

Abstract
As a popular sample preparation approach, filter-aided sample preparation (FASP) has been widely used in proteomic analysis. However, several limitations have been noted, including sample loss during filtration, repetitive centrifugation steps, and the possibility of breakage of filtration membrane. Extraction bias among different sample preparation strategies presents another challenge. To overcome these limitations and address remaining challenges, we developed a novel surfactant and chaotropic agent assisted sequential extraction/on-pellet digestion (SCAD) protocol. The new strategy resulted in higher protein yield and improved peptide recovery and protein coverage compared to two conventional sample preparation methods (FASP and urea). In combination of three strategies, more than 10,000 distinct protein groups were identified with 1% FDR from MDA-MB-231 cells without any prefractionation. This in-depth proteome analysis was accomplished by optimization of protein extraction, enzymatic digestion, LC gradient, and peptide sequencing method. Ingenuity Pathways Analysis (IPA) of proteins exclusively identified in SCAD revealed several crucial signaling pathways that regulate breast cancer progression. SCAD also enabled an unbiased extraction of different categories of proteins (membrane, intracellular, nuclear) associated with tumorigenesis, which integrates the advantages of FASP and urea extraction. This novel strategy expedites comprehensive protein identification, which is applicable for biomarker discovery in various types of cancers.
Keywords: Protein extraction, enzymatic digestion, protein identification, SCAD, FASP, urea, peptide sequencing, LFQ, iBAQ, breast cancer
INTRODUCTION
With the advent of soft ionization techniques and tremendous advancements in mass spectrometry (MS) instrumentation, modern MS has provided unprecedented insight into the composition of the proteome.1,2 Protein extraction is often the first step and bottleneck of the overall experimental workflow that can significantly affect the experimental outcome and proteome coverage. Only the extracted proteins are accessible for subsequent sample preparation and fractionation steps, and consequently, they are the only ones to be detected in the MS. Over the past few decades, numerous protein extraction and digestion protocols have been developed.3–11 Sodium dodecyl sulfate (SDS) is a strong denaturant that is widely used in various types of biological research. It is, however, incompatible with MS analysis. Wisniewski et al. developed the FASP method that allows the sample to be first dissolved in SDS, and then be subsequently removed before enzymatic digestion.3 Since then, the method has been successfully applied and derivatized to the preparation of samples from different sources such as cultured cells and fresh, frozen, FFPE tissues.3,4,8,12,13 Despite the tremendous success of this strategy, some caveats have been reported.14,15 Breakage of the filtration membrane occurs at times during the ultracentrifugation step, resulting in sample loss. The capacity of the filtration unit only allows a limited amount of sample to be loaded onto the device. Moreover, the peptide yield is also dramatically reduced when the loading material exceeds maximum capacity. The extensive centrifugation steps for the buffer exchange are time-consuming, and the filtration unit is the prerequisite device for the experiment, yet not every lab has access to it. In-solution digestion is another popular sample preparation approach that uses chaotropic denaturant such as urea to disrupt the noncovalent interactions between the macromolecules thus enhancing the protein solubility. Fewer steps are needed for the protein digestion and the chaotropic agent can be easily removed before the MS analysis, making it a favorable choice for many laboratories.16–19 However, the method is limited by the low extraction efficiency of membrane proteins, which restricts its application for biological scaffolds as well as glycoproteins.20
Although various MS-based sample preparation techniques are available to obtain protein profiles, fewer studies have focused on the quantitative bias introduced by different protein extraction strategies. Using the protein fraction from mitochondria of rat liver, Leon et al. evaluated the performance of several in-solution and filter-aided digestion protocols.21 Their study revealed some method-dependent quantification variations; however, the research scope was limited to mitochondrial proteins with approximately 330 proteins quantified, which hindered its application to other organelles and species. Glatter et al. compared the global proteome extraction differences between in-solution and FASP-based sample preparation strategies.22 Both bacteria and human cells were investigated in the study where the inconsistency of extraction efficiency was observed between two sample types. Although the results provide more insights into the organism specific quantitative bias from different sample preparation methods, the LC-MS method was not optimized for maximizing the peptide identification (ID) and quantification, rendering less than half of the proteins that were identified from the entire proteome.
In the peptide-centric proteomic analysis, the quantification accuracy of the corresponding proteins heavily relies on the completeness of peptide detection.23 In order to improve the number of identified peptides, multidimensional fractionation techniques are commonly employed.7,24,25 With extensive offline fractionations, Kim et al. presented the first draft map of the human proteome.26 Using a modified offline high-pH fractionation technique, Bekker-Jensen et al. demonstrated that proteomic analysis can reach a similar depth as the next-generation RNA-seq technology.25 These approaches reduced the complexity of peptide mixtures and enabled deep analysis of cellular proteomes. Substantial increments of instrument time and sample consumption are needed to compensate for the increased number of protein IDs, which not only reduce the throughput of the experiment, but also impede the automation of the entire procedure. With the improving performance of LC systems and mass spectrometers, the analytical detection gap between single-shot and multidimensional proteomics has been remarkably reduced.17,18,27,28 The depth of a single shot proteomic analysis is largely affected by the quality of peptide separation.29 Using a 30 cm capillary LC column (1.7 μm beads) coupled to an Orbitrap Fusion MS instrument, Coon and colleagues reported comprehensive analysis of the yeast proteome within an hour.18,28 Optimization of fragmentation and other MS parameters is also crucial for maximizing peptide detection. Tu et al. compared four MS2 fragmentations on an Orbitrap Fusion and noticed a 20% difference in peptide IDs.30 By carefully tuning MS parameters such as dynamic exclusion (DE) and m/z detection window, Pirmoradian et al. reported the detection of 5354 proteins, which is close to 50% of the proteome coverage17·
In this study, we developed a surfactant and chaotropic agent assisted sequential extraction/on-pellet digestion (SCAD) protocol, which provides high protein yield and peptide recovery with minimal sample consumption. The peptide assay was performed after desalting, which took into consideration the sample loss during protein extraction/digestion and provided better assessment of sample amount loaded onto the column for quantitative analysis. In this work, the SCAD method was extensively evaluated in comparison to two widely used sample preparation strategies, in solution digestion (urea) and FASP. For maximizing the proteome coverage of a single injection, different LC gradients were investigated, and the peptide sequencing approach was optimized on the Orbitrap Fusion Lumos instrument. Finally, the quantitative protein extraction bias among three different methods were evaluated. Compared to other methods, SCAD represents an improved protocol for enhanced proteomics by combining the unique features from the other methods and rendering a more comprehensive coverage of different categories of proteins.
