

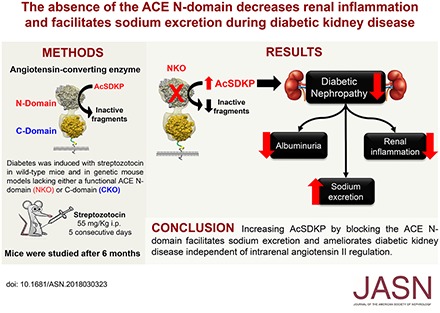
Abstract
Background
Recent evidence emphasizes the critical role of inflammation in the development of diabetic nephropathy. Angiotensin-converting enzyme (ACE) plays an active role in regulating the renal inflammatory response associated with diabetes. Studies have also shown that ACE has roles in inflammation and the immune response that are independent of angiotensin II. ACE’s two catalytically independent domains, the N- and C-domains, can process a variety of substrates other than angiotensin I.
Methods
To examine the relative contributions of each ACE domain to the sodium retentive state, renal inflammation, and renal injury associated with diabetic kidney disease, we used streptozotocin to induce diabetes in wild-type mice and in genetic mouse models lacking either a functional ACE N-domain (NKO mice) or C-domain (CKO mice).
Results
In response to a saline challenge, diabetic NKO mice excreted 32% more urinary sodium compared with diabetic wild-type or CKO mice. Diabetic NKO mice also exhibited 55% less renal epithelial sodium channel cleavage (a marker of channel activity), 55% less renal IL-1β, 53% less renal TNF-α, and 53% less albuminuria than diabetic wild-type mice. This protective phenotype was not associated with changes in renal angiotensin II levels. Further, we present evidence that the anti-inflammatory tetrapeptide N-acetyl-seryl-asparyl-lysyl-proline (AcSDKP), an ACE N-domain–specific substrate that accumulates in the urine of NKO mice, mediates the beneficial effects observed in the NKO.
Conclusions
These data indicate that increasing AcSDKP by blocking the ACE N-domain facilitates sodium excretion and ameliorates diabetic kidney disease independent of intrarenal angiotensin II regulation.
Keywords: angiotensin II, AcSDKP, diabetic nephropathy, Angiotensin-converting enzyme, sodium transporters, ENaC cleavage
Visual Abstract
Diabetic kidney disease is characterized by hypertension, albuminuria, and a progressive decline in renal function leading to ESRD.1 Recent studies indicate a critical role of inflammation in the development of diabetic kidney injury.2 This inflammatory response leads to defective sodium handling and improper renal compensatory mechanisms that contribute to hypertension3,4 which, in turn, further damages the kidney and worsens the progression of diabetic nephropathy.5 At present, the molecular mechanisms explaining sodium retention of the diabetic nephron remain poorly understood.
Patients and animal models with both type 1 and type 2 diabetes retain sodium and have increased total exchangeable body sodium.6,7 Angiotensin II is a major player in the establishment of a sodium retentive state because it activates key sodium transporters in the kidney.8–10 However, the absence of systemic renin-angiotensin system (RAS) activation in diabetes,11,12 together with studies showing that whole-kidney angiotensin II levels can increase, decrease, or remain unchanged during the progression of diabetic nephropathy,13–19 suggest that other mechanisms might also be involved in controlling sodium homeostasis during diabetes.
Modern studies have broadened the known roles of the RAS by showing that angiotensin-converting enzyme (ACE) has angiotensin II independent roles in inflammation and the immune response.20–22 ACE is a dipeptidyl carboxypeptidase with two similar, but not identical, catalytic domains—the N-terminal and the C-terminal domains—that can process a broad diversity of substrates besides angiotensin I. The repertoire of ACE substrates varies from tripeptides to peptides of 42 amino acids.22 However, the kinetics of enzymatic activity also differ for each domain. For example, the conversion of Ang I to Ang II is predominately catalyzed by the C-domain, whereas the degradation of the tetrapeptide N-acetyl-seryl-asparyl-lysyl-proline (AcSDKP) is mediated almost exclusively by the N-domain catalytic site.22 Indeed, substantial evidence indicates that the ACE N-domain is the primary enzyme responsible for the degradation of AcSDKP in vivo because mice lacking a functional ACE N-domain show an increase of AcSDKP in plasma and urine.22,23
AcSDKP is produced from the precursor protein thymosin β4, a G-actin–sequestering peptide that maintains the pool of monomeric actin within cells, and it is highly expressed in higher eukaryotes. The production of thymosin β4-derived AcSDKP involves successive enzymatic hydrolysis mediated by meprin-α and prolyl oligopeptidase (POP).24 AcSDKP has gained attention due to its anti-inflammatory and antifibrotic effects in vivo.23,25 Several studies have demonstrated that chronic administration of AcSDKP prevents kidney fibrosis and improves renal insufficiency in rodent models of diabetes.26–29 Indeed, a recent study shows that mice lacking thymosin β4 have lower levels of renal AcSDKP and show an exacerbated kidney injury in response to angiotensin II infusion.30 However, whether this tetrapeptide has a role in the abnormal sodium handling observed during diabetic nephropathy is unknown. In this study, we induced diabetes with streptozotocin (STZ) in wild-type (WT) mice and in genetic mouse models lacking either a functional ACE N- (NKO) or C-domain (CKO). We found that NKO mice displayed an enhanced natriuretic response to saline loading, less epithelial sodium channel (ENaC) subunit cleavage (a marker of activation), and less amiloride-sensitive sodium excretion compared with diabetic WT or CKO mice. The improved sodium excretion was independent of renal angiotensin II variations, and it was associated with a substantial decrease in renal inflammatory cytokines such as IL-1β and TNF-α, less proteinuria, and less renal damage. In addition, we observed that blocking the production of AcSDKP eliminates the protective effects observed in diabetic NKO mice. These findings indicate that blocking the ACE N-domain improves diabetic kidney disease through a mechanism that involves renal accumulation of AcSDKP and it is independent of changes in renal angiotensin II levels.
