

Abstract
Synaptic NMDA receptors activating nuclear calcium-driven adaptogenomics control a potent body-own neuroprotective mechanism, referred to as acquired neuroprotection. Viral vector-mediated gene transfer in conjunction with stereotactic surgery has previously demonstrated the proficiency of several nuclear calcium-regulated genes to protect in vivo against brain damage caused by toxic extrasynaptic NMDA receptor signaling following seizures or stroke. Here we used noninvasive nose-to-brain administration of Activin A and SerpinB2, two secreted nuclear calcium-regulated neuroprotectants, for post-injury treatment of brain damage following middle cerebral artery occlusion (MCAO) in C57BL/6N mice. The observed reduction of the infarct volume was comparable to the protection obtained by intracerebroventricular injection of recombinant Activin A or SerpinB2 or by stereotactic delivery 3 weeks prior to the injury of a recombinant adeno-associated virus containing an expression cassette for the potent neuroprotective transcription factor Npas4. These results establish post-injury, nose-to-brain delivery of Activin A and SerpinB2 as effective and possibly clinically applicable treatments of acute and chronic neurodegenerative conditions.
Keywords: neuroprotection, stroke, nasal delivery
Graphical Abstract
A novel non-invasive neuroprotective treatment has been developed that uses intranasal application of Activin A and SerpinB2. This therapeutic approach reduces brain damage in a mouse stroke model, and it may also be beneficial in chronic neurodegenerative diseases.
Introduction
Neurons possess an intrinsic mechanism that can increase their ability to survive harmful conditions. The buildup of this protective shield, which is known as acquired neuroprotection, is initiated by synaptic activity and requires calcium signaling to the nucleus and the activation of gene transcription.1, 2, 3 The survival-enhancing genes activated by this process and collectively referred to as activity-regulated inhibitor of death (AID) genes include regulators of gene transcription, such as Atf3, Npas4, Nr4a1, Gadd45β, Gadd45γ, and Bcl6, but also secreted proteins encoded, for example, by inhba or serpinb2.2, 3 The mechanisms underlying increased resilience are not fully understood for all genes, but shielding mitochondria against excitotoxic damage induced by extrasynaptic NMDA receptors and protecting structural integrity and cellular bioenergetics are emerging common themes.2, 4, 5, 6, 7, 8, 9
Overexpression of each AID gene using recombinant adeno-associated viruses (rAAVs) provides effective protection against apoptotic cell death in cultured hippocampal neurons and neurodegeneration in vivo induced by kainate-induced seizures.1, 2 Moreover, expression of Atf3 and Inhba, also known as Activin A, reduces brain damage caused by ischemic conditions following middle cerebral artery occlusion (MCAO) in the mouse.8, 10 However, so far, the design of the in vivo neuroprotection experiments involved a pre-injury stereotactic delivery to the appropriate brain regions of rAAVs carrying AID gene expression cassettes.2, 8, 10 This therapy regime is unsuitable for a clinical application, which led us to develop an alternative strategy. Here we used AID gene product-containing liquid drops applied to the nasal epithelium of the mouse to transfer neuroprotective proteins to the brain. The efficacy of noninvasive nose-to-brain delivery of AID gene products was assessed using a mouse model of permanent distal MCAO that induces cortical infarcts.11 We provide a proof-of-principle study that demonstrates a robust protection against MCAO-induced brain damage achieved by post-injury, nose-to-brain delivery of the two secreted AID gene products, Activin A and SerpinB2.
Results
Selection of Activin A and SerpinB2
For the study, we selected two genes, inhba and serpinb2, whose expression using rAAVs had previously been shown to provide effective neuroprotection both in cultured neurons and in vivo.2, 8, 10 The gene product of inhba, inhibin β-A, forms a homodimer known as Activin A. Activin A is a member of the transforming growth factor β (TGF-β) superfamily of growth and differentiation factors that act on cells through type I and II Activin A receptors.12, 13, 14 The gene serpinb2 (also known as plasminogen activator inhibitor type 2 [PAI-2]) encodes a serine protease inhibitor of the serpin family.15, 16 SerpinB2 is an inhibitor of extracellular urokinase plasminogen activator,17 however, many other activities and functions have been suggested.18, 19, 20 Secreted AID gene products were chosen because we reasoned that the efficacy of putative therapeutic compounds administered via a noninvasive nose-to-brain delivery route may be higher if their target molecules reside in the extracellular space or on the cell surface rather than in intracellular compartments.
Neuroprotection after Intracerebroventricular Injection of Recombinant Activin A and SerpinB2
We first established the neuroprotective potential of recombinant Activin A and recombinant SerpinB2. We found that mice injected into the left ventricle with 100 ng of either Activin A or SerpinB2 within 10 min after the MCAO had significantly less brain damage compared to the control mice (Figures 1A–1D). The observed reduction of the infarct volume was similar to that obtained in mice that were stereotactically injected into the cortex 3 weeks prior to MCAO with rAAVs containing expression cassettes for Npas4 (rAAV-Npas4) (Figures 2A–2D), which has potent neuroprotective activities.2, 5, 21, 22 It is also comparable to the reduction in stroke volume obtained using rAAV-mediated expression of Activin A.8 This experiment also revealed that not every AID gene provides effective protection in the MCAO model of acute neurodegeneration. We found that the expression of the AID gene Gadd45β using rAAV-Gadd45β failed to produce effects that were significantly different from the controls, i.e., infection with rAAV-GFP or injection of PBS (Figures 2C and 2D).
Figure 1.

Intracerebroventricular Injection of Recombinant Activin A or SerpinB2 Protects against MCAO-Induced Brain Damage
(A and B) Time frame of the experiment and schematic illustration of stereotactic intracerebroventricular injections. (A) Within 10 min after MCAO (left middle cerebral artery), mice were stereotactically injected with 100 ng recombinant human Activin A or 100 ng recombinant human SerpinB2. Control mice were injected with aCSF. Infarct volumes were assessed 7 days post-MCAO using silver staining. (B) Stereotactic intracerebroventricular injections were given unilaterally with a single point shot to the left ventricle. (C) Examples of silver-stained coronal brain sections of mice 7 days after MCAO, which had been stereotactically injected within 10 min after the onset of MCAO or sham surgery with the indicated recombinant human proteins or aCSF. Representative images from the same anterior and posterior distance of the brain are shown. Infarct areas appear in light gray; borders to the healthy tissue are indicated with a white line. Scale bar, 0.25 cm. (D) Infarct volumes in whole ischemic brains of mice that were subjected to MCAO or sham surgery and received an intracerebroventricular injection of 100 ng of either recombinant Activin A or recombinant SerpinB2 in the left ventricle; aCSF was used as a control. Infarct sizes were determined 7 days after MCAO (sham aCSF, n = 9; sham Activin A, n = 4: sham SerpinB2, n = 9; MCAO aCSF, n = 22; MCAO Activin A, n = 16; and MCAO SerpinB2, n = 16). Data are expressed as mean ± SD. Statistical analysis was done using non-parametric one-way ANOVA with Bonferroni post-test; statistically significant differences are indicated with asterisks (*p < 0.05 and ***p < 0.001).