EXPERIMENTAL DESIGN
Sample Preparation
Cell Culture.
MDA-MB-231 cells were maintained in DMEM supplemented with 10% fetal bovine serum (Gibco, Gaithersburg, MD), in a 37 °C humid cell incubator with 5% CO2 1 × 107 cells were seeded into a 15 cm dish and cultured for 3 days. The cells were harvested followed by trypsin digestion and washed with PBS three times. The cell pellets were stored at −80 °C until use.
Protein Extraction and Digestion.
For SCAD, 10 μL of cells (6 × 107/ml) were dissolved in 30 μL of buffer solution (4% SDS, 50 mM Tris buffer) and incubated at 95 °C for 10 min. Cell lysate was sonicated for 15 min. The protein extract was reduced with 10 mM dithiothreitol (DTT) for 30 min at room temperature and alkylated with 50 mM iodoacetic acid (IAA) for an additional 30 min in the dark. SDS was removed by two rounds of precipitation. For the first time, cold acetone (−20 °C) was added to a final concentration of 80% (v/v), and the protein was precipitated overnight at −20 °C. For the second round, 80% acetone/water (v/v) was added, followed by incubation at −20 °C for 2 h. The sample was centrifuged at 16,000 × g for 15 min, and the pellet was air-dried at room temperature. 8M urea was added to the pellet, and on-pellet digestion was performed with Lys-C/trypsin (1:100, Promega) for 4 h at 37 °C. 50 mM Tris buffer was added to the urea solution along with trypsin (1:100) for overnight digestion. The reaction was quenched with 1% TFA and desalted with Sep-Pak C18 cartridge (Waters). Concentrations of peptide mixture were measured by peptide assay (Thermo). Samples were lyophilized and stored in −80 °C until use.
For FASP, 10 μL of cells were dissolved in 30 μL of buffer solution (4% SDS, 50 mM Tris buffer, 0.1 M DTT) and incubated at 95 °C for 5 min. The experiments were conducted following the previously published procedure.3 In brief, protein lysate was loaded onto a 30k centrifugal filter unit and underwent ultrafiltration at 14,000 × g. Urea was used to remove the SDS, and the sample was alkylated with 0.05 M iodocacetamide. Proteins were subjected to on-membrane trypsin digestion (1:50), quenched with 1% TFA, and desalted with a Sep-Pak C18 cartridge (Waters).
For in-solution digestion with urea, 10 μL of cells were lysed in lysis buffer containing 8 M urea, 50 mM Tris-HCl, 30 mM NaCl, and protease inhibitor tablet. The experiments followed the protocol described previously.19 In brief, protein extract was reduced with DTT, alkylated with IAA, and subjected to digestion with trypsin (1:50). The samples were quenched with 1% TFA and desalted with a Sep-Pak C18 cartridge (Waters).
MS Acquisition.
Peptide mixtures were reconstituted in 0.1% FA and 3% ACN and loaded onto either the 15 or 30 cm fabricated column (C18, 75 μm). The columns were filled with 1.7 μm Ethylene Bridged Hybrid packing materials (130 Å, Waters) for improved resolving power of online LC separation. Different gradient times from 90 to 180 min were tested for peptide IDs. Samples were injected onto a Dionex UltiMate 3000 UPLC system coupled to the Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Fisher Scientific, San Jose, CA).
Universal Method.
The instrument was operated in data-dependent acquisition (DDA) mode. MS scans were recorded in the orbitrap (OT) in the range of m/z 400–1500 at resolution of 120 K, followed by MS/MS acquisition in the linear ion trap (IT). Top speed algorithm was initiated with the cycle time of 3 s and an isolation width of 1.6 m/z. Normalized collisional energy (NCE) of 28% (HCD) or 35% (CID) was selected for fragmentation. 4 × 105 and 4 × 103 were selected as the automatic gain control (AGC) targets for MS1 and MS2 scans, respectively. The maximum injection time was set to be 50 and 300 ms for MS1 and MS2 scans, respectively.
OT-HCD Method.
MS scans were acquired in profile mode in the range of m/z 300–1500 at resolution of 60 K, followed by selection of the 15 most intense ions for HCD fragmentation with an isolation width of 1 m/z. 2 × 105 and 1 × 104 were selected as the AGC targets for MS and MS/MS scans, respectively. The maximum injection time was set to 100 ms for both MS and MS/MS scans. Tandem mass spectra were acquired with a NCE of 30.
Data Analysis
Proteome Discoverer 2.1 and MaxQuant were used to perform peptide identification and quantification. The data was searched against the Swiss-prot human protein database (isoform included, December 2015, 42129). For MS1 scans, a precursor ion mass tolerance of 10 ppm was used, and two missed cleavages were allowed. Fragment ion tolerance was set to 0.02 Da for the Orbitrap MS2 detections and 0.5 Da for the IT detections. The variable modifications included methionine oxidation and N-terminal protein acetylation, whereas carbamidomethylation of cysteine residues was set as fixed modification. The false discovery rate (FDR) was set at 0.01 for both peptide and protein identification using the target-decoy strategy.31 The minimal number of unique peptide(s) per protein was 1. Quantification was performed using MaxQuant (1.6.0.1) with MaxLFQ algorithm.32 The “match between run” option was enabled to maximize the number of quantification events across samples.
All statistical analyses were accomplished by Perseus.33 GOBP, GOCC, GOMC, and KEGG were used for protein annotations. For hierarchical clustering of different sample preparation methods, a minimum of one-third of valid values were required. MaxLFQ intensities were normalized using z-score analysis and both column and row clustering were generated based on Euclidean distance using the average linkage method. Enrichment analysis was performed using Fisher’s exact test with a Benjamini-Hochberg FDR threshold of 0.02. Intensity-based absolute quantitation (iBAQ) values were calculated based on the summed intensities of all unique peptides for a protein divided by the number of theoretical tryptic peptides between 6 and 30 amino acids in length.34 Proteins exclusively identified by each extraction method were explored by Ingenuity Pathways Analysis (IPA; Ingenuity Systems, Redwood City, CA) to reveal signaling networks and biological processes.
