

Abstract
With each new microarray or RNA-seq experiment, massive transcriptomic information is generated with the purpose to produce a list of candidate genes for functional analyses. Yet an effective strategy remains elusive to prioritize the genes on these candidate lists. In this review, we outline a prioritizing strategy by taking a step back from the bench and leveraging the rich range of public databases. This in silico approach provides an economical, less biased, and more effective solution. We discuss the publicly available online resources that can be used to answer a range of questions about a gene. Is the gene of interest is expressed in the system of interest (using expression databases)? Where else is this gene expressed (using added-value transcriptomic resources)? What pathways and processes is the gene involved in (using enriched gene pathway analysis and mouse knockout databases)? Is this gene correlated with human diseases (using human disease variant databases)? Using mouse fetal testis as an example, our strategies identified 298 genes annotated as expressed in the fetal testis. We cross-referenced these genes to existing microarray data and narrowed the list down to cell type specific candidates (35 for Sertoli cells, 11 for Leydig cells, and 25 for germ cells). Our strategies can be customized so that allows researchers to effectively and confidently prioritize genes for functional analysis.
Keywords: databases, knowledgebases, ontology, GO terms, expression atlas, enrichment, trancriptomics, data-mining, gene prioritization, candidate gene, sex determination, testis, ovary
Introduction
The process of sex determination is complex, involving multiple pathways that shape the identity of the gonads. Identifying the genes that make up these pathways is like solving a complex puzzle. We propose that by using public databases investigators can efficiently identify new candidate genes for their involvement in gonadogenesis. While scientists have gathered a number of missing puzzle pieces, unexplained cases of disorders of sexual development (DSD) and infertility clearly indicate that many key pathways have not yet been placed in the picture [Ono, and Harley, 2013]. In order to fill in the missing pieces, the field has used mouse models to generate numerous transcriptomic studies that reveal variations in gene expression between sexes, cell types, developmental stages and different genotypes [Jameson et al., 2012; McClelland et al., 2015; Beverdam, and Koopman, 2006; Nef et al., 2005; Bouma et al., 2010; Bouma et al., 2007; Albrecht, and Eicher, 2001; Rolland et al., 2011; Coveney et al., 2008; Munger et al., 2013]. Each of these studies had the goal to identify new genes that control key processes in sex determination of the gonads.
Over time the strategies used to generate new candidate genes has changed with the availability of different technologies. Initial cDNA array screens in the 1990s and early 2000s compared male and female gonads to search for genes enriched in either testis or ovary, with the assumption that enriched genes in one sex were important for development of that fate [Wertz, and Herrmann, 2000; Bowles et al., 2000; McClive et al., 2003; Menke, and Page, 2002]. With the advent of microarrays, comparing gene expression between multiple groups, such as testis and ovary at different developmental time-points, became feasible. This approach allowed the field to identify genes that were expressed at key developmental time-points, and begun the process of fitting together in pathways that were initiated by marshaling transcriptions factors such as SOX9 [Bouma et al., 2010; Coveney et al., 2008; Grimmond et al., 2000; Nef et al., 2005; Munger et al., 2009]. Subsequently, to move beyond male and female comparisons and to gain more cell type specific information, scientists generated transcriptomic data from enriched cell populations isolated from cell type-specific reporter mice. This strategy was used to isolate somatic cells (Sertoli, Leydig, and others) and germ cells to identify key differences between lineages in addition to sexes [Nef et al., 2005; Beverdam, and Koopman, 2006; McClelland et al., 2015; Rolland et al., 2011; Inoue et al., 2015; Bouma et al., 2007]. This approach was used in the GUDMAP (GenitoUrinary Development Molecular Anatomy Project) Consortium-backed microarray of four key cell types (supporting, germ, interstitial, and endothelial cells), aiming at differences in cell population gene expression over a developmental time-course [Jameson et al., 2012]. Over the past two decades, transcriptomic studies identified numerous factors and sex-linked genes that are critical for gonadogenesis. Efforts by other groups continued to fill in the puzzle with the advent of RNA-seq and whole exome sequencing. RNA-seq allowed gene expression to be assayed at a greater sensitivity in a more unbiased way than microarrays on whole gonads and sorted cells [McClelland et al., 2015; Inoue et al., 2015; Lindeman et al., 2015]. In tandem, the decreasing cost for next-gen sequencing has made whole exome sequencing of DSD patients economically feasible [for review see Ostrer, 2014; Ono, and Harley, 2013]. The function of genes from mutations identified in DSD patients can then be confirmed in the mouse model in complete knockout or exact recapitulations of the mutated human gene. One common feature of these approaches is that they produce an extensive list of candidate genes. The key question then becomes: how do we prioritize the list of candidate genes and determine which ones to pursue for further functional analyses?
Although the way we identify candidate genes continues to evolve, the way in which we prioritize candidate genes for further functional analysis remains rudimentary: the most obvious candidates were selected, based on whether they fit into existing pathways with known roles in gonadogenesis. From experience we know that transcription factors or sex-chromosome linked genes are good candidates for functional analyses. Traditionally, scientists tried to prioritize genes by targeting genes with a sexually dimorphic expression pattern, profiling gene expression over a developmental time-course, and then performing in situ hybridization or immunohistochemistry to the cell type in which the gene was expressed [McClive et al., 2003; Rolland et al., 2011; Menke, and Page, 2002; Bouma et al., 2004]. The gene with the most robust sex-specific expression, the best working in situ hybridization probe (or antibody), and some background information in PubMed goes forward as the candidate gene. This prioritization problem is not new: genes identified in early cDNA screens were present in subsequent microarrays, and found to be critical for gonadogenesis 20–25 years after identification. The problem was not the identification of these genes, the issue was to prioritize them or identify them as a good candidate for further functional analysis. Confronted by a list that contains hundreds of genes, the “gene prioritization problem” looms bigger than ever. We argue that current methods of selecting a candidate gene are unnecessarily expensive, ineffective and biased in today’s information rich world. We propose that the community could better prioritize genes for functional validation by utilizing publically available gene expression and transcriptomic databases. These resources represent a valuable pool of information that can be used freely by any scientist to move projects and hypotheses forward.
In this review, we discuss the development and use of gene ontology (GO) terms by databases and the key features of a “good” gene list before describing four key resources: expression atlases and databases, added-value transcriptomic databases, pathway or GO term enrichment analysis, and mouse knockout or human disease variant databases. We describe how to utilize these databases to prioritize genes, and discuss the strengths and limitations of each resource. Finally, we demonstrate how to use these resources as a discovery tool. As an example, we analyze “testis” annotated genes in the Eurexpress expression database. Although the reanalysis of existing data sets and in silico discovery is still a fairly new concept in the field of sex determination, we argue that by capitalizing on the wide range of publically available databases, scientists can prioritize genes in a candidate list without exhausting research dollars and time chasing ghosts.
I. Online tools for generating and analyzing the genes-of-interest list
While enormous amounts of functional data are available in published literature, each paper typically reports on a limited number of genes and developmental stages. However, there are numerous publically available databases that host a wide range of expression and transcriptomic data covering murine embryonic development. To increase utility of individual databases, most core databases are linked with specialist databases to create ways to springboard between resources. For example, Mouse Genome Informatics (MGI; http://www.informatics.jax.org/; Table 1) provides a comprehensive data page linking to most major resources for each gene. However, it is not yet possible to perform a bulk query and retrieve a set of metrics or a list of genes from a core database. How the user implements the strategies outlined in this paper will depend on the nature of the question and what type data are used. If the investigator has already had data from a patient or mouse model, prioritizing genes by focusing on key biological processes or signaling pathways might be the first step. Conversely, if the investigators are searching for new candidates, then reanalyzing published transcriptomic data may provide new genes of interest, followed by validating the expression patterns in the existing expression databases. In this section, we aim to guide the investigator through how each type of database works and what can be gained from each one so the investigator can customize their search to solve their “gene prioritization problem”.
Table 1: List of resources that can be used to prioritize candidate genes.
MGI is often considered the one stop shop when investigating a new gene. By using resources like the GO Consortium and AmiGO2 to understand the specifics on gene ontology the investigator can use tools like DAVID and GSEA to their advantage to uncover interesting processes and pathways present in the candidate gene list. These enrichment tools are underpinned by data from knowledgebases such as Entrez Gene, ENSEMBL, UniProt and TRANSFAC. The EMBL-EBI Expression Atlas provides a rich resource of reanalyzed transcriptomic data in mouse and human. The relevance of candidate genes in human disease can then be investigated using too such as OMIM and HGMD. URLs are current as on September 2016.
| Resource | Functional Description | URL |
|---|---|---|
| MGI (Mouse Genome Informatics) |
MGI is the international database resource for the laboratory mouse, providing integrated genetic, genomic, and biological data to facilitate the study of human health and disease. |
http://www.informatics.jax.org |
| Gene Ontology (GO) Consortium |
The GO Consortium develops up-to-date, comprehensive, computational models of biological systems, from the molecular level to larger pathways, cellular and organism-level systems. |
Homepage: http://geneontology.org/ Descriptors: http://geneontology.org/page/ontology-documentation http://geneontology.org/page/development |
| AmiGO2 | Searchable interface of GO Consortium | http://amigo.geneontology.org/amigo/landing |
| DAVID (Database for Annotation, Visualization and Integrated Discovery) |
DAVID is a comprehensive set of functional annotation tools for investigators to understand biological meaning behind arge list of genes. |
https://david.ncifcrf.gov/ |
| GSEA (Gene Set Enrichment Analysis) |
GSEA is a computational tool that determines whether an set of genes shows statistically significant, concordant differences between two biological states. |
http://software.broadinstitute.org/gsea/index.jsp |
| Entrez Gene |
Gene provides a record including nomenclature, Reference Sequences (RefSeqs), maps, pathways, variations,phenotypes, and links to genome-, phenotype-, and locus-specific resources. |
https://www.ncbi.nlm.nih.gov/gene/ |
| ENSEMBL | Ensembl is a genome browser for vertebrate genomes that supports research in comparative genomics, evolution, sequence variation and transcriptional regulation. |
http://useast.ensembl.org/index.html |
| UniProt | UniProt is a comprehensive, high-quality and freely accessible resource of protein sequence and functional information |
http://www.uniprot.org/ |
| TRANSFAC | TRANSFARC houses data on eukaryotic transcription factors, their experimentally-proven binding sites, consensus binding sequences (positional weight matrices) and regulated genes. |
http://www.gene-regulation.com/pub/databases.html (last updated 2005) |
| EMBL-EBI Gene Expression Atlas (GEA) |
Gene expression data displayed in this resource is re-analyzed in-house to detect genes showing interesting baseline and differential expression patterns in different cell types and organs, in addition to different developmental stages, disease states and biological/experimental conditions |
https://www.ebi.ac.uk/gxa/home |
| Online Mendelian Inheritance in Man (OMIM) |
OMIM is a comprehensive online collection of records on human disease genes and genetic phenotypes. |
https://omim.org/ |
| UK10K Project | UK10K is an example of one of the many smaller efforts to understand the link between low-frequency and rare genetic changes, and human disease |
http://www.uk10k.org |
| Human Gene Mutation Database (HGMD, from Cardiff University) |
HGMD is a collection of germline mutations in nuclear genes that underlie, or are associated with, human inherited disease. -HGMD Cardiff: unlicensed and limited access (no access to mutations identified in the last 2.5 years) -HGMD Professional: licensed through Qiagen and complete access to entire collection |
http://www.hgmd.cf.ac.uk/ac/index.php |
| Human Gene Mutation Database (HGMD) Professional |
https://www.qiagenbioinformatics.com/products/human-gene-mutation-database/ | |
| IKMC (International Knockout Mouse Consortium) IMPC (International Mouse Phenotyping Consortium) IMSR (International Mouse Strain Resource) |
-IKMC generates targeted ES-cells of all known protein coding mouse genes and companion Cre driver lines - IMPC generates the mouse strains and perform standardized phenotyping. - All gene trap alleles are housed at Jax (Jackson Laboratories) - ISMR is a searchable catalogue of over 2000 strains |
IKMC: access through IKMC/IMPC hub IMPC: www.knockoutmouse.org IKMC/IMPC web portals were merged to create a central hub: http://www.mousephenotype.org Jax: https://www.jax.org IMSR: www.findmice.org; |
| DMDD (Deciphering the Mechanisms of Developmental Disorders) |
Results from the Consortium to study knock-out lines where embryonic development is compromised. |
https://dmdd.org.uk |
| HGNC (HUGO Gene Nomenclature Committee) Multi-Symbol Checker |
This tool to check submitted gene names in a list against HUGO verified names and their known synonyms, |
http://www.genenames.org/cgi-bin/symbol_checker |
The Construction of a Gene Ontology Framework
The Gene Ontology (GO) framework [Ashburner et al., 2000] is an example of a flexible and rapidly developing textual framework for the molecular functions of gene products, including their sub-cellular localization, the biological processes in which they function, and the processes in which they participate during embryogenesis. The GO Consortium (http://geneontology.org/; Table 1), initially a collaboration between the Mouse Genome Informatics, FlyBase and Saccharomyces Genome databases, was formed to create descriptions of biological processes and establish standards for the community-wide organization of a set of frameworks in order to enable the integration of data from different model organisms [Ashburner et al., 2000; Blake et al., 2000; Ringwald et al., 2000; Ball et al., 2000; Consortium, 1999]. The structured language laid out by the Gene Ontology Consortium to describe the properties of gene products are called GO terms. GO terms are catalogued in a central thesaurus hosted by the GO Consortium (http://geneontology.org/page/ontology-documentation; Table 1). Databases using GO terms have these terms as set outputs (or answers). Therefore, it is important to understand how GO frameworks are constructed, the limitations of GO terms and how GO terms are utilized by different databases.
There are three central themes defined by GO terms based on the function of the protein. 1) Terms that describe the Cellular Component, the parts of a cell or its extracellular environment. 2) Terms that describe the Molecular Function, the basic functions of a gene product at the molecular level. 3) Terms that describe Biological Process, which is defines as the molecular events necessary for the function of cells, tissues, organs, and organisms [Ashburner et al., 2000]. Within the ontology framework each term has defined relationships to one or more other terms in the same classification, and sometimes to terms in other classifications; some terms are parent terms to a host of more specific terms like a branching tree to describe things in greater and greater detail. For an example of GO annotation, the gene product “Sox9” can be described by the Molecular Function term “transcription”, the Biological Process terms “cell differentiation” and “system development”, and the Cellular Component term “nucleus”. To look up terms for specific genes, a brief overview can be found in the Gene Ontology Classifications section of MGI and more detail can be obtained by querying AmiGO2 (http://amigo.geneontology.org/amigo/landing; Table 1).
The consistent use of GO terms for cellular and biological processes and for anatomical structures throughout development is essential for the success, development and maintenance of biological databases. Creating and maintaining ontologies seem straightforward; however, the creation and maintenance of ontologies in the context of developmental biology poses some interesting problems. The subtleties between different ontology terms used to by the Biological Process Ontology Guidelines for Development to describe development and differentiation of lineages and tissues can be found in the GO Consortium resources (http://geneontology.org/page/development). The goal of ontology is to represent knowledge in a computer-interpretable manner and that can (hopefully) be used to cross-reference data among different databases. The success of a database and the ontology depends greatly on the needs of the end user of the database, the developmental biologist wants to know about the genes and proteins involved and to map the expression of the genes associated with a tissues and cell populations at a different developmental stages. For example for the process of differentiation of one cell type into a different cell type, there are multiple ways this process could be represented by the GO terms. Consider these three similar yet distinct terms that could be used to describe the differentiation process (GO term in italics):
[cell type] cell fate commitment —
The process whereby the developmental fate of a cell becomes restricted such that it will develop into a [cell type] cell.