Figure 2.

Viral Vector-Mediated Expression of Npas4 Protects against MCAO-Induced Brain Damage
(A and B) Time frame of the experiment and schematic illustration of stereotactic intracerebral injections. (A) MCAO (left middle cerebral artery) was 3 weeks after stereotactic injections into the left cortex. Infarct volumes were assessed 7 days post-MCAO using silver staining. (B) Stereotactic injections into the left cortex were done by a double line shot into the primary somatosensory cortex barrel field (S1BF) and into primary somatosensory cortex forelimb region (S1FL). (C) Examples of silver-stained coronal brain sections of mice subjected to MCAO or sham surgery 3 weeks after stereotactic injection of rAAV-hrGFP, rAAV-Npas4-FLAG, rAAV-Gadd45β-FLAG, or PBS into the left cortex. Representative images corresponding to the same anterior and posterior distance of the brain are shown. Infarct areas appear in light gray; borders to the healthy tissue are indicated with a white line. Transgene expression was verified using immunohistochemistry (data not shown). Scale bar, 0.25 cm. (D) Infarct volumes in whole ischemic brains of mice subjected to stereotactic injection of the indicated rAAVs 3 weeks before MCAO or sham surgery. PBS was used as a control. Infarct sizes were determined 7 days after MCAO (sham PBS, n = 5; sham GFP, n = 7; sham Npas4, n = 6; sham Gadd45β, n = 5; MCAO PBS, n = 8; MCAO GFP, n = 6; MCAO Npas4, n = 8; and MCAO Gadd45β, n = 6). Data are expressed as mean ± SD. Statistical analysis was done using non-parametric one-way ANOVA with Bonferroni post-test; statistically significant differences are indicated with an asterisk (*p < 0.05; n.s., not significant).
Detection of Recombinant Proteins in Brain Lysates after Intracerebroventricular Injection
To investigate whether recombinant proteins stereotactically injected into the ventricles diffuse into the brain tissue, we analyzed brain lysates using immunoblot analysis. Whether or not a protein is detectable in brain lysates depends primarily on the amount of protein delivered by intracerebroventricular injection, in addition to the penetration of the protein into the parenchyma, efficacy of the diffusion process, and degradation of the protein. Given the protein concentrations available to us (see the Materials and Methods) and the limits of the recommended intracerebroventricular injection volumes for mice, we were able to deliver 5 μg GFP, 3 μg SerpinB2, but only 1.3 μg Activin A.
Using immunoblot analysis of brain lysates prepared 1 hr after stereotactic delivery of the proteins, GFP and SerpinB2 were readily detectable in lysates from the cortex and in lysates from the brain tissue containing striatum, putamen, basal forebrain, thalamus, and hypothalamus (Figures 3A and 3B). We were unable to detect Activin A using this type of experiment. This is most likely due to the small amounts of Activin A that we were able to inject into the ventricles, although we cannot rule out the possibility that, in addition, Activin A is more rapidly degraded than GFP or SerpinB2 after intracerebroventricular injection. Nevertheless, these experiments show that proteins stereotactically injected into the ventricles can diffuse into the brain parenchyma and intact proteins remain detectable for at least 1 hr after the injections.
Figure 3.

Detection of Recombinant Proteins in Brain Lysates after Intracerebroventricular Injection
(A and B) Immunoblot analysis of recombinant GFP (A) and recombinant FLAG-tagged SerpinB2 (B) in lysates obtained from the cortex and in lysates obtained from brain tissue containing striatum, putamen, basal forebrain, thalamus, and hypothalamus. Representative immunoblots using anti-GFP antibodies (A) or anti-FLAG antibodies (B) are shown; tubulin served as a loading control. Lysates were prepared 1 hr after intracerebroventricular injection of recombinant proteins (GFP; n = 3; and SerpinB2; n = 2) or aCSF (n = 5). As positive controls, in lanes 1 and 6, 5 ng recombinant GFP (A) and 14 ng recombinant SerpinB2 (B) were directly loaded onto the gel. The lysates used in lanes 2 and 4 and the lysates used in lanes 3 and 5 in both (A) and (B) are from the same mouse brain. All lysates were analyzed in at least three independent immunoblot experiments.
Neuroprotection after Intranasal Application of Recombinant Activin A and SerpinB2
The use of stereotactic injections of either recombinant protein or viruses seems unsuitable for a potential clinical application of neuroprotective gene products. We therefore set up a noninvasive, intranasal delivery method to supply Activin A and SerpinB2 to the brain. Several examples in the literature document successful use of the nose-to-brain delivery route for peptides and proteins. This includes the neurotrophic factors BDNF, CNTF, and NT-4, but also erythropoietin, insulin, and oxytocin.23, 24, 25, 26 Although mechanistically not fully understood, molecules applied to the nasal epithelium are thought to enter the brain by traveling along olfactory nerve axons that pass through the holes in the cribriform plate to reach their target structure in the brain, the glomeruli layer of the olfactory bulb.27, 28 Not every study that describes effects on brain functions of intranasally applied compounds includes data on the detection of the delivered compounds in the brain.29, 30 This is most likely due to limits in the detection levels given the tools available.
We encountered a similar problem. We failed to detect recombinant human Activin A or Serpin B2 in different brain regions such as olfactory bulb and cortex at various times (5 min to 1 hr) after intranasal delivery. The methods that we applied include immunoblot analysis, immunocytochemistry, and ELISA using either antibodies that recognize the human version of Activin A or SerpinB2 or antibodies to the His-Tag of Activin A. However, we could detect a robust neuroprotective activity of recombinant human Activin A or SerpinB2 in the brain after intranasal application (Figure 4). A total volume of 20 μL protein solution (see below) was applied in drops of 2 μL to the left naris every 2 min. The animals received 3 rounds of treatment with 20 μL protein solution each, starting 10 min after the MCAO followed by additional intranasal applications at 24 and 48 hr post-injury (Figures 4A and 4B). The protein solutions contained or lacked the absorption enhancer Tetradecyl-β-D-maltoside (TDM) (for details, see the Materials and Methods). TDM is thought to lead to a more permeable apical membrane architecture of nasal epithelial cells and to alterations of tight junction structure.31 No apparent toxicity or behavioral abnormalities were observed in any of the mice during the 7 days of the experiment.