RESULTS AND DISCUSSION
Development of the SCAD Method
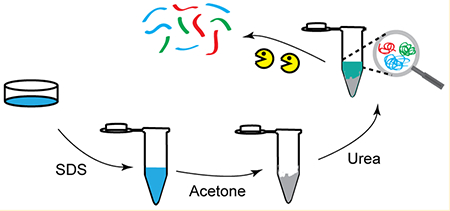
Although FASP has gained increasing popularity in the proteomics field, several limitations have hindered the protein extraction including breakage of the filtration membrane, relatively low digestion efficiency and sample yield, and the need for repetitive centrifugation steps. Furthermore, extraction bias among different sample preparation strategies has been observed. To circumvent these issues, we developed surfactant and chaotropic agent assisted sequential extraction/on-pellet digestion (SCAD) protocol (Figure 1). The filtration device was replaced by acetone precipitation to avoid the extensive centrifugation process. (Preservation of water-soluble proteins is discussed in Supporting Information and Figure S-1.) The protein yield and digestion efficiency were enhanced by the on-pellet digestion. Sequential extraction with urea enhanced the comprehensiveness and unbiased enrichment of proteins in various categories, including detergent insoluble proteins (DIP),17,35,36 while the peptide assay provides a more accurate estimation of the amount of peptide mixture loaded onto the column for quantitative analysis. Different gradient time and peptide sequencing techniques were investigated to maximize the peptide and protein IDs, and enrichment analysis was performed to evaluate the extraction bias among different sample preparation strategies.
Figure 1.

Workflow of the SCAD strategy. Proteins were first solubilized by SDS and then subjected to precipitation to remove the detergent. The pellet was reconstituted in urea, and on-pellet digestion was performed. The concentration of the peptide mixture was measured after desalting to ensure accurate estimation of peptide amount.
SCAD Improves Protein Extraction, Protein Coverage, and Peptide Recovery
The overall efficiency of protein extraction heavily depends on the sample preparation strategy by which the proteomic analysis is conducted. Accurate quantification relies on protein extraction from the cells. To compare the efficiency of different extraction methods, 10 μL of the cells (6 × 107/mL) was used for each method, and three technical replicates were performed. As exemplified in Figure 2A, the SCAD method enabled the extraction of the highest amount of peptides (600 μg) from merely 10 μL of cells, which clearly outperformed the other two methods. An almost 3-fold increase in peptide yield was observed when compared with FASP. Although the increase was less pronounced in comparison to the urea method, an elevated level of 20% was observed. Previous studies reported that FASP exhibited decreased performance when protein load was higher than 100 μg.14 To evaluate the performance of SCAD at different amount of starting materials, two other cell amounts (2 and 5 μL) were also tested to better determine the extraction efficiency of SCAD. In line with previous report, lower amount of starting material resulted in higher yield for FASP, whereas a decrease of yield was observed for higher load (Figure S-2). The yield of SCAD, however, remains unchanged with the increase of starting material. Taken together, the SCAD method exhibits superior performance for protein extraction compared to existing methods. This approach is especially valuable when the experimental material is limited. The distinct differences of extracted protein quantities among three sample preparation protocols also testified the impact of sample preparation on protein quantification.
Figure 2.

Yield of each sample preparation strategy measured by peptide assay (A), comparison of sequence coverage achieved (B), number of peptides (C), and protein groups (D) under different LC gradient times with OT-HCD.
For the peptide-centric label-free quantification, accurate estimation of protein abundance depends on the efficiency of tryptic peptide recovery, which is reflected by the number of unique peptide IDs, the protein sequence coverage, and the number of total identified MS/MS spectra (PSM). To evaluate the performance of different sample preparation protocols, 1 μg of digested cell lysate from each of the different methods was analyzed with various lengths of chromatographic gradient (90, 120, and 180 min) by HCD fragmentation and MS2 acquisition in the OT. As expected, 180 min rendered a higher number of unique peptide IDs compared to the other two gradient times, with the highest number of identified unique peptides being 40380 on average for SCAD. On the contrary, lower numbers of unique peptide IDs were observed for the 90 min gradient, with the lowest number of unique peptide IDs being 18818 from the FASP method. It is obvious from the results that, at every gradient time, more unique peptides were identified by the new method compared to FASP, and a comparable or slightly better result was observed in comparison to the urea method. It is worth noting that the percentage difference of the unique peptide IDs between SCAD and FASP was significantly enlarged when the gradient time increased from 90 to 180 min (51% vs 77%), indicating that when the peptide mixture was fully separated by the online LC system, more unique peptides were generated by the SCAD approach. SDS and urea are both excellent denaturant, under the synergic effect of both, proteins would be more completely denatured and the tertiary structure would be exposed and subjected to the proteolytic digestion. The action of on-pellet digestion also boosted the peptide recovery, which rendered the highest digestion efficiency and protein coverage among all three methods. An alternative way to evaluate the proteome coverage is to calculate the average amino acid sequence coverage per protein ID. As shown in Figure 2B, the average sequence coverage for SCAD is 30%, which is significantly higher than the average sequence coverage for FASP (15%) and urea (25%).
Acquisition rate of the mass analyzer is often the bottleneck of deep proteome analysis. If the peptide eluting rate exceeds the sequencing speed of the method, the less abundant, coeluting peptides may be skipped for fragmentation in the DDA mode. As a result, the corresponding proteins will not be identified. Given the same digestion efficiency of two sample preparation strategies, equal number of proteins can be identified using the same gradient. If the digestion efficiency of one strategy is higher than the other, more unique peptides will be generated from the same protein. These peptides can be fragmented and identified if they are fully separated by the LC system. Alternatively, the more abundant peptides will have priority to be fragmented in DDA, and when they are coeluting with other peptides, those peptides present at low abundance might be “missed” during the acquisition. In contrast, fewer unique peptides will be generated from each protein in the method with lower digestion efficiency, rendering a less complex peptide mixture. These peptides can be better separated, and even peptides at lower abundance could be selected for fragmentation, leading to detection of the corresponding protein. Under this circumstance, fewer unique peptides will be detected in total while more protein groups are identified. As illustrated in Figures 2C,D, in a 90 min gradient, 52% more peptide IDs were detected by SCAD (28558) and urea (27990) in comparison to FASP (18818), while the highest number of proteins were observed by FASP. This could lead to the misinterpretation of the data that the lower digestion efficiency protocol generates more protein IDs. To circumvent the issue, the optimization of gradient time and fragmentation method is needed for comprehensive peptide sequencing. By extending the gradient profile from 90 to 180 min, the difference between FASP and the other two methods was significantly minimized, and similar numbers of identified proteins were achieved.
