), respectively. After labeling, cells were mixed and injected at 5 × 106 per mouse through intravenous injection into a recipient WT mouse on chow diet or on Western diet for 2 or 8 wk as indicated. After 16 h, the percentage of labeled CD11b+ donor cells within stromal vascular fraction (SVF) was quantified by flow cytometry. Results shown were averages of 3 separate experiments. B: quantitation of Ly6Chi monocytes present in blood leukocytes isolated from WT and Acat1−M/−M mice under chow or Western diet for 8 wk as indicated. C and D: analysis of integrin expressions of Ly6Chi monocytes isolated from WT and Acat1−M/−M mice on chow or on Western diet as indicated. Ly6Chi monocytes were isolated from mouse blood, and the expression levels of integrin CD29 (C) and CD11b (D) were quantified by flow cytometry (n = 15–20 per group). MFI, mean fluorescence intensity. *P < 0.05; **P < 0.01. E: gating strategy employed for flow cytometry analysis of Ly6Chi blood monocytes. After excluding cell debris, blood leukocytes (CD45+) were identified as 2 populations, CD11b+Ly6G− cells and CD11b+Ly6G+ cells (which correspond to neutrophils). After excluding the neutrophil population, the CD11b+Ly6G− cells were further selected for CD11b+CD115+ monocytes. CD45+Ly6G−CD11b+CD115+ monocytes were analyzed for Ly6C expression. CD45+Ly6G−CD11b+CD115+Ly6Chi monocytes were then selected and analyzed for the expression CD29 and CD11b. FSC-A, forward scatter area; SSC-A, side scatter area. F: flow cytometry analysis of CD29 and CD11b expression on Ly6Chi monocytes of WT and Acat1−M−M mice with isotype controls. Mice were on 8-wk Western diet. n = 15–20 Mice per genotype.
The altered phenotype of ATM lacking Acat1, as demonstrated in Figs. 5 and 6, may affect the overall inflammatory state of AT in these mice. To test this possibility, we prepared cell homogenates from gonadal and brown adipose tissues from WT and Acat1−M/−M mice under chow and Western diets and compared the mRNA levels of multiple genes known to be associated with adipose inflammation (18, 51, 57, 61). The results of the real-time PCR experiments showed that, as expected, in WAT of Acat1−M/−M mice under Western diet (but not chow diet) condition, multiple genes for various inflammatory cytokines, most notably CCL2 (monocyte chemotactic protein-1, MCP-1), CCL5 (regulated upon activation, normally T-expressed, and presumably secreted, RANTES), and CCL7 (MCP-3), were all significantly reduced (Fig. 7, WAT). Six other proinflammatory genes were also reduced, although the degrees of suppression were less obvious. Overall, these changes are consistent with the interpretation that Acat1−M/−M produces a less proinflammatory phenotype in WAT. Interestingly, under Western diet, the expression of adiponectin, an adipokine known to have antidiabetic action, was not reduced in WAT from Acat1−M/−M mice. Results of parallel experiments showed that under Western diet, the inflammatory gene expressions of BAT from Acat1−M/−M mice were also moderately reduced; however, except for CCL2, the differences between the two genotypes were not significant (Fig. 7, BAT). We noted that the data presented in Fig. 7, BAT, show that F4/80 mRNA is slightly elevated in Acat1−M/−M Western, whereas the data presented in Fig. 5, A and B, show that there is no difference in BAT F4/80 IHC staining between WT Western and Acat1−M/−M Western. We suspect that in Fig. 5, the IHC antibody stain (brown color) in tissue might be partially masked by the brown color of BAT. In future studies, a more appropriate approach would be to use immunofluorescence stain.
Fig. 7.

Depleting myeloid Acat1 influences inflammatory gene profiles of white adipose tissue (WAT) and brown adipose tissue (BAT) isolated from mice fed a Western diet. mRNA expression levels of various M1-like genes associated with inflammation were analyzed in gonadal WAT and BAT by RT-PCR. Tissues were collected from male wild-type (WT) or myeloid-specific Acat1 knockout (Acat1−M/−M) mice fed a chow or Western diet for 8 wk. Results are expressed as means ± SE. n = 8–10 Per group. CCL, C-C motif chemokine; Cxcl, C-X-C motif chemokine; Igfbp3, IGF-binding protein 3; IL1rl1, IL-1 receptor-like 1; IL4ra, IL-4 receptor α-chain; inos, inducible nitric oxide synthase; Lpsbp, LPS-binding protein; Saa3, serum amyloid A3; Soc3, suppressor of cytokine signaling 3; Timp1, tissue inhibitor of metalloproteinases 1; Tnfa, TNF-α. *P < 0.05; **P < 0.01.
Taken together, these results suggest that myeloid-specific abrogation of Acat1 attenuates diet-induced inflammatory response mainly in WAT, whereas the response in BAT to Acat1−M/−M seems secondary in nature. We next asked whether genes involved in anti-inflammatory response were also altered in our system and performed additional RT-PCR experiments to test this possibility. The results (Fig. 8) showed that under Western diet, certain genes involved in anti-inflammatory responses, including Arg1 and Erg2 but not other genes tested here, were downregulated; Acat1−M/−M restored the expression of Arg1, but the effect did not reach statistical significance. The results of Figs. 7 and 8 together implicate that blocking myeloid ACAT1 mainly suppresses the genes involved in inflammatory responses in WAT in vivo. The effect of Acat1−M/−M on promoting the expression of certain M2-like markers in ATM, shown in Fig. 5, E–H, may occur at the posttranscription level.
Fig. 8.