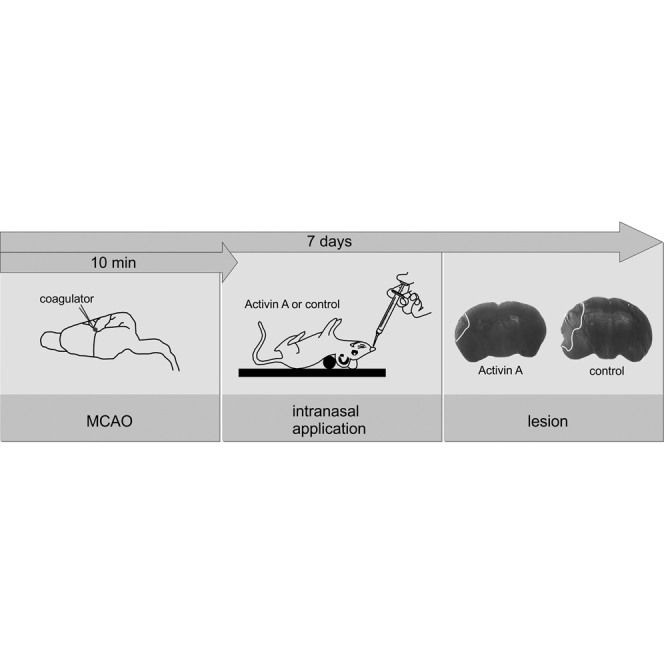
Figure 4.

Intranasal Delivery of Recombinant Activin A or SerpinB2 Protects against MCAO-Induced Brain Damage
(A and B) Time frame of the experiment (A) and schematic illustration of intranasal protein application in a supine position (B). (A) Recombinant proteins (1 μg each of GFP, Activin A, SerpinB2, and ΔNpas4 or a combination of 1 μg Activin A and 1 μg SerpinB2) were delivered 10 or 60 min after MCAO (left middle cerebral artery) to the left naris (see the Materials and Methods for details). Intranasal application was repeated 24 and 48 hr post-MCAO. Infarct volumes were assessed 7 days post-MCAO using silver staining. (C) Examples of silver-stained coronal brain sections of mice that received intranasal administration of GFP, Activin A, or SerpinB2 10 min after the onset of MCAO. The presence of TDM in the nasally applied protein solution is indicated. Representative images corresponding to the same anterior and posterior distance of the brain are shown. Infarct areas appear in light gray; borders to the healthy tissue are indicated with a white line. Scale bar, 0.25 cm. (D) Infarct volumes in whole ischemic brains of mice subjected to intranasal delivery of the indicated recombinant proteins 10 or 60 min after the onset of MCAO. The presence of TDM in the nasally applied protein solution is indicated. Infarct sizes were determined 7 days after MCAO (GFP, 10 min, TDM, n = 12; GFP, 10 min, n = 9; GFP, 60 min, TDM, n = 6; Activin A, 10 min, TDM, n = 11; Activin A, 10 min, n = 14; Activin A, 60 min, TDM, n = 6; SerpinB2, 10 min, TDM, n = 10; SerpinB2, 10 min, n = 11; SerpinB2, 60 min, TDM, n = 6; Activin A + SerpinB2, 10 min, TDM, n = 8; and ΔNpas4, 10 min, TDM, n = 7). Data are expressed as mean ± SD. Statistical analysis was done using non-parametric one-way ANOVA with Bonferroni post-test; statistically significant differences are indicated with asterisks (*p < 0.05 and **p < 0.01; n.s., not significant).
For each nasal application, 1 μg each of recombinant Activin A, recombinant SerpinB2, recombinant GFP, or recombinant human ΔNpas4 was used. Human ΔNpas4 is a functionally inactive fragment of ΔNpas4 that, in addition to recombinant GFP, served as a negative control. We found that Activin A and SerpinB2, but not GFP or ΔNpas4, nasally applied in TDM-containing solutions significantly reduced the MCAO-induced brain damage (Figures 4C and 4D). Nasal application of a combination of 1 μg Activin A and 1 μg SerpinB2 in TDM-containing solution using the standard treatment regime (i.e., delivery at 10 min, 24 hr, and 48 hr post-injury) also provided robust protection against stroke-induced brain damage (Figure 4D). However, the combination treatment was statistically not significantly more effective than application of either Activin A or SerpinB2 alone (Figure 4D). Protein solutions that lacked TDM failed to reduce the infarct volume, indicating the importance of an absorption enhancer for efficient protein delivery to the brain after nasal application (Figures 4C and 4D).
Since a small fraction of nasally applied protein can enter the bloodstream, we investigated the possibility that Activin A or SerpinB2 acts systemically to reduce infarct volumes. However, tail vein injections of 0.5 μg Activin A or SerpinB2, which largely exceeds the amount of protein expected to enter the bloodstream after nasal application of 1 μg Activin A or SerpinB2, failed to produce protection against MCAO-induced brain damage (data not shown). Thus, it seems likely that nose-to-brain delivery rather than a systemic site of action is responsible for neuroprotection afforded by nasally applied Activin A or SerpinB2.
To investigate the width of the therapeutic time window, we shifted the initial application of Activin A or SerpinB2 from 10 to 60 min, leaving unchanged the subsequent applications at 24 and 48 hr post-trauma. This regime rendered the treatment ineffective (Figure 4D). These results demonstrate that a post-injury nose-to-brain delivery of Activin A or SerpinB2 can provide robust protection against MCAO-induced brain damage but that the therapeutic window for this treatment may be very narrow.
Discussion
Our findings provide proof of principle for a neuroprotective treatment in mice using intranasal application of the recombinant neuroprotective proteins Activin A and SerpinB2. Given the simple, non-invasive nature of the delivery method and the use of endogenous factors virtually identical in mice and humans, our results hold great promise for a clinical application.