Methods
Animal Models
Mice harboring point mutations that inactivate either the amino- (NKO) or the carboxy- (CKO) terminal catalytic domain of ACE were generated as described previously.31,32 Briefly, NKO mice express a full-length ACE protein in which site-directed mutagenesis was used to change histidines 395 and 399 to encode for lysine. These specific amino acid changes inactivate the enzymatic activity of the ACE N-terminal catalytic site by mutating the two zinc-binding histidines. A similar approach was used to create the CKO strain. In these mice, histidines 993 and 997, responsible for the zinc-binding and catalytic activity of the C-terminal enzymatic site, were mutated to encode for lysine. The C- and N-domain–specific substrates—Hip-His-Leu and N-acetyl-Ser-Asp-acetyl-Lys-Pro, respectively— were used to evaluate the separate activity of each ACE domain.31,32 The genetic background of NKO, CKO, and control WT mice used in this study is C57BL/6J.
Diabetic Model Induced by STZ
All protocols using animals were approved by the Cedars-Sinai Institutional Animal Care and Use Committee and conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Eight- to 12-week-old male NKO, CKO, or WT littermates were used. Diabetes was induced by five consecutive daily intraperitoneal (i.p.) injections of STZ (Sigma, St. Louis, MO; 55 mg/kg, freshly prepared in 0.05 M sodium citrate buffer, pH 4.5) after a 6-hour fast.33 Nondiabetic groups received citrate buffer. A week after the last STZ injection, blood glucose levels were measured, and mice that failed to reach hyperglycemia (>200 mg/dl) were excluded from the study. Because of the relatively mild diabetic nephropathy observed in C57BL/6J mice, all experimental groups were followed for 6 months to assure the development of significant diabetic kidney injury. To evaluate salt sensitivity, a subgroup of diabetic WT mice was exposed to a high sodium diet (4% w/w NaCl in the food; Harlan Laboratories, Placentia, CA) for 3 weeks starting 5 months after the onset of diabetes. Blood glucose levels were monitored using the Contour Blood Glucose Monitoring System (Bayer HealthCare LLC, Tarrytown, NY). Plasma aldosterone was assessed using a commercial ELISA kit (Cayman Chemical, Ann Arbor, MI). Systolic BP was measured in previously trained conscious mice using the tail-cuff method (Visitech BP2000 system; Visitech Systems Inc., Apex, NC). BP was measured 20 times for each time point. Twenty-four-hour food and water intake as well as urine and sodium excretion were assessed using metabolic cages. Urinary albumin concentration was measured by ELISA (Albuwell M; Exocell, Philadelphia, PA). Creatinine was assessed by an enzymatic method (Creatinine LiquiColor Test; Stanbio, Boerne, TX). Urinary prostasin and furin were assessed by western blot in 24-hour urine samples. For this, urine samples were concentrated five-fold for nondiabetic and 20-fold for diabetic animals using centrifugal filters with a molecular mass cut-off of 10 kD (Amicon Ultra 10 K Centrifugal Filter Devices; Merck Millipore Ltd.). An amount of concentrated urine equivalent to 10 µg creatinine was denatured, resolved, transferred into polyvinylidene difluoride (PVDF) membranes (Millipore Immobilon-FL; EMD Millipore, Billerica, MA), and then probed with specific antibodies against prostasin and furin. After washing, membranes were incubated with the appropriate fluorochrome-labeled secondary antibody. Supplemental Table 1 catalogs the amounts assayed, vendors, and dilutions for each antibody. Urinary sodium and potassium were determined by flame photometry (model 2655–10; Cole-Parmer). Urinary AcSDKP was measured using an ELISA commercial kit (SPI-BIO Inc., Massy, France), as described previously.34
To inhibit the release of AcSDKP from its precursor, thymosin β4, an additional group of NKO and WT diabetic was were treated with the POP inhibitor S17092 (a generous gift from Servier, Suresnes, France). S17092 was administrated daily by i.p. injection at a dose of 10 mg/kg dissolved in 100 µl of a mixture of 30% DMSO and 70% PBS prepared every day. Nontreated mice received 100 µl of vehicle. S17092 administration began after 5 months of diabetes and continued for 1 month, until the animals were euthanized after 6 months of diabetes. Kidneys were removed and preserved for further analysis.
Saline Challenge
The diuretic and natriuretic responses to extracellular fluid volume expansion were examined as previously described.35 Briefly, mice were acutely anesthetized with isoflurane, injected i.p. with a volume of 37°C 0.9% NaCl equivalent to 10% of their body weight, and placed immediately in metabolic cages without food. Urine was collected hourly over 5 hours. Excretion results were expressed as the percentage of the sodium and volume load injected. After the saline challenge, mice were allowed a 2-day recovery period before euthanasia.
GFR Measurements
GFR was determined in conscious mice using a transcutaneous detector (NIC-Kidney; MediBeacon GmbH, Mannheim, Germany) by monitoring fluorescent intensity for 90 minutes after a single intravenous bolus of FITC-sinistrin (15 mg/100 g body wt; Fresenius Kabi Austria GmbH, Linz, Austria).36–39 To determine GFR, the t1/2 time of FITC-sinistrin was calculated using a three-compartment model according to the manufacturer’s instructions.38
Measurement of Renal Cytokines and Intrarenal RAS Components
For the measurement of renal cytokines, half of the snap-frozen left kidney was homogenized in Radioimmunoprecipitation assay buffer containing 1× protease inhibitor cocktail (Roche, Indianapolis, IN), 1 mM PMSF, 10 μg/ml pepstain A, and 5 mM disodium EDTA. After centrifugation at 19,000 × g for 30 minutes at 4°C, the supernatant was collected and the protein concentration was determined using a bicinchoninic acid protein assay. IL-1β, TNF-α, IL-6, and TGF-β were measured using ELISA kits in whole-kidney lysate (eBioscience, San Diego, CA) and expressed as picograms of cytokine per milligram of kidney protein. Renal ACE and renin expression were assessed by western blot. Briefly, RIPA homogenates were denatured, resolved, transferred into PVDF membranes, and then probed with specific antibodies against ACE and renin (Supplemental Table 1). Renal angiotensin II was determined with an enzyme immunoassay (Peninsula Laboratories International Inc., San Carlos, CA). For this, half of the snap-frozen left kidney was weighed and homogenized in ice-cold methanol. After centrifugation at 12,000 × g for 10 minutes at 4°C, the collected supernatant was dried by centrifugal evaporation. Dried pellets were rehydrated with the buffer provided by the kit. Angiotensin II was measured following manufacturer’s instruction as previously described and expressed as femtomoles of angiotensin II per gram of wet kidney.36
Sodium Transporter Analysis
Half of the snap-frozen right kidney was homogenized in a 5% sorbitol containing 0.5 mM disodium EDTA, 5 mM histidine-imidazole buffer, 0.2 mM PMSF, 9 μg/ml aprotinin, and 5 μl/ml of a phosphatase inhibitor cocktail (P2850; Sigma-Aldrich), pH 7.5. Kidney extracts were denatured, resolved, transferred into PVDF membranes, and then probed with specific antibodies against Na+/H+ exchanger isoform 3 (NHE3), Na+-K+-2Cl− cotransporter isoform 2 (NKCC2), NKCC2 phosphorylated at Thr 96 and 101, NaCl cotransporter (NCC), NCC phosphorylated at Ser 71, and α, β, and γ subunits of the ENaC. β-actin was measured to verify uniform protein loading (Supplemental Table 1). Immunoblots were scanned with the Odyssey Infrared Imaging System and quantitated with the Image Studio Lite Ver 5.2 (Li-COR, Lincoln, NE). Values were normalized to the mean intensity of the nondiabetic WT group defined as 1.0.