[cell type] cell fate specification —
The process whereby a cell becomes capable of differentiating autonomously into a [cell type] cell in an environment that is neutral with respect to the developmental pathway. Upon specification, the cell fate can be reversed.
[cell type] cell fate determination —
The process whereby a cell becomes capable of differentiating autonomously into a [cell type] cell regardless of its environment; upon determination, the cell fate cannot be reversed.
Each of these terms, although similar on first glance, encapsulates important information about the cell and the process that takes place, and they are not interchangeable. Knowing what pertinent GO terms mean in detail and how those GO terms are nested within other GO terms is critical to extract important information about function of a gene product.
GO terms can be a powerful tool to describe biology and make findings accessible across data formats and platforms. The core item the investigator needs to begin is a “good” candidate gene list, but what defines a “good” list?
Key features of a “good” candidate gene list
Lists of ‘interesting’ genes, which may range in size from hundreds to thousands of genes, can reveal patterns, signaling cascades and processes important for organogenesis. The analysis of these data sets has gradually become the responsibility of the biologist, rather than the bioinformatician, as more biologist-friendly tools become available. In addition to analyzing new primary data, reanalysis and inclusion of published data should be considered to bolster a new analysis. Before considering how to analyze the genes-of-interest list, the quality of the list itself should be assessed. Realistically, any correlations and directions indicated by a list should be confirmed by bench work and functional analysis. Regardless of this, as a general rule a ‘good’ gene list has a few key features [modified from Huang et al., 2009a]:
A reasonable number of biological (not technical) replicates and the reproducibility of the list before analysis, either from independent experiments or by statistical testing (appropriate p-values and test parameter should be chosen for each experiment), are important considerations, especially when re-analyzing data.
The genes-of-interest list contains a series (not just one or two) bona fide marker genes (i.e. Sox9, Amh, and Dhh for the fetal testis) that would be expected as a result for the given experiment or analysis. When looking at reprocessed data, it is important to remember that the statistical approaches for microarray and RNA-seq analysis have changed over time; this is especially important when processing data from pre-2012. Simple reprocessing of experiments using newer analysis methodology may result in subtle or more dramatic changes in the final genes of interest list compared to the original analysis. If using reprocessed data, checking for known marker genes remains a simple, yet effective metric to determine if the analysis and data is clean.
The number of genes on the list is big enough for pathway analysis (around 100 genes) but not so big that it is not comparable (around 2000–3000 genes). The size of a list can affect the ontologies that are selected for, especially for smaller lists. As a test, the enriched terms should appear in the queried list and not in a random list of approximately the same size (number of genes).
Expression atlases and databases
The first question the investigator often asks is in which cell type/s a gene is expressed during gonadogenesis. In the past, in situ hybridization on embryonic gonads was used to validate the expression of candidate genes from mRNA expression studies, and determine in which cell type the gene was expressed. This first pass analysis should now be considered redundant for many developmental biologists with the completion of large-scale in situ hybridization atlases that aim to capture gene expression throughout the murine embryo on a gene-by-gene basis. Several databases used high-throughput robotic technology to conduct RNA in situ hybridization on sectioned or whole murine tissues [Geffers et al., 2012]. The consortiums then manually mapped and annotated gene expression patterns on standardized images using anatomical GO terms (Fig. 1A; [Reymond et al., 2002; Visel et al., 2004; Gitton et al., 2002]). These databases can play a critical role in validation of the quality of a gene list, taking an in-depth look at many genes in a list with a focus on a specific process or pathway, or can be used as a discovery tool to uncover new candidate genes and expression patterns (discussion on using these databases as a discovery tool can be found in Section II)
Fig. 1: Overview and examples of gene entries in the Eurexpress, Gene Expression Atlas, and OMIM databases.
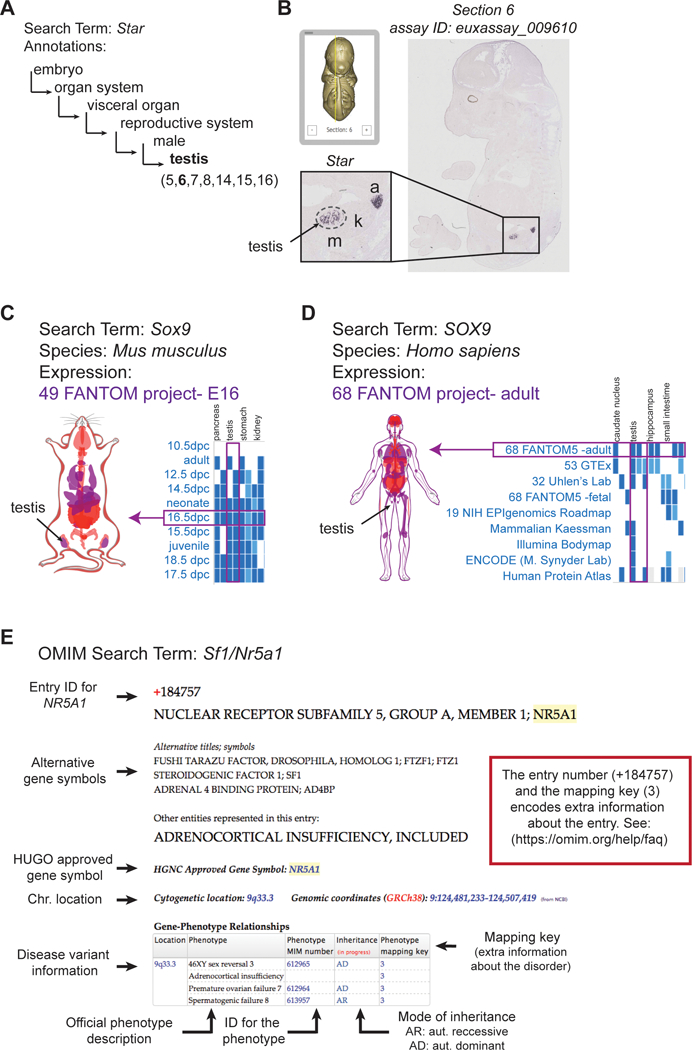
(A) In situ hybridization images of 14.5 dpc embryos are compiled in the Eurexpress Transcriptome Atlas Database for Mouse Embryo (http://www.eurexpress.org). Expression observed in an image is annotated and encoded as text that can be queried. The more general anatomical terms branch into more specific terms. For example, the nested terms that describe the testis are displayed. (B) The Eurexpress viewer scrolls through sections in a “virtual embryo environment” and zoom into regions as similar to a digital microscope. For the gene Star, the testis can be visualized in the 6th section. The testis region is demarcated by a dashed line; a, adrenal; k, kidney; m, mesonephros. (C, D) The Gene Expression Atlas houses easy to access transcriptomic data for a wide variety of species from all major consortia and sequencing efforts. For the gene Sox9, detailed information are available on expression in the (C) mouse and (D) human. Data is presented in a tabular format that can be sorted using a number of parameters (graphics shown here are part of the full table). Selecting data from a project in the table, such as 16.5 dpc 49 FANTOM (C, in mouse) or 68 FANTOM (D, in human), highlights the organs (in our case, the testis) that are included in that dataset in purple on the interactive body-map. (E) The OMIM database (Online Mendelian Inheritance in Man) catalogues human disease variants. Entries can be either by phenotype or gene. In the example of a search for the gene NR5A1 (also known as SF1), there are many alternative gene symbols that have been used historically; however, the disease information is catalogued under the HUGO approved symbol (NR5A1). Information on the location of the gene, all phenotypes caused by mutations in NR5A1 are listed in a table with ID numbers for each phenotype and additional information on inheritance. The entry number and mapping key encode additional information about the disorder.
In the race to map the expression pattern to the transcriptome throughout development, specialized knowledgebases, such as the Allen Brain Atlas (ABA) ([Lein et al., 2007; Jones et al., 2009]; http://brain-map.org/), which catalogues gene expression in brain sections throughout development and adulthood, have developed alongside with more general whole embryo atlases, such as Eurexpress (mainly whole mid-gestation murine embryos; http://www.eurexpress.org/ee/; [Diez-Roux et al., 2011]) and the fledgling 3D Atlas of Human Embryo Development developed by the Academic Medical Centre in the Netherlands (Carnegie Stage 7–23 (15–60 days); http://www.3dembryoatlas.com/; [de Bakker et al., 2016]). Specialized databases cataloguing expression of genes in specific murine tissues such as the ABA, the Gene Expression Nervous System Atlas (GENSAT; the nervous system; http://www.gensat.org/; [Gong et al., 2003]), the GenitoUrinary Development Molecular Anatomy Project (GUDMAP; the gonads, reproductive tract, kidney and urinary tract; http://www.gudmap.org/; [Brunskill et al., 2008]) and FaceBase (curated craniofacial data from mouse, human and zebrafish; https://www.facebase.org; [Van Otterloo et al., 2016]) have now been integrated into broader databases. These collaborations bring together specialist expression data with that from projects on the whole embryo such as the Edinburgh Mouse Atlas of Gene Expression (EMAGE; [Hill et al., 2004]; http://www.emouseatlas.org/emage/) and the Mouse Genome Informatics (MGI) Gene Expression Database (GXD; [Christiansen et al., 2006]; http://www.informatics.jax.org/expression.shtml). Generally, each database uses a set of standard anatomical ontology to annotate gene expression in each image (for details see: [Hayamizu et al., 2015]).
As an example database, the Eurexpress transcriptome atlas catalogues annotated RNA in situ hybridization expression patterns for approximately 18,000 Mus musculus protein-coding genes at 14.5 days post coitus (14.5 dpc; http://www.eurexpress.org). Using Eurexpress sections taken throughout the entire embryo, annotated gene expression patterns can be visualized in an online viewer. This atlas aims to achieve complete representation of all embryonic tissues throughout the 24 representative sagittal sections. For example, the gene Star is expressed in the adrenal and fetal Leydig cells at 14.5 dpc; in the Eurexpress assay it is annotated as expressed in the testis on section 5–8 and 14–16. Flipping through the online slide deck and zooming in, expression of Star is clearly detected in the adrenal and testis in section 6 (Fig. 1B). The quality of the in situ hybridization in Eurexpress is high but not always consistent. The Eurexpress Project reports that 18% of genes tested were not detected at 14.5 dpc; whether this is due to ineffective probe design, the experimental conditions, or the gene is not expressed in the 14.5 dpc embryo is unknown. Therefore, a negative result does not necessarily mean that a gene is not expressed in vivo; false negatives cannot be excluded. In our experience, among all the genes annotated in the testis, 83% of the in situ hybridization results with annotated testis expression were considered “publication quality” (see Section II). However, in some cases we found that expression was often detected even though it was not annotated. If the structure of interest is not annotated as “expressed” or the assay is classified as “not detected”, we recommend the readers to look through the entire slide deck.
Eurexpress and other expression databases allow basic and advanced queries based on annotated anatomy, gene name, gene symbol, template, and gene sequence. Entries in the Eurexpress database are linked to other databases, such as the ABA [Jones et al., 2009; Lein et al., 2007], EMAGE [Christiansen et al., 2006; Ringwald et al., 1994], and the GENSAT [Gong et al., 2003], and to informational resources such as Entrez Gene (https://www.ncbi.nlm.nih.gov/gene/), ENSEMBL (http://useast.ensembl.org/index.html), and MGI. The key drawback of these atlases is the lack of the ability to process a batch of genes generated by approaches such as microarrays and RNA-seq. The lack of an option to do bulk queries on a gene list, instead of on individual genes, in many of these databases means that the biologist has to manually go through digital slide decks for each gene to find the structures of interest. Although structures typically occur in the same 3–5 slice window of the 24 slice slide deck, each slice image must then be manually downloaded at high resolution and processed. Currently, the most efficient way of examining batches of genes within most expression databases is to automatically extract information from the webpage rather than displaying it to the investigator using a third party program. This way the queried image files for each gene can be downloaded as a batch; the images can then be scrolled through on a host computer. The introduction of a tool that allows users submit lists of genes and return batch searches within the database will make accessing the information stored within expression databases far easier and increase the utility of expression databases for biologists. Until this kind of tool is implemented, the investigator has two choices: 1) harvest/extract the image files from the database website by writing an automation script or, 2) go through the browser manually. Even if the investigator choses the more laborious manual approach, there is great value to be gained from trawling through expression atlases.
Transcriptomic databases (for non-bioinformaticians)
The abundance of transcriptomic data stored in the Gene Expression Omnibus (GEO, a public functional genomics data repository) provides many opportunities for the investigator; however, many biologists do not have the programming skills to exploit this resource. Luckily, there are some useful databases that can assist the biologist querying transcriptomic data. Transcriptomic microarray data covering various stages of gonad development and a variety of enriched cell types has been generated by a number of groups [McClelland et al., 2015; Inoue et al., 2015; Jameson et al., 2012]. These data complement the spatiotemporal expression data in databases like Eurexpress. It is now standard for all the raw data for these types of studies to be deposited in GEO, so they are available for reanalysis. However, mining published transcriptomic data (such as microarray and RNA-seq) can be challenging without appropriate analysis frameworks. Many “at the bench researchers” lack the expertise to extensively mine the RNA-seq and microarray data stored in GEO. This means researchers are not able to fully utilize published data in their field.
To use data from primary archives, a certain level of expertise is needed — raw or processed data must be downloaded, and then the data can be analyzed independently, or in combination with other data. For microarray data many biologists have some expertise in using software programs such as Partek to analyze and reanalyze data from CEL files; however RNA-seq data is more complicated to analyze. Added-value databases make the biological content of the expression data more accessible to non-bioinformaticians. These tools aggregate data stored in repositories such as GEO by extracting relevant information from the raw primary data, and therefore allow the user to ask biological questions through a user-friendly interface. For example, the user can determine in which samples their gene-of-interest is expressed, or which genes are differentially expressed between the two samples without having to handle the raw data directly.
One of the key added-value aggregators of transcriptomic information is the Gene Expression Atlas (http://www.ebi.ac.uk/gxa; [Kapushesky et al., 2009]), which provides information about gene expression in different cell types and organs, in addition to different developmental stages, disease states and biological/experimental conditions. This atlas has expression data from a large number of species including all common model organisms. The user can query individual genes looking for differential gene expression by gene names or by searching for genes correlated with an attribute such as cell types. For example, Sox9/SOX9 expression can be queried in a general (to pull up all available species) or in a specific species (Fig. 1C & D). In mouse, expression data is available for a variety of embryonic and adult stages from the FANTOM 49 Consortium project and other individual projects. Examining expression in the testis reveals expression during embryonic development, specifically at E16 or 16.5 dpc expression in the testis, pancreas, kidney and other organs (purple in Fig. 1C). In human, expression data is available for a number of Consortiums, including FANTOM 65, which examines expression in adult tissues. SOX9 is expressed in the testis, skin and a number of distinct brain regions (purple in Fig. 1D).
Pathway analysis: GO term enrichment analysis
Pathway analysis is one of the most biologist-accessible ways to look for patterns in candidate gene lists. Many similar publicly available analysis software and tools that were developed in the early 2000s can be used to functionally analyze large genes-of-interest lists [Huang et al., 2009b; Khatri et al., 2012]. However, many of these databases are no longer fully updated and maintained; for this reason, out of the freely available tools we recommend DAVID (Database for Annotation, Visualization and Integrated Discovery; Table 1; [Huang et al., 2007; Huang et al., 2009b]) and GSEA (Gene Set Enrichment Analysis; Table 1; [Subramanian et al., 2005]). DAVID is currently upgrading: v6.8 with updated knowledgebase will be available on October 17, 2016 (data here is analyzed in v6.8Beta), and v6.7 will be available for continued use until January 15, 2017. GSEA and DAVID use the same core approach of searching a genes-of-interest list, and then systematically map the list against a bank of GO terms in order to identify the most overrepresented or enriched terms out of all the linked terms that associate with the genes on the list. This kind of enrichment analysis strategy allows investigators to identify biological patterns and processes that may be relevant to their area of study that would never be discovered by looking at the list with the naked eye. Knowledgebases, such as DAVID and GSEA, draw on different repositories (including NCBI, https://www.ncbi.nlm.nih.gov/) and UniProt (http://www.uniprot.org/)), therefore hosting multiple GO terms for a single gene in an attempt to increase the comprehensiveness of the query output. A single gene can be mapped to many different and redundant terms, just as a single term maps to many genes. DAVID deals with this GO term redundancy by clustering and classifying the redundant terms into themed “annotation clusters” that can be searched by the user.