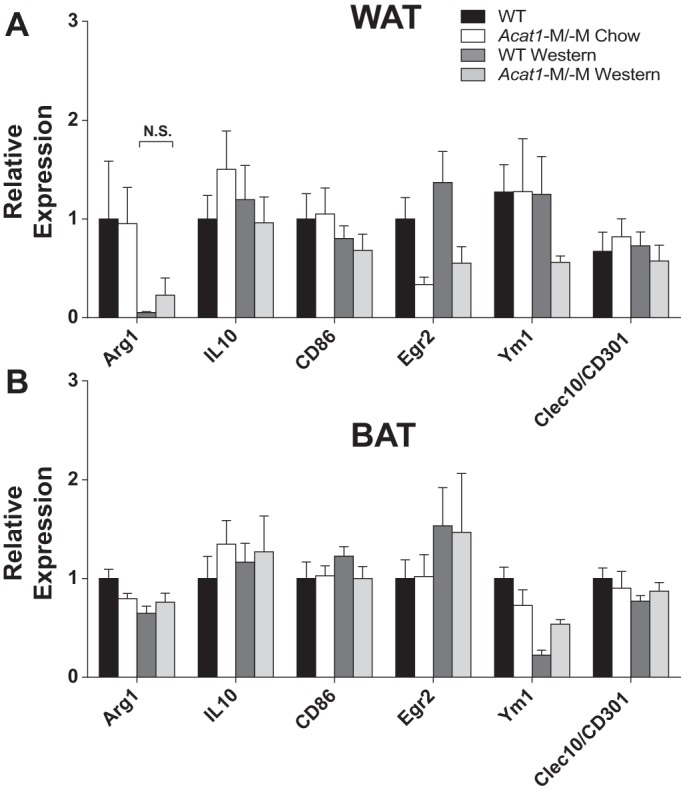
Depleting myeloid Acat1 does not influence the expressions of M2-like genes in white (WAT) and brown adipose (BAT) tissues isolated from mice fed a Western diet. mRNA expression levels of M2-like/anti-inflammatory genes were analyzed in gonadal WAT (A) and BAT (B) by RT-PCR. Tissues were collected from male wild-type (WT) or myeloid-specific Acat1 knockout (Acat1−M/−M) mice fed a chow or Western diet for 8 wk. Results are expressed as means ± SE. n = 8–10 Per group. Arg1, arginase-1; Clec10, C-type lectin domain family 10; Egr2, early growth response 2; N.S., not significant; Ym1, chitinase 3-like 3.
Inhibiting ACAT1 diminishes activation of multiple inflammatory genes in response to LPS in a macrophage cell line.
Cells in the myeloid lineage include monocytes, macrophages, dendritic cells, neutrophils, and eosinophils as well as microglia in the central nervous system. These cell types have all been implicated in the inflammatory process. We suspect that the less proinflammatory effects of Acat1−M/−M observed in Figs. 5–7 are at least in part through inhibiting ACAT1 in macrophages. We employed the mouse RAW 264.7 cell line to test this possibility. This cell line has been employed as a model cell for macrophages by many investigators. In our previous study (21), we had also used this cell line and showed that it behaves very similar to peritoneal macrophage in responses to ACAT1 blockage. Lipopolysaccharide (LPS) is an endotoxin from gram-negative bacteria and has been used extensively to induce inflammation in vitro and in vivo. Adding LPS to RAW 264.7 causes rapid activation of the proinflammatory transcription factors nuclear factor-κB and the activating protein-1; these effects lead to subsequent transcriptional activation of numerous genes associated with inflammatory responses (4, 44). We first performed pilot experiments and found that, consistent with results reported by others, LPS added to RAW 264.7 quickly stimulated the expressions of five genes associated with inflammatory response, iNOS, MCP-1, CCL5, CCL7, and COX2, in a concentration-dependent (1–100 ng/ml) and time-dependent manner (4–8 h; results not shown). To produce ACAT1 blockage in macrophages, we employed a small-molecule ACAT1-specific inhibitor, K604. In various cell types, when added directly to growth media at submicromolar concentrations, K604 enters the cells quickly and inhibits ACAT1 by >80% without significantly inhibiting ACAT2 (22). We treated RAW 264.7 grown in medium and 10% heat-inactivated bovine serum with K604 (at 0.5 µM) or with vehicle only for 18 h and then exposed these cells without or with LPS (at 1 ng/ml) for 8 or 24 h. Figure 9A shows the timeline of this experiment. Afterward, cells were harvested (at 8th hour) to monitor gene expressions of iNOS, MCP-1, CCL5, CCL7, and COX2 by performing RT-PCRs. The results showed that, without LPS, the expressions of iNOS, MCP-1, CCL5, CCL7, and COX2 in cells treated without or with K604 were all comparable. LPS led to large inductions of all five genes; pretreating cells with K604 led to a small but significant degree of suppression for all five genes examined. Quantitation of results shown in Fig. 9B showed that the percentage suppression by K604 was 44% for iNOS, 27% for MCP-1, 37% for CCL5, 38% for CCL7, and 34% for COX2. To validate the results of the RT-PCR experiment, after cells were not treated or treated with LPS for 24 h, we monitored the protein content expressions of iNOS and COX2 by performing Western blot analyses of cell homogenates. (We monitored the expressions of iNOS and COX2 in cell homogenates only, for fear that on LPS activation, significant amounts of MCP-1, CCL5, and CCL7 might be secreted out of the cells into the medium.) The results of Western blot analyses (Fig. 9) showed that without LPS, the expressions of iNOS and COX2 proteins in cells treated without or with K604 were very low. LPS treatment for 24 h led to very large increases in protein contents for both iNOS and COX2; treating cells with K604 led to significant suppression for both iNOS and COX2; the percentage suppression by K604 was 67% for iNOS and 64% for COX2. Taken together, these results show that in RAW 264.7, on acute activation with LPS, blocking ACAT1 with K604 suppresses the upregulation of multiple proinflammatory genes.
Fig. 9.