To estimate the in vivo activity of ENaC, we performed an amiloride test as described before.40 Briefly, the urinary bladder was emptied by abdominal massage; mice were injected with 100 µl saline and placed in metabolic cages. Four hours after injection, the urinary bladder was emptied again and urine was combined with the urine sampled in the urine collector of the metabolic cage. The 4-hour urine collection was analyzed for sodium, potassium, and creatinine as described above. This measurement was considered baseline sodium and potassium excretion. Two days later, the same procedure was performed with an i.p. injection of amiloride (5 µg/g body wt in 100 µl 0.9% NaCl). The difference between baseline- and amiloride-induced sodium and potassium excretion was calculated for each mouse and considered a surrogate of ENaC activity.
Histologic Examination
Half of the right kidney was fixed with 10% buffered formalin and embedded in paraffin. For the assessment of tubulointerstitial fibrosis in the cortex, 4-μm-thick sections of renal tissues were deparaffinized, rehydrated, and stained with Masson’s trichrome. The analysis of mesangial expansion was performed using periodic acid–Schiff staining. Whole-slide scanning at ×40 magnification was performed by Aperio ScanScope AT Turbo (Leica Biosystems, Wetzlar, Germany). Total cortical area and fibrotic area from a whole-slide image were measured using Aperio ImageScope software. For immunohistochemical analysis, 4-μm-thick sections of paraffin-embedded renal tissues were stained for IL-1β, IL-6, TNF-α, TGF-β, the endothelium marker CD31, and the macrophage marker F4/80 as previously described.41 Briefly, after deparaffinization and rehydration, renal sections were treated for antigen retrieval using the Protease XXIV kit (BioGenex, Fremont, CA). Quenching of endogenous peroxidase activity was achieved by incubating the sections for 60 minutes in 30% hydrogen peroxide in methanol. After washing them with distilled water, sections were incubated with the avidin/biotin blocking kit (BioGenex, Fremont, CA) after an incubation with normal horse serum during 30 minutes (Abcam, Cambridge, MA). After that, sections were incubated with background block (Cell Marque, Rocklin, CA) for 10 minutes and then incubated with the corresponding primary antibody overnight at 4°C. Supplemental Table 2 catalogs the amounts assayed, vendors, and dilutions for each antibody. Sections were then incubated for 30 minutes at room temperature with the secondary antibody according to the primary antibody host (Cell Marque or BioGenex). A horseradish peroxidase detection system was used for diaminobenzydine color development (Cell Marque). Samples were counterstained with hematoxylin. Immunohistochemical analysis was performed in each animal on 20 consecutive microscopic fields at ×40 magnification using a light microscope, Nikon E400 (Nikon Instrument Group, Melville, NY). The immunolocalization of renal IL-1β, TNF-α, IL-6, and TGF-β was expressed as percentage of positive staining per area. Macrophage infiltration was expressed as total number of F4/80-positive cells per area. The immunolocalization of CD31 was expressed as percentage of positive staining per glomerulus by evaluating 20 glomeruli per sample.
Primary Culture of Renal Tubular Epithelial Cells
Primary tubular cells were cultured from collagenase-digested cortical fragments of kidneys isolated from WT, NKO, and CKO 4-week-old male mice as described before.42 Briefly, renal cortices were dissected in ice-cold dissection solution (RPMI with 15 mmol/L HEPES, 1% [v/v] BSA, pH 7.4, and osmolality 300 mosmol/kg H2O) and sliced into 1-mm pieces. The fragments were digested with dissection solution containing 0.1% (w/v) type-2 collagenase and 100 µg/ml soybean trypsin inhibitor at 37°C for 40 minutes. After digestion, the supernatant was sieved through two nylon sieves (pore size 250 and 70 µm). Tubular fragments retained in the 70-µm sieve were resuspended in culture media (DMEM, 5 mM glucose supplemented with 1% FBS, 15 mmol/l HEPES, 2 mmol/L L-glutamine, 50 nmol/L hydrocortisone, 5 µg/ml insulin, 5 µg/ml transferrin, 50 nmol/L selenium, 0.55 mmol/L sodium pyruvate, nonessential amino acids, 100 IU/ml penicillin, and 100 µg/ml streptomycin buffered to pH 7.4 and osmolality adjusted to 300 mosmol/kg H2O with NaCl), seeded onto collagen-coated six-well plates, and left unstirred for 48 hours at 37°C and 5% CO2 in a humidified incubator. The culture media was replaced every 2 days. After 7 days, cells were organized as a confluent monolayer and were stimulated with culture media containing 30 mM glucose and 300 mosmol/kg H2O with or without lisinopril (10 μM), losartan (100 μM), S17092 (50 µM), or AcSDKP (10−10 to 10−6 M) according to protocol. Multiple inflammatory markers were screened in the culture media using a Membrane-Based Array Kit (RayBiotech, Norcross, GA). IL-1β levels were measured using an ELISA kit. To determine the intracellular levels of AcSDKP, tubular cells were homogenized in RIPA buffer containing 0.5 mM disodium EDTA and 1 μM lisinopril. After centrifugation at 20,000 × g for 15 minutes at 4°C, a volume of the supernatant was added to ice-cold methanol and centrifuged at 12,000 × g for 10 minutes at 4°C. The supernatant was dried by centrifugal evaporation. Dried pellets were rehydrated with the buffer provided by the kit. AcSDKP was measured using an ELISA commercial kit. To evaluate the abundance of proximal tubular cells, cells were detached with Accutase (Innovative Cell Technologies, San Diego, CA), stained with an antibody against megalin (a marker of proximal tubular cells), and analyzed by flow cytometry.