When submitting a query to a pathway analysis tool, the lists of genes can be in a number of formats. DAVID’s flexible input allows for a broad range of identifiers to be uses as initial search criteria. However, it is recommended that unique universal gene identifiers are used as inputs so that redundant gene names are not confused. GSEA also has a series of curated gene lists that cover a curated range of gene sets drawn from chemical and genetic perturbation experiments from PubMed, in addition to genes that share conserved cis-regulatory motifs [Xie et al., 2005] and transcription factor targets (using TRANSFAC, BioBase licensed through Qiagen; http://www.gene-regulation.com/pub/databases.html; [Matys et al., 2003; Wingender et al., 2000]). In both GSEA and DAVID the output or results of the search can be viewed and exported in a variety of ways. Be aware that these tools will restart to the main page after a period of inactivity, regardless of which step the analysis is paused at (you can set the timeout in GSEA, but for DAVID it is automatically set at 20 min inactivity). Both tools have features that link pathways and genes to disease associations, in addition to more protein-based tools that highlight protein functional domains and motifs.
For enrichment analysis it is important to remember that size matters: a larger gene list generally results in higher statistical confidence and more significant P-values in lowly enriched terms and more specific ontological terms at the ends of the ontological branches. Conversely, the broader and more general terms ontological terms are less enriched. The effect of list size on the absolute enrichment P-values means that it is not recommended that the users directly compare the absolute enrichment P-values across gene lists [Huang et al., 2009c]. In cases where well under 100 genes are in the genes-of-interest list, tools like DAVID can still be used. But using the statistical P-values as metrics of significance must be used cautiously, as the statistical power behind the enrichment analysis is limited by the small number of genes [Huang et al., 2009a]. Searching such a small list will produce a very focused list of ontologies and annotations that can be thoroughly explored by the user; however, the statistics produced by the software are largely meaningless.
The basis of enrichment analysis is that there are differences between the biological processes in an abnormal or perturbed state (or in the case of developmental biology often a different cell type or time point or genotype). The assumption is that the co-functioning genes (or related genes, which can be determined by looking at GO terms) should be enriched together and that these terms will therefore be selected as a relevant or significant group [Huang et al., 2009b; Huang, and Yao, 2010; Huang et al., 2007; Huang et al., 2009a]. The degree of enrichment depends on the background of gene expression, or the noise. The background is a key factor that can influence the conclusions drawn from the data and the certainty with which we can say genes are enriched [Huang et al., 2009a]. The background must be set up to perform the comparison in tools such as DAVID and should be carefully considered. The background gene set for analysis should only include the genes that have a chance of being selected. For this reason, choosing the whole genome when the whole genome is not represented on the Affymetrix Chip can skew the data. Similarly, when analyzing RNA-seq data, including all genes in the genome instead of discarding those genes for which no counts were recorded will skew the data [Huang et al., 2009a]. Carefully selecting a background should be a priority for each study. Customized Chip background lists are available for all commonly used microarray platforms and individualized background lists can be easily imported to meet the user’s individual needs.
Mouse knockout databases
In order to build a comprehensive functional catalogue of the mammalian genome, a collaboration was launched to create a comprehensive library of knock-out/conditional allele mouse models for researchers to utilize. In mice, this project has been spearheaded by the International Knockout Mouse Consortium (IKMC; https://www.mousephenotype.org/; [Bradley et al., 2012]). The aim of the IKMC is to generate targeted ES-cells of all known protein coding genes in mice and companion Cre driver lines [Rosen et al., 2015]. The current design used for targeting vector allows the production of reporter, conditional and knockout alleles, and provides researchers with flexibility in the design of their experiments and the ability to complement CRISPR/Cas9 strategies [Rosen et al., 2015]. The International Mouse Phenotyping Consortium (IMPC; www.knockoutmouse.org; [Ring et al., 2015]) builds on the work of the IKMC to generate the mouse strains and perform standardized phenotyping. The production of mouse strains from these ES cells are tracked and this information is freely available to the research community. The IKMC/IMPC web portals were merged to create a central hub (http://www.mousephenotype.org; [Rosen et al., 2015]) that has detailed information about the available resources including a catalogue of the targeting vectors, targeted alleles, ES cell clones, and mutant mouse strains generated and links out to other repositories. All gene trap alleles are housed at Jax (http://www.informatics.jax.org/allele). In addition, the International Mouse Strain Resource (IMSR; www.findmice.org; [Eppig et al., 2015; Eppig, and Strivens, 1999]) has a searchable catalogue of over 2000 Cre strains produced by the scientific community that links to the MGD and the repository holding the material.
At least 30% of the targeted knockouts generated in mouse by programs such as the IKMC and IMPC result in embryonic or perinatal death [Adams et al., 2013]. This lead to the inception of the Wellcome Trust-funded research program Deciphering the Mechanisms of Developmental Disorders (DMDD; https://dmdd.org.uk; [Mohun et al., 2013; Adams et al., 2013; Wilson et al., 2016]) that aims to characterize these lethal mutants further. The DMDD focuses on phenotyping embryonic lethal mutants to shed light on the genetic regulation of tissue differentiation, organ formation and embryo morphogenesis [Adams et al., 2013]. This resource is designed for developmental biologists and clinicians; it also complements existing United Kingdom clinical programs focused on better understanding low-frequency and rare genetic changes leading to human disease, such as, the Deciphering Developmental Disorders (https://www.ddduk.org) and UK10K (http://www.uk10k.org) projects.
Human disease variant databases
Online Mendelian Inheritance in Man (OMIM; http://www.omim.org/; [Amberger et al., 2011; Amberger et al., 2009; Hamosh et al., 2005]) is a resource, published since the 1960s, aimed at cataloguing known human disease variants and improving disease classification with a focus on diseases that have a significant genetic basis. One of the goals of the OMIM is to develop a standard nomenclature for features of a disorder (traits) through the Human Phenotype Ontology [Amberger et al., 2011]. OMIM is a curated resource based exclusively on the biomedical literature that links to genomic databases and model organism information, as well as other clinical resources. OMIM serves both molecular biologists and healthcare providers by classifying disorders and biological variation reported in the literature[Amberger et al., 2011]. Entries in OMIN can be classified under the phenotype or the gene. For example, the gene NR5A1 (also known as SF1), resides on Chromosome 9q33.3 (Fig. 1E). Mutations in this gene result in four known phenotypes: 46XY sex reversal, premature ovarian failure, spermatogenic failure, and adrenocortical insufficiency (Fig. 1E). The allelic variants associated with each phenotype listed for a gene are briefly described under the gene entry. More detailed information about the disorder is provided in a separate descriptive entry corresponding to the phenotype (not a unique locus), this entry has a separate identifier. The entry number and mapping key encode additional information about the disorder; more information about the assignment of different MIM numbers can be found at http://www.omim.org/help/faq.
OMIM now facilitates a series of more advanced search options, such as retrieval of similar concepts, clinical or anatomical features. Currently the user is still restricted to querying a single gene at a time through the OMIM interface. However, OMIM does actively encourage the large-scale mining of its repository; API (Application Program Interface) access to the entire OMIM repository (updated nightly) is freely accessible for individual research use with a reasonable fair-use license signed upon download (https://omim.org/api/). Once downloaded, tools such as “R” can be used to query batches of genes using gene names. For example, users can query all the genes on their genes-of-interest list against the OMIM database and pull out features such as the gene name, MIM number and OMIM description into a searchable Excel file. The OMIM descriptions can then be searched as a batch for pathologies and disorders of interest.
The Human Gene Mutation Database (HGMD) represents the other comprehensive collection of mutations that underlie or are associated with human inherited disease. From its inception the HGMD (which is run out of Cardiff University, Wales, UK) was part of a commercial agreement meaning it runs two versions. The online HGMD version that is “free” for academics (http://www.hgmd.cf.ac.uk/ac/index.php; [Stenson et al., 2008; Cooper et al., 2006]) is available via the Cardiff University and a licensed version of the database, “HGMD Professional” (see Table 1) is available through QIAGEN. Newly added mutational information is available to paid users for 2.5 years from the date of initial inclusion in the database before it can be accessed in the free academic resource [Stenson et al., 2009]. Therefore, we recommend using OMIM if you do not have access to the HGMD Professional version of the database.
II. Using databases as a discovery tool to construct a genes-of-interest list for testis development
In addition to interrogate genes-of-interest lists, databases can also be used as a discovery tool to identify new candidate genes. By querying annotated “testis expressed” genes generated by the Eurexpress expression screen, we identified a pseudo-candidate list with genes expressed in the testis at 14.5 dpc. This list contains 298 entries with 289 protein-coding genes, 6 unannotated transcripts, and 3 microRNAs (see Table S1). Among the 289 annotated protein encoding genes, 34 of them are still listed under Rik ID numbers (numbered genes ending with ‘Rik’ are annotated genes without a canonical name yet) although the gene has subsequently been renamed. To ensure the remaining 255 genes are listed under the current approved gene names, we ran them through the HGNC (HUGO Gene Nomenclature Committee) Multi-Symbol Checker (http://www.genenames.org/cgi-bin/symbol_checker). This tool checks all the names in the submitted list against HUGO verified names and their known synonyms. This tool does not cross reference Rik IDs, we therefore cross-referenced the 34 genes under Rik IDs in the MGI database to locate the current gene identifier (Table S1). As many of the databases began cataloguing entries over 15 years ago, not all gene are listed under the current HUGO approved name. This means that the investigator may overlook entries that remain curated under defunct synonyms of IDs. Once we confirmed/identified the gene names for all 295 genes (289 protein-encoding genes + 6 unannotated transcripts), we obtained the in situ hybridization images, and score them on an arbitrary metric of 1–4 (1-very high publication quality; 2-high publication quality; 3-moderate quality (requires confirmation); 4-low quality (not publication quality)). For every gene that scored a 1 or 2 (142 genes; Table S2), we downloaded a representative full embryo image containing the testis and cropped a 600 × 600 pixel section containing the testis in Photoshop. In most cases the first testis of the pair appeared in slice 3–7 of the slide deck (before the kidney) whereas the second testis appeared between slide 11–21 (after the kidney).
Table S1: A full list of 298 entries under the annotation of expressed in the “testis” from the Eurexpress database.
This table contains the complete list of entries listed in the Eurexpress databases under the anatomical annotation of “testis” for embryos at 14.5 dpc on May 1, 2016. Non-HUGO approved symbols listed in Eurexpress have been replaced with HUGO approved symbols for data analysis.
| HUGO approved gene name |
Non-HUGO symbols listed in Eurexpress |
HUGO approved name |
|---|---|---|
| 1700011H14Rik | 1700011H14Rik | * |
| Aatf | Apoptosis Antagonizing Transcription Factor | |
| Abhd17c | 2210412D01Rik | Abhydrolase Domain Containing 17C |
| Abl2 | ABL Proto-Oncogene 2, Non-Receptor Tyrosine Kinase | |
| Abtb2 | Ankyrin Repeat And BTB (POZ) Domain Containing 2 | |
| Acsbg1 | Acyl-Coa Synthetase Bubblegum Family Member 1 | |
| Adamts16 | ADAM Metallopeptidase With Thrombospondin Type 1 Motif 16 | |
| Adamts2 | ADAM Metallopeptidase With Thrombospondin Type 1 Motif 2 | |
| Adh1 | Alcohol Dehydrogenase 1A (Class I), Alpha Polypeptide | |
| Ajap1 | Adherens Junctions Associated Protein 1 | |
| Akr1cl | 4921521F21Rik | Aldo-Keto Reductase Family 1, Member C-Like |
| Amh | Anti-Mullerian Hormone | |
| Anxa11 | Annexin A11 | |
| Anxa4 | Annexin A4 | |