Inhibiting acyl-CoA:cholesterol acyltransferase 1 (ACAT1) with small-molecule ACAT1-specific inhibitor K604 reduces LPS-mediated inflammatory response in RAW 264.7 macrophages. A: timeline of K604 and LPS treatment of RAW 264.7 macrophages. B: effects of ACAT1 inhibition on M1-like/proinflammatory genes in RAW 264.7 maintained in DMEM + 10% calf serum for 18 h and then treated with or without 1 ng/ml LPS for 8 h. CCL, C-C motif chemokine; MCP1, monocyte chemotactic protein-1. C: effects of ACAT1 inhibition on inducible nitric oxide synthetase (iNOS) and cyclooxygenase 2 (COX2) protein levels in RAW 264.7 cells without or with 1 ng/ml LPS for 24 h. D: quantitative analysis by densitometry of results represented in C. Protein levels were normalized by using β-tubulin as the loading control, and the data were normalized to control (DMSO treated) samples. Data in D represent the average from 2 independent experiments (1 with a sample size of 3 and the other with a sample size of 2). Results are expressed as means ± SE. n = 5–6 Per group. *P < 0.05; **P < 0.01; ***P < 0.001.
Blocking ACAT1 in cholesterol-loaded macrophages does not increase total cellular free cholesterol content.
In a mouse model for diet-induced obesity, Prieur et al. (41) showed that macrophages in adipose tissue accumulated a significant amount of unesterified cholesterol inside the cells and became cholesterol-loaded cells (i.e., foam cells). In cholesterol-loaded macrophages and other immune cells, the lack of lipid efflux transporters such as ATP-binding cassette subfamily A member 1 (ABCA1) and ATP-binding cassette subfamily G member 1 (ABCG1) causes the accumulation of free cholesterol inside the cells; the buildup of cholesterol at the plasma membranes and other membranes of these cells rendered them highly proinflammatory (53). In contrast, our results presented in Figs. 5–9 show that unlike the ABCA1/ABCG1 blockage, ACAT1 blockage causes macrophages to become less proinflammatory in vivo and in vitro. Based on this comparison, we suspect that ACAT1 blockage may not cause an increase in cellular free cholesterol content. We employed two cell culture systems, bone marrow-derived macrophages (BMDM) isolated from WT and Acat1−M/−M mice as well as RAW 264.7-treated without or with K604, to test our prediction. To produce a cholesterol loading condition, these cells were treated with saturating concentration (100 µg/ml) of acetyl low-density lipoprotein (AcLDL) for 48 h. Afterward, cells were harvested for cellular free cholesterol content analyses. The results showed that in both BMDM (Fig. 10A) and RAW 264.7 (Fig. 10B), cholesterol loading by AcLDL caused a large increase in cellular free cholesterol contents (by ≥200%). However, blocking ACAT1 through Acat1 KO or through K604 inhibition does not cause detectable increase in cellular free cholesterol concentration in these cells. These results are in concordance with the early findings by Yamauchi and colleagues (60), who showed that blocking ACAT1 promotes sterol efflux by increasing the ABCA1 protein as well as by increasing the cholesterol pool destined for sterol efflux.
Fig. 10.

Acyl-CoA:cholesterol acyltransferase 1 (ACAT1) blockage does not alter the free (unesterified) cellular cholesterol content of macrophages treated with or without acetylated low-density lipoprotein (AcLDL). A: free cholesterol levels (measured by using Wako kit) of wild-type (WT) or myeloid-specific Acat1 knockout (A1KO) bone marrow-derived macrophages (BMDM) maintained in DMEM + 10% calf serum media supplemented with 20% L929 cell-conditioned media (contains macrophage colony-stimulating factor) for 7 days and then treated with 100 µg/ml AcLDL for 48 h (with or without simultaneously treating with K604 at 0.5 µM from a 5 mM stock in DMSO). Cells without K604 received DMSO at 0.1%. B: free cholesterol content of RAW 264.7 macrophages grown in DMEM + 10% heat-inactivated calf serum for 24 h and then treated without K604 or AcLDL (control, CT), with K604, with AcLDL, or with AcLDL and K604 for 48 h as indicated. AcLDL was used at 100 µg/ml; K604 was used at 0.5 µM from a 5 mM stock in DMSO; the cells without K604 treatment received DMSO only at 0.1%. Results are expressed as means ± SE. n = 5–6 Per group. *P < 0.05.
Acat1−M/−M does not affect systemic immunity on acute virus infection.
The results presented in Figs. 2–9 suggest ACAT1 in macrophages as a potential therapeutic target for treating diet-induced obesity and other related metabolic diseases. However, we were concerned that Acat1−M/−M may produce a mechanistic side effect by compromising innate immune response, especially on acute viral infection. We tested this possibility by infecting mice with vaccinia virus (VV-WR strain). The result showed that 10 days after virus infection, the viral titers (Fig. 11A) and the titers of serum antibodies neutralizing VV-WR (Fig. 11B) were very similar between these two genotypes, suggesting that Acat1 deletion in monocytes/macrophages does not compromise systemic immunity on acute virus infection.
Fig. 11.