Statistical Analyses
All statistical analyses were performed using GraphPad Prism 7.04 (GraphPad Software, San Diego, CA). Data are presented as individual dot plots and the mean±SEM. Differences among the three genotypes with a single factor were compared by one-way ANOVA. Differences between nondiabetic and diabetic mice within the same genotype and the differences between diabetic WT and mutant mice were compared by two-way ANOVA. For all tests, a two-tailed P value <0.05 was considered statistically significant.
Results
Diabetic Mice Lacking a Functional ACE N-Domain Display an Enhanced Natriuretic Response to Saline Challenge
Diabetes was induced in NKO, CKO, and WT littermates using low doses of STZ. Mice were followed for 6 months. At the end of the protocol, no significant differences in blood glucose, body weight, kidney-to–body weight ratio, or BP were observed among diabetic groups. Also, food and water intake and urine excretion were similar among diabetic groups (Table 1). To evaluate the natriuretic response to an extracellular volume expansion, we performed a saline challenge. Diabetic WT mice displayed an impaired natriuretic response that was 50% of that observed in nondiabetic WT mice (Figure 1, A and B). After 5 hours, diabetic WT eliminated 28%±3%, whereas nondiabetic WT excreted 48%±5% of injected sodium (P<0.01, Figure 1B). Interestingly, diabetic NKO mice exposed to equal treatment excreted a greater amount of sodium in the same period of time, evidencing a more rapid natriuretic response compared with diabetic WT mice (Figure 1, A and B). At the 2- and 3-hour time points, the sodium excretion was significantly lower in diabetic WT compared with diabetic NKO mice (P<0.05, Figure 1A). At the end of the protocol, diabetic WT eliminated 28%±3%, whereas diabetic NKO excreted 37%±4% of injected sodium (P<0.05, Figure 1B). Diabetic CKO mice displayed a similar response to diabetic WT mice (Figure 1, A and B). No significant differences were observed in urine volume excretion among all experimental groups exposed to the saline challenge (Figure 1, C and D). No salt sensitivity was evidenced in this model of diabetic nephropathy because diabetic WT mice exposed to a high-salt diet display no increase in BP (Supplemental Figure 1).
Table 1.
Phenotypic characteristics of WT, NKO, and CKO mice after 6 mo of diabetes induced by STZ
| Parameter | WT | NKO | CKO | |||
|---|---|---|---|---|---|---|
| Non-D | Diabetic | Non-D | Diabetic | Non-D | Diabetic | |
| Blood glucose, mg/dl | 129±10 | 545±16a | 132±5 | 510±20a | 133±4 | 524±16a |
| Body weight, g | 29±2 | 26±1 | 28±1 | 26±2 | 29±1 | 26±1 |
| Kidney-to-body weight, mg/g | 5.5±0.3 | 7.0±0.5b | 5.0±0.3 | 7.3±0.5b | 5.3±0.2 | 7.5±0.4b |
| Systolic BP, mm Hg | 102±6 | 104±4 | 105±7 | 101±4 | 106±7 | 96±6 |
| Food intake, g/d | 7.3±0.8 | 10.4±0.4b | 6.7±0.6 | 10.7±0.6c | 6.8±0.7 | 9.9±0.7b |
| Water intake, ml/d | 2.2±0.9 | 20±1a | 2.3±0.2 | 18±3a | 2.4±0.4 | 18±2a |
| Urine excretion, ml/d | 2.6±0.4 | 15±1a | 2.3±0.4 | 14±2a | 2.1±0.5 | 15±1c |
Mice not treated with STZ (Non-D) were used as controls. Values represent mean±SEM.
P<0.001 versus corresponding Non-D group.
P<0.05 versus corresponding Non-D group.
P<0.01 versus corresponding Non-D group.
Figure 1.

Diabetic mice lacking a functional ACE N-domain display a more rapid natriuretic response compared with diabetic WT and CKO mice. Mice were challenged with an i.p. bolus of warmed saline equivalent to 10% of their body weight and placed in metabolic cages for hourly urine collection. Plots represent the (A) fraction of injected sodium and (C) volume excreted and the (B) accumulated excretion of sodium and (D) volume over a 5-hour collection period. Data are expressed as mean±SEM; n=5–8 per group. *P<0.05; **P<0.01 versus corresponding nondiabetic (Non-D); #P<0.05 diabetic WT versus diabetic NKO; ##P<0.01 diabetic WT versus Non-D WT.
The Intrarenal RAS Is Suppressed in STZ-Induced Diabetic Mice
Our first approach to understand the enhanced natriuretic response of NKO diabetic mice was to evaluate the intrarenal RAS, a major regulator of renal sodium handling. In nondiabetic mice, renal levels of ACE and angiotensin II were similar between WT, NKO, and CKO mice (Figure 2, A and B). Renal expression of renin was also indistinguishable between WT and NKO nondiabetic mice. However, in nondiabetic CKO mice, renal renin was overexpressed compared with nondiabetic WT mice (2.1±0.2-fold increase, P<0.01, Figure 2C). Although the ACE C-domain is the main site of angiotensin II synthesis in vivo, this compensatory overexpression of renin in CKO mice restores the physiologic levels of angiotensin II through the less efficient N-domain.32 After 6 months of diabetes, intrarenal ACE expression and angiotensin II levels were significantly decreased in all diabetic groups compared with their respective nondiabetic control mice (Figure 2, A and B). No significant changes in renal expression of renin were observed after the induction of diabetes (Figure 2C). In total, these findings suggest that the classic components of the intrarenal RAS are suppressed and, presumably, not involved in the differential natriuretic response of diabetic NKO mice.
Figure 2.

Diabetes induced by STZ suppresses renal levels of ACE and angiotensin. (A) ACE expression was assessed by western blot and (B) angiotensin II was measured by ELISA in whole-kidney homogenates from nondiabetic (Non-D) and diabetic mice. (C) Renin expression was evaluated by western blot in whole-kidney homogenate; n=5–7. Immunoblots were performed with a constant amount of protein per lane; β-actin was used as loading control. Only blots from two representative samples are shown. Non-D WT group was considered as 1. *P<0.05; **P<0.01; ***P<0.001. Data are represented as individual values for each mouse (dots). Horizontal bars represent the mean±SEM.