| Ap3b2 | Adaptor Related Protein Complex 3 Beta 2 Subunit | |
| Apc | APC, WNT Signaling Pathway Regulator | |
| Apex1 | Apurinic/Apyrimidinic Endodeoxyribonuclease 1 | |
| Apln | Apelin | |
| Arl6ip2 | Atlastin Gtpase 2 | |
| Art5 | ADP-Ribosyltransferase 5 | |
| Ash2l | ASH2 Like Histone Lysine Methyltransferase Complex Subunit | |
| Aspa | Aspartoacylase | |
| Atp1b1 | Atpase Na+/K+ Transporting Subunit Beta 1 | |
| Atp6v1b1 | Atpase H+ Transporting V1 Subunit B1 | |
| Atp6v1e1 | Atpase H+ Transporting V1 Subunit E1 | |
| Atp8b2 | Atpase Phospholipid Transporting 8B2 | |
| Bbx | BBX, HMG-Box Containing | |
| BC034902 | BC034902 | * |
| Bex2 | Brain Expressed X-Linked 2 | |
| Brca1 | BRCA1, DNA Repair Associated | |
| Btf3 | Basic Transcription Factor 3 | |
| Bub1 | BUB1 Mitotic Checkpoint Serine/Threonine Kinase | |
| Bub1b | BUB1 Mitotic Checkpoint Serine/Threonine Kinase B | |
| Camkk2 | Calcium/Calmodulin Dependent Protein Kinase Kinase 2 | |
| Casp6 | Caspase 6 | |
| Cd34 | CD34 Molecule | |
| Cdca7l | Cell Division Cycle Associated 7 Like | |
| Cdk5rap1 | CDK5 Regulatory Subunit Associated Protein 1 | |
| Cenpj | Centromere Protein J | |
| Cenpo | 2810429O05Rik | Centromere Protein O |
| Cep68 | BC027174 | Centrosomal Protein 68 |
| Clca1 | Chloride Channel Accessory 1 | |
| Clca4 | Chloride Channel Accessory 4 | |
| Clcn2 | Chloride Voltage-Gated Channel 2 | |
| Cldn13 | Claudin 13 | |
| Clmn | Calmin | |
| Cobll1 | Cordon-Bleu WH2 Repeat Protein Like 1 | |
| Col4a1 | Collagen Type IV Alpha 1 Chain | |
| Col9a3 | Collagen Type IX Alpha 3 Chain | |
| Copb2 | Coatomer Protein Complex Subunit Beta 2 | |
| Csrp1 | Cysteine And Glycine Rich Protein 1 | |
| Ctnna2 | Catenin Alpha 2 | |
| Cx3cl1 | C-X3-C Motif Chemokine Ligand 1 | |
| Cxadr | Coxsackie Virus And Adenovirus Receptor | |
| Cxcl12 | C-X-C Motif Chemokine Ligand 12 | |
| Cxxc6 | Tet Methylcytosine Dioxygenase 1 | |
| Cyp11a1 | Cytochrome P450 Family 11 Subfamily A Member 1 | |
| Cyp51 | Cytochrome P450 Family 51 Subfamily A Member 1 | |
| D930030O05Rik | D930030O05Rik | * |
| Daam2 | Dishevelled Associated Activator Of Morphogenesis 2 | |
| Dcun1d5 | 4833420K19Rik | DCN1, Defective In Cullin Neddylation 1, Domain Containing 5 |
| Ddx41 | DEAD-Box Helicase 41 | |
| Ddx48 | Eukaryotic Translation Initiation Factor 4A3 | |
| Dnmt3b | DNA Methyltransferase 3 Beta | |
| Dppa3 | Developmental Pluripotency Associated 3 | |
| Dppa5 | Developmental Pluripotency Associated 5 | |
| Drg1 | Developmentally Regulated GTP Binding Protein 1 | |
| Dsp | Desmoplakin | |
| E430025E21Rik | * | |
| Eif2b1 | Eukaryotic Translation Initiation Factor 2B Subunit Alpha | |
| Eif2s3y | Eukaryotic Translation Initiation Factor 2, Subunit 3, Structural Gene Y-Linked |
|
| Elavl2 | ELAV Like RNA Binding Protein 2 | |
| Eps8 | Epidermal Growth Factor Receptor Pathway Substrate 8 | |
| Eps8l2 | EPS8 Like 2 | |
| Ets1 | ETS Proto-Oncogene 1, Transcription Factor | |
| Etv5 | ETS Variant 5 | |
| Fads1 | Fatty Acid Desaturase 1 | |
| Fam214a | BC031353 | Family With Sequence Similarity 214, Member A |
| Fasn | Fatty Acid Synthase | |
| Fastkd2 | 2810421I24Rik | FAST Kinase Domains 2 |
| Fbrsl1 | 2410025L10Rik | Fibrosin-Like 1 |
| Fiz1 | FLT3 Interacting Zinc Finger 1 | |
| Fosl2 | FOS Like 2, AP-1 Transcription Factor Subunit | |
| Foxn2 | Forkhead Box N2 | |
| Frzb | Frizzled-Related Protein | |
| Fthfd | Aldehyde Dehydrogenase 1 Family Member L1 | |
| Fzd3 | Frizzled Class Receptor 3 | |
| Gas6 | Growth Arrest Specific 6 | |
| Gem | GTP Binding Protein Overexpressed In Skeletal Muscle | |
| Gjb2 | Gap Junction Protein Beta 2 | |
| Gmnn | Geminin, DNA Replication Inhibitor | |
| Gnl3 | G Protein Nucleolar 3 | |
| Gpc4 | Glypican 4 | |
| Gpr37 | G Protein-Coupled Receptor 37 | |
| Gpr56 | Adhesion G Protein-Coupled Receptor G1 | |
| Gprk5 | G Protein-Coupled Receptor Kinase 5 | |
| Gramd1b | GRAM Domain Containing 1B | |
| Grrp1 | Family With Sequence Similarity 110 Member D | |
| Gsta4 | Glutathione S-Transferase Alpha 4 | |
| Gstm1 | Glutathione S-Transferase Mu 1 | |
| Gstm4 | Glutathione S-Transferase Mu 4 | |
| Gstm5 | Glutathione S-Transferase Mu 5 | |
| Gstm7 | Glutathione S-Transferase, Mu 7 | |
| Gtf2b | General Transcription Factor IIB | |
| Gtf2e2 | General Transcription Factor IIE Subunit 2 | |
| Gtf3c3 | General Transcription Factor IIIC Subunit 3 | |
| Gucy1a3 | Guanylate Cyclase 1 Soluble Subunit Alpha | |
| Gucy1b3 | Guanylate Cyclase 1 Soluble Subunit Beta | |
| Hadhsc | Hydroxyacyl-Coa Dehydrogenase | |
| Hat1 | Histone Acetyltransferase 1 | |
| Heatr1 | B130016L12Rik | HEAT Repeat Containing 1 |
| Hmg20a | High Mobility Group 20A | |
| Hmga1 | High Mobility Group AT-Hook 1 | |
| Hoxd9 | Homeobox D9 | |
| Hs6st1 | Heparan Sulfate 6-O-Sulfotransferase 1 | |
| Hsd17b7 | Hydroxysteroid 17-Beta Dehydrogenase 7 | |
| Hsd17b7 | Hydroxysteroid 17-Beta Dehydrogenase 7 Pseudogene 2 | |
| Hsd3b3 | Hydroxy-Delta-5-Steroid Dehydrogenase, 3 Beta- And Steroid Delta- Isomerase 3 |
|
| Hsp90aa1 | Heat Shock Protein 90 Alpha Family Class A Member 1 | |
| Hspa1a | Heat Shock Protein Family A (Hsp70) Member 1A | |
| Hspa2 | Heat Shock Protein Family A (Hsp70) Member 2 | |
| Iah1 | 4833421E05Rik | Isoamyl Acetate-Hydrolyzing Esterase 1 Homolog |
| Idh1 | Isocitrate Dehydrogenase (NADP(+)) 1, Cytosolic | |
| Idi1 | Isopentenyl-Diphosphate Delta Isomerase 1 | |
| Ier5l | Immediate Early Response 5 Like | |
| Ifi27 | D12Ertd647e | Interferon, Alpha-Inducible Protein 27 |
| Ifitm7 | ||
| Immt | Inner Membrane Mitochondrial Protein | |
| Irf1 | Interferon Regulatory Factor 1 | |
| Itga9 | Integrin Subunit Alpha 9 | |
| Itgb8 | Integrin Subunit Beta 8 | |
| Kcnt1 | Potassium Sodium-Activated Channel Subfamily T Member 1 | |
| Kctd14 | Potassium Channel Tetramerization Domain Containing 14 | |
| Kif2c | Kinesin Family Member 2C | |
| Kirrel2 | Kin Of IRRE Like 2 (Drosophila) | |
| L3mbtl | L(3)Mbt-Like 1 (Drosophila) | |
| Lba1 | Tetratricopeptide Repeat And Ankyrin Repeat Containing 1 | |
| Lin28 | Lin-28 Homolog A | |
| Liph | Lipase H | |
| Lisch7 | Lipolysis Stimulated Lipoprotein Receptor | |
| LOC620538 | * | |
| LOC673219 | * | |
| Lyar | Ly1 Antibody Reactive | |
| Mafg | MAF Bzip Transcription Factor G | |
| Mcm8 | Minichromosome Maintenance 8 Homologous Recombination Repair Factor |
|
| Melk | Maternal Embryonic Leucine Zipper Kinase | |
| Mettl25 | BC067068 | Methyltransferase Like 25 |
| Mmp8 | Matrix Metallopeptidase 8 | |
| mmu-miR-291a-5p | ^ | |
| mmu-miR-291b-5p | ^ | |
| mmu-miR-696 | ^ | |
| Mod1 | Chromobox 1 | |
| Mphosph6 | M-Phase Phosphoprotein 6 | |
| Mpp3 | Membrane Palmitoylated Protein 3 | |
| Mrpl23 | Mitochondrial Ribosomal Protein L23 | |
| Mrpl24 | Mitochondrial Ribosomal Protein L24 | |
| Mrps27 | Mitochondrial Ribosomal Protein S27 | |
| Mtif2 | Mitochondrial Translational Initiation Factor 2 | |
| Mybbp1a | MYB Binding Protein 1a | |
| Mybl1 | MYB Proto-Oncogene Like 1 | |
| Nat10 | N-Acetyltransferase 10 | |
| Ndufa9 | NADH:Ubiquinone Oxidoreductase Subunit A9 | |
| Ndufab1 | NADH:Ubiquinone Oxidoreductase Subunit AB1 | |
| Ndufs1 | NADH:Ubiquinone Oxidoreductase Core Subunit S1 | |
| Ndufs3 | NADH:Ubiquinone Oxidoreductase Core Subunit S3 | |
| Ndufv1 | NADH:Ubiquinone Oxidoreductase Core Subunit V1 | |
| Nepro | BC027231 | Nucleolus And Neural Progenitor Protein |
| Nfe2 | Nuclear Factor, Erythroid 2 | |
| Npr1 | Natriuretic Peptide Receptor 1 | |
| Nr4a1 | Nuclear Receptor Subfamily 4 Group A Member 1 | |
| Nrg1 | Neuregulin 1 | |
| Nsmaf | Neutral Sphingomyelinase Activation Associated Factor | |
| Nts | Neurotensin | |
| Nupl1 | Nucleoporin 58 | |
| Nutf2 | Nuclear Transport Factor 2 | |
| Orc5l | Origin Recognition Complex Subunit 5 | |
| Osbpl10 | Oxysterol Binding Protein Like 10 | |
| Parm1 | 9130213B05Rik | Prostate Androgen-Regulated Mucin-Like Protein 1 |
| Pcanap6 | Solute Carrier Family 45 Member 3 | |
| Pcmtd1 | A030012M09Rik | Protein-L-Isoaspartate (D-Aspartate) O-Methyltransferase Domain Containing 1 |
| Pcsk6 | Proprotein Convertase Subtilisin/Kexin Type 6 | |
| Pdgfa | Platelet Derived Growth Factor Subunit A | |
| Pdzd4 | PDZ Domain Containing 4 | |
| Per2 | Period Circadian Clock 2 | |
| Pgpep1 | Pyroglutamyl-Peptidase I | |
| Pink1 | PTEN Induced Putative Kinase 1 | |
| Pip5k2a | Phosphatidylinositol-5-Phosphate 4-Kinase Type 2 Alpha | |
| Pla2g5 | Phospholipase A2 Group V | |
| Plekha1 | Pleckstrin Homology Domain Containing A1 | |
| Plekha2 | Pleckstrin Homology Domain Containing A2 | |
| Polr2h | RNA Polymerase II Subunit H | |
| Polr2l | RNA Polymerase II Subunit L | |
| Polr3f | RNA Polymerase III Subunit F | |
| Por | Cytochrome P450 Oxidoreductase | |
| Por | Porcupine Homolog (Drosophila) | |
| Pou6f1 | POU Class 6 Homeobox 1 | |
| Ppm1a | Protein Phosphatase, Mg2+/Mn2+ Dependent 1A | |
| Ppt1 | Palmitoyl-Protein Thioesterase 1 | |
| Prkar2b | Protein Kinase Camp-Dependent Type II Regulatory Subunit Beta | |
| Prlr | Prolactin Receptor | |
| Prlr | Prolactin Receptor | |
| Prss15 | Lon Peptidase 1, Mitochondrial | |
| Psma1 | Proteasome Subunit Alpha 1 | |
| Ptpro | Protein Tyrosine Phosphatase, Receptor Type O | |
| Ptpro | Protein Tyrosine Phosphatase, Receptor Type U | |
| Rad52b | RAD52 Motif Containing 1 | |
| Rbbp7 | RB Binding Protein 7, Chromatin Remodeling Factor | |
| Rcc2 | Regulator Of Chromosome Condensation 2 | |
| Rcl1 | RNA Terminal Phosphate Cyclase Like 1 | |
| Reep2 | BC020184 | Receptor Accessory Protein 2 |
| Ren1 | ||
| Rgs11 | Regulator Of G-Protein Signaling 11 | |
| Rhpn1 | Rhophilin Rho Gtpase Binding Protein 1 | |
| Rmdn2 | Regulator Of Microtubule Dynamics 2 | |
| Rnf138 | Ring Finger Protein 138 | |
| Rnf181 | 2500002L14Rik | Ring Finger Protein 181 |
| Rnf213 | D11Ertd759e | Ring Finger Protein 213 |
| Rnf34 | Ring Finger Protein 34 | |
| Rnmt | RNA Guanine-7 Methyltransferase | |
| Rpp30 | Ribonuclease P/MRP Subunit P30 | |
| Rps27l | Ribosomal Protein S27 Like | |
| Sc4mol | Methylsterol Monooxygenase 1 | |
| Scarb1 | Scavenger Receptor Class B Member 1 | |
| Schip1 | Schwannomin Interacting Protein 1 | |
| Scrn2 | Secernin 2 | |
| Sct | Secretin | |
| Sec11l1 | SEC11 Homolog A, Signal Peptidase Complex Subunit | |
| Senp8 | SUMO/Sentrin Peptidase Family Member, NEDD8 Specific | |
| Serpinb6a | Serine (Or Cysteine) Peptidase Inhibitor, Clade B, Member 6a | |
| Sgk3 | Serum/Glucocorticoid Regulated Kinase Family Member 3 | |
| Sil1 | SIL1 Nucleotide Exchange Factor | |
| Slc16a1 | Solute Carrier Family 16 Member 1 | |
| Slc20a1 | Solute Carrier Family 20 Member 1 | |
| Slc25a31 | Solute Carrier Family 25 Member 31 | |
| Slc25a39 | D11Ertd333e | Solute Carrier Family 25, Member 39 |
| Slc29a1 | Solute Carrier Family 29 Member 1 (Augustine Blood Group) | |
| Slc38a1 | Solute Carrier Family 38 Member 1 | |
| Slc44a1 | Solute Carrier Family 44 Member 1 | |
| Slc7a5 | Solute Carrier Family 7 Member 5 | |
| Smarca4 | SWI/SNF Related, Matrix Associated, Actin Dependent Regulator Of Chromatin, Subfamily A, Member 4 |
|
| Smoc1 | SPARC Related Modular Calcium Binding 1 | |
| Snrpn | Small Nuclear Ribonucleoprotein Polypeptide N | |
| Snw1 | SNW Domain Containing 1 | |
| Sostdc1 | Sclerostin Domain Containing 1 | |
| Sox2 | SRY-Box 2 | |
| Sox4 | SRY-Box 4 | |
| Spice1 | D16Ertd480e | Spindle And Centriole Associated Protein 1 |
| Srfbp1 | Serum Response Factor Binding Protein 1 | |
| Srpx2 | Sushi Repeat Containing Protein, X-Linked 2 | |
| Star | Steroidogenic Acute Regulatory Protein | |
| Star | Guanylate Cyclase 2C | |
| Star | Steroidogenic Acute Regulatory Protein | |
| Stc1 | Stanniocalcin 1 | |
| Stc2 | Stanniocalcin 2 | |
| Stk25 | Serine/Threonine Kinase 25 | |
| Stxbp2 | Syntaxin Binding Protein 2 | |
| Tacr3 | Tachykinin Receptor 3 | |
| Tacstd1 | Epithelial Cell Adhesion Molecule | |
| Taf7 | TATA-Box Binding Protein Associated Factor 7 | |
| Tbc1d8 | TBC1 Domain Family Member 8 | |
| Tcf21 | Transcription Factor 21 | |
| Tde1 | Serine Incorporator 3 | |
| Tdrd12 | 2410004F06Rik | Tudor Domain Containing 12 |
| Tesc | Tescalcin | |
| Tex2 | Testis Expressed 2 | |
| Tex261 | Testis Expressed 261 | |
| Tgif | TGFB Induced Factor Homeobox 1 | |
| Timm8b | Translocase Of Inner Mitochondrial Membrane 8 Homolog B | |
| Tle6 | Transducin Like Enhancer Of Split 6 | |
| Tmcc3 | Transmembrane And Coiled-Coil Domain Family 3 | |
| Tmem22 | Solute Carrier Family 35 Member G2 | |
| Tmem35 | Transmembrane Protein 35A | |
| Tmem64 | 9630015D15Rik | Transmembrane Protein 64 |
| Tmem86a | 1810054O13Rik | Transmembrane Protein 86A |
| Tpd52 | Tumor Protein D52 | |
| Tpr | Translocated Promoter Region, Nuclear Basket Protein | |
| Trim71 | Tripartite Motif Containing 71 | |
| Trip13 | Thyroid Hormone Receptor Interactor 13 | |
| Trpv2 | Transient Receptor Potential Cation Channel Subfamily V Member 2 | |
| Trrap | Transformation/Transcription Domain Associated Protein | |
| Ttc39c | 2810439F02Rik | Tetratricopeptide Repeat Domain 39C |
| Txnrd2 | Thioredoxin Reductase 2 | |
| Tyro3 | TYRO3 Protein Tyrosine Kinase | |
| Ube2t | Ubiquitin Conjugating Enzyme E2 T | |
| Uchl1 | Ubiquitin C-Terminal Hydrolase L1 | |
| Uchl5 | Ubiquitin C-Terminal Hydrolase L5 | |
| Usf1 | Upstream Transcription Factor 1 | |
| Utrn | Utrophin | |
| Wdr10 | Intraflagellar Transport 122 | |
| Wdr21 | DDB1 And CUL4 Associated Factor 4 | |
| Wdr31 | WD Repeat Domain 31 | |
| Wdr36 | WD Repeat Domain 36 | |
| Xrcc6 | X-Ray Repair Cross Complementing 6 | |
| Yars2 | Tyrosyl-Trna Synthetase 2 | |
| Zdhhc24 | Zinc Finger DHHC-Type Containing 24 | |
| Zfp451 | Zinc Finger Protein 451 | |
| Zfp516 | Zinc Finger Protein 516 | |
indicates that a transcript but no gene is annotated (6 genes).
indicates a pre-miRNA transcript.