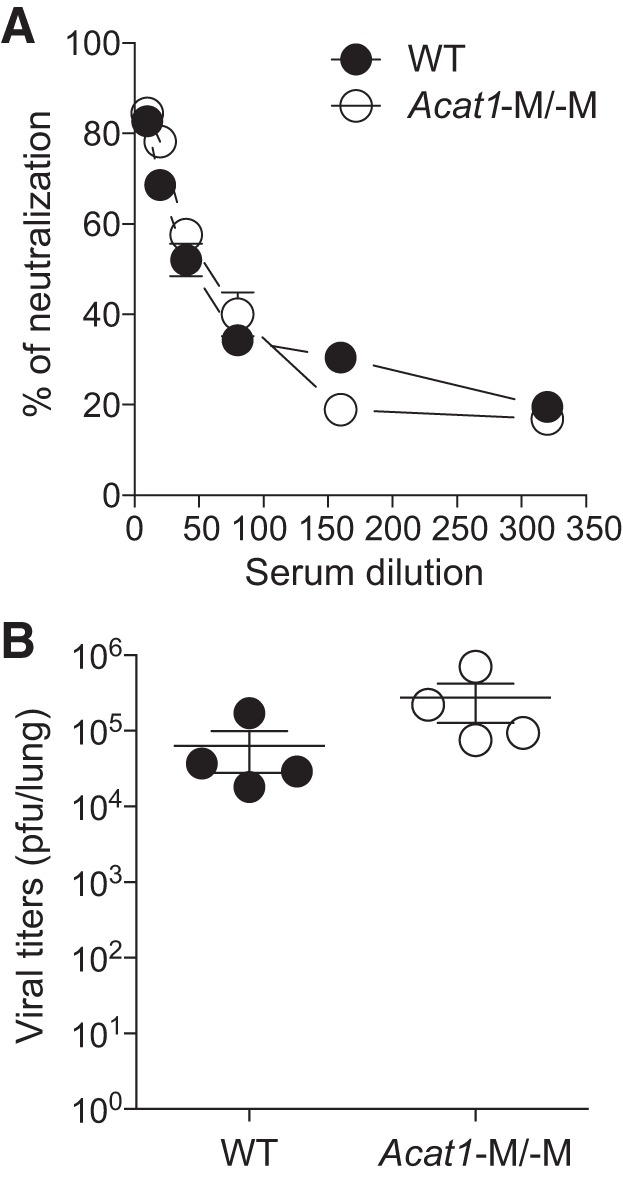
Myeloid-specific Acat1 knockout (Acat1−M/−M) does not affect systemic immune responses on acute vaccinia virus infection. At day 10 postinfection of 1,000 plaque-forming units (pfu) of vaccinia virus (Western Reserve strain), the virus load per lung was measured by plaque assay (A), and the serum from wild-type (WT) or Acat1−M/−M mice was tested by using the neutralization assay (B). In A, the data are represented in log scale. Results are means ± SE. Four mice in each group were used. *P < 0.05.
DISCUSSION
Here, we use a well-characterized myeloid-specific Acat1 KO mouse model (21, 50) to evaluate the roles of monocyte/macrophage ACAT1 in diet-induced obesity. The results show that on a Western diet, Acat1−M/−M causes mice to be more resistant to diet-induced obesity. Additional studies show that Acat1−M/−M causes two major alterations in activated monocytes/macrophages. First, it decreases the expression of CD29 on Ly6Chi monocytes. This alteration can cause inflammatory monocytes/macrophages to interact less with the activated endothelium that lines the adipose tissue, resulting in less monocyte/macrophage infiltration into the inflamed adipose tissue. In addition, Acat1−M/−M causes ATM to polarize toward an anti-inflammatory, M2-like phenotype and reduces the expressions of various inflammatory genes in WAT. Cell culture experiments show that inhibiting ACAT1 in a macrophage-like cell line significantly diminishes the upregulations of multiple inflammatory genes in response to LPS activation. In addition, inhibiting ACAT1 in cholesterol-loaded macrophages maintained in serum containing medium does not increase total cellular free cholesterol concentration. Unlike global Acat1 KO, myeloid Acat1 KO does not cause leukocytosis (Fig. 1 and Ref. 21). In addition, myeloid Acat1 KO does not compromise systemic immunity on acute viral infection (Fig. 11). Overall, our current study shows that inactivating the gene that encodes the enzyme important for cholesterol storage in macrophages can alter adipose tissue biology, reduces inflammatory responses, and leads to resistance to diet-induced obesity, without compromising acute virus infection.
At present, it is not clear how Acat1−M/−M can lead to lower expression of CD29 and cause anti-inflammatory phenotype in monocytes/macrophages. Although ACAT1 inhibition does not increase total cellular free cholesterol content, it may cause increase in cholesterol content in certain internal membranes, such that it eventually leads to the lower expression of key receptor(s) at the plasma membrane, which controls the expression of CD29 and other macromolecules that affect macrophage responses to inflammation. Opposing the action of ACAT are cholesteryl-ester hydrolases, which liberate cholesterol from cholesteryl esters, as reviewed in Ref. 49. Bie and colleagues (3) created a novel transgenic mouse by overexpressing one of the cholesteryl-ester hydrolase genes in macrophages. Interestingly, when fed with Western diet, these mice under LDL receptor KO background show lower ATM content and exhibit improved insulin sensitivity. The beneficial phenotypes observed in this transgenic mouse may also be caused by an increase in local cholesterol content in the cell membranes of macrophages. This hypothesis is consistent with the findings that treating macrophages with high-density lipoprotein or apolipoprotein A1, which promotes cholesterol efflux, significantly decreased the expressions of a range of chemotactic cytokines through disruption of lipid raft structure in cell membranes (6, 24).
LPS has been used extensively to induce inflammation in various model systems in vitro and in vivo. In mice, LPS is considered as the triggering inflammatory factor causative of the onset of insulin resistance, obesity, and diabetes (7). There are several membrane receptors and several soluble proteins that can all bind to LPS, including scavenger receptors, β2-integrins, the glycosylphosphatidylinositol-linked protein CD14 (14), and, most importantly, Toll-like receptors 2 (TLR2) and 4 (TLR4; Ref. 27). The TLRs belong to the family of pattern-recognition receptors. These receptors can use exogenous materials derived from pathogens as well as endogenous materials from damaged tissues and dead cells as ligands. Importantly, the interaction between LPS and the TLRs can occur in various membrane compartments that include plasma membrane, endosomes, and lysosomes (27). Blocking ACAT1 is expected to alter the cholesterol content in these membranes. It is thus possible that ACAT1 blockage acts by altering the interaction between LPS and its receptors, thereby attenuating the ability of LPS to mediate its signaling activity that leads to nuclear factor-κB and activating protein-1. We are currently testing this and other possibilities in the laboratory.