The Enhanced Natriuretic Response of Diabetic NKO Mice Is Associated with Lower ENaC Activity
The rate at which a saline load is excreted is a function of GFR and renal sodium transporter activity.43 We measured GFR using a transcutaneous GFR method as described previously.36–39 At the end of the protocol, GFR in diabetic groups tended to increase (NS) compared with nondiabetic littermates (WT: 1.28±0.08; NKO: 1.24±0.09; CKO: 1.30±0.06-fold increase versus nondiabetic; Figure 3A). No differences were observed among diabetic groups. Although renal angiotensin II levels and GFR were not determined during the saline challenge, our current findings suggest that these parameters might not mediate the enhanced natriuretic response of diabetic NKO mice.
Figure 3.

Lower ENaC activity in diabetic NKO mice compared with diabetic WT and CKO mice after 6 months of diabetes. (A) The assessment of GFR was performed using a transcutaneous detector after a single intravenous bolus of FITC-sinistrin; n=4–5. (B) Sodium transporter expression was analyzed in kidney homogenates from nondiabetic (Non-D) and diabetic mice. Immunoblots were performed with a constant amount of protein per lane; β-actin was used as loading control. Relative abundance from each group is displayed below the corresponding blot. Non-D WT group was considered as 1. Only blots from four representative samples are shown; n=5–7. (C) Difference (Δ) in urine [Na+]/[creatinine] (top panel) and [K+]/[creatinine] (bottom panel) between vehicle (0.9% saline) and amiloride injection (5 µg/g body wt in 100 µl 0.9% NaCl). Urine was collected for 4 hours; n=3–5. *P<0.05; **P<0.01; ***P<0.001 versus corresponding Non-D. Data are represented as individual values for each mouse (dots). Horizontal bars represent the mean±SEM.
To further understand the molecular mechanisms underlying the differential sodium handling of diabetic NKO mice, we performed a sodium transporter profile by immunoblot analysis. In nondiabetic mice, the sodium transporter abundance and phosphorylation revealed no significant differences between WT, NKO, and CKO mice (Figure 3B). After 6 months of diabetes, we found no significant changes of total NHE3 and NKCC2 expression in any experimental group (Figure 3B). On the other hand, NKCC2 phosphorylation was significantly increased in all diabetic mice compared with nondiabetic controls. No differences in NKCC2 phosphorylation were observed between WT, NKO, and CKO diabetic mice (Figure 3B). To evaluate distal sodium transporters, we measured NCC and ENaC expression in total kidney homogenates. After 6 months of diabetes, the abundance and phosphorylation of NCC were significantly increased in diabetic mice compared with nondiabetic controls (Figure 3B). No differences were observed between diabetic groups. Cleavage of α and γ ENaC subunits is associated with increased channel activation.44 The abundances of full-length and cleaved forms of αENaC were increased in diabetic WT (full length [FL]: 3.2±0.4; cleaved [CL]: 3.1±0.4-fold increase; P<0.001; Figure 3B) and diabetic CKO (FL: 2.6±0.3; CL: 2.9±0.3-fold increase; P<0.01; Figure 3B) compared with nondiabetic littermates. However, no significant increase was observed in diabetic NKO mice (FL: 1.5±0.1; CL: 1.4±0.4-fold increase versus nondiabetic NKO; P=NS; Figure 3B). Similarly, the expression of βENaC was significantly increased in diabetic WT mice (1.7±0.2-fold increase, P<0.05, Figure 3B) compared with nondiabetic controls, whereas no significant differences were observed in diabetic NKO mice. No differences were observed in full-length γENaC. However, the cleaved form of γENaC was significantly increased in diabetic WT and CKO (WT: 3.0±0.4-fold increase, P<0.001 and CKO: 2.9±0.4-fold increase, P<0.01; Figure 3B) compared with nondiabetic groups. Again, diabetic NKO mice displayed no significant increase of cleaved γENaC compared with nondiabetic littermates (Figure 3B).
To determine whether the lower expression of ENaC subunits in diabetic NKO mice was associated with lower ENaC activity in vivo, we performed an amiloride test. Here, the difference between baseline and the amiloride-induced natriuretic response is a surrogate of ENaC activity in vivo.40 As shown in Figure 3C, the natriuretic response of diabetic WT mice was significantly higher compared with nondiabetic controls (3.0±0.5-fold increase, P<0.05; Figure 3C), indicating higher ENaC activity in diabetic versus nondiabetic WT mice. However, the amiloride-sensitive sodium excretion was not significantly higher in NKO diabetic mice versus nondiabetic controls (1.7±0.4-fold increase, P=0.69; Figure 3C, upper panel) confirming a lower ENaC activity in diabetic NKO mice. In addition, diabetic WT mice retained a significant amount of potassium in response to amiloride, adding further evidence to the increased ENaC activity of this experimental group (Figure 3C, lower panel). Plasma aldosterone, the main regulator of ENaC, was similar among all experimental groups, suggesting no role for this hormone in the lower ENaC activity of diabetic NKO mice (Supplemental Figure 2).
Diabetic NKO Mice Display Lower Levels of Renal Inflammation, Fibrosis, and Albuminuria Compared with Diabetic WT Mice
To evaluate whether the enhanced natriuretic response of diabetic NKO mice was associated with lower kidney damage, we measured renal levels of proinflammatory cytokines, tubulointerstitial fibrosis, macrophage infiltration, and microalbuminuria. After 6 months of diabetes, ELISA analysis of kidney homogenates revealed that all diabetic groups displayed higher levels of renal IL-1β, TNF-α, and IL-6 compared with nondiabetic littermates (Figure 4, A–C). However, diabetic NKO showed significantly lower content of IL-1β and TNF-α compared with diabetic WT mice (IL-1β: 114±26 versus 256±30 pg/mg kidney, P<0.05; TNF-α: 64±10 versus 137±20 pg/mg kidney, P<0.05; Figure 4, A and B). No significant differences were observed in renal IL-6 levels between diabetic groups (Figure 4C). Immunohistochemical analysis added further data regarding the localization of these inflammatory cytokines in kidney samples. Significant positive staining for IL-1β (Figure 4D), TNF-α (Figure 4E), and IL-6 (Figure 4F) was observed in renal tubules of diabetic mice, suggesting that tubular epithelial cells might be an important source of inflammatory cytokines during diabetic nephropathy. Quantification of immunohistochemical analysis revealed that renal levels of IL-1β, TNF-α, and IL-6 were significantly increased in diabetic WT and CKO mice compared with nondiabetic controls. No significant increase of these cytokines was observed in diabetic NKO mice (Supplemental Figure 3, A–C). It is worth mentioning that IL-6 appears to decrease in diabetic NKO by immunohistochemical analysis but not by ELISA, indicating that other sources, not evidenced by histology, might contribute to renal IL-6 accumulation. Further analysis of renal injury showed that diabetic NKO mice displayed significantly lower levels of tubulointerstitial fibrosis (0.48%±0.09% versus 0.9%±0.1% of total kidney area, P<0.05; Figure 5A), TGF-β (357±41 versus 743±119 pg/mg kidney, P<0.05; Figure 5B, Supplemental Figure 3D), and renal macrophage infiltration (36±8 versus 75±9 F4/80-positive cells per field, P<0.01; Figure 5C) compared with diabetic WT mice. The levels of tubulointerstitial fibrosis, TGF-β, and macrophage infiltration of diabetic CKO mice were indistinguishable from diabetic WT mice (Figure 5).