Mechanism of Neuroprotection
While the mechanism by which SerpinB2 increases neuronal survival is not fully understood,2 neuroprotection by Activin A involves the reduction of toxic extrasynaptic NMDA receptor signaling.8 MCAO, the disease model chosen for this study, is considered a prototypical acute neurodegenerative condition mediated by toxic extrasynaptic NMDA receptor signaling.32, 33, 34, 35 The pathomechanism involves reverse operation of neuronal and glial glutamate transporters in ischemic conditions36 as well as glutamate release by the cystine-glutamate antiporter,37 both processes leading to excessively high levels of extracellular glutamate and stimulation of extrasynaptic NMDA receptor.4, 34 Increased extrasynaptic NMDA receptor signaling also contributes significantly to structural disintegration and loss of neurons in several other severely disabling neurological disorders, including Alzheimer’s disease, Huntington’s disease, and amyotrophic lateral sclerosis.34 Activin A-containing therapeutics counteract the deleterious effects of extrasynaptic NMDA receptor signaling.8 They help maintain the structural and functional integrity of neurons, and they may, therefore, also slow down the progressive cognitive decline associated with a range of chronic neurodegenerative diseases.34
Therapeutic Time Window and Clinical Applications
The value of any neuroprotective therapy depends on whether its application falls into a time window in which the intervention is effective. In this study, Activin A or SerpinB2 was applied within 10 min after the MCAO to provide effective neuroprotection in a model of acute neurodegeneration in mice. This time point is likely to fall outside the time window a patient suffering a stroke would generally receive professional medical attention. However, given that the treatment involves presumably safe compounds and a very simple means of delivery that can be carried out by a lay person, it would not be unreasonable that Activin A/SerpinB2 nasal drops become part of a standard medicine cabinet, making the treatment immediately available when needed, in particular to high-risk stroke patients.
In our experiments, intranasal application of Activin A or SerpinB2 at 60 min after the onset of MCAO failed to reduce brain damage. However, Activin A may have the potential to be neuroprotective when administered hours after injury.38 Using CD-1 mice and focal cerebral ischemia and reperfusion, intracerebroventricular injection of Activin A 6 hr after transient blockade of the middle cerebral artery has been reported to significantly reduce the stroke volume assessed 3 days after the onset of ischemia.38 Although those results cannot be directly compared with our data due to differences in the stroke model, mouse strains, and delivery routes used, they raise hopes that, at least in principle, the therapeutic window of Activin A neuroprotective treatment can be in the range of several hours. In light of possible clinical applications of Activin A and/or SerpinB2, it is, therefore, an important goal of future studies to extend the therapeutic window. Several parameters may be optimized, such as the dose applied and the repetition pattern. In addition, it is worth exploring ways of facilitating nose-to-brain delivery and, at the same time, protecting the compounds from degradation en route. One possibility in this context is the use of nanoparticles or liposomes as vehicles, which may improve pharmacokinetic properties and bioavailability of the compounds.39 Provided that the data obtained by studying additional parameters of Activin A and SerpinB2 application in mice faithfully reflect the situation in humans, such information may help predict therapeutic benefits in realistic clinical settings where the start of treatment after disease onset is variable.
Finally, in addition to their possible value in stroke therapy, Activin A and/or SerpinB2 may be used to treat slowly progressing, chronic neurodegenerative conditions, in particular those that involve increased toxic extrasynaptic NMDA receptor signaling, such as Alzheimer’s disease, Huntington’s disease, and amyotrophic lateral sclerosis (ALS).34, 40 In chronic neurodegenerative diseases, treatment with nasally applied Activin A and/or SerpinB2 should start immediately after diagnosis. Although it is not known whether Activin A or SerpinB2 has neuroregenerative activities and can restore neuronal structures and functions already lost during the course of the disease, they may prevent or slow down further deterioration and cognitive decline.
Materials and Methods
Approval of the Study and Compliance with ARRIVE Guidelines
The study was approved by the animal care committee, Regierungspräsidium Karlsruhe, Referat 35, Germany, Aktenzeichen G217/11 and G202/12. The authors confirm that all experiments conform to all relevant regulatory standards. Experimental design and reporting standards comply with ARRIVE guidelines and recommendations from an NIH-sponsored workshop. Biostatistical and biometrical planning was guided by the Department of Medical Biometry at the Institute of Medical Biometry and Informatics at Heidelberg University. A priori power calculations of animal numbers were done prior to starting the study. Sample sizes were calculated using SAS version 9.1 proc power to ensure adequate power of key experiments in detecting pre-specified effect size.
Animals
C57BL/6N male mice (8 weeks ± 5 days old) from Charles River Laboratories (Sulzfeld, Germany) were housed in groups (maximum of 3 animals/group), in the animal facility at Heidelberg University, in standard cages (15 × 21 × 13.5 cm) on a normal 12:12-hr light:dark cycle with ad libitum access to water and food. Each cage contained nesting material. At the start of the experiments, mice weighed 25 ± 1 g. Animals were randomly allocated to treatment groups. A total of 237 mice was investigated: 76 mice were subjected to intracerebroventricular injection of recombinant proteins, to investigate protection against MCAO-induced brain damage; 51 mice were subjected to intracerebral injections of rAAVs, to investigate protection against MCAO-induced brain damage; 10 mice were subjected to intracerebroventricular injection of recombinant proteins followed by immunoblot analysis of the recombinant proteins using brain lysates; and 100 mice were subjected to intranasal administration of recombinant proteins, to investigate protection against MCAO-induced brain damage. All mice subjected to analysis of MCAO-induced brain damage (n = 227) were euthanized on day 7 post-MCAO. The treatment of mice and data analysis were performed without knowledge of the treatment group.
rAAVs
The vectors used to construct and package rAAVs have been described previously.1 The rAAV cassette for mRNA expression contains a cytomegalovirus (CMV) and chicken β-actin hybrid promoter. The following rAAVs were generated and confirmed by DNA sequencing: rAAV-hrGFP, rAAV-Npas4-FLAG, and rAAV-Gadd45β-FLAG. The rAAV vectors were generated by standard molecular biology techniques and verified by sequencing. Viral particles were produced and purified as described previously.1 Mice were stereotactically injected with rAAVs at a dose of 1–2 × 109 genomic particles in a volume of 2 μL into the left cortex.
Recombinant Proteins, Anesthetic, Reagents, and Antibodies
Recombinant Proteins
For stereotactic intracerebroventricular injection, recombinant human Activin A (amino acids 311–426, catalog cyt-569, Prospec, East Brunswick, NJ) and recombinant human SerpinB2 (amino acids 1–415, catalog TP303139, OriGene, Herford, Germany) were used. For intranasal delivery, recombinant GFP (Aequorea Victoria GFP full-length protein, amino acids 1–238, catalog ab116434, Abcam, Cambridge, UK) was used. Recombinant Activin A from Prospec was not used for intranasal delivery because it led to an irritation of the nasal epithelium with increased salivation. Recombinant human Activin A (amino acids 311–426, catalog 338-AC-050, R&D Systems, Wiesbaden, Germany) was used for intranasal delivery. Stereotactic intracerebroventricular injections of Activin A from both commercial sources yielded similar protection against MCAO-induced brain damage. Recombinant fragment of human Npas4 (ΔNpas4, amino acids 704–802, catalog H00266743-Q01, Novus, Lingen, Germany) was used.