We also find that the size of the white adipocyte present in the ACAT1−M/−M mouse is significantly smaller than that in the WT mouse; the difference in size between these two genotypes occurs in both chow diet and Western diet. This is an unexpected finding. Since the paucity of ATM as well as its polarization toward the M2-like phenotype only occur when ACAT1−M/−M mice were on Western diet, not on chow diet, these results cannot satisfactorily explain the difference in fat cell size between these two genotypes. Other studies had shown that decreased ATM alone might not necessarily lead to decreased adipocyte size. For example, studies showed that mice lacking monocyte chemotactic protein-1 (MCP-1) or C-C chemokine receptor type 2, which play important roles in monocyte migration into the inflamed tissue (10), expressed less ATM and improved insulin sensitivity when fed with Western diet; however, unlike Acat1−M/−M mice reported here, these mice have similar fat mass and similar adipocyte size as the WT mice (26, 55). In our system, the smaller white adipocyte (WA) size correlates well with better insulin sensitivity observed in the Acat1−M/−M mice because these alterations occur in both chow and Western diets. Interactions between the ATM and WA are complex. ATM and WA may affect adaptive thermogenesis in WA, as reported in Refs. 35 and 42. Therefore, it is possible that Acat1−M/−M may cause subtle change(s) in ATM to increase thermogenesis in WA, which leads to smaller WA size and better insulin sensitivity. On the other hand, the concept of controlling adaptive thermogenesis in WA through interaction between WA and ATM has recently been challenged (15, 43), and further investigations are needed to resolve the discrepancy in results from different laboratories. An additional limitation of our current system is that we use Acat1−M/−M as our genetic system but focus on the effect of macrophages in adipose tissue biology. However, myeloid cell lineage includes macrophages, eosinophils, neutrophils, and microglia in the central nervous system. It is possible that the difference in fat cell size may be caused by lack of Acat1 in myeloid cells other than the macrophages. At present, little is known about the contribution of eosinophils or neutrophils or microglia in adipose tissue biology. Further investigations are required to find the mechanistic explanation to account for the fat cell size difference between the WT and Acat1−M/−M mice.
In summary, we show that inactivating ACAT1 in the myeloid cell lineage improves insulin sensitivity and suppresses diet-induced obesity. Our current data suggest that it is the combination of lower ATM with M2-like, anti-inflammatory phenotype coupled with reduced fat cell size that leads to improved insulin sensitivity and resistance to diet-induced obesity. In developed countries, type 2 diabetes and obesity are major health problems. Our findings reported here suggest ACAT1 in the myeloid cell lineage as a potential new candidate for preventive/therapeutic treatment for insulin resistance and obesity in humans who do not carry certain monogenic mutations in apolipoprotein A1, high-density lipoproteins, or other macromolecules that affect the cellular cholesterol removal process. The current study examined the effects of Western diet feeding for ≤16 wk. The results (Fig. 2B) show that Acat1−M−M improved fasting glucose, improved insulin resistance, and protected against weight gain during the obesity development stage (i.e., high-fat feeding for 8–10 wk); when high-fat feeding continued for ≥12 wk, Acat1−M/−M no longer protected against weight gain. These data suggest that as an antiobesity treatment, Acat1−M/−M may best serve as a target for prevention therapy. Further studies are needed to determine whether inhibiting ACAT1 in macrophages can serve as a possible target for therapeutic intervention.
GRANTS
This work was supported by NIH Grants R01-AG-037609 and R01-HL-060306 (to T.-Y. Chang and C. C. Y. Chang), R01-DK-080756 and U24-DK-093000 (to J. K. Kim), and P20-GM-113132 (to D. Madden). E. M. Melton is supported by NIH Postdoctoral Fellowship 1-F32-HL-124953.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
L.-H.H., E.M.M., H.L., P.S., C.C.Y.C., and T.-Y.C. conceived and designed research; L.-H.H., E.M.M., H.L., P.S., D.J., C.-Y.T., T.M., H.H., and R.H.F. performed experiments; L.-H.H., E.M.M., H.L., P.S., D.J., C.-Y.T., T.M., H.S., H.H., R.H.F., J.K.K., E.U., C.C.Y.C., and T.-Y.C. analyzed data; L.-H.H., E.M.M., H.L., P.S., D.J., C.-Y.T., H.S., R.H.F., J.K.K., E.U., C.C.Y.C., and T.-Y.C. interpreted results of experiments; L.-H.H., E.M.M., H.L., P.S., D.J., H.H., and J.K.K. prepared figures; L.-H.H., E.M.M., J.K.K., and T.-Y.C. drafted manuscript; H.L., T.M., J.K.K., E.U., C.C.Y.C., and T.-Y.C. edited and revised manuscript; L.-H.H., E.M.M., H.L., P.S., D.J., C.-Y.T., T.M., H.S., H.H., R.H.F., J.K.K., E.U., C.C.Y.C., and T.-Y.C. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Gustav Lienhard for advice throughout the course of this study and thank Drs. Dean Madden, Brent Berwin, and Patricia Ernst and members of the Chang laboratory for helpful discussion. We also thank Brent Berwin for providing the L929 cells. Hyperinsulinemic-euglycemic clamp and metabolic cage studies were performed at the University of Massachusetts Mouse Metabolic Phenotyping Center.