Figure 4.

Diabetic NKO mice display lower levels of renal inflammatory cytokines. Kidneys from diabetic WT, NKO, and CKO mice and their respective nondiabetic (Non-D) controls were collected after 6 months of diabetes. Whole-kidney homogenates were assessed for (A) IL-1β, (B) TNF-α, and (C) IL-6 by ELISA. Results were expressed as picograms of cytokine per milligram of total kidney protein. n=5–8 per group. *P<0.05; **P<0.01. Data are represented as individual values for each mouse (dots). Horizontal bars represent the mean±SEM. Immunolocalization of renal (D) IL-1β, (E) TNF-α, and (F) IL-6 was performed by immunohistochemistry; n=5. Twenty fields per sample were analyzed. Original magnification, ×40. Immunohistochemistry quantification in Supplemental Figure 3.
Figure 5.

Diabetic NKO mice display lower interstitial fibrosis and macrophage infiltration compared with diabetic WT. (A) Interstitial fibrosis in cortex was evaluated using Masson’s trichrome staining and expressed as the percentage of fibrotic area of the total cortical area from a whole-slide image. (B) TGF-β was assessed by immunohistochemistry (quantification in Supplemental Figure 3) and ELISA in whole-kidney homogenate and expressed as picograms of TGF-β per milligram of kidney protein. (C) Macrophage infiltration was assessed using F4/80 immunostaining and expressed as F4/80-positive cells per field. Fifteen fields per sample were analyzed. Original magnification, ×40. n=5 per group. *P<0.05; **P<0.01. Data are represented as individual values for each mouse (dots). Horizontal bars represent the mean±SEM.
Microalbuminuria was observed in all diabetic groups (Figure 6A). However, diabetic NKO mice displayed significantly lower levels of urinary albumin (66±11 versus 139±17 µg albumin per milligram creatinine, P<0.01; Figure 6A) compared with diabetic WT mice. No statistical differences were observed between diabetic CKO and WT mice. In addition, diabetic WT displayed higher levels of urinary prostasin (4.2±0.6-fold increase, P<0.001; Figure 6B) and furin (5.7±0.7-fold increase, P<0.001; Figure 6C) compared with nondiabetic controls. In NKO diabetic mice, the levels of these serine proteases remained significantly lower compared with diabetic WT mice (P<0.01). The higher abundance of urinary proteins observed in diabetic WT and CKO mice was associated with significant glomerular damage. Mesangial expansion and glomerular size were higher in diabetic WT and CKO compared with diabetic NKO mice (Figure 6, D–F). Finally, glomerular CD31 expression (a marker of endothelial cells) was significantly compromised in all diabetic groups (WT: 89%; NKO: 33%; CKO: 53% reduction versus nondiabetic control; P<0.01; Supplemental Figure 4). Notably, the suppression of CD31 expression was far more pronounced in diabetic WT and CKO mice compared with diabetic NKO mice (P<0.05, Supplemental Figure 4).
Figure 6.

Diabetic NKO mice display less albumin and serine proteases in the urine compared with diabetic WT. (A) Microalbuminuria was measured by ELISA and expressed as micrograms of albumin per milligram of creatinine. n=5–9 per group. Urinary (B) prostasin and (C) furin were assessed by western blot in urine samples concentrated five-fold for nondiabetic and 20-fold for diabetic animals using centrifugal filters with a molecular mass cut-off of 10 kD. Immunoblots were performed with an amount of concentrated urine equivalent to 10 µg of creatinine; n=6. (D) Representative images of periodic acid–Schiff staining. Original magnification, ×40. For the quantification of (E) mesangial area and (F) glomerular size using periodic acid–Schiff staining, all glomeruli (80–120 glomeruli) from whole-slide scans were evaluated using ImageJ software; n=5. *P<0.05; **P<0.01; ***P<0.001. Data are represented as individual values for each mouse (dots). Horizontal bars represent the mean±SEM.
Inhibition of AcSDKP Production Eliminates the Beneficial Effects Observed in Diabetic NKO Mice
To determine whether AcSDKP mediated the anti-inflammatory and the enhanced natriuretic response of diabetic NKO mice, we treated an additional group of diabetic WT and NKO mice with the POP inhibitor S17092 to block the production of AcSDKP. The treatment started after 5 months of diabetes and lasted for 1 month. Diabetic mice receiving vehicle were used as controls. Nondiabetic NKO mice displayed higher levels of urinary AcSDKP compared with nondiabetic WT mice (2.4±0.4-fold increase, P<0.01; Figure 7A). This difference was maintained after the induction of diabetes. The accumulation of AcSDKP in the urine of NKO mice evidences less degradation compared with WT mice. No accumulation of urinary AcSDKP was observed in CKO mice (Supplemental Figure 5). The chronic treatment with S17092 reduced urinary AcSDKP in diabetic NKO mice (52%±8% reduction versus diabetic NKO+vehicle, P<0.05; Figure 7A). No changes were observed in diabetic WT mice treated with S17092 (Figure 7A). A saline challenge revealed that the enhanced natriuretic response observed in diabetic NKO mice was blunted by treatment with S17092. Five hours after the saline load, diabetic NKO mice treated with vehicle excreted 33%±3% of injected sodium, whereas NKO treated with S17092 excreted 23%±1% (P<0.05; Figure 7B). The impairment of sodium excretion induced by S17092 was accompanied by a higher expression of both the full-length and the cleaved fragment of αENaC, together with higher abundance of βENaC and cleaved γENaC (P<0.05 versus diabetic NKO treated with vehicle; Figure 7C).