We also investigated the missed cleavage rate between different strategies. Interestingly, although FASP rendered the lowest peptide IDs as shown above, it generates the lowest missed cleavage rate (Figure S-3). In contrast, the highest missed cleavage rate was observed in the SCAD method. It was noted that the missed cleavage rate of SCAD method was 22%, which was comparable or even better than previous literatures.14,37 Previous work by Wisniewski et al. reported an increase of missed cleavage rate with the increase of urea concentration.14 We envisioned that similar trend could be observed in our study. By reducing the urea concentration, lower missed cleavage may be achieved, which will further boost the peptide/protein recovery. Investigation of the effect of urea concentration on missed cleavage rate of SCAD is currently underway.
Comparison and Optimization of Peptide Sequencing Techniques
We next evaluated how different fragmentation techniques could affect protein detection and identification. In order to decipher the method-dependent extraction bias among the three different approaches, it is of great importance to identify and quantify as many proteins as possible. To accomplish this goal, we first set up the experiments to evaluate the performance of two mass analyzers, IT or OT, in combination with HCD or CID fragmentation to achieve maximal protein IDs. Four tandem mass acquisition strategies are available from Fusion Lumos instrument settings. The three most commonly used methods, OT-HCD, IT-HCD, and IT-CID, were investigated in the current study. Since the SCAD method generates the highest number of tryptic peptides compared to the other two strategies, 1 μg of tryptic peptides extracted by SCAD was analyzed in a 90 min gradient. Eliuk et al. developed the universal method to maximize the peptide identification without prior knowledge of analyte abundance and sample complexity.38 As a starting point, we first compared the HCD Orbitrap method optimized in our lab with the Universal methods introduced by Thermo. As shown in Figure 3A, more peptide IDs, PSMs, and MS/MS were given by ITHCD (an average of 28581 peptides, 37765 PSMs, 95083 MS/MS) compared to IT-CID (25131, 32751, 78710). The difference in performance noted between these two fragmentation methods could be attributed to the better fragmentation efficiency, shorter collisional time, and less idle time between each module of the instrument by IT-HCD. Among these three MS2 acquisition techniques, OT-HCD (28558, 37798, 82585) achieved similar results in peptide IDs and PSM numbers compared to IT-HCD, which clearly outperforms that of IT-CID. Notably, lower number of tandem mass spectra (higher success rate) was acquired by the OT-HCD compared to IT-HCD, illustrating the advantage of high mass accuracy and better spectral quality in OT for peptide identification. Similar outcomes were also reported by previous publications,30,39 further supporting our observations.
Figure 3.

Comparison of number of peptides, PSMs, and MS/MS (A) and protein groups (B) with different peptide sequencing approaches under 90 min gradient; number of proteins with optimized IT-HCD under 180 min gradient (C); Venn diagram showing overlap of proteins among different sample preparation strategies (D).
To further improve the protein IDs without extending the gradient time, a longer LC column of 30 cm packed with 1.7 μm beads was evaluated. The temperature of the column was maintained at 60 °C throughout the analysis with a column heater in order to counteract the pressure increase induced by the longer column. By applying this strategy, a significant increase of 23% more peptide IDs and 13% more PSMs were observed using the same OT-HCD method in a 90 min gradient. The increased peptide number also led to the identification of 15% more proteins compared to the same OTHCD method with a 15 cm column.
In shotgun proteomic experiments, the performance of peptide sequencing is affected by the analyte amounts. In order to maximize peptide identifications independent of the abundance of analytes, the universal method is programmed to use longer MS2 acquisition time. If the analytes are present at low abundance in the peptide mixture, the extended tandem MS acquisition allows the target analytes to be collected for longer period to ensure spectral quality. In the current study, the sample amount of the analytes was predetermined; therefore, the duty cycle was impeded by the utilization of longer MS2 acquisition time. Isolation window is another key factor that affects the number of peptide IDs. Using a larger isolation window enables higher ion influx to fill the trap, while a smaller isolation window minimizes the interference from coeluted peptides. For the present study, different MS2 acquisition times (15, 35, 50, 100, and 300 ms) were examined. The best result was achieved with 35 ms (Figure S-4). By decreasing the maximum injection time from 300 to 35 ms, the number of peptide IDs and PSMs was increased by 13%. We further compared different isolation windows (0.6, 1, and 1.6 m/z). Although 1.6 m/z generated the highest number of tandem MS spectra, 1.0 m/z rendered the highest number of peptide IDs and PSMs (Figure S-5). Dynamic exclusion (DE) is another critical parameter that staves off the oversampling of the high abundance precursors. After comparing three DE times (15, 30, and 45 s), slightly improved peptide IDs were observed with 45 s DE time (Figure S-6).
Upon tuning various parameters, we optimized the universal method by reducing the MS2 injection time to 35 ms and constraining the isolation window to 1 m/z to balance between the scan rate and sensitivity of instrument method to enable maximal peptide IDs. Using this optimized IT-HCD method in conjunction with a 30 cm column, the results clearly outperformed the other four strategies with regard to peptide IDs, PSMs, and MS/MS (38500, 47640, 102958). More impressively, this strategy greatly improved the number of protein IDs. In comparison to the OT-HCD with a 15 cm column in the 180 min LC-MS run (5411), the 90 min gradient time of optimized IT-HCD with a 30 cm column identified even a slightly larger number of proteins (5587), with an identification rate of 62 proteins per minute. As the resolving power of LC separation is proportional to the gradient time, two additional experiments with longer gradient time (120 and 180 min) were also evaluated. As shown in Figure S-7, in a single LC-MS run, 6280 and 7131 protein groups were identified from these two gradient times. These numbers increased to 7360 and 8360, respectively, in a triplicate analysis. Peptide mixtures extracted by urea and FASP methods were also investigated using the same instrument method. We identified, on average, 7068 and 7023 protein groups in a single LC-MS run, and by merging three replicates together, these numbers increased to 8311 and 7958, respectively (Figure 3C). By comparing the proteome coverage among the three different extraction strategies, a high percentage of overlap (>64%) was observed, and a total of 6534 protein groups were detected by all three methods. Collectively, we obtained a filtered data set (1% FDR) of 10099 proteins (Figure 3D). This created the current largest proteome data set of breast cancer cells without any fractionation step (7875 proteins reported in the recent literature by using 270 min LC separation40).