REFERENCES
- 1.Accad M, Smith SJ, Newland DL, Sanan DA, King LE Jr, Linton MF, Fazio S, Farese RV Jr. Massive xanthomatosis and altered composition of atherosclerotic lesions in hyperlipidemic mice lacking acyl CoA:cholesterol acyltransferase 1. J Clin Invest 105: 711–719, 2000. doi: 10.1172/JCI9021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Basu SK, Goldstein JL, Anderson GW, Brown MS. Degradation of cationized low density lipoprotein and regulation of cholesterol metabolism in homozygous familial hypercholesterolemia fibroblasts. Proc Natl Acad Sci USA 73: 3178–3182, 1976. doi: 10.1073/pnas.73.9.3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bie J, Zhao B, Song J, Ghosh S. Improved insulin sensitivity in high fat- and high cholesterol-fed Ldlr−/− mice with macrophage-specific transgenic expression of cholesteryl ester hydrolase: role of macrophage inflammation and infiltration into adipose tissue. J Biol Chem 285: 13630–13637, 2010. doi: 10.1074/jbc.M109.069781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bognar E, Sarszegi Z, Szabo A, Debreceni B, Kalman N, Tucsek Z, Sumegi B, Gallyas F Jr. Antioxidant and anti-inflammatory effects in RAW264.7 macrophages of malvidin, a major red wine polyphenol. PLoS One 8: e65355, 2013. doi: 10.1371/journal.pone.0065355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buhman KF, Accad M, Farese RV. Mammalian acyl-CoA:cholesterol acyltransferases. Biochim Biophys Acta 1529: 142–154, 2000. doi: 10.1016/S1388-1981(00)00144-X. [DOI] [PubMed] [Google Scholar]
- 6.Bursill CA, Castro ML, Beattie DT, Nakhla S, van der Vorst E, Heather AK, Barter PJ, Rye KA. High-density lipoproteins suppress chemokines and chemokine receptors in vitro and in vivo. Arterioscler Thromb Vasc Biol 30: 1773–1778, 2010. doi: 10.1161/ATVBAHA.110.211342. [DOI] [PubMed] [Google Scholar]
- 7.Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, Waget A, Delmée E, Cousin B, Sulpice T, Chamontin B, Ferrières J, Tanti JF, Gibson GR, Casteilla L, Delzenne NM, Alessi MC, Burcelin R. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56: 1761–1772, 2007. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- 8.Chang CC, Huh HY, Cadigan KM, Chang TY. Molecular cloning and functional expression of human acyl-coenzyme A:cholesterol acyltransferase cDNA in mutant Chinese hamster ovary cells. J Biol Chem 268: 20747–20755, 1993. [PubMed] [Google Scholar]
- 9.Chang TY, Li BL, Chang CC, Urano Y. Acyl-coenzyme A:cholesterol acyltransferases. Am J Physiol Endocrinol Metab 297: E1–E9, 2009. doi: 10.1152/ajpendo.90926.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Charo IF, Taubman MB. Chemokines in the pathogenesis of vascular disease. Circ Res 95: 858–866, 2004. doi: 10.1161/01.RES.0000146672.10582.17. [DOI] [PubMed] [Google Scholar]
- 11.Chawla A, Nguyen KD, Goh YP. Macrophage-mediated inflammation in metabolic disease. Nat Rev Immunol 11: 738–749, 2011. doi: 10.1038/nri3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fantin VR, Wang Q, Lienhard GE, Keller SR. Mice lacking insulin receptor substrate 4 exhibit mild defects in growth, reproduction, and glucose homeostasis. Am J Physiol Endocrinol Metab 278: E127–E133, 2000. doi: 10.1152/ajpendo.2000.278.1.E127. [DOI] [PubMed] [Google Scholar]
- 13.Fazio S, Major AS, Swift LL, Gleaves LA, Accad M, Linton MF, Farese RV Jr. Increased atherosclerosis in LDL receptor-null mice lacking ACAT1 in macrophages. J Clin Invest 107: 163–171, 2001. doi: 10.1172/JCI10310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fenton MJ, Golenbock DT. LPS-binding proteins and receptors. J Leukoc Biol 64: 25–32, 1998. doi: 10.1002/jlb.64.1.25. [DOI] [PubMed] [Google Scholar]
- 15.Fischer K, Ruiz HH, Jhun K, Finan B, Oberlin DJ, van der Heide V, Kalinovich AV, Petrovic N, Wolf Y, Clemmensen C, Shin AC, Divanovic S, Brombacher F, Glasmacher E, Keipert S, Jastroch M, Nagler J, Schramm KW, Medrikova D, Collden G, Woods SC, Herzig S, Homann D, Jung S, Nedergaard J, Cannon B, Tschöp MH, Müller TD, Buettner C. Alternatively activated macrophages do not synthesize catecholamines or contribute to adipose tissue adaptive thermogenesis. Nat Med 23: 623–630, 2017. doi: 10.1038/nm.4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science 327: 656–661, 2010. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han MS, Jung DY, Morel C, Lakhani SA, Kim JK, Flavell RA, Davis RJ. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science 339: 218–222, 2013. doi: 10.1126/science.1227568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hotamisligil GS. Role of endoplasmic reticulum stress and c-Jun NH2-terminal kinase pathways in inflammation and origin of obesity and diabetes. Diabetes 54, Suppl 2: S73–S78, 2005. doi: 10.2337/diabetes.54.suppl_2.S73. [DOI] [PubMed] [Google Scholar]