Most analysis was performed on a list of 295 genes excluding the 3 pre-miRNAs or 289 genes when including only known protein-coding transcripts.
Table S2: 142 Genes Annotated As Expressed In The “Testis” Ranked 1 Or 2 (Publication Quality) In The Eurexpress Databases.
Of the 295 transcript entries (excluding the 3 pre-miRNAs ) in the Eurexpress database, 142 were considered publication quality. We scored the in situ hybridization images for each of the genes on a metric of 1–4. 1-very high publication quality; 2-high publication quality; 3-moderate quality (reconsider as publication quality); 4-low quality (not publication quality). Genes scoring a 1 or 2 are listed in this table. These data were obtained from Eurexpress Transcriptome Atlas Database for Mouse Embryo (http://www.eurexpress.org) on May 1, 2016.
| Gene | Chromosome | Gene Name |
|---|---|---|
| Acsbg1 | 9 | Acyl-Coa Synthetase Bubblegum Family Member 1 |
| Adamts16 | 13 | A Disintegrin-Like And Metallopeptidase (Reprolysin Type) With Thrombospondin Type 1 Motif, 16 |
| Adamts2 | 11 | A Disintegrin-Like And Metallopeptidase (Reprolysin Type) With Thrombospondin Type 1 Motif, 2 |
| Adh1 | 3 | Alcohol Dehydrogenase 1 (Class I) |
| Ap3b2 | 7 | Adaptor-Related Protein Complex 3, Beta 2 Subunit |
| Apc | 18 | Adenomatosis Polyposis Coli |
| Apex1 | 14 | Apurinic/Apyrimidinic Endonuclease 1 |
| Apln | X | Apelin |
| Arl6ip2 | 17 | Atlastin Gtpase 2 |
| Atp6v1e1 | 6 | Atpase, H+ Transporting, Lysosomal V1 Subunit E1 |
| Atp8b2 | 3 | Atpase, Class I, Type 8b, Member 2 |
| Bex2 | X | Brain Expressed X-Linked 2 |
| Bub1 | 2 | Bub1, Mitotic Checkpoint Serine/Threonine Kinase |
| Cd34 | 1 | Cd34 Antigen |
| Cdca7l | 12 | Cell Division Cycle Associated 7 Like |
| Cdk5rap1 | 2 | Cdk5 Regulatory Subunit Associated Protein 1 |
| Clca1 | 3 | Chloride Channel Accessory 1 |
| Clca4 | 3 | Chloride Channel Accessory 4a/4b |
| Clcn2 | 16 | Chloride Channel, Voltage-Sensitive 2 |
| Col9a3 | 2 | Collagen, Type Ix, Alpha 3 |
| Copb2 | 9 | Coatomer Protein Complex, Subunit Beta 2 (Beta Prime) |
| Ctnna2 | 6 | Catenin (Cadherin Associated Protein), Alpha 2 |
| Cxadr | 16 | Coxsackie Virus And Adenovirus Receptor |
| Cxcl12 | 6 | Chemokine (C-X-C Motif) Ligand 12 |
| Cyp11a1 | 9 | Cytochrome P450, Family 11, Subfamily A, Polypeptide 1 |
| Cyp51 | 5 | Cytochrome P450, Family 51 |
| Rnf213 | 11 | Ring Finger Protein 213 (Rnf213) |
| Ifi27 | 12 | Interferon, Alpha-Inducible Protein 27 (Ifi27 ) |
| Spice1 | 16 | Spindle And Centriole Associated Protein 1 (Spice1 ) |
| Daam2 | 17 | Dishevelled Associated Activator Of Morphogenesis 2 |
| Dppa3 | 6 | Developmental Pluripotency-Associated 3 |
| Dsp | 13 | Desmoplakin |
| Elavl2 | 4 | Elav (Embryonic Lethal, Abnormal Vision, Drosophila)-Like 2 (Hu Antigen B) |
| Ets1 | 9 | E26 Avian Leukemia Oncogene 1, 5' Domain |
| Etv5 | 16 | Ets Variant 5 |
| Fads1 | 19 | Fatty Acid Desaturase 1 |
| Fthfd | 6 | Aldehyde Dehydrogenase 1 Family, Member L1 |
| Gas6 | 8 | Growth Arrest Specific 6 |
| Gem | 4 | Gtp Binding Protein (Gene Overexpressed In Skeletal Muscle) |
| Gjb2 | 14 | Gap Junction Protein, Beta 2 |
| Gnl3 | 14 | Guanine Nucleotide Binding Protein-Like 3 (Nucleolar) |
| Gpc4 | X | Glypican 4 |
| Adgrg1 | 8 | Adhesion G Protein-Coupled Receptor G1 (Adgrg1 ) |
| Gramd1b | 9 | Gram Domain Containing 1b |
| Grrp | 4 | Glycine/Arginine Rich Protein 1 |
| Gsta4 | 9 | Glutathione S-Transferase, Alpha 4 |
| Gstm1 | 3 | Glutathione S-Transferase, Mu 1 |
| Gstm4 | 3 | Glutathione S-Transferase, Mu 4 |
| Gstm7 | 3 | Glutathione S-Transferase, Mu 7 |
| Gtf2b | 3 | General Transcription Factor Iib |
| Gtf2e2 | 8 | General Transcription Factor Ii E, Polypeptide 2 (Beta Subunit) |
| Gtlf3b | 11 | N-Acetyltransferase Domain Containing 1 |
| Gucy1a3 | 3 | Guanylate Cyclase 1, Soluble, Alpha 3 |
| Hat1 | 2 | Histone Aminotransferase 1 |
| Hmg20a | 9 | High Mobility Group 20a |
| Hmga1 | 17 | High Mobility Group At-Hook 1 |
| Hoxd9 | 2 | Homeobox D9 |
| Hs6st1 | 1 | Heparan Sulfate 6-O-Sulfotransferase 1 |
| Hsd17b7 | 1 | Hydroxysteroid (17-Beta) Dehydrogenase 7 |
| Hsd3b3 | 3 | Hydroxy-Delta-5-Steroid Dehydrogenase, 3 Beta- And Steroid Delta-Isomerase 3 |
| Hsp90aa1 | 12 | Heat Shock Protein 90, Alpha (Cytosolic), Class A Member 1 |
| Hspa1a | 17 | Heat Shock Protein 1a |
| Idi1 | 13 | Isopentenyl-Diphosphate Delta Isomerase |
| Ifitm7 | 16 | Interferon Induced Transmembrane Protein 7 |
| Irf1 | 11 | Interferon Regulatory Factor 1 |
| Itga9 | 9 | Integrin Alpha 9 |
| Kcnt1 | 2 | Potassium Channel, Subfamily T, Member 1 |
| L3mbtl | 2 | L(3)Mbt-Like (Drosophila) |
| Trank1 | 9 | Tetratricopeptide Repeat And Ankyrin Repeat Containing 1 (Trank1 ) |
| Liph | 16 | Lipase, Member H |
| Lsr | 7 | Lipolysis Stimulated Lipoprotein Receptor (Lsr) |
| Melk | 4 | Maternal Embryonic Leucine Zipper Kinase |
| Mmp8 | 9 | Matrix Metallopeptidase 8 |
| Me1 | 9 | Malic Enzyme 1, Nadp(+)-Dependent, Cytosolic (Me1) |
| Mphosph6 | 8 | M Phase Phosphoprotein 6 |
| Mrpl23 | 7 | Mitochondrial Ribosomal Protein L23 |
| Mybbp1a | 11 | Myb Binding Protein (P160) 1a |
| Npr1 | 3 | Natriuretic Peptide Receptor 1 |
| Nrg1 | 8 | Neuregulin 1 |
| Nts | 10 | Neurotensin |
| Nupl1 | 14 | Nucleoporin Like 1 |
| Osbpl10 | 9 | Oxysterol Binding Protein-Like 10 |
| Slc45a3 | 1 | Solute Carrier Family 45, Member 3 (Slc45a3) |
| Pcsk6 | 7 | Proprotein Convertase Subtilisin/Kexin Type 6 |
| Pdzd4 | X | Pdz Domain Containing 4 |
| Pgpep1 | 8 | Pyroglutamyl-Peptidase I |
| Pla2g5 | 4 | Phospholipase A2, Group V |
| Plekha1 | 7 | Pleckstrin Homology Domain Containing, Family A (Phosphoinositide Binding Specific) Member 1 |
| Polr3f | 2 | Polymerase (Rna) Iii (Dna Directed) Polypeptide F |
| Por | 5 | P450 (Cytochrome) Oxidoreductase |
| Ppt1 | 4 | Palmitoyl-Protein Thioesterase 1 |
| Prkar2b | 12 | Protein Kinase, Camp Dependent Regulatory, Type Ii Beta |
| Prlr | 15 | Prolactin Receptor |
| Psma1 | 7 | Proteasome (Prosome, Macropain) Subunit, Alpha Type 1 |
| Rad52b | 11 | Rad52 Motif 1 |
| Rcc2 | 4 | Regulator Of Chromosome Condensation 2 |
| Rcl1 | 19 | Rna Terminal Phosphate Cyclase-Like 1 |
| Ren1 | 1 | Renin 1 Structural |
| Rgs11 | 17 | Regulator Of G-Protein Signaling 11 |
| Rhpn1 | 15 | Rhophilin, Rho Gtpase Binding Protein 1 |
| Rnf138 | 18 | Ring Finger Protein 138 |
| Msmo1 | 8 | Methylsterol Monoxygenase 1 (Msmo1 ) |
| Scarb1 | 5 | Scavenger Receptor Class B, Member 1 |
| Schip1 | 3 | Schwannomin Interacting Protein 1 |
| Scrn2 | 11 | Secernin 2 |
| Sct | 7 | Secretin |
| Serpinb6a | 13 | Serine (Or Cysteine) Peptidase Inhibitor, Clade B, Member 6a |
| Slc20a1 | 2 | Solute Carrier Family 20, Member 1 |
| Slc25a31 | 3 | Solute Carrier Family 25 (Mitochondrial Carrier; Adenine Nucleotide Translocator), Member 31 |
| Slc29a1 | 17 | Solute Carrier Family 29 (Nucleoside Transporters), Member 1 |
| Slc38a1 | 15 | Solute Carrier Family 38, Member 1 |
| Slc7a5 | 8 | Solute Carrier Family 7 (Cationic Amino Acid Transporter, Y+ System), Member 5 |
| Smoc1 | 12 | Sparc Related Modular Calcium Binding 1 |
| Snrpn | 7 | Small Nuclear Ribonucleoprotein N |
| Sostdc1 | 12 | Sclerostin Domain Containing 1 |
| Sox2 | 3 | Sry (Sex Determining Region Y)-Box 2 |
| Stc1 | 14 | Stanniocalcin 1 |
| Stc2 | 11 | Stanniocalcin 2 |
| Stxbp2 | 8 | Syntaxin Binding Protein 2 |
| Tacr3 | 3 | Tachykinin Receptor 3 |
| Epcam | 17 | Epithelial Cell Adhesion Molecule (Epcam) |
| Tbc1d8 | 1 | Tbc1 Domain Family, Member 8 |
| Tcf21 | 10 | Transcription Factor 21 |
| Tcl1 | 12 | T Cell Lymphoma Breakpoint 1 |
| Serinc3 | 2 | Serine Incorporator 3 (Serinc3 ) |
| Tesc | 5 | Tescalcin |
| Tex2 | 11 | Testis Expressed Gene 2 |
| Tle6 | 10 | Transducin-Like Enhancer Of Split 6 |
| Tmcc3 | 10 | Transmembrane And Coiled Coil Domains 3 |
| Tmem35 | X | Transmembrane Protein 35a |
| Tpd52 | 3 | Tumor Protein D52 |
| Trip13 | 13 | Thyroid Hormone Receptor Interactor 13 |
| Trrap | 5 | Transformation/Transcription Domain-Associated Protein |
| Tyro3 | 2 | Tyro3 Protein Tyrosine Kinase 3 |
| Ube2t | 1 | Ubiquitin-Conjugating Enzyme E2t |
| Uchl1 | 5 | Ubiquitin Carboxy-Terminal Hydrolase L1 |
| Yars2 | 16 | Tyrosyl-Trna Synthetase 2 (Mitochondrial) |
| Zdhhc24 | 19 | Zinc Finger, Dhhc Domain Containing 24 |
| Zfp451 | 1 | Zinc Finger Protein 451 |
| Zfp516 | 18 | Zinc Finger Protein 516 |
We first identified previously described marker genes and validated their expression. We compared the list of 289 protein-encoding genes and 6 unannotated transcripts (a total of 295) from the Eurexpress database to published microarray data for the genes enriched in five key testicular cell types (supporting or Sertoli cells, germ cells, interstitial cells, Leydig cells, and endothelial cells; Table S3) at 13.5 dpc [Jameson et al., 2012]. For the supporting or Sertoli cell-enriched genes, among 295 testis-expressing genes from the Eurexpress database and 491 genes from the Sertoli cell enriched genes from the Jameson et al microarray, 35 genes were found in both datasets (Fig. 2A), including 5 known Sertoli cell genes Amh, Ptgds, Etv5, Tyro3 and Col9a3 (Fig. 2B). Sixteen of the 35 genes were of publication quality (Fig. 2C). For the male germ cell genes, 25 genes were shared by both the Eurexpress database and the Jameson et al microarray (Fig. 3A), including Sox2, Dazl, and Dppa3 (Fig. 3B). Among the 25 genes, 10 were of publication quality in situ hybridization images from the Eurexpress database (Fig. 3C).
Table S3: List of genes represented in both the list of Eurexpress testis annotated genes and the gene lists from Jameson et al., 2012 microarray.
To validate the expression of previously reported genes expressed in Sertoli, germ, interstitial, Leydig and endothelial cells, we compared the list of 295 entries from the Eurexpress database to published Jameson et al microarray data. Genes in both datasets are listed by cell type in this table. (295 entries is 289 protein-encoding genes and 6 unannotated transcripts). These data were obtained from Eurexpress Transcriptome Atlas Database for Mouse Embryo (http://www.eurexpress.org) on May 1, 2016.
| Supporting (Sertoli) cells |
Leydig cells | Mixed interstitial cells | Germ cells | Endothelial cells |
|---|---|---|---|---|
| Gene name | Gene name | Gene name | Gene name | Gene name |
| Adamts16 | Cyp11a1 | Clca1 | Abl2 | Acsbg1 |
| Amh | Fads1 | Cxcl12 | Ap3b2 | Nr4a1 |
| Atp8b2 | Gramd1b | Gucy1b3 | Bub1b | |
| Bex2 | Hsd17b7 | Itga9 | Cdk5rap1 | |
| Camkk2 | Prkar2b | Itgb8 | Ddx41 | |
| Col9a3 | Prlr | Nrg1 | Dppa3 | |
| Ctnna2 | Ren1 | Sostdc1 | Elavl2 | |
| Cxadr | Scarb1 | Stc1 | Epcam | |
| Dsp | Sct | Tacr3 | Gnl3 | |
| Eps8 | Star | Gtf3c3 | ||
| Fzd3 | Stc1 | Irf1 | ||
| Gjb2 | Liph | |||
| Gpr37 | Mybbp1a | |||
| Gstm7 | Nsmaf | |||
| Lsr | Pip4k2a | |||
| Mybl1 | Pou6f1 | |||
| Nfe2 | Rcc2 | |||
| Npr1 | Rcl1 | |||
| Pdgfa | Rdm1 | |||
| Pgpep1 | Srfbp1 | |||
| Pla2g5 | Taf7 | |||
| Ppt1 | Tet1 | |||
| Schip1 | Trip13 | |||
| Sgk3 | Uchl1 | |||
| Sil1 | Wdr31 | |||
| Slc20a1 | ||||
| Slc38a1 | ||||
| Slc7a5 | ||||
| Stc2 | ||||
| Tbc1d8 | ||||
| Tesc | ||||
| Tmcc3 | ||||
| Tpd52 | ||||
| Trank1 | ||||
| Tyro3 |
Fig. 2: Sertoli cell-expressed genes identified from “testis” annotated entries in the Eurexpress database.