We then extended our analysis to interrogate unique signaling pathways uncovered by different extraction strategies. As indicated in Figure 4, IPA revealed key signaling pathways that regulate proliferation, differentiation, invasion, and metastasis of breast cancer cells (highlighted in red), such as IL-7 Signaling Pathway, EGF Signaling, FGF Signaling, GMCSF Signaling, PI3K/AKT Signaling, and p53 Signaling. All of these were identified in the preparation using the SCAD method (P-value < 0.05), while only a few of them passed the significance threshold using the other two methods, highlighting the superior performance of the SCAD method in regard to extracting signaling molecules that regulate crucial cellular processes inside the cancer cells.
Figure 4.

IPA analysis of protein-coding genes exclusively identified by each extraction method. Top 20 IPA pathways are plotted with their respective P values. Pathways marked in red are key signaling pathways implicated in breast cancer research.
Quantitative Protein Profiling
After the in-depth characterization of the protein IDs, we moved on to perform quantitative protein profiling and attempted to elucidate the extraction bias introduced by different experimental designs. Here, MaxQuant LFQ method was utilized to analyze the data from the three different extraction methods.41,42 We used hierarchical clustering to categorize the proteins into different groups based on their expression profiles. As illustrated in Figure 5, the proteins can be well classified into four distinct clusters. For the set of proteins that were better extracted by FASP and SCAD methods, various types of membrane proteins were enriched. In contrast, the enriched proteins using the urea and SCAD methods were localized in cytoplasm or bound to nuclear component. The FASP method showed enrichment of proteins involved in cell-cell interaction (Figure S-8), and no specific subcellular distributions were identified for the proteins that have higher extraction efficiency in FASP and urea. In line with the previous publication,22 the detergent based FASP method showed better enrichment of membrane proteins as compared to the urea method, whereas the nuclear and cellular proteins were significantly enriched by chaotropic buffers like urea. By conducting serial extractions, the SCAD method combines the advantages of FASP and urea for higher extraction efficiency of membrane and intracellular and nuclear proteins, resulting in a more comprehensive and unbiased protein extraction method for quantitative analysis.
Figure 5.

Heatmap visualization of protein expression profiles of different sample preparation strategies. A dendrogram of three extraction methods with three technical replicates was shown at the top. Protein expression values were z-score normalized prior to clustering. Hierarchical clustering of protein expression profiles identified four groups. Gene ontology enrichment analysis was performed for each protein group and cellular components of proteins, which had higher extraction efficiency in the SCAD and FASP methods compared with the urea method (right, upper) or higher expressions in the urea and SCAD methods versus FASP (right, lower).
Upon completing the comparison of relative protein expression, we extended our evaluation to investigate the difference introduced by different strategies on the absolute protein abundance. Here, we used iBAQ to infer the copy number of the proteins inside the cells, and iBAQ values were calculated for every quantified protein. Ribosomes are the primary sites of protein synthesis inside the cells. The abnormality of ribosomal proteins (RPs) are usually associated with increased risk of cancer.43 In the present study, we quantified 160 RPs in total (Table S-1). As indicated in Figure 6A, the SCAD method rendered the highest iBAQ values of RPs. A slightly lower iBAQ values were detected for the urea method. Among all three strategies, FASP generated the lowest number of RPs, which covered only 40% of iBAQ intensity compared to SCAD. The results demonstrated the enhanced performance of SCAD for the extraction of RPs, which correlate well with our finding in Figure 5. Notably, seven proteins were only detected in the SCAD and urea methods, including RPL39 (Figure S-9). Recent studies have shown that RPL39 plays an important role in the breast cancer initiation and metastasis.44 Therefore, an appropriate extraction method is the key to ensuring comprehensive recovery of RPs for putative biomarker discovery. Metastasis is often the fatal step of cancer progression.45,46 Minn et al. identified 54 genes that mediate breast cancer metastasis to lung. Among these, 26 protein-coding genes were quantified in our study, including four functionally validated genes (MMP1, CXCL1, PTGS2, and ID1). Increased expression of matrix metalloproteinase-1 (MMP1) has been clinically associated with invasiveness of various types of cancer, including breast, pancreatic, and malignant melanoma.47,48 A more than 2-fold increase of iBAQ intensity was observed when comparing the SCAD method to the FASP method, highlighting the superior performance of SCAD in terms of reflecting the copy number of proteins inside the cell (Figure S-10). Extracellular matrix (ECM) and ECM-associated proteins are essential components of multicellular scaffold.20 Their dysregulation triggers biomechanical changes that promote the metastasis cascade.46 Characterizations of ECM molecules are, in general, challenging due to the poor solubility in the extraction buffer. In the present study, we investigated how well these ECM molecules can be extracted by different sample preparation strategies. As shown in Figure 6B and Table S-2, 136 ECM molecules were quantified in total, and SCAD afforded a 10% increase in iBAQ values over the other two methods. A slightly higher extraction efficiency was observed for FASP over that of urea mainly due to the enrichment preference of membrane proteins. Dysregulation of histone proteins, in particular, PTMs of histones, is another hallmark of cancer.49 Thus, the iBAQ values of histone proteins were also investigated. As exemplified in Figure S-11, SCAD and urea methods exhibited better extraction efficiency of histone proteins compared to FASP. More than 2-fold increases of histone proteins were detected in SCAD in comparison to FASP (Table S-3), which is in good agreement with our results shown above.
Figure 6.

iBAQ protein abundance of ribosomal proteins (A) and ECM and ECM-associated proteins (B).