Figure 7.

Inhibition of AcSDKP synthesis eliminates the beneficial effects observed in diabetic NKO mice. (A) AcSDKP was assessed by ELISA in urine samples of WT and NKO diabetic mice receiving either S17092 or vehicle and their respective nondiabetic (Non-D) controls; n=5–8. (B) Mice were challenged with an i.p. bolus of warmed saline equivalent to 10% of their body weight and placed in metabolic cages for hourly urine collection. Plots represent the fraction of injected sodium (left panel) and the accumulated excretion of sodium (right panel) over a 5-hour collection period. *P<0.05; **P<0.01. n=3–5. Data are represented as individual values for each mouse (dots). Horizontal bars represent the mean±SEM. (C) The full-length (FL) and the cleaved (CL) active portions of the ENaC α and γ subunits as well as βENaC were analyzed in kidney homogenates. Immunoblots were performed with a constant amount of protein per lane; β-actin was used as loading control. Relative abundance from each group is displayed below the corresponding blot. Non-D WT group was considered as 1. *P<0.05; **P<0.01; ***P<0.001 versus WT (Non-D) controls; #P<0.05 versus diabetic NKO+vehicle. n=3–8 per group.
Further analysis of renal inflammation revealed that diabetic NKO mice exposed to the S17092 treatment showed higher levels of renal IL-1β and TNF-α compared with diabetic NKO receiving vehicle. Renal IL-1β increased from 125±32 pg/mg kidney in diabetic NKO receiving vehicle to 281±27 pg/mg kidney in diabetic NKO treated with S17092 (P<0.05; Figure 8A). TNF-α also increased from 69±13 to 123±11 pg/mg kidney, although this change was NS (P=0.09; Figure 8B). In addition, tubulointerstitial fibrosis and macrophage infiltration were increased in diabetic NKO treated with S17092 (Supplemental Figure 6). Microalbuminuria increased from 86±13 to 221±41 µg albumin per milligram creatinine (P<0.05; Figure 8C). Even more importantly, data analysis revealed a significant negative correlation between urine albumin and AcSDKP (r=−0.631, P<0.01; Figure 8D). Finally, diabetic NKO treated with S17092 displayed higher levels of urinary prostasin (2.1±0.1-fold increase, P<0.001; Figure 8E) and furin (2.4±0.3-fold increase, P<0.05; Figure 8F) compared with diabetic NKO treated with vehicle. Both serine proteases reached urinary levels that were indistinguishable from diabetic WT mice. As observed for albumin, there is a significant negative correlation between urine serine proteases and AcSDKP (Supplemental Figure 7). In total, these data demonstrate that blocking AcSDKP production using the POP inhibitor S17092 eliminates the protective phenotype of diabetic NKO mice.
Figure 8.

Inhibition of AcSDKP synthesis increases inflammation and microalbuminuria in diabetic NKO mice. Whole-kidney homogenates were assessed for (A) IL-1β and (B) TNF-α by ELISA. Data are expressed as picograms of cytokine per milligram of total kidney protein. (C) Microalbuminuria was measured by ELISA and expressed as micrograms of albumin per milligram of creatinine. (D) Urinary albumin from (C) was plotted against urinary AcSDKP from Figure 7A, combining WT and NKO diabetic mice receiving either vehicle or S17092. Plots indicate a negative correlation between albumin and AcSDKP in the urine. Urinary (E) prostasin and (F) furin were assessed by western blot in urine samples concentrated five-fold for nondiabetic and 20-fold for diabetic animals using centrifugal filters with a molecular mass cut-off of 10 kD. Immunoblots were performed with an amount of concentrated urine equivalent to 10 µg of creatinine. Diabetic WT group was considered as 1. n=5–7 per group. *P<0.05; ***P<0.001. Data are represented as individual values for each mouse (dots). Horizontal bars represent the mean±SEM.
Lacking a Functional ACE N-Domain Blunts the Release of IL-1β from Renal Tubular Epithelial Cells in Response to High Glucose
To further explore the mechanism by which the ACE N-domain regulates the inflammatory response associated with diabetic nephropathy, we cultured primary renal tubular epithelial cells.42 The characterization of these cells revealed that 75% derive from proximal tubules (megalin positive; Supplemental Figure 8). The other 25% of cells might be either proximal tubular cells that lost megalin from the cell surface or epithelial cells that belong to other portions of the nephron. We performed a comprehensive analysis of cytokines in the culture media using a commercial cytokine array kit. Among 23 inflammatory markers evaluated, IL-1β was the most abundant cytokine released by renal tubular cells exposed to high glucose (Supplemental Figure 9). ELISA confirmed that epithelial cells exposed to high glucose released significantly higher levels of IL-1β compared with nonstimulated cells (from 6±1 to 49±6 pg/ml, P<0.001; Figure 9A). Interestingly, this response was blunted by the ACE inhibitor lisinopril but not by the angiotensin II type 1 (AT1) receptor blocker losartan, excluding the AT1 receptor as a mediator of these effects (Figure 9A). The absence of response to 30 mM mannitol stimulation eliminated an osmotic effect. To determine which ACE domain was involved in this response, a primary culture of renal tubular epithelial cells from NKO and CKO mice was established. We found that the IL-1β response of tubular cells from NKO mice was significantly lower compared with WT cells (59%±6% reduction, P<0.01; Figure 9B). No differences were observed between WT and CKO tubular cells (Figure 9B). Further analysis of the intracellular AcSDKP levels showed that NKO tubular cells accumulate higher levels of this tetrapeptide compared with WT and CKO cells. Also, the intracellular AcSDKP abundance was not affected by high glucose (Supplemental Figure 10). To evaluate whether AcSDKP mediates the inhibition of IL-1β synthesis in NKO tubular cells, we studied an additional group of tubular cells treated with S17092 to block the synthesis of AcSDKP. Now, the response of NKO tubular cells to high glucose was indistinguishable from equivalently treated WT tubular cells (Figure 9C). These studies indicate that blocking the ACE N-domain and the consequent accumulation of AcSDKP contribute to lower renal inflammation during diabetes through an inhibitory effect on IL-1β production or release from renal tubular cells. Finally, we performed the opposite experiment by treating high-glucose–stimulated WT tubular cells with different concentrations of pure AcSDKP (from 10−10 to 10−6 M) for 24 hours. This time, AcSDKP failed to blunt IL-1β production in WT tubular cells (Figure 9D). Although the reason for this unexpected finding is unknown, we hypothesize that: (1) AcSDKP is rapidly degraded by WT ACE even before exerting its anti-inflammatory effects, (2) other ACE N-domain substrates/products mediate the regulation of IL-1β synthesis, or (3) exogenous AcSDKP added to the cell culture fails to reach its target. In any case, increasing AcSDKP by blocking the ACE N-domain seems to be therapeutically more advantageous than AcSDKP administration.