Anesthetic
Tribromethanol (Avertin) was prepared by dissolving 0.5 g 2,2,2 Tribromethanol (catalog T48402, Sigma-Aldrich, Steinheim, Germany) in 1.0 mL 2-methyl-2-butanol (catalog 152463, Sigma-Aldrich, Steinheim, Germany). The solution was heated to 40°C while stirring vigorously. Distilled water was added to the stirring solution to a final volume of 40 mL followed by sterile filtration of the solution. The solution was kept protected from light at 4°C and was used no longer than 14 days. Mice were given 0.5 mL/25 g body weight (corresponding to 250 mg/kg body weight) by intraperitoneal injection.
For stereotactic rAAV injections, a solution was prepared consisting of 0.5 mL SEDIN (1 mg/mL; ALVETRA, Neumünster, Germany), 4.5 mL 0.9% NaCl (B. Braun, Melsungen, Germany), 1.0 mL Midazolam 5 mg/mL (Hameln Pharma Plus, Hameln, Germany), and 1.0 mL Fentanyl-Janssen (0.05 mg/mL, Janssen-Cilag, Neuss, Germany). The solution was sterile filtered and mice were given 0.175 mL/25 g body weight by intraperitoneal injection. Antagonist solution consisted of 0.5 mL ATIPAZOLE (5 mg/mL; Prodivet Pharmaceuticals, Raeren, Belgium), 5.0 mL Flumazenil Kabi (0.1 mg/mL; Fresenius Kabi Deutschland, Bad Homburg, Germany), and 3.0 mL Naloxon Inresa (0.4 mg/mL; Inresa Arzneimittel, Freiburg, Germany). The solution was sterile filtered and mice were given 0.21 mL/25 g body weight by subcutaneous injection. For euthanasia, 300 mg/kg Narcoren (16 mg/100 mL; Boehringer Ingelheim, Ingelheim am Rhein, Germany) was injected.
Reagents
TDM (catalog 15826), TritionX-100 (catalog T9284), protease inhibitor cocktail (catalog P8340), silver nitrate (catalog 10220), and 25% ammonium hydroxide (catalog 30501-M) were from Sigma-Aldrich (Steinheim, Germany). We used sodium desoxycholate monohydrate (catalog B20759, Thermo Fisher Kandel, Karlsruhe, Germany); dodecylsulfate-sodium-salt (catalog 20765.03, Serva, Heidelberg, Germany); sodium-di-hydrogen-phosphate-hydrate (catalog Z12298, Grüssing, Filsum, Germany); and sodium citrate dihydrate (catalog A935748, Merck, Darmstadt, Germany). Lithium carbonate (catalog A6327, 0500), hydrochinon (catalog A6289, 1000), and 37% formaldehyde (catalog 131328, 1211) were from AppliChem (Darmstadt, Germany). Bio-Rad protein assay (catalog 500-0006, Bio-Rad, Munich, Germany) and ECL Western Blotting Detection Reagents (catalog RPN 2209, GE Healthcare) were also used.
Antibodies
Rabbit anti-FLAG antibody (catalog F7425) and mouse anti-tubulin (catalog T9026) were from Sigma-Aldrich (Steinheim, Germany), and rabbit anti-GFP (catalog A6455) was from Molecular Probes (Thermo Fisher Scientific, Dreieich, Germany). Secondary antibodies were from Dianova (Munich, Germany): horseradish peroxidase (HRP)-conjugated goat anti-rabbit immunoglobulin G (IgG) (catalog 111-035-144) and goat anti-mouse HRP (catalog 115 035 003).
Stereotactic Injections
Stereotactic Intracerebral Injection into the Cortex of Mice
C57BL/6N male mice (8 weeks ± 5 days old) weighing 25 ± 1 g were randomly grouped and anesthetized with a mixture of Sedin, Midazolam, and Fentanyl-Janssen, and they were placed on a rodent stereotactic frame on a heat pad, temperature controlled by an ATC1000 DC rectal thermometer (World Precision Instruments, Berlin, Germany). RAAVs (rAAV-hrGFP, rAAV-Npas4-FLAG, and rAAV-Gadd45β-FLAG) were infused into the left cortex (coordinates relative to bregma: first site, anteroposterior (AP) 0.2 mm, mediolateral (ML) 2.0, and dorsoventral (DV) −2.0; second site, AP 0.2, ML 2.0, and DV −1.8; third site, AP 0.2, ML 3.0, and DV −4.0; and fourth site, AP 0.2, ML 3.0, and DV −3.5.) using an Ultra Micro Pump III (World Precision Instruments, Berlin, Germany) to drive a 10-μL Nanofil syringe (World Precision Instruments, Berlin, Germany). A total volume of 2 μL containing 1–2 × 109 genomic particles of rAAVs was injected at a rate of 200 nL/min, after which the needle was left in place at each injection site for 2 min to prevent backflow before needle withdrawal. Control mice were injected with the same volume of PBS using the same method. After stereotactic injections, mice were allowed to recover from anesthesia by subcutaneous application of a mixture with ATIPAZOLE, Flumazenil, and Naloxon, and they were returned to their home cages when they were fully awake. At 3 weeks after stereotactic delivery of rAAVs, animals were subjected to MCAO.
Stereotactic Intracerebroventricular Injection of Mice and Subsequent MCAO
Immediately after MCAO and still anesthetized from the distal MCAO, C57BL/6N male mice (8 weeks ± 5 days old) weighing 25 ± 1 g were placed on a rodent stereotactic frame on a heat pad, temperature controlled by an ATC1000 DC rectal thermometer (World Precision Instruments, Berlin, Germany). Recombinant Activin A (100 ng/2 μL) (Prospec, East Brunswick, NJ) and recombinant SerpinB2 (100 ng/2 μL) (OriGene, Herford, UK) were infused into the left ventricle (coordinates relative to bregma: AP 0.1 mm, ML 0.9, and DV −3.0) using an Ultra Micro Pump III (World Precision Instruments, Berlin, Germany) to drive a 10-μL Nanofil syringe (World Precision Instruments, Berlin, Germany). Recombinant proteins were dissolved in artificial cerebrospinal fluid (aCSF) to a final concentration of 100 ng/2 μL. The recombinant proteins were injected at a rate of 400 nL/min, after which the needle was left in place for 5 min to equilibrate the fluid in the ventricle and to prevent backflow before withdrawal. Control mice were injected with the same volume of aCSF by the same method. After stereotaxic injection, mice were allowed to recover from anesthesia, and they were returned to their home cages once fully awake.