CONCLUSIONS
In this study, a sequential extraction method that combines the surfactant (SDS) with chaotropic agent (urea) was developed. We took advantage of the exceptional solubilizing power of SDS and replaced the ultrafiltration device with acetone precipitation to overcome the caveats reported for the use of the FASP approach. An on-pellet digestion workflow was utilized to boost peptide recovery as well as protein identifications (especially the low solubility proteins like membrane proteins and nuclear proteins). The solubility of DIP was also improved by urea. Using this sample preparation strategy, we were able to extract more than 600 μg of proteins from 10 μL of MDA-MB-231 cell, which increased the protein yield by 3-fold compared to the FASP method and clearly outperformed the urea method. The peptide recovery and protein coverage were also significantly improved. After optimizing the gradient profile and peptide sequencing technique, more than 10,000 proteins were identified in combination of three strategies, rendering the highest number of protein IDs for breast cancer cells with no prefractionation steps. The enrichment analysis of cellular components demonstrated comprehensive extraction of proteins in various categories (i.e., membrane, intracellular, and nuclear proteins) achieved by the SCAD method, which combines the unique features of both FASP and urea methods. The absolute quantities of different types of proteins were probed by iBAQ, underscoring the improved extraction efficiency of the SCAD method over the other two strategies. Several crucial signaling pathways regulating breast cancer progression were revealed by IPA analysis of proteins exclusively identified using the SCAD approach. A number of RPs were only detected using the SCAD and urea extraction methods. The results highlight the importance of selecting an appropriate sample preparation method for proper biological applications.
Looking forward, enhanced proteome recovery could facilitate the observation of various types of PTMs, including phosphorylation, glycosylation, and methylation. It was reported that changes in glycosylation of ECM proteins maybe involved in the modulation of the invasiveness and chemoresistance in cancer cells.50 Protein methylation also plays fundamental roles in diverse biological processes of cancer.49,51 Given the enhanced extraction efficiency of ECM, RP, and histone proteins, the preservation of PTM-containing peptides is also expected. Therefore, this novel strategy not only expedites comprehensive protein identification but also has the potential to promote PTM detection when the experimental material is limited (e.g., glycosylation and methylation) and thus may find widespread applicability for biomarker discovery in various types of cancers.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported in part by the National Institutes of Health (NIH) grants R01AG052324 (to LL), R01 DK071801 (to LL), and R01 CA213293 (to WX). The Orbitrap instruments were purchased through the support of an NIH shared instrument grant (NIH-NCRR S10RR029531) and Office of the Vice Chancellor for Research and Graduate Education at the University of Wisconsin-Madison. LL acknowledges a Vilas Distinguished Achievement Professorship and Janis Apinis Professorship with funding provided by the Wisconsin Alumni Research Foundation and University of Wisconsin-Madison School of Pharmacy. We thank Kellen DeLaney in the Li Lab for editorial help during revision
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.8b00197.
Protein yield of each precipitation strategy measured by peptide assay and number of protein groups with optimized IT-HCD under 90 min gradient (15 cm column); efficiency of peptide recovery at different starting amounts of cells for SCAD and FASP methods; percentage of identified peptides with missed cleavages; number of peptides, PSMs, MS/MS over a 90 min gradient at maximum injection time of 15, 35, 50, 100, and 300 ms; number of peptides, PSMs, MS/MS over a 90 min gradient using precursor isolation window of 0.6,1.0, and 1.6 m/z; number of peptides, PSMs, MS/MS over a 90 min gradient at dynamic exclusion time of 15, 30, and 45 s; number of protein groups for OT-HCD with 180 min gradient in comparison to optimized ITHCD with different gradient times, 90, 120, and 180 min; gene ontology enrichment analysis (cellular components) of proteins that had higher extraction efficiency in the FASP methods; iBAQ intensity of seven ribosomal proteins that were only detected in the SCAD and urea methods; iBAQ intensity of matrix metal-loproteinase-1 (MMP1) with three different sample preparation strategies; iBAQ intensity of histone proteins with three different sample preparation strategies (PDF)
Identified ribosomal proteins (XLSX)
Identified ECM proteins (XLSX)
Identified histone proteins (XLSX)
Accession Codes
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD008967.52 Username: reviewer26135@ebi.ac.uk. Password: jKw7EqUX.
The authors declare no competing financial interest.
REFERENCES
- (1). Aebersold R; Mann M Mass-Spectrometric Exploration of Proteome Structure and Function. Nature 2016, 537 (7620), 347–355. [DOI] [PubMed] [Google Scholar]
- (2). Aebersold R; Mann M Mass Spectrometry-Based Proteomics. Nature 2003, 422 (6928), 198–207. [DOI] [PubMed] [Google Scholar]
- (3). Wiśniewski JR; Zougman A; Nagaraj N; Mann M Universal Sample Preparation Method for Proteome Analysis. Nat. Methods 2009, 6 (5), 359–362. [DOI] [PubMed] [Google Scholar]
- (4). Shevchenko A; Tomas H; Havliš J; Olsen JV; Mann M In-Gel Digestion for Mass Spectrometric Characterization of Proteins and Proteomes. Nat. Protoc 2007, 1 (6), 2856–2860. [DOI] [PubMed] [Google Scholar]