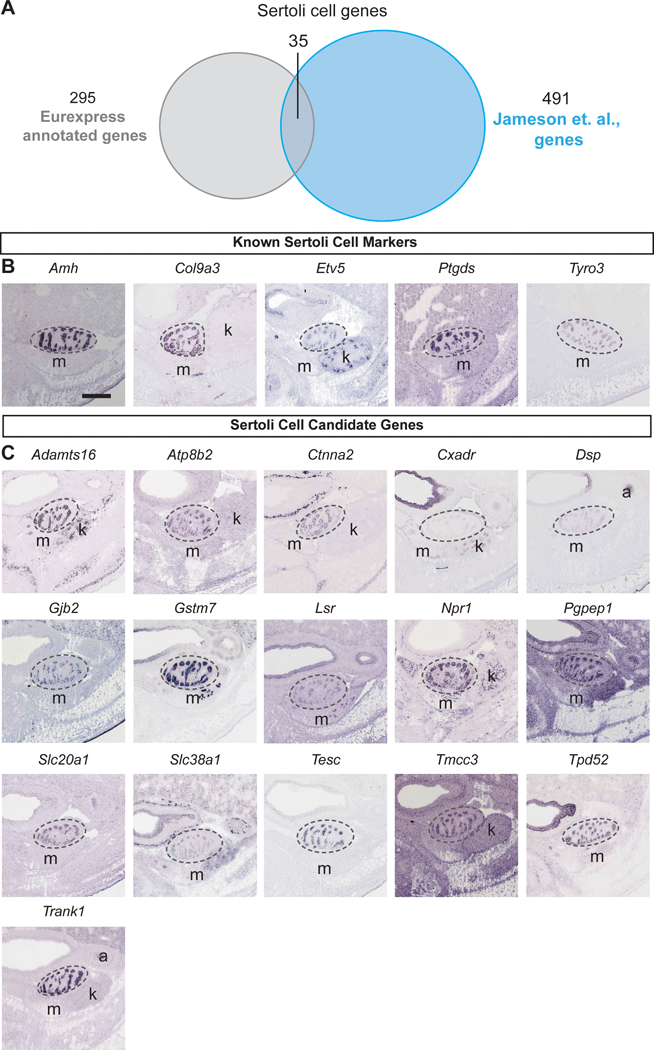
(A) The list of 295 entries annotated as testis-expressed in the Eurexpress database were compared to 491 Sertoli cell expressed genes from a microarray of sorted cells at 13.5 dpc (Jameson et. al., 2012). 35 genes were represented in both sources (see Supp Table 2). (B) Out of these 35 genes, 5 known Sertoli cell marker genes were identified: Amh, Col9a3, Etv5, Ptgds, and Tyro3. 16 novel Sertoli cell candidate genes with a quality ranking of 1 or 2 were annotated in the Eurexppress database: Adamts16, Atp8b2, Ctnna2, Cxadr, Dsp, Gjb2. Gstm7, Lsr, Mpr1, Pgepep1, Slc20a1, Slc38a1, Tesc, Tmcc3, Tpd52a and Trank1. The testis region is demarcated by a dashed line. a: adrenal; k: kidney; m: mesonephros. Scale bar: 0.5 mm.
Fig. 3: Germ cell-expressed genes identified from “testis” annotated entries in the Eurexpress database.
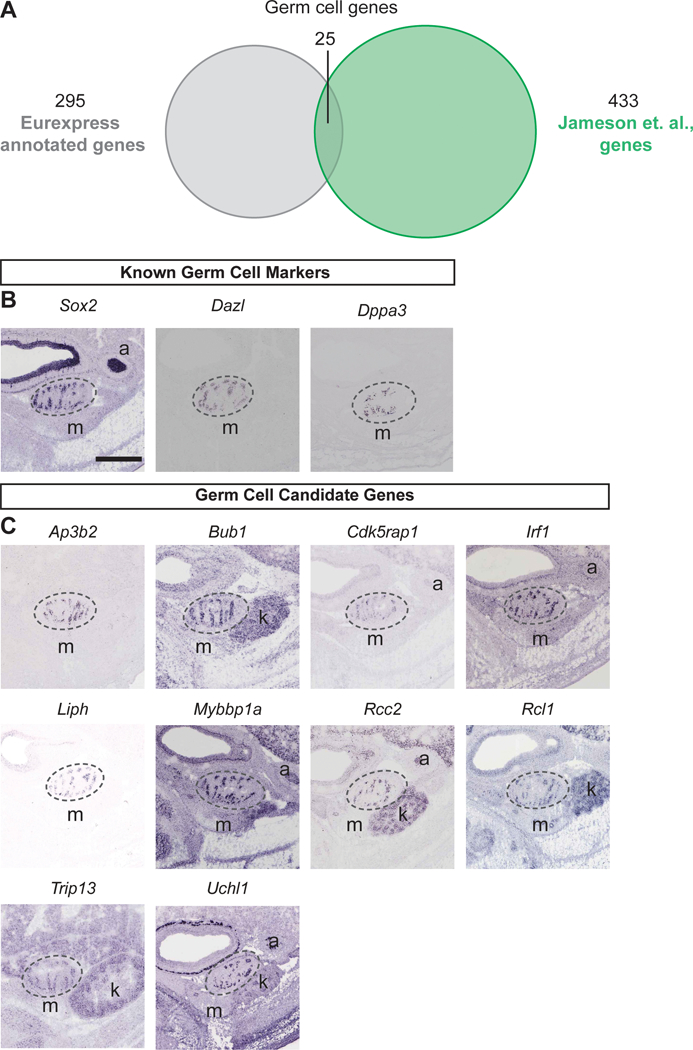
(A) The list of 295 entries annotated as testis-expressed in the Eurexpress database were compared to 433 germ cell-expressed genes from a microarray of sorted cells at 13.5 dpc (Jameson et. al., 2012). 25 genes were represented in both sources (see Supp Table 2). (B) These genes included the known germ cell markers Sox2 and Dppa3. The germ cell marker Dazl was also included as a positive control. In the Eurexpress database, there were 10 novel germ cell genes that were also represented in the microarray: Ap3b2, Bub1, Cdkrap1, Irf1, Liph, Mybbp1a, Rcc2, Rcl1, Trip13 and Uchl1. The testis region is demarcated by a dashed line. a: adrenal; k: kidney; m: mesonephros. Scale bar: 0.5 mm.
The testis interstitium houses heterogeneous populations of cells including vasculature, steroidogenic fetal Leydig cells, and non-steroidogenic interstitial cells. In the Jameson et al microarray [2012], a Mafb-eGFP line was used to isolate both steroidogenic and non-steroidogenic interstitial cells and a Flk-mCherry line was used to isolate endothelial cells. However, expression of steroidogenic Leydig cell genes was found in cells isolated from both these lines. As a result, a mixed interstitial cell list without steroidogenic genes and a steroidogenic Leydig cell list were generated by the authors. Of the 130 mixed interstitial cell genes identified in the Jameson et al microarray [2012], 9 genes were also represented in the Eurexpress database (Fig. 4A). In addition to 4 publication quality in situ hybridization images for known interstitial marker genes (Fig. 4B), we identified images for 5 of the 9 overlapping interstitial cell genes (Fig. 4C). RNA-seq data on sorted non-steroidogenic interstitial cells and fetal Leydig cells at 12.5 dpc suggests that the expression of 3 of these genes (Clca1, Itga9 and Nrg1) may be in fetal Leydig cells [McClelland et al., 2015]. The Jameson et al microarray [2012] produced a list of Leydig cell-specific genes, 11 of which were also represented in the Eurexpress database (Fig. 4D); 8 of them are of publication quality (Fig. 4E).
Fig. 4: Interstitial and fetal Leydig cell-expressed genes identified from “testis” annotated entries in the Eurexpress database.
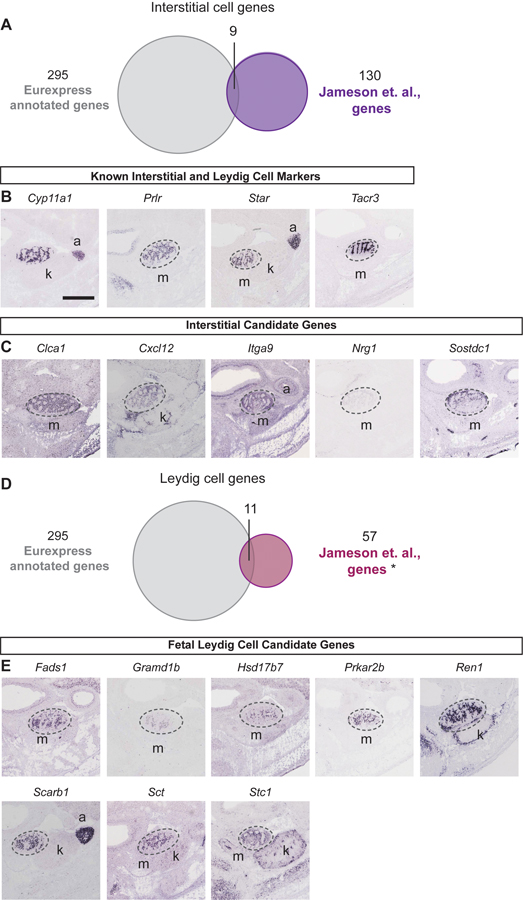
(A) The list of 295 entries annotated as testis-expressed in the Eurexpress database were compared to 130 interstitial cell-expressed genes from a microarray of sorted cells at 13.5 dpc (Jameson et. al., 2012). 9 genes were represented in both sources (see Supp Table 2). (C) Among these genes, 4 known interstitial/Leydig cell marker genes were identified: Cyp11a1, Prlr, Star and Tacr3. (C) high quality in situ hybridization images were available for 5 of the 9 overlapping interstitial genes: Clca1, Cxcl12, Itga9, Nrg1 and Sostdc1. (D) 11 genes were represented in both the Leydig cell microarray and the Eurexpress database (see Supp Table 2); (E) high quality in situ hybridization images were available for 8 genes: Fads1, Gramd1b, Hsd17b7, Prkar2b, Ren1, Scarb1, Sct and Stc1. . The testis region is demarcated by a dashed line. a: adrenal; k: kidney; m: mesonephros. Scale bar: 0.5 mm.
In addition to verifying microarray and other transcriptomic data, the data from expression atlases can be used to identify novel putative markers of the different cell lineages. The resolution of the Eurexpress images is sufficient for characterizing the subdivision of organs or mapping regional differences within structurally complex organs [Diez-Roux et al., 2011; McClelland et al., 2015; Yang, and Chen, 2014]. We subsequently examined the images with a quality score of a 1 or 2 that were not represented in the Jameson et al microarray and categorized them based on testis structures. Inside the testis cords, we found 28 genes with putative Sertoli cell expression and 24 genes with putative expression in germ cells. Twenty-six genes were found in the interstitium, and 9 genes were expressed in the entire testis, and 3 genes were expressed putatively in the vasculature. For the genes expressed in the testis cords, we could not determine with certainty whether the expression was localized to the Sertoli cells or the germ cells. Likewise, interstitium-expressed genes could be expressed in the fetal Leydig cells or in non-steroidogenic interstitial cells.
This list of candidate genes (295 genes) can be considered as a small randomized data set similar to an experiment examining the expression patterns of genes in a 14.5 dpc testis. As a first pass for the following pathway analysis, we used the “Tissue Expression” feature in DAVID to get a quick look at a list of reported expression patterns in different tissues for each gene [Huang et al., 2009a]. Using this tool we were able to interrogate expression patterns of 162 genes and determined that 55 genes had expression reported in the fetal or adult testis (Table S4). We then queried the complete list of genes in DAVID (V6.8Beta) to search for enriched associations among these genes. As expected, because this gene list was randomly assembled, not a complete representation of gene expression at 14.5 dpc and not directly testing a hypothesis, there were few strongly enriched processes or pathways. Subsequently, as an example, we performed pathway enrichment analysis on the list of 289 protein-encoding genes to look for enriched clusters; we identified an overrepresentation of genes involved in Glutathione metabolism (6 genes) and transcriptional regulation (39 genes) in the gene list (Table S5). When looking at a newly generated gene list, an initial submission to DAVID after validating expression of a few genes in the list can provide directions for specific niche searches. Once a pathway or process is identified as being enriched using DAVID, expression of other pathway components can be validated using expression databases and transcriptomic resources. These pathways can then be queried in OMIM to look for association with human disease. For example, from RNA-seq data profiling the interstitial cells of the fetal testis, “neuroactive ligand interaction” is overrepresented by analysis using DAVID. By identifying the receptor/ligand pairs in different testicular cell populations, a putative model could be constructed. Several components of this model had a known association with disorders of sex development [Diez-Roux et al., 2011; McClelland et al., 2015]). By reconstructing the pathways in silico, expression of the gene Frem2/FREM2 (Fras1 related extracellular matrix protein 2) and its family member Fras1 (Fraser syndrome 1 homolog), known DSD genes (OMIM:219000), were identified in the developing gonad [McClelland et al., 2015; Jadeja et al., 2005].
Table S4: “Tissue Expression” feature in DAVID identifies 55 genes with expression annotated in the testis.