Stereotactic Intracerebroventricular Injection of Mice and Subsequent Immunoblot Analysis
C57BL/6N male mice (8 weeks ± 5 days old) weighing 25 ± 1 g were anesthetized with Tribromethanol, and they were placed on a rodent stereotactic frame on a heat pad, temperature controlled by an ATC1000 DC rectal thermometer (World Precision Instruments, Berlin, Germany). To stay within the range of volume for intracerebroventricular injections recommended by the Institutional Animal Care and Use Committee of the NIH (i.e., 10 μL for a 25-g mouse), we infused 5 μg recombinant GFP (5 μL of a 1 μg/μL protein solution; Abcam, Cambridge, UK), 3 μg recombinant SerpinB2 (8 μL of a 0.308 μg/μL protein solution; OriGene, Herford, UK), and 1.3 μg recombinant Activin A (10 μL of a 0.130 μg/μL protein solution; Prospec, East Brunswick, NJ) into the left ventricle (coordinates relative to bregma: AP 0.1 mm, ML 0.9, and DV −3.0) using an Ultra Micro Pump III (World Precision Instruments, Berlin, Germany) driving a 10-μL Nanofil syringe (World Precision Instruments, Berlin, Germany). The recombinant proteins were injected at a rate of 400 nL/min, after which the needle was left in place for 5 min to equilibrate the fluid in the ventricle and to prevent backflow before withdrawal. Control mice were injected with 5 μL aCSF using the same method. Brain lysates were prepared 1 hr after stereotactic injections.
MCAO and Assessment of Stroke Volume
MCAO was carried out as described previously.10, 41 The treatment induced a permanent distal occlusion of the MCA. C57BL/6N male mice (8 weeks ± 5 days old) were anesthetized by intraperitoneal injection of 500 μL Tribromethanol (250 mg/kg body weight) and placed in a recumbent position. The animals were allowed to breathe spontaneously and were not ventilated. An incision was made from the left eye to the ear. When the temporal muscle was removed by electrocoagulation, the left MCA was visible through the semitranslucent temporal surface of the skull. After a small burr hole was made in the temporal bone with dental drill, the inner layer of the skull was removed with fine forceps, and the dura mater was opened carefully to expose the MCA. Care was taken to avoid damage to the brain tissue.
NaCl solution (0.9%) was present in the area surrounding the MCA. A microbipolar electrocoagulator ERBE ICC 200 (Erbe Elektromedizin, Tübingen, Germany) was used to permanently occlude the MCA. During surgical procedures, rectal temperature was maintained at 37°C ± 0.5°C with an ATC1000 DC temperature-controlled heat plate (World Precision Instruments, Berlin, Germany). After the incision was closed, mice were allowed to recover from anesthesia and returned to their home cages where the temperature was maintained at 37°C by placing the cage on an HT 50 S heat plate (Minitüb, Tiefenbach, Germany). In these conditions, animals were maintained homeothermic until full recovery from anesthesia. Sham-operated mice were subjected to the identical procedures without the MCA occlusion.
On day 7 after MCAO, animals were sacrificed under deep anesthesia with Narcoren and perfused intracardially with 20 mL NaCl solution (0.9%). The brains were removed from the skull and were immediately frozen on dry ice. Six consecutive 20-μm-thick coronal cryosections were cut every 400 μm and subjected to determination of total infarct volume using a silver staining technique.42 The silver-stained sections were scanned at 1,200 dpi, and infarct area was measured by using ImageJ software (NIH Image). Surgery was performed and ischemic damage was measured by an investigator who had no knowledge of the treatment group; rAAV or recombinant protein was applied by stereotactic injection or intranasal delivery.
Intranasal Delivery of Proteins
For intranasal administration of recombinant GFP (Abcam, Cambridge, UK), recombinant human Activin A (R&D Systems, Wiesbaden, Germany), recombinant human SerpinB2 (OriGene, Herford, UK), a combination of recombinant human Activin A and recombinant human SerpinB2, or recombinant human ΔNpas4 (Novus, Lingen, Germany), mice were under Tribromethanol anesthesia resulting from the MCAO procedure. Mice were placed in a supine position and a pad was inserted under the dorsal neck to extend the head back toward the supporting surface. During intranasal application procedures, rectal temperature was maintained at 37°C ± 0.5°C with an ATC1000 DC temperature-controlled heat plate (World Precision Instruments, Berlin, Germany). For each treatment, 1 μg each of Activin A, SerpinB2, GFP, or ΔNpas4 or a combination of 1 μg Activin A and 1 μg SerpinB2 in a volume of 20 μL with or without TDM was applied; we used TDM concentrations effective in enhancing nose-to-brain delivery in the range of 0.14%–0.25%. The mice received 3 rounds of treatment, starting 10 min after the MCAO followed by additional intranasal applications at 24 and 48 hr post-injury. The total volume of 20 μL protein solution was administered to the left naris within 20 min by applying 2-μL drops with a small pipette every 2 min. Mice remained another 20 min in the same position, after which they were put back into a natural resting position before returning to their home cages once fully awake.
Immunoblotting
Tissue lysates were prepared with radioimmunoprecipitation assay (RIPA) buffer (150 mM sodium chloride, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mM Tris-HCl [pH 8.0]) containing 1% protease inhibitor cocktail. Protein concentrations in the lysates were measured using the Bio-Rad protein assay. Lysates containing 40 μg total protein were analyzed by immunoblot analysis using standard protocols. Protein samples were separated by SDS-PAGE and transferred onto nitrocellulose membranes. Membranes were blocked with PBS containing 5% non-fat dry milk and 0.1% Tween 20, and they were subsequently incubated with rabbit anti-FLAG antibody or rabbit anti-GFP antibody diluted 1:500 and 1:10,000, respectively, in PBS containing 0.1% Tween 20 and 5% BSA. Tubulin detected using a mouse anti-tubulin antibody served as a protein loading control. Following incubation with appropriate secondary antibodies (HRP-conjugated goat anti-rabbit IgG or HRP-conjugated goat anti-mouse), signals were visualized using ECL Western Blotting Detection Reagents.
Statistical Analysis
Data are expressed as mean ± SD. Statistical tests were performed with GraphPad Prism 5.0 software. Statistical significance was assessed using non-parametric one-way ANOVA with Bonferroni post-test to compare all pairs of columns. Significance level alpha was set to 0.05 (95% confidence intervals).