- (5). Giansanti P; Tsiatsiani L; Low TY ew; Heck AJR Six Alternative Proteases for Mass Spectrometry-Based Proteomics beyond Trypsin. Nat. Protoc 2016, 11 (5), 993–1006. [DOI] [PubMed] [Google Scholar]
- (6). Wolters DA; Washburn MP; Yates JR An Automated Multidimensional Protein Identification Technology for Shotgun Proteomics. Anal. Chem 2001, 73 (23), 5683–5690. [DOI] [PubMed] [Google Scholar]
- (7). Davis S; Charles PD; He L; Mowlds P; Kessler BM; Fischer R Expanding Proteome Coverage with CHarge Ordered Parallel Ion aNalysis (CHOPIN) Combined with Broad Specificity Proteolysis. J. Proteome Res 2017, 16 (3), 1288–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8). Erde J; Loo RRO; Loo JA Enhanced FASP (eFASP) to Increase Proteome Coverage and Sample Recovery for Quantitative Proteomic Experiments. J. Proteome Res 2014, 13, 1885–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9). Duan X; Young R; Straubinger RM; Page B; Cao J; Wang H; Yu H; Canty JM; Qu J A Straightforward and Highly Efficient Precipitation/on-Pellet Digestion Procedure Coupled with a Long Gradient Nano-LC Separation and Orbitrap Mass Spectrometry for Label-Free Expression Profiling of the Swine Heart Mitochondrial Proteome. J. Proteome Res 2009, 8 (6), 2838–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10). Ye M; Pan Y; Cheng K; Zou H Protein Digestion Priority Is Independent of Protein Abundances. Nat. Methods 2014, 11 (3), 220–222. [DOI] [PubMed] [Google Scholar]
- (11). Pitteri SJ; JeBailey L; Faca VM; Thorpe JD; Silva MA; Ireton RC; Horton MB; Wang H; Pruitt LC; Zhang Q; et al. Integrated Proteomic Analysis of Human Cancer Cells and Plasma from Tumor Bearing Mice for Ovarian Cancer Biomarker Discovery. PLoS One 2009, 4 (11), e7916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12). Erde J; Loo RRO; Loo JA Improving Proteome Coverage and Sample Recovery with Enhanced FASP (eFASP) for Quantitative Proteomic Experiments. Methods Mol. Biol 2017, 1550, 11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13). Chandramouli K; Qian P-Y Proteomics: Challenges, Techniques and Possibilities to Overcome Biological Sample Complexity. Hum. Hum. Genomics Proteomics 2009, 2009, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14). Wisniewski JR Quantitative Evaluation of Filter Aided Sample Preparation (FASP) and Multienzyme Digestion FASP Protocols. Anal. Chem 2016, 88 (10), 5438–5443. [DOI] [PubMed] [Google Scholar]
- (15). Feist P; Hummon AB Proteomic Challenges: Sample Preparation Techniques for Microgram-Quantity Protein Analysis from Biological Samples. Int. J. Mol. Sci 2015, 16 (2), 3537–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16). Zhou J-Y; Chen L; Zhang B; Tian Y; Liu T; Thomas SN; Chen L; Schnaubelt M; Boja E; Hiltke T; et al. Quality Assessments of Long-Term Quantitative Proteomic Analysis of Breast Cancer Xenograft Tissues. J. Proteome Res 2017, 16 (12), 4523–4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17). Pirmoradian M; Budamgunta H; Chingin K; Zhang B; Astorga-Wells J; Zubarev RA Rapid and Deep Human Proteome Analysis by Single-Dimension Shotgun Proteomics. Mol. Cell. Proteomics 2013, 12 (11), 3330–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18). Hebert AS; Richards AL; Bailey DJ; Ulbrich A; Coughlin EE; Westphall MS; Coon JJ The One Hour Yeast Proteome. Mol. Cell. Proteomics 2014, 13 (1), 339–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19). Liu F; Ma F; Wang Y; Hao L; Zeng H; Jia C; Wang Y; Liu P; Ong IM; Li B; et al. PKM2Methylation by CARM1 Activates Aerobic Glycolysis to Promote Tumorigenesis. Nat. Cell Biol 2017, 19 (11), 1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20). Naba A; Clauser KR; Ding H; Whittaker CA; Carr SA; Hynes RO The Extracellular Matrix: Tools and Insights for The “omics” era. Matrix Biol. 2016, 49, 10–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21). León IR; Schwammle V; Jensen ON; Sprenger RR Quantitative Assessment of In-Solution Digestion Efficiency Identifies Optimal Protocols for Unbiased Protein Analysis. Mol. Cell. Proteomics 2013, 12 (10), 2992–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22). Glatter T; Ahrné E; Schmidt A Comparison of Different Sample Preparation Protocols Reveals Lysis Buffer-Specific Extraction Biases in Gram-Negative Bacteria and Human Cells. J. Proteome Res 2015, 14 (11), 4472–4485. [DOI] [PubMed] [Google Scholar]
- (23). Duncan MW; Aebersold R; Caprioli RM The Pros and Cons of Peptide-Centric Proteomics. Nat. Biotechnol 2010, 28 (7), 659–664. [DOI] [PubMed] [Google Scholar]
- (24). Lundberg E; Fagerberg L; Klevebring D; Matic I; Geiger T; Cox J;Älgenäs C; Lundeberg J; Mann M; Uhlen M Defining the Transcriptome and Proteome in Three Functionally Different Human Cell Lines. Mol. Syst. Biol 2010, 6 (450), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25). Bekker-Jensen DB; Kelstrup CD; Batth TS; Larsen SC; Haldrup C; Bramsen JB; Sørensen KD; Høyer S; Ørntoft TF; Andersen CL; et al. An Optimized Shotgun Strategy for the Rapid Generation of Comprehensive Human Proteomes. Cell Syst. 2017, 4 (6), 587–599.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26). Kim MS; Pinto SM; Getnet D; Nirujogi RS; Manda SS; Chaerkady R; Madugundu AK; Kelkar DS; Isserlin R; Jain S; et al. A Draft Map of the Human Proteome. Nature 2014, 509 (7502), 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27). Thakur SS; Geiger T; Chatterjee B; Bandilla P; Fröhlich F; Cox J; Mann M Deep and Highly Sensitive Proteome Coverage by LC-MS/MS Without Prefractionation. Mol. Cell. Proteomics 2011, 10 (8), M110–003699.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28). Senko MW; Remes PM; Canterbury JD; Mathur R; Song Q; Eliuk SM; Mullen C; Earley L; Hardman M; Blethrow JD; et al. Novel Parallelized Quadrupole/linear Ion Trap/orbitrap Tribrid Mass Spectrometer Improving Proteome Coverage and Peptide Identification Rates. Anal. Chem 2013, 85 (24), 11710–11714. [DOI] [PubMed] [Google Scholar]