162 of the Eurexpress genes had annotated expression in the DAVID “Tissue Expression” tool. Of these 162 genes, 55 had expression documented in the testis. These 55 genes are listed in this table. DAVID was accessed on September 12, 2016
| Anti-Mullerian Hormone(Amh) |
| ADP-Ribosyltransferase 5(Art5) |
| G Protein-Coupled Receptor Kinase 5(Grk5) |
| Glutathione S-Transferase, Mu 5(Gstm5) |
| Homeobox D9(Hoxd9) |
| Hydroxysteroid (17-Beta) Dehydrogenase 7(Hsd17b7) |
| Heat Shock Protein 2(Hspa2) |
| Heat Shock Protein 90, Alpha (Cytosolic), Class A Member 1(Hsp90aa1) |
| Ly1 Antibody Reactive Clone(Lyar) |
| Myeloblastosis Oncogene-Like 1(Mybl1) |
| Phospholipase A2, Group V(Pla2g5) |
| POU Domain, Class 6, Transcription Factor 1(Pou6f1) |
| Prolactin Receptor(Prlr) |
| Secretin(Sct) |
| Scavenger Receptor Class B, Member 1(Scarb1) |
| Steroidogenic Acute Regulatory Protein(Star) |
| Stanniocalcin 1(Stc1) |
| Syntaxin Binding Protein 2(Stxbp2) |
| Testis Expressed Gene 2(Tex2) |
| Testis Expressed Gene 261(Tex261) |
| Utrophin(Utrn) |
| Proteasome (Prosome, Macropain) Subunit, Alpha Type 1(Psma1) |
| Serine Incorporator 3(Serinc3) |
| Coatomer Protein Complex, Subunit Beta 2 (Beta Prime)(Copb2) |
| Apoptosis Antagonizing Transcription Factor(Aatf) |
| Ring Finger Protein 138(Rnf138) |
| Sclerostin Domain Containing 1(Sostdc1) |
| Ring Finger Protein 181(Rnf181) |
| RNA (Guanine-7-) Methyltransferase(Rnmt) |
| Solute Carrier Family 25, Member 39(Slc25a39) |
| PTEN Induced Putative Kinase 1(Pink1) |
| NADH Dehydrogenase (Ubiquinone) 1, Alpha/Beta Subcomplex, 1(Ndufab1) |
| Bobby Sox Homolog (Drosophila)(Bbx) |
| Aldo-Keto Reductase Family 1, Member C-Like(Akr1cl) |
| WD Repeat Domain 31(Wdr31) |
| Solute Carrier Family 25 (Mitochondrial Carrier; Adenine Nucleotide Translocator), Member 31(Slc25a31) |
| Kinesin Family Member 2C(Kif2c) |
| Interferon Induced Transmembrane Protein 7(Ifitm7) |
| Fatty Acid Desaturase 1(Fads1) |
| Inner Membrane Protein, Mitochondrial(Immt) |
| Mitochondrial Translational Initiation Factor 2(Mtif2) |
| Ring Finger Protein 34(Rnf34) |
| Intraflagellar Transport 122(Ift122) |
| Lin-28 Homolog A (C. Elegans)(Lin28a) |
| Calmin(Clmn) |
| Zinc Finger Protein 451(Zfp451) |
| Ets Variant 5(Etv5) |
| Heat Shock Protein 1A(Hspa1a) |
| Calcium/Calmodulin-Dependent Protein Kinase Kinase 2, Beta(Camkk2) |
| Centrosomal Protein 68(Cep68) |
| Basic Transcription Factor 3(Btf3) |
| General Transcription Factor IIB(Gtf2b) |
| GRAM Domain Containing 1B(Gramd1b) |
| Lipase, Member H(Liph) |
| Ring Finger Protein 213(Rnf213) |
Table S5: Example of annotation clustering from DAVID describing two enriched processes, Glutathione metabolism and transcriptional regulation.
Querying the Eurexpress list of genes using the annotation clustering tool in DAVID can define related processes. For example, the processes of Glutathione metabolism and transcriptional regulation were over represented in the Eurexpress gene list. DAVID was accessed on September 12, 2016.
| Category | Term | Count | PValue | List Total | Fold Enrichment |
|---|---|---|---|---|---|
| INTERPRO | IPR003081:Glutathione S-transferase, Mu class | 4 | 9.17E-05 | 275 | 40.58805195 |
| INTERPRO | IPR004046:Glutathione S-transferase, C-terminal | 5 | 0.001125184 | 275 | 10.76198347 |
| INTERPRO | IPR004045:Glutathione S-transferase, N-terminal | 5 | 0.001125184 | 275 | 10.76198347 |
| GOTERM_BP_DIRECT | GO:0018916~nitrobenzene metabolic process | 3 | 0.001306243 | 249 | 50.02710843 |
| GOTERM_MF_DIRECT | GO:0004364~glutathione transferase activity | 5 | 0.001350374 | 250 | 10.20451613 |
| GOTERM_MF_DIRECT | GO:0043295~glutathione binding | 4 | 0.001854759 | 250 | 15.817 |
| KEGG_PATHWAY | mmu00480:Glutathione metabolism | 6 | 0.002019562 | 128 | 6.569318182 |
| INTERPRO | IPR010987:Glutathione S-transferase, C-terminal-like | 5 | 0.003592444 | 275 | 7.892121212 |
| GOTERM_BP_DIRECT | GO:0042178~xenobiotic catabolic process | 3 | 0.004438218 | 249 | 28.5869191 |
| GOTERM_BP_DIRECT | GO:0006749~glutathione metabolic process | 5 | 0.004473115 | 249 | 7.411423472 |
| KEGG_PATHWAY | mmu00980:Metabolism of xenobiotics by cytochrome P450 | 5 | 0.006215115 | 128 | 6.690972222 |
| KEGG_PATHWAY | mmu00982:Drug metabolism - cytochrome P450 | 5 | 0.007815539 | 128 | 6.272786458 |
| UP_SEQ_FEATURE | domain:GST N-terminal | 4 | 0.008489343 | 255 | 9.420130719 |
| UP_SEQ_FEATURE | domain:GST C-terminal | 4 | 0.019907987 | 255 | 6.892778575 |
| KEGG_PATHWAY | mmu05204:Chemical carcinogenesis | 5 | 0.06493113 | 128 | 3.272758152 |
| INTERPRO | IPR012336:Thioredoxin-like fold | 5 | 0.122225117 | 275 | 2.630707071 |
| GOTERM_BP_DIRECT | GO:0008152~metabolic process | 10 | 0.291498366 | 249 | 1.378157257 |
| Annotation Cluster 2 | Enrichment Score: 2.202438644893318 | ||||
| Category | Term | Count | PValue | List Total | Fold Enrichment |
| UP_KEYWORDS | Nucleus | 86 | 2.44E-04 | 281 | 1.432714499 |
| UP_KEYWORDS | Transcription | 42 | 8.85E-04 | 281 | 1.694999913 |
| UP_KEYWORDS | Transcription regulation | 39 | 0.002932861 | 281 | 1.626656414 |
| GOTERM_BP_DIRECT | GO:0006351~transcription, DNA-templated | 40 | 0.012933994 | 249 | 1.466801787 |
| UP_KEYWORDS | DNA-binding | 33 | 0.014036324 | 281 | 1.541673065 |
| GOTERM_MF_DIRECT | GO:0003677~DNA binding | 37 | 0.041367714 | 250 | 1.372971261 |
| GOTERM_BP_DIRECT | GO:0006355~regulation of transcription, DNA-templated | 39 | 0.080401663 | 249 | 1.285281442 |
Tools like DAVID rely on querying what is known about the function of a gene. As a result, although powerful in uncovering functionality, this approach cannot make inferences or uncover unknown functionality. This limitation is illustrated by the example of Pdgfa (platelet-derived growth factor-alpha), which encodes the PDGF-A and is annotated in GO for the molecular function of platelet-derived growth factor receptor binding. However, although it is annotated for the biological processes of lung alveolar development (GO:0048286), salivary gland morphogenesis (GO:0060683) and bone development (GO:0060348; among others), it is not annotated for the biological process of male gonad development (GO:0008584). To a researcher in the field of sex determination and germ cell development, it may be obvious that Pdgfa should be involved in male gonad development, as we know this pathway is important for gonadogenesis [Brennan et al., 2003; Cool et al., 2011; Schmahl et al., 2008]. However without appropriate annotation for this biological process, the program cannot computationally retrieve Pdgfa as associated with testicular development. The tool is only as complete and up-to date as the GO terms it uses.
III. The power of annotation: the case of the missing ovary
If 298 entries are annotated as expressed in the testis in the Eurexpress database, we would assume that the same discovery capacity should be found in querying the ovary. When queried under the anatomy search for “ovary”, only 28 entries were retrieved. These numbers of genes are far less than the numbers of gene catalogued from the microarray data [Jameson et al., 2012; Nef et al., 2005; Liu et al., 2010]. We have discussed that many genes are annotated in several biological processes, yet we know that some biological processes are better studied than others. This should be a consideration when interpreting query results relying on GO terms, as some processes and gene families are more thoroughly annotated than the others largely due to historical reasons or the size of the research field. Similarly, the expression of genes annotated in some organs, for example the ovary, depends on semi-experts being able to accurately identify an organ in a tissue section [Diez-Roux et al., 2011]. In the 298 entries annotated for the testis, 11 (3.7%) genes were annotated incorrectly, where expression was actually in the neighboring pancreas or kidney. In an organ like the ovary, which doesn’t develop apparent distinguishing features like testis cords, it is a lack of annotation in databases that hampers researchers, rather than a lack of expression of ovarian genes.
In some cases, a lack of annotation can become a serious problem that skews results. All GO terms tend to be weighted equally in enrichment tools whereas in reality certain ontologies are better described than others [Huang et al., 2009b]. This is often more of a problem in emerging or smaller fields. A better-described ontology, based on gene families or specific processes where there is more available data, tend to have a higher chance of being associated with any list. The search is only as good as the ontology and the annotation behind it.
Conclusion
We are surrounded by transcriptomic data telling us what genes go up and what genes go down. Thanks to the affordability of transcriptomics, we have more data than we could have ever imaged. Each microarray or RNA-seq experiment produces an extensive list of candidate genes. What we haven’t had is a way to prioritize the genes on these lists for functional analysis. In this review we have provided strategies to narrow down the genes-of-interest list and identify which genes in a candidate list to pursue for further functional analyses. The key frameworks used by databases revolve around standardization of terms; both gene ontology (GO) terms and anatomical descriptions. Understanding how GO term descriptors work and are used by different databases is a critical first step for the researcher. Coupled with an understanding of GO terms, we provide a strategy using 4 types of databases to help researchers prioritize candidate genes. 1) Expression databases can be used to verify the expression of genes of interest in the different organs throughout development. 2) After confirming expression of the genes of interest, investigators can use databases like DAVID and GSEA to uncover interesting processes and pathways present in the candidate gene list. Focused searches that expand out from these pathways can identify candidate genes that may be involved in central processes in the system of interest. 3) Resources such as that EMBL-EBI Expression Atlas that annotate reanalyzed transcriptomic data can provide important information about gene expression patterns across developmental time points, during disease states, and in different species including human. 4) Finally, the importance of a gene in human disease can then be investigated by querying knowledgebases such as OMIM and HGMD. Using this approach a candidate gene list can be whittled down to a selection of promising candidates that fit the investigator’s criteria for strong candidate for functional analysis.
This prioritizing approach requires the investigator to move between multiple databases. Luckily, there are new efforts underway to centralize the data housed by different Consortiums, and to better store and annotate the kind of visual data that developmental biologists generate. The best example of this evolving database and framework is the Edinburgh Mouse Atlas Project (EMAP), which aims to build a 3-D anatomical atlas of mouse embryo development using a flexible anatomical ontology. The ontology uses a thesaurus of alternative anatomical terminologies that was built upon the Kaufman “Atlas of Mouse Development” and allows flexibility within queries. This framework, by enabling visual tracing of progenitors and derivatives for each structure or organ, aims to provide the kind of fate tracing map that are missing in other databases. The structures in the digital model are anchored by key anatomical terms, and the data are then associated with that tissue ‘lineage’ within the spatial framework. This kind of framework provides information about lineages belonging to structures, like an organ, over time. The EMAP database currently links out to, and draws data from Eurexpress, and both EMAP and Eurexpress link out to interface with resources such as MGI.
Ultimately, the community needs these resources to become more centralized. To this end, the MGI Gene Expression Database (GXD) project and the Edinburgh Mouse Atlas (EMA) project are working together to develop a comprehensive resource called the Mouse Gene Expression Information Resource (MGEIR) (for more information see: http://www.emouseatlas.org/emage/about/mgeir.html). MGEIR aims to provide a central resource that will house text-and spatial-based gene expression data. Gene expression data will be acquired from the literature and by direct data submission from external labs/consortia. This resource merges ‘spatial’ in situ hybridization data into the EMAGE’s 3-D format and flexible anatomical framework with the ‘non-spatial’ expression data (i.e. RT-PCR) collected in the GXD [Christiansen et al., 2006; de Boer et al., 2009; Hill et al., 2004; Geffers et al., 2012]. The development of MGIER and the creation of centralized databanks will provide the investigators with an amazing array of information at the click of a mouse.
The next big challenge for expression databases will be the onslaught of single cell RNA-seq data; this new technology offers an unprecedented amount of detail about the transcriptome of any tissue. It’s reasonable to expect that in the next few years, high quality single cell datasets will be available for many organs, including the developing mouse and human gonad. How these datasets are integrated into expression databases will be a challenge for the use of ontologies. The flexible anatomical design, used by databases like EMAP, may provide enough flexibility to trace progenitor cell populations and population derivatives for each tissue or organ (for a detailed overview of current eMouseAtlas capabilities see: [Armit et al., 2015]). How tissue ‘lineages’ are managed within the spatial framework of the expression atlas will provide a challenge for database custodians.
The next time when the investigator is facing a “gene prioritization problem”, we encourage the investigator to step away from the bench and use the tools and databases described here to move their projects and hypotheses forward. Publically available data can be used to efficiently narrow down a candidate gene list to produce strong candidates for functional analysis. Using databases, genes can be selected that have known expression in the organ of interest and are involved in important signaling pathways or biological processes. The genes of interest can represented in other transcriptomic studies or associated with human disease. So, before you decide to a make a genetically modified mouse, sit down at your desk and put the other type of mouse to work.
Table 2: Expression databases and resources that can be used to prioritize candidate genes.
Individual specialty resources such as ABA, GENSAT and GUDMAP provide specialized data and resources to their respective communities. Broader resources looking at embryonic development include Eurexpress and GenePaint. The data from both these resources feeds into the larger EMAP project and the MGI-GXD. The development of the MGEIR database, which is a collaboration between the EMAP and GXD projects, will provide a centralized hub for murine gene expression data.
| Expression databases | Stages/Tissue | Aim and Scope of the Database | URL |
|---|---|---|---|
| ABA (Allen Brain Atlas) |
Embryonic to adult brain and spinal cord |
Database of gene expression patterns in the mouse brain and spinal cord. |
http://brain-map.org/ For mouse: http://mouse.brain-map.org/ |
| GENSAT (Gene Expression Nervous System Atlas) |
- 10.5/15.5 dpc heads - P7, adult brain |
Map the expression of genes in the central nervous system of the mouse, using both in situ hybridization and transgenic mouse techniques. The EGFP BAC-transgenic mice created by this project are available to the scientific community (278 new Cre driver lines). |
http://www.gensat.org/ |
| GUDMAP (GenitoUrinary Development Molecular Anatomy Project) |
- 14.5 dpc whole embryos |
Consortium of laboratories working to provide the scientific and medical community with tools to facilitate research into the GU system. |
http://www.gudmap.org/ |
| FaceBase | - various mouse, human and zebrafish |
Consortium generating data in support of advancing research into craniofacial development and malformation. Comprehensive Includes data from high-throughput genetic, molecular, biological, imaging and computational techniques. |
https://www.facebase.org |
| Eurexpress | - 14.5 dpc whole embryos |
A transcriptome atlas database for mouse embryo. | http://www.eurexpress.org/ee/ |
| GenePaint | - 10.5/14.5 dpc whole embryos - 15.5, P7, adult (P57) brain |
Digital atlas of gene expression profiles in the mouse. | http://www.genepaint.org/ |
| The e-Mouse Atlas Project (EMAP) which includes: EMA (The e-Mouse Atlas) EMAGE (Edinburgh Mouse Atlas of Gene Expression) eHistology (Online Atlas of Mouse Development) |
- 1 cell – 18.5 dpc embryo - P0-P57 |
EMA, the e-Mouse Atlas. A 3-D anatomical atlas of mouse embryo development including detailed histology. EMAGE, the e-Mouse Atlas of Gene Expression. Anatomy is reconstructed from serial sections of single embryos at each representative developmental stage enabling 3D graphical display and analysis of in situ expression data eHistology resource, allows interactive exploration of cellular- resolution color images detailing mouse development with annotations from the Kaufman “Atlas of Mouse Development”. |
EMA: http://www.emouseatlas.org/emap/ema/ EMAGE: http://www.emouseatlas.org/emage/ eHistology: http://www.emouseatlas.org/emap/eHistology/ |
| MGI-GXD (Mouse Genome Informatics Gene Expression Database) |
- 1 cell – 18.5 dpc embryo - adult |
GXD collects and integrates the gene expression information in MGI. Its primary emphasis is on endogenous gene expression during mouse development. It integrates different types of data and provides links to other resources to place the data into the larger biological and analytical context. |
http://www.informatics.jax.org/expression.shtml |
| MGEIR (Mouse Gene Expression Information Resource) |
Pulls data from GXD and EMAP |
MGEIR is a collaboration between the GXD project and the EMAP project. | Under development |
| 3D Atlas of Human Embryo Development |
Morphological data from Carnegie Stage 7–23 |
This resource is a morphological (not an expression) atlas. It has 3D fully reconstructed human embryos covering the phase of organogenesis, between Carnegie stage 7 (15–17 days old embryo) and 23 (56–60 days). |
http://www.3dembryoatlas.com/ |
Acknowledgements
We thank members of the Yao Lab for helpful discussions on the concepts in this paper. The concept for this review was born out of work done by KSM during her time in the Koopman Lab, University of Queensland, Australia. All work contributing to this review was completed in the Yao Lab, NIEHS/NIH, USA. KSM would like to thank Peter Koopman and Josephine Bowles for their encouragement and support. This research was supported by the Intramural Research Program (ES102965 to HHCY) of the NIH, National Institute of Environmental Health Sciences. The in situ hybridization data in this review is drawn exclusively from the Eurexpress Transcriptome Atlas Database in May 2016.