Author Contributions
H.B. conceived the project. H.B. and B.B. designed the experiments. B.B. and U.W. performed all the experiments. H.B., B.B., and U.W. analyzed and interpreted the data. H.B. and B.B. wrote the paper.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgments
We thank Dr. Thomas Bruckner, Department of Medical Biometry, Institute of Medical Biometry and Informatics at Heidelberg University, for his help with the biostatistical and biometrical planning of the study and Jing Yan for his help with the design of the figures. This work was supported by the Deutsche Forschungsgemeinschaft (DFG) (BA 1007/4-1), a European Research Council (ERC) Advanced Grant (233024), the Sonderforschungsbereich (SFB) 1134 of the DFG, and the DFG Forschergruppe FOR 2289. H.B. is a member of the Excellence Cluster CellNetworks at Heidelberg University.
References
- 1.Zhang S.-J., Steijaert M.N., Lau D., Schütz G., Delucinge-Vivier C., Descombes P., Bading H. Decoding NMDA receptor signaling: identification of genomic programs specifying neuronal survival and death. Neuron. 2007;53:549–562. doi: 10.1016/j.neuron.2007.01.025. [DOI] [PubMed] [Google Scholar]
- 2.Zhang S.J., Zou M., Lu L., Lau D., Ditzel D.A., Delucinge-Vivier C., Aso Y., Descombes P., Bading H. Nuclear calcium signaling controls expression of a large gene pool: identification of a gene program for acquired neuroprotection induced by synaptic activity. PLoS Genet. 2009;5:e1000604. doi: 10.1371/journal.pgen.1000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bading H. Nuclear calcium signalling in the regulation of brain function. Nat. Rev. Neurosci. 2013;14:593–608. doi: 10.1038/nrn3531. [DOI] [PubMed] [Google Scholar]
- 4.Hardingham G.E., Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bas-Orth C., Bading H. The divergence-convergence model of acquired neuroprotection. Mech. Dev. 2013;130:396–401. doi: 10.1016/j.mod.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 6.Qiu J., Tan Y.-W., Hagenston A.M., Martel M.A., Kneisel N., Skehel P.A., Wyllie D.J., Bading H., Hardingham G.E. Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat. Commun. 2013;4:2034. doi: 10.1038/ncomms3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahlgren H., Bas-Orth C., Freitag H.E., Hellwig A., Ottersen O.P., Bading H. The nuclear calcium signaling target, activating transcription factor 3 (ATF3), protects against dendrotoxicity and facilitates the recovery of synaptic transmission after an excitotoxic insult. J. Biol. Chem. 2014;289:9970–9982. doi: 10.1074/jbc.M113.502914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lau D., Bengtson C.P., Buchthal B., Bading H. BDNF reduces toxic extrasynaptic NMDA receptor signaling via synaptic NMDA receptors and nuclear-calcium-induced transcription of inhba/Activin A. Cell Rep. 2015;12:1353–1366. doi: 10.1016/j.celrep.2015.07.038. [DOI] [PubMed] [Google Scholar]
- 9.Depp C., Bas-Orth C., Schroeder L., Hellwig A., Bading H. Synaptic activity protects neurons against calcium-mediated oxidation and contraction of mitochondria during excitotoxicity. Antioxid. Redox Signal. 2017 doi: 10.1089/ars.2017.7092. Published online November 14, 2017. [DOI] [PubMed] [Google Scholar]
- 10.Zhang S.J., Buchthal B., Lau D., Hayer S., Dick O., Schwaninger M., Veltkamp R., Zou M., Weiss U., Bading H. A signaling cascade of nuclear calcium-CREB-ATF3 activated by synaptic NMDA receptors defines a gene repression module that protects against extrasynaptic NMDA receptor-induced neuronal cell death and ischemic brain damage. J. Neurosci. 2011;31:4978–4990. doi: 10.1523/JNEUROSCI.2672-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lubjuhn J., Gastens A., von Wilpert G., Bargiotas P., Herrmann O., Murikinati S., Rabie T., Marti H.H., Amende I., Hampton T.G., Schwaninger M. Functional testing in a mouse stroke model induced by occlusion of the distal middle cerebral artery. J. Neurosci. Methods. 2009;184:95–103. doi: 10.1016/j.jneumeth.2009.07.029. [DOI] [PubMed] [Google Scholar]
- 12.Krieglstein K., Zheng F., Unsicker K., Alzheimer C. More than being protective: functional roles for TGF-β/activin signaling pathways at central synapses. Trends Neurosci. 2011;34:421–429. doi: 10.1016/j.tins.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 13.Rodríguez-Martínez G., Velasco I. Activin and TGF-β effects on brain development and neural stem cells. CNS Neurol. Disord. Drug Targets. 2012;11:844–855. doi: 10.2174/1871527311201070844. [DOI] [PubMed] [Google Scholar]
- 14.Link A.S., Zheng F., Alzheimer C. Activin Signaling in the pathogenesis and therapy of neuropsychiatric diseases. Front. Mol. Neurosci. 2016;9:32. doi: 10.3389/fnmol.2016.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Law R.H., Zhang Q., McGowan S., Buckle A.M., Silverman G.A., Wong W., Rosado C.J., Langendorf C.G., Pike R.N., Bird P.I., Whisstock J.C. An overview of the serpin superfamily. Genome Biol. 2006;7:216. doi: 10.1186/gb-2006-7-5-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Silverman G.A., Whisstock J.C., Askew D.J., Pak S.C., Luke C.J., Cataltepe S., Irving J.A., Bird P.I. Human clade B serpins (ov-serpins) belong to a cohort of evolutionarily dispersed intracellular proteinase inhibitor clades that protect cells from promiscuous proteolysis. Cell. Mol. Life Sci. 2004;61:301–325. doi: 10.1007/s00018-003-3240-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawano T., Morimoto K., Uemura Y. Partial purification and properties of urokinase inhibitor from human placenta. J. Biochem. 1970;67:333–342. doi: 10.1093/oxfordjournals.jbchem.a129257. [DOI] [PubMed] [Google Scholar]
- 18.Lyons-Giordano B., Loskutoff D., Chen C.S., Lazarus G., Keeton M., Jensen P.J. Expression of plasminogen activator inhibitor type 2 in normal and psoriatic epidermis. Histochemistry. 1994;101:105–112. doi: 10.1007/BF00269356. [DOI] [PubMed] [Google Scholar]
- 19.Gross T.J., Sitrin R.G. The THP-1 cell line is a urokinase-secreting mononuclear phagocyte with a novel defect in the production of plasminogen activator inhibitor-2. J. Immunol. 1990;144:1873–1879. [PubMed] [Google Scholar]