- (29). Shishkova E; Hebert AS; Coon JJ Now, More Than Ever, Proteomics Needs Better Chromatography. Cell Syst. 2016, 3 (4), 321–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30). Tu C; Li J; Shen S; Sheng Q; Shyr Y; Qu J Performance Investigation of Proteomic Identification by HCD/CID Fragmentations in Combination with High/Low-Resolution Detectors on a Tribrid, High-Field Orbitrap Instrument. PLoS One 2016, 11 (7), e0160160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31). Elias JE; Haas W; Faherty BK; Gygi SP Comparative Evaluation of Mass Spectrometry Platforms Used in Large-Scale Proteomics Investigations. Nat. Methods 2005, 2 (9), 667–675. [DOI] [PubMed] [Google Scholar]
- (32). Cox J; Hein MY; Luber CA; Paron I; Nagaraj N; Mann M Accurate Proteome-Wide Label-Free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ. Mol. Cell. Proteomics 2014, 13 (9), 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33). Tyanova S; Temu T; Sinitcyn P; Carlson A; Hein MY; Geiger T; Mann M; Cox J The Perseus Computational Platform for Comprehensive Analysis of (Prote)omics Data. Nat. Methods 2016, 13 (9), 731–740. [DOI] [PubMed] [Google Scholar]
- (34). Schwanhausser B; Busse D; Li N; Dittmar G; Schuchhardt J; Wolf J; Chen W; Selbach M Global Quantification of Mammalian Gene Expression Control. Nature 2011, 473 (7347), 337–342. [DOI] [PubMed] [Google Scholar]
- (35). Chen Y; Li Y; Zhong J; Zhang J; Chen Z; Yang L; Cao X; He QY; Zhang G; Wang T Identification of Missing Proteins Defined by Chromosome-Centric Proteome Project in the Cytoplasmic Detergent-Insoluble Proteins. J. Proteome Res 2015, 14 (9), 3693–3709. [DOI] [PubMed] [Google Scholar]
- (36). Zhang W; Chen X; Yan Z; Chen Y; Cui Y; Chen B; Huang C; Zhang W; Yin X; He Q-Y; et al. Detergent-Insoluble Proteome Analysis Revealed Aberrantly Aggregated Proteins in Human Preeclampsia Placentas. J. Proteome Res 2017, 16 (12), 4468–4480. [DOI] [PubMed] [Google Scholar]
- (37). Chiva C; Ortega M; Sabidä E Influence of the Digestion Technique, Protease, and Missed Cleavage Peptides in Protein Quantitation. J. Proteome Res 2014, 13 (9), 3979–3986. [DOI] [PubMed] [Google Scholar]
- (38). Eliuk S; Soltero N; Remes P; Scheffler K; Senko M; Zabrouskov V; Fisher T; Jose SA “ Universal “ Data-Dependent Mass Spectrometry Method That Eliminates Time-Consuming Method Optimization for Achieving Maximal Identifications from Each Sample. Thermo Sci 2015, 49014. [Google Scholar]
- (39). Espadas G; Borràs E; Chiva C; Sabidó E Evaluation of Different Peptide Fragmentation Types and Mass Analyzers in Data-Dependent Methods Using an Orbitrap Fusion Lumos Tribrid Mass Spectrometer. Proteomics 2017, 17 (9), 1600416. [DOI] [PubMed] [Google Scholar]
- (40). Sacco F; Silvestri A; Posca D; Pirrò S; Gherardini PF; Castagnoli L; Mann M; Cesareni G Deep Proteomics of Breast Cancer Cells Reveals That Metformin Rewires Signaling Networks Away from a Pro-Growth State. Cell Syst. 2016, 2 (3), 159–171. [DOI] [PubMed] [Google Scholar]
- (41). Cox J; Mann M MaxQuant Enables High Peptide Identification Rates, Individualized P.p.b.-Range Mass Accuracies and Proteome-Wide Protein Quantification. Nat. Biotechnol 2008, 26 (12), 1367–1372. [DOI] [PubMed] [Google Scholar]
- (42). Cox J; Neuhauser N; Michalski A; Scheltema RA; Olsen JV; Mann M Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment. J. Proteome Res 2011, 10 (4), 1794–1805. [DOI] [PubMed] [Google Scholar]
- (43). Goudarzi KM; Lindström MS Role of Ribosomal Protein Mutations in Tumor Development (Review). Int. J. Oncol 2016, 48 (4), 1313–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44). Dave B; Granados-Principal S; Zhu R; Benz S; Rabizadeh S; Soon-Shiong P; Yu K-D; Shao Z; Li X; Gilcrease M; et al. Targeting RPL39 and MLF2 Reduces Tumor Initiation and Metastasis in Breast Cancer by Inhibiting Nitric Oxide Synthase Signaling. Proc. Natl. Acad. Sci. U. S. A 2014, 111 (24), 8838–8843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45). Minn AJ; Gupta GP; Siegel PM; Bos PD; Shu W; Giri DD; Viale A; Olshen AB; Gerald WL; Massague J Genes That Mediate Breast Cancer Metastasis to Lung. Nature 2005, 436 (7050), 518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46). Venning FA; Wullkopf L; Erler JT Targeting ECM Disrupts Cancer Progression. Front. Oncol 2015, 5, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47). Liu H; Kato Y; Erzinger SA; Kiriakova GM; Qian Y; Palmieri D; Steeg PS; Price JE The Role of MMP-1 in Breast Cancer Growth and Metastasis to the Brain in a Xenograft Model. BMC Cancer 2012, 12 (1), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48). McGowan PM; Duffy MJ Matrix Metalloproteinase Expression and Outcome in Patients with Breast Cancer: Analysis of a Published Database. Ann. Oncol 2008, 19 (9), 1566–1572. [DOI] [PubMed] [Google Scholar]
- (49). Chervona Y; Costa M Histone Modifications and Cancer: Biomarkers of Prognosis? Am. J. Cancer Res 2012, 2 (5), 589–597. [PMC free article] [PubMed] [Google Scholar]
- (50). Freire-de-Lima L Sweet and Sour: The Impact of Differential Glycosylation in Cancer Cells Undergoing Epithelial—Mesenchymal Transition. Front. Oncol 2014, 4 (March), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51). Wang Q; Wang K; Ye M Strategies for Large-Scale Analysis of Non-Histone Protein Methylation by LC-MS/MS. Analyst 2017, 142 (19), 3536–3548. [DOI] [PubMed] [Google Scholar]
- (52). Vizcaíno JA; Csordas A; Del-Toro N; Dianes JA; Griss J; Lavidas I; Mayer G; Perez-Riverol Y; Reisinger F; Ternent T; et al. 2016. Update of the PRIDE Database and Its Related Tools. Nucleic Acids Res 2016, 44 (D1), D447–D456. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.