- 19.Hu Z, Molloy MJ, Usherwood EJ. CD4+ T-cell dependence of primary CD8+ T-cell response against vaccinia virus depends upon route of infection and viral dose. Cell Mol Immunol 13: 82–93, 2016. doi: 10.1038/cmi.2014.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang LH, Gui J, Artinger E, Craig R, Berwin BL, Ernst PA, Chang CC, Chang TY. Acat1 gene ablation in mice increases hematopoietic progenitor cell proliferation in bone marrow and causes leukocytosis. Arterioscler Thromb Vasc Biol 33: 2081–2087, 2013. doi: 10.1161/ATVBAHA.112.301080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang LH, Melton EM, Li H, Sohn P, Rogers MA, Mulligan-Kehoe MJ, Fiering SN, Hickey WF, Chang CC, Chang TY. Myeloid acyl-CoA:cholesterol acyltransferase 1 deficiency reduces lesion macrophage content and suppresses atherosclerosis progression. J Biol Chem 291: 6232–6244, 2016. doi: 10.1074/jbc.M116.713818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ikenoya M, Yoshinaka Y, Kobayashi H, Kawamine K, Shibuya K, Sato F, Sawanobori K, Watanabe T, Miyazaki A. A selective ACAT-1 inhibitor, K-604, suppresses fatty streak lesions in fat-fed hamsters without affecting plasma cholesterol levels. Atherosclerosis 191: 290–297, 2007. doi: 10.1016/j.atherosclerosis.2006.05.048. [DOI] [PubMed] [Google Scholar]
- 23.Imhof BA, Aurrand-Lions M. Adhesion mechanisms regulating the migration of monocytes. Nat Rev Immunol 4: 432–444, 2004. doi: 10.1038/nri1375. [DOI] [PubMed] [Google Scholar]
- 24.Iqbal AJ, Barrett TJ, Taylor L, McNeill E, Manmadhan A, Recio C, Carmineri A, Brodermann MH, White GE, Cooper D, DiDonato JA, Zamanian-Daryoush M, Hazen SL, Channon KM, Greaves DR, Fisher EA. Acute exposure to apolipoprotein A1 inhibits macrophage chemotaxis in vitro and monocyte recruitment in vivo. eLife 5: e15190, 2016. doi: 10.7554/eLife.15190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Janssen MJ, van Voorst F, Ploeger GE, Larsen PM, Larsen MR, de Kroon AI, de Kruijff B. Photolabeling identifies an interaction between phosphatidylcholine and glycerol-3-phosphate dehydrogenase (Gut2p) in yeast mitochondria. Biochemistry 41: 5702–5711, 2002. doi: 10.1021/bi025550j. [DOI] [PubMed] [Google Scholar]
- 26.Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, Kitazawa S, Miyachi H, Maeda S, Egashira K, Kasuga M. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest 116: 1494–1505, 2006. doi: 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11: 373–384, 2010. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 28.Kim HJ, Higashimori T, Park SY, Choi H, Dong J, Kim YJ, Noh HL, Cho YR, Cline G, Kim YB, Kim JK. Differential effects of interleukin-6 and -10 on skeletal muscle and liver insulin action in vivo. Diabetes 53: 1060–1067, 2004. doi: 10.2337/diabetes.53.4.1060. [DOI] [PubMed] [Google Scholar]
- 29.Koliwad SK, Streeper RS, Monetti M, Cornelissen I, Chan L, Terayama K, Naylor S, Rao M, Hubbard B, Farese RV Jr. DGAT1-dependent triacylglycerol storage by macrophages protects mice from diet-induced insulin resistance and inflammation. J Clin Invest 120: 756–767, 2010. doi: 10.1172/JCI36066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kusunoki J, Hansoty DK, Aragane K, Fallon JT, Badimon JJ, Fisher EA. Acyl-CoA:cholesterol acyltransferase inhibition reduces atherosclerosis in apolipoprotein E-deficient mice. Circulation 103: 2604–2609, 2001. doi: 10.1161/01.CIR.103.21.2604. [DOI] [PubMed] [Google Scholar]
- 31.Lee E, Jung DY, Kim JH, Patel PR, Hu X, Lee Y, Azuma Y, Wang HF, Tsitsilianos N, Shafiq U, Kwon JY, Lee HJ, Lee KW, Kim JK. Transient receptor potential vanilloid type-1 channel regulates diet-induced obesity, insulin resistance, and leptin resistance. FASEB J 29: 3182–3192, 2015. doi: 10.1096/fj.14-268300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lindner A, Santilli D, Hodgett J, Nerlinger C. Effects of 5-fluorouracil on the hematopoietic system of the mouse. Cancer Res 20: 497–502, 1960. [PubMed] [Google Scholar]
- 33.Murano I, Barbatelli G, Parisani V, Latini C, Muzzonigro G, Castellucci M, Cinti S. Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. J Lipid Res 49: 1562–1568, 2008. doi: 10.1194/jlr.M800019-JLR200. [DOI] [PubMed] [Google Scholar]
- 34.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, Locati M, Mantovani A, Martinez FO, Mege JL, Mosser DM, Natoli G, Saeij JP, Schultze JL, Shirey KA, Sica A, Suttles J, Udalova I, van Ginderachter JA, Vogel SN, Wynn TA. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41: 14–20, 2014. [Correction in Immunity 41: 339–340, 2014.] 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nguyen KD, Qiu Y, Cui X, Goh YP, Mwangi J, David T, Mukundan L, Brombacher F, Locksley RM, Chawla A. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature 480: 104–108, 2011. doi: 10.1038/nature10653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, Vats D, Morel CR, Goforth MH, Subramanian V, Mukundan L, Ferrante AW, Chawla A. Alternative M2 activation of Kupffer cells by PPARδ ameliorates obesity-induced insulin resistance. Cell Metab 7: 496–507, 2008. doi: 10.1016/j.cmet.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oh DY, Morinaga H, Talukdar S, Bae EJ, Olefsky JM. Increased macrophage migration into adipose tissue in obese mice. Diabetes 61: 346–354, 2012. doi: 10.2337/db11-0860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol 72: 219–246, 2010. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- 40.Patankar YR, Mabaera R, Berwin B. Differential ASC requirements reveal a key role for neutrophils and a noncanonical IL-1β response to Pseudomonas aeruginosa. Am J Physiol Lung Cell Mol Physiol 309: L902–L913, 2015. doi: 10.1152/ajplung.00228.