- 20.Borstnar S., Vrhovec I., Svetic B., Cufer T. Prognostic value of the urokinase-type plasminogen activator, and its inhibitors and receptor in breast cancer patients. Clin. Breast Cancer. 2002;3:138–146. doi: 10.3816/CBC.2002.n.018. [DOI] [PubMed] [Google Scholar]
- 21.Choy F.C., Klarić T.S., Koblar S.A., Lewis M.D. The Role of the Neuroprotective Factor Npas4 in Cerebral Ischemia. Int. J. Mol. Sci. 2015;16:29011–29028. doi: 10.3390/ijms161226144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Woitecki A.M., Müller J.A., van Loo K.M., Sowade R.F., Becker A.J., Schoch S. Identification of Synaptotagmin 10 as Effector of NPAS4-Mediated Protection from Excitotoxic Neurodegeneration. J. Neurosci. 2016;36:2561–2570. doi: 10.1523/JNEUROSCI.2027-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alcalá-Barraza S.R., Lee M.S., Hanson L.R., McDonald A.A., Frey W.H., 2nd, McLoon L.K. Intranasal delivery of neurotrophic factors BDNF, CNTF, EPO, and NT-4 to the CNS. J. Drug Target. 2010;18:179–190. doi: 10.3109/10611860903318134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thorne R.G., Pronk G.J., Padmanabhan V., Frey W.H., 2nd Delivery of insulin-like growth factor-I to the rat brain and spinal cord along olfactory and trigeminal pathways following intranasal administration. Neuroscience. 2004;127:481–496. doi: 10.1016/j.neuroscience.2004.05.029. [DOI] [PubMed] [Google Scholar]
- 25.Renner D.B., Svitak A.L., Gallus N.J., Ericson M.E., Frey W.H., 2nd, Hanson L.R. Intranasal delivery of insulin via the olfactory nerve pathway. J. Pharm. Pharmacol. 2012;64:1709–1714. doi: 10.1111/j.2042-7158.2012.01555.x. [DOI] [PubMed] [Google Scholar]
- 26.Neumann I.D., Maloumby R., Beiderbeck D.I., Lukas M., Landgraf R. Increased brain and plasma oxytocin after nasal and peripheral administration in rats and mice. Psychoneuroendocrinology. 2013;38:1985–1993. doi: 10.1016/j.psyneuen.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 27.Ozsoy Y., Gungor S., Cevher E. Nasal delivery of high molecular weight drugs. Molecules. 2009;14:3754–3779. doi: 10.3390/molecules14093754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lochhead J.J., Thorne R.G. Intranasal delivery of biologics to the central nervous system. Adv. Drug Deliv. Rev. 2012;64:614–628. doi: 10.1016/j.addr.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 29.Rodríguez Cruz Y., Mengana Támos Y., Muñoz Cernuda A., Subirós Martines N., González-Quevedo A., Sosa Testé I., García Rodríguez J.C. Treatment with nasal neuro-EPO improves the neurological, cognitive, and histological state in a gerbil model of focal ischemia. Sci. World J. 2010;10:2288–2300. doi: 10.1100/tsw.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang J.P., Liu H.J., Wang Z.L., Cheng S.M., Cheng X., Xu G.L., Liu X.F. The dose-effectiveness of intranasal VEGF in treatment of experimental stroke. Neurosci. Lett. 2009;461:212–216. doi: 10.1016/j.neulet.2009.06.060. [DOI] [PubMed] [Google Scholar]
- 31.Arnold J.J., Ahsan F., Meezan E., Pillion D.J. Correlation of tetradecylmaltoside induced increases in nasal peptide drug delivery with morphological changes in nasal epithelial cells. J. Pharm. Sci. 2004;93:2205–2213. doi: 10.1002/jps.20123. [DOI] [PubMed] [Google Scholar]
- 32.Khoshnam S.E., Winlow W., Farzaneh M., Farbood Y., Moghaddam H.F. Pathogenic mechanisms following ischemic stroke. Neurol. Sci. 2017;38:1167–1186. doi: 10.1007/s10072-017-2938-1. [DOI] [PubMed] [Google Scholar]
- 33.Durukan A., Tatlisumak T. Acute ischemic stroke: overview of major experimental rodent models, pathophysiology, and therapy of focal cerebral ischemia. Pharmacol. Biochem. Behav. 2007;87:179–197. doi: 10.1016/j.pbb.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 34.Bading H. Therapeutic targeting of the pathological triad of extrasynaptic NMDA receptor signaling in neurodegenerations. J. Exp. Med. 2017;214:569–578. doi: 10.1084/jem.20161673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dirnagl U. Pathobiology of injury after stroke: the neurovascular unit and beyond. Ann. N Y Acad. Sci. 2012;1268:21–25. doi: 10.1111/j.1749-6632.2012.06691.x. [DOI] [PubMed] [Google Scholar]
- 36.Rossi D.J., Oshima T., Attwell D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature. 2000;403:316–321. doi: 10.1038/35002090. [DOI] [PubMed] [Google Scholar]
- 37.Soria F.N., Pérez-Samartín A., Martin A., Gona K.B., Llop J., Szczupak B., Chara J.C., Matute C., Domercq M. Extrasynaptic glutamate release through cystine/glutamate antiporter contributes to ischemic damage. J. Clin. Invest. 2014;124:3645–3655. doi: 10.1172/JCI71886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mukerji S.S., Rainey R.N., Rhodes J.L., Hall A.K. Delayed activin A administration attenuates tissue death after transient focal cerebral ischemia and is associated with decreased stress-responsive kinase activation. J. Neurochem. 2009;111:1138–1148. doi: 10.1111/j.1471-4159.2009.06406.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sonvico F., Clementino A., Buttini F., Colombo G., Pescina S., Stanisçuaski Guterres S., Raffin Pohlmann A., Nicoli S. Surface-Modified Nanocarriers for Nose-to-Brain Delivery: From Bioadhesion to Targeting. Pharmaceutics. 2018;10:34. doi: 10.3390/pharmaceutics10010034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parsons M.P., Raymond L.A. Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron. 2014;82:279–293. doi: 10.1016/j.neuron.2014.03.030. [DOI] [PubMed] [Google Scholar]
- 41.Martinou J.C., Dubois-Dauphin M., Staple J.K., Rodriguez I., Frankowski H., Missotten M., Albertini P., Talabot D., Catsicas S., Pietra C. Overexpression of BCL-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischemia. Neuron. 1994;13:1017–1030. doi: 10.1016/0896-6273(94)90266-6. [DOI] [PubMed] [Google Scholar]
- 42.Herrmann O., Baumann B., de Lorenzi R., Muhammad S., Zhang W., Kleesiek J., Malfertheiner M., Köhrmann M., Potrovita I., Maegele I. IKK mediates ischemia-induced neuronal death. Nat. Med. 2005;11:1322–1329. doi: 10.1038/nm1323. [DOI] [PubMed] [Google Scholar]