References
- Ono M, Harley VR: Disorders of sex development: new genes, new concepts. Nat Rev Endocrinol 9:79–91 (2013). [DOI] [PubMed] [Google Scholar]
- Jameson SA, Natarajan A, Cool J, Defalco T, Maatouk DM, Mork L, et al. : Temporal transcriptional profiling of somatic and germ cells reveals biased lineage priming of sexual fate in the fetal mouse gonad. PLoS Genet 8:e1002575 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClelland KS, Bell K, Larney C, Harley VR, Sinclair AH, Oshlack A, et al. : Purification and Transcriptomic Analysis of Mouse Fetal Leydig Cells Reveals Candidate Genes for Specification of Gonadal Steroidogenic Cells. Biol Reprod 92:145–145 (2015). [DOI] [PubMed] [Google Scholar]
- Beverdam A, Koopman P: Expression profiling of purified mouse gonadal somatic cells during the critical time window of sex determination reveals novel candidate genes for human sexual dysgenesis syndromes. Hum Mol Genet 15:417–431 (2006). [DOI] [PubMed] [Google Scholar]
- Nef S, Schaad O, Stallings NR, Cederroth CR, Pitetti JL, Schaer G, et al. : Gene expression during sex determination reveals a robust female genetic program at the onset of ovarian development. Dev Biol 287:361–377 (2005). [DOI] [PubMed] [Google Scholar]
- Bouma GJ, Hudson QJ, Washburn LL, Eicher EM: New Candidate Genes Identified for Controlling Mouse Gonadal Sex Determination and the Early Stages of Granulosa and Sertoli Cell Differentiation. Biol Reprod 82:380–389 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouma GJ, Affourtit JP, Bult CJ, Eicher EM: Transcriptional profile of mouse pre-granulosa and Sertoli cells isolated from early-differentiated fetal gonads. Gene Expr Patterns 7:113–123 (2007). [DOI] [PubMed] [Google Scholar]
- Albrecht KH, Eicher EM: Evidence that Sry is expressed in pre-Sertoli cells and Sertoli and granulosa cells have a common precursor. Dev Biol 240:92–107 (2001). [DOI] [PubMed] [Google Scholar]
- Rolland AD, Lehmann KP, Johnson KJ, Gaido KW, Koopman P: Uncovering Gene Regulatory Networks During Mouse Fetal Germ Cell Development. Biol Reprod 84:790–800 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coveney D, Ross AJ, Slone JD, Capel B: A microarray analysis of the XX Wnt4 mutant gonad targeted at the identification of genes involved in testis vascular differentiation. Gene Expr Patterns 8:529–537 (2008). [DOI] [PubMed] [Google Scholar]
- Munger SC, Natarajan A, Looger LL, Ohler U, Capel B: Fine time course expression analysis identifies cascades of activation and repression and maps a putative regulator of mammalian sex determination. PLoS Genet 9:e1003630 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertz K, Herrmann BG: Large-scale screen for genes involved in gonad development. Mech Dev 98:51–70 (2000). [DOI] [PubMed] [Google Scholar]
- Bowles J, Bullejos M, Koopman P: A subtractive gene expression screen suggests a role for vanin‐1 in testis development in mice. Genesis 27:124–135 (2000). [PubMed] [Google Scholar]
- McClive PJ, Hurley TM, Sarraj MA, van den Bergen JA, Sinclair AH: Subtractive hybridisation screen identifies sexually dimorphic gene expression in the embryonic mouse gonad. Genesis 37:84–90 (2003). [DOI] [PubMed] [Google Scholar]
- Menke DB, Page DC: Sexually dimorphic gene expression in the developing mouse gonad. Gene Expr Patterns 2:359–367 (2002). [DOI] [PubMed] [Google Scholar]
- Grimmond S, Van Hateren N, Siggers P, Arkell R, Larder R, Soares MB, et al. : Sexually dimorphic expression of protease nexin-1 and vanin-1 in the developing mouse gonad prior to overt differentiation suggests a role in mammalian sexual development. Hum Mol Genet 9:1553–1560 (2000). [DOI] [PubMed] [Google Scholar]
- Munger SC, Aylor DL, Syed HA, Magwene PM, Threadgill DW, Capel B: Elucidation of the transcription network governing mammalian sex determination by exploiting strain-specific susceptibility to sex reversal. Genes Dev 23:2521–2536 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Shima Y, Miyabayashi K, Tokunaga K, Sato T, Baba T, et al. : Isolation and characterization of fetal Leydig progenitor cells of male mice. Endocrinology :en20151773 (2015). [DOI] [PubMed]
- Lindeman RE, Gearhart MD, Minkina A, Krentz AD, Bardwell VJ, Zarkower D: Sexual cell-fate reprogramming in the ovary by DMRT1. Curr Biol 25:764–771 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrer H: Disorders of sex development (DSDs): an update. J Clin Endocrinol Metab 99:1503–1509 (2014). [DOI] [PubMed] [Google Scholar]
- Bouma GJ, Hart GT, Washburn LL, Recknagel AK, Eicher EM: Using real time RT-PCR analysis to determine multiple gene expression patterns during XX and XY mouse fetal gonad development. Gene Expr Patterns 5:141–149 (2004). [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. : Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25:25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake JA, Eppig JT, Richardson JE, Davisson MT: The Mouse Genome Database (MGD): expanding genetic and genomic resources for the laboratory mouse. The Mouse Genome Database Group. Nucleic Acids Res 28:108–111 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringwald M, Eppig JT, Kadin JA, Richardson JE: GXD: a Gene Expression Database for the laboratory mouse: current status and recent enhancements. The Gene Expresison Database group. Nucleic Acids Res 28:115–119 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball CA, Dolinski K, Dwight SS, Harris MA, Issel-Tarver L, Kasarskis A, et al. : Integrating functional genomic information into the Saccharomyces genome database. Nucleic Acids Res 28:77–80 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium TF: The FlyBase database of the Drosophila Genome Projects and community literature. -PubMed -NCBI. Nucleic Acids Res (1999). [DOI] [PMC free article] [PubMed]
- Huang DW, Sherman BT, Lempicki RA: Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57 (2009a). [DOI] [PubMed] [Google Scholar]
- Geffers L, Herrmann B, Eichele G: Web-based digital gene expression atlases for the mouse. Mamm Genome 23:525–538 (2012). [DOI] [PubMed] [Google Scholar]
- Reymond A, Marigo V, Yaylaoglu MB, Leoni A, Ucla C, Scamuffa N, et al. : Human chromosome 21 gene expression atlas in the mouse. Nature 420:582–586 (2002). [DOI] [PubMed] [Google Scholar]
- Visel A, Thaller C, Eichele G: GenePaint.org: an atlas of gene expression patterns in the mouse embryo. Nucleic Acids Res 32:D552–6 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitton Y, Dahmane N, Baik S, Altaba ARI, Neidhardt L, Scholze M, et al. : A gene expression map of human chromosome 21 orthologues in the mouse. Nature 420:586–590 (2002). [DOI] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, et al. : Genome-wide atlas of gene expression in the adult mouse brain. Nature 445:168–176 (2007). [DOI] [PubMed] [Google Scholar]
- Jones AR, Overly CC, Sunkin SM: The Allen Brain Atlas: 5 years and beyond. Nat Rev Neurosci 10:821–828 (2009). [DOI] [PubMed] [Google Scholar]
- Diez-Roux G, Banfi S, Sultan M, Geffers L, Anand S, Rozado D, et al. : A high-resolution anatomical atlas of the transcriptome in the mouse embryo. PLoS Biol 9:e1000582 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bakker BS, de Jong KH, Hagoort J, de Bree K, Besselink CT, de Kanter FEC, et al. : An interactive three-dimensional digital atlas and quantitative database of human development. Science 354:aag0053–aag0053 (2016). [DOI] [PubMed] [Google Scholar]
- Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, et al. : A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature 425:917–925 (2003). [DOI] [PubMed] [Google Scholar]
- Brunskill EW, Aronow BJ, Georgas K, Rumballe B, Valerius MT, Aronow J, et al. : Atlas of gene expression in the developing kidney at microanatomic resolution. Dev Cell 15:781–791 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Otterloo E, Williams T, Artinger KB: The old and new face of craniofacial research: How animal models inform human craniofacial genetic and clinical data. Dev Biol 415:171–187 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill DP, Begley DA, Finger JH, Hayamizu TF, McCright IJ, Smith CM, et al. : The mouse Gene Expression Database (GXD): updates and enhancements. Nucleic Acids Res 32:D568–71 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen JH, Yang Y, Venkataraman S, Richardson L, Stevenson P, Burton N, et al. : EMAGE: a spatial database of gene expression patterns during mouse embryo development. Nucleic Acids Res 34:D637–41 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayamizu TF, Baldock RA, Ringwald M: Mouse anatomy ontologies: enhancements and tools for exploring and integrating biomedical data. Mamm Genome 26:422–430 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringwald M, Baldock R, Bard J, Kaufman M, Eppig JT, Richardson JE, et al. : A database for mouse development. Science 265:2033–2034 (1994). [DOI] [PubMed] [Google Scholar]
- Kapushesky M, Emam I, Holloway E, Kurnosov P, Zorin A, Malone J, et al. : Gene Expression Atlas at the European Bioinformatics Institute. Nucleic Acids Res 38:gkp936–D698 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA: Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37:1–13 (2009b). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri P, Sirota M, Butte AJ: Ten Years of Pathway Analysis: Current Approaches and Outstanding Challenges. PLOS Comput Biol 8:e1002375 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, et al. : DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res 35:W169–75 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. : Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. PNAS 102:15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Lu J, Kulbokas EJ, Golub TR, Mootha V, Lindblad-Toh K, et al. : Systematic discovery of regulatory motifs in human promoters and 3’ UTRs by comparison of several mammals. Nature 434:338–345 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V, Fricke E, Geffers R, Gössling E, Haubrock M, Hehl R, et al. : TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic Acids Res 31:374–378 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingender E, Chen X, Hehl R, Karas H, Liebich I, Matys V, et al. : TRANSFAC: an integrated system for gene expression regulation. Nucleic Acids Res 28:316–319 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA: Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37:1–13 (2009c). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Yao HH: Diverse functions of Hedgehog signaling in formation and physiology of steroidogenic organs. Mol Reprod Dev 77:489–496 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley A, Anastassiadis K, Ayadi A, Battey JF, Bell C, Birling M-C, et al. : The mammalian gene function resource: the International Knockout Mouse Consortium. Mamm Genome 23:580–586 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen B, Schick J, Wurst W: Beyond knockouts: the International Knockout Mouse Consortium delivers modular and evolving tools for investigating mammalian genes. Mamm Genome 26:456–466 (2015). [DOI] [PubMed] [Google Scholar]
- Ring N, Meehan TF, Blake A, Brown J, Chen C- K, Conte N, et al. : A mouse informatics platform for phenotypic and translational discovery. Mamm Genome 26:413–421 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eppig JT, Motenko H, Richardson JE, Richards-Smith B, Smith CL: The International Mouse Strain Resource (IMSR): cataloging worldwide mouse and ES cell line resources. Mamm Genome 26:448–455 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eppig JT, Strivens M: Finding a mouse: the International Mouse Strain Resource (IMSR). Trends Genet 15:81–82 (1999). [DOI] [PubMed] [Google Scholar]
- Adams D, Baldock R, Bhattacharya S, Copp AJ, Dickinson M, Greene NDE, et al. : Bloomsbury report on mouse embryo phenotyping: recommendations from the IMPC workshop on embryonic lethal screening. Dis Model Mech 6:571–579 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohun T, Adams DJ, Baldock R, Bhattacharya S, Copp AJ, Hemberger M, et al. : Deciphering the Mechanisms of Developmental Disorders (DMDD): a new programme for phenotyping embryonic lethal mice. Dis Model Mech 6:562–566 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson R, McGuire C, Mohun T, DMDD Project: Deciphering the mechanisms of developmental disorders: phenotype analysis of embryos from mutant mouse lines. Nucleic Acids Res 44:D855–61 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amberger J, Bocchini C, Hamosh A: A new face and new challenges for Online Mendelian Inheritance in Man (OMIM®). Hum Mutat 32:564–567 (2011). [DOI] [PubMed] [Google Scholar]
- Amberger J, Bocchini CA, Scott AF, Hamosh A: McKusick’s Online Mendelian Inheritance in Man (OMIM). Nucleic Acids Res 37:D793–6 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA: Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res 33:D514–7 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson PD, Ball E, Howells K, Phillips A, Mort M, Cooper DN: Human Gene Mutation Database: towards a comprehensive central mutation database. J Med Genet 45:124–126 (2008). [DOI] [PubMed] [Google Scholar]
- Cooper DN, Stenson PD, Chuzhanova NA: The Human Gene Mutation Database (HGMD) and its exploitation in the study of mutational mechanisms. Curr Protoc Bioinformatics Chapter 1:Unit 1.13–1.13.20 (2006). [DOI] [PubMed]
- Stenson PD, Mort M, Ball EV, Howells K, Phillips AD, Thomas NS, et al. : The Human Gene Mutation Database: 2008 update. Genome Med 1:13 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Chen J: Developmental programs of lung epithelial progenitors: a balanced progenitor model. Wiley interdisciplinary reviews Developmental biology 3:331–347 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadeja S, Smyth I, Pitera JE, Taylor MS, van Haelst M, Bentley E, et al. : Identification of a new gene mutated in Fraser syndrome and mouse myelencephalic blebs. Nat Genet 37:520–525 (2005). [DOI] [PubMed] [Google Scholar]
- Brennan J, Tilmann C, Capel B: Pdgfr-α mediates testis cord organization and fetal Leydig cell development in the XY gonad. Genes Dev 17:800–810 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cool J, DeFalco TJ, Capel B: Vascular-mesenchymal cross-talk through Vegf and Pdgf drives organ patterning. Proc Natl Acad Sci U S A 108:167–172 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmahl J, Rizzolo K, Soriano P: The PDGF signaling pathway controls multiple steroid-producing lineages. Genes Dev 22:3255–3267 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CF, Liu C, Yao HH: Building pathways for ovary organogenesis in the mouse embryo. Curr Top Dev Biol 90:263–290 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer BA, Ruijter JM, Voorbraak FPJM, Moorman AFM: More than a decade of developmental gene expression atlases: where are we now? Nucleic Acids Res 37:7349–7359 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armit C, Richardson L, Hill B, Yang Y, Baldock RA: eMouseAtlas informatics: embryo atlas and gene expression database. Mamm Genome 26:431–440 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]