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prieur X, Mok CY, Velagapudi VR, Núñez V, Fuentes L, Montaner D, Ishikawa K, Camacho A, Barbarroja N, O’Rahilly S, Sethi JK, Dopazo J, Orešič M, Ricote M, Vidal-Puig A. Differential lipid partitioning between adipocytes and tissue macrophages modulates macrophage lipotoxicity and M2/M1 polarization in obese mice. Diabetes 60: 797–809, 2011. doi: 10.2337/db10-0705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rao RR, Long JZ, White JP, Svensson KJ, Lou J, Lokurkar I, Jedrychowski MP, Ruas JL, Wrann CD, Lo JC, Camera DM, Lachey J, Gygi S, Seehra J, Hawley JA, Spiegelman BM. Meteorin-like is a hormone that regulates immune-adipose interactions to increase beige fat thermogenesis. Cell 157: 1279–1291, 2014. doi: 10.1016/j.cell.2014.03.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reitman ML. How does fat transition from white to beige? Cell Metab 26: 14–16, 2017. doi: 10.1016/j.cmet.2017.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Remppis A, Bea F, Greten HJ, Buttler A, Wang H, Zhou Q, Preusch MR, Enk R, Ehehalt R, Katus H, Blessing E. Rhizoma coptidis inhibits LPS-induced MCP-1/CCL2 production in murine macrophages via an AP-1 and NFκB-dependent pathway. Mediators Inflamm 2010: 194896, 2010. doi: 10.1155/2010/194896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rosenson RS, Brewer HB Jr, Davidson WS, Fayad ZA, Fuster V, Goldstein J, Hellerstein M, Jiang XC, Phillips MC, Rader DJ, Remaley AT, Rothblat GH, Tall AR, Yvan-Charvet L. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation 125: 1905–1919, 2012. doi: 10.1161/CIRCULATIONAHA.111.066589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rőszer T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediators Inflamm 2015: 816460, 2015. doi: 10.1155/2015/816460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rudel LL, Lee RG, Cockman TL. Acyl coenzyme A: cholesterol acyltransferase types 1 and 2: structure and function in atherosclerosis. Curr Opin Lipidol 12: 121–127, 2001. doi: 10.1097/00041433-200104000-00005. [DOI] [PubMed] [Google Scholar]
- 48.Sakashita N, Miyazaki A, Chang CC, Chang TY, Kiyota E, Satoh M, Komohara Y, Morganelli PM, Horiuchi S, Takeya M. Acyl-coenzyme A:cholesterol acyltransferase 2 (ACAT2) is induced in monocyte-derived macrophages: in vivo and in vitro studies. Lab Invest 83: 1569–1581, 2003. doi: 10.1097/01.LAB.0000095687.17383.39. [DOI] [PubMed] [Google Scholar]
- 49.Sekiya M, Osuga J, Igarashi M, Okazaki H, Ishibashi S. The role of neutral cholesterol ester hydrolysis in macrophage foam cells. J Atheroscler Thromb 18: 359–364, 2011. doi: 10.5551/jat.7013. [DOI] [PubMed] [Google Scholar]
- 50.Shibuya Y, Chang CC, Huang LH, Bryleva EY, Chang TY. Inhibiting ACAT1/SOAT1 in microglia stimulates autophagy-mediated lysosomal proteolysis and increases Aβ1-42 clearance. J Neurosci 34: 14484–14501, 2014. doi: 10.1523/JNEUROSCI.2567-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest 116: 1793–1801, 2006. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tabas I. Consequences of cellular cholesterol accumulation: basic concepts and physiological implications. J Clin Invest 110: 905–911, 2002. doi: 10.1172/JCI0216452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tall AR, Yvan-Charvet L. Cholesterol, inflammation and innate immunity. Nat Rev Immunol 15: 104–116, 2015. doi: 10.1038/nri3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Warner GJ, Stoudt G, Bamberger M, Johnson WJ, Rothblat GH. Cell toxicity induced by inhibition of acyl coenzyme A:cholesterol acyltransferase and accumulation of unesterified cholesterol. J Biol Chem 270: 5772–5778, 1995. doi: 10.1074/jbc.270.11.5772. [DOI] [PubMed] [Google Scholar]
- 55.Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, Charo I, Leibel RL, Ferrante AW Jr. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest 116: 115–124, 2006. doi: 10.1172/JCI24335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112: 1796–1808, 2003. doi: 10.1172/JCI200319246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest 115: 1111–1119, 2005. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112: 1821–1830, 2003. doi: 10.1172/JCI200319451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yagyu H, Kitamine T, Osuga J, Tozawa R, Chen Z, Kaji Y, Oka T, Perrey S, Tamura Y, Ohashi K, Okazaki H, Yahagi N, Shionoiri F, Iizuka Y, Harada K, Shimano H, Yamashita H, Gotoda T, Yamada N, Ishibashi S. Absence of ACAT-1 attenuates atherosclerosis but causes dry eye and cutaneous xanthomatosis in mice with congenital hyperlipidemia. J Biol Chem 275: 21324–21330, 2000. doi: 10.1074/jbc.M002541200. [DOI] [PubMed] [Google Scholar]
- 60.Yamauchi Y, Chang CC, Hayashi M, Abe-Dohmae S, Reid PC, Chang TY, Yokoyama S. Intracellular cholesterol mobilization involved in the ABCA1/apolipoprotein-mediated assembly of high density lipoprotein in fibroblasts. J Lipid Res 45: 1943–1951, 2004. doi: 10.1194/jlr.M400264-JLR200. [DOI] [PubMed] [Google Scholar]
- 61.Ye L, Kleiner S, Wu J, Sah R, Gupta RK, Banks AS, Cohen P, Khandekar MJ, Boström P, Mepani RJ, Laznik D, Kamenecka TM, Song X, Liedtke W, Mootha VK, Puigserver P, Griffin PR, Clapham DE, Spiegelman BM. TRPV4 is a regulator of adipose oxidative metabolism, inflammation, and energy homeostasis. Cell 151: 96–110, 2012. doi: 10.1016/j.cell.2012.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]