Figure 9.

Lacking a functional ACE N-domain blunts the release of IL-1β from renal tubular epithelial cells in response to high glucose. (A) A primary cell culture of renal tubular epithelial cells from WT mice was established. After 7 days of culture, the glucose concentration of the culture media was increased from 5 (low) to 30 mM (high) for 24 hours, with or without lisinopril (Lis, 10 μM) or losartan (Los, 100 μM). (B) Renal tubular epithelial cells obtained from WT, NKO, and CKO mice were exposed to low and high glucose. (C) Renal tubular epithelial cells from WT and NKO were exposed to low, high, and high glucose plus S17092 (50 µM). (D) Renal tubular epithelial cells from WT mice were exposed to low; high; and high glucose plus 10−10, 10−8, or 10−6 M AcSDKP. IL-1β was evaluated in the culture media by ELISA. *P<0.05; **P<0.01; ***P<0.001 versus WT low glucose; ##P<0.01. Each dot represents an individual experiment. Horizontal bars represent the mean±SEM.
Discussion
In this study, WT mice made diabetic with STZ develop an early stage of diabetic nephropathy characterized by sodium retention in response to a sodium challenge (Figure 1). However, the sodium retentive state observed in STZ-treated mice is not accompanied by hypertension or salt sensitivity, indicating that other renal or systemic compensatory mechanisms involved in BP regulation remain functional in the early stage of diabetic nephropathy. The excretion rate of a saline load and the control of BP are functions of GFR and renal sodium transporter/channel activity.43 This study showed that all diabetic groups display the typical hyperfiltration of early diabetic nephropathy.45 This increase in GFR, in the long term, will eliminate enough sodium to prevent any increase in BP, even under a high-sodium diet. However, our acute sodium overload approach and the assessment of sodium transporters reveal that tubular function of diabetic mice is already compromised at early stages of diabetic kidney disease. Although this sodium retentive state is considered a compensatory response to the copious water and sodium losses driven by hyperfiltration,46 it might be excessive and predispose to hypertension as diabetic nephropathy progresses. Indeed, studies in both humans and animal models have demonstrated that excessive sodium retention and the consequent volume expansion, together with the decline in renal function and GFR observed during the progression of diabetic kidney disease, are key contributors to hypertension.6,47,48 Even more importantly, high BP, which is twice as prevalent in patients with diabetes compared with the nondiabetic population,6 dramatically accelerates the progression of renal disease.
In C57Bl/6j mice, STZ-induced diabetes leads to a mild diabetic nephropathy. This is a useful animal model to investigate the initial and subtle abnormalities, both functional and structural, of the diabetic kidney that impair normal sodium handling. Here, we observed that diabetic mice lacking a functional N-domain can excrete a sodium overload more rapidly than WT diabetic mice (Figure 1). This improved sodium metabolism seems to be a consequence of the lower abundance and activity of the distal sodium transporter ENaC (Figure 3). The activity of ENaC was assessed by cleavage of the α and γ subunits and the natriuretic response to amiloride. This transporter is critical for the final adjustment of sodium excretion and BP regulation. The analysis of other renal sodium transporters such as NHE3, NKCC2, or NCC revealed no significant differences between WT, NKO, or CKO diabetic mice, discarding a potential role of these transporters and pointing toward ENaC as a key mediator of our observations.
The key question that arises from our current results is how the absence of the ACE N-domain modifies the expression/activation of ENaC and facilitates sodium excretion. Renin-angiotensin-aldosterone system overactivation and the consequent intrarenal accumulation of angiotensin II has been classically proposed as the main mechanism controlling sodium handling.49 Moreover, ACE inhibitors are the standard treatment for hypertension associated with diabetic kidney disease.50,51 However, whole-kidney angiotensin II levels in diabetes can increase, decrease, or even remain unchanged during the progression of diabetic nephropathy.13–19 As previously described,19 there was a significant reduction of ACE expression in the kidneys of STZ-treated mice that correlates with a suppression of renal angiotensin II levels (Figure 2). Although similar findings were described in other experimental models of diabetes, the mechanism behind this response remains elusive.52,53 The concept of RAS suppression goes against the beneficial effects of RAS blockade in diabetic nephropathy, suggesting that, at least in this model, the regulation of the renal RAS during diabetes might be more complicated than generally appreciated. Notably, the downregulation of renal ACE and angiotensin II was similar in WT, NKO, and CKO diabetic mice, suggesting that ACE might contribute to the sodium retentive state associated with diabetic nephropathy through mechanisms that go beyond the well known ACE/angiotensin II/AT1 receptor axis. Because most commonly prescribed ACE inhibitors are virtually non–domain selective, it is very difficult to determine whether the renoprotective effect is due to the inhibition of one domain or the other.54 This can only be achieved with a long-term treatment using highly selective ACE inhibitors such as RXP407 (N-domain blocker) or RXPA 380 (C-domain blocker).54 AT1 receptor blockers have also demonstrated high efficacy in treating hypertension, prolonging renal survival, and decreasing microalbuminuria associated with diabetic kidney disease. Indeed, current clinical studies cannot discriminate whether ACE inhibitors or AT1 receptor blockers are more suitable for diabetic nephropathy.55 Because we have not included an AT1 receptor blocker in our studies, we cannot add further data to solve this clinical controversy. However, we found that nontraditional components of the RAS, such as the ACE N-domain and AcSDKP, emerge as potential therapeutic targets to improve kidney function during diabetes.
ACE can process a wide diversity of substrates besides angiotensin I. Among these, AcSDKP has gained attention for its anti-inflammatory and antifibrotic properties in several pathologic conditions, including diabetic kidney disease.27,56 The degradation of AcSDKP is mediated almost exclusively by the ACE N-domain catalytic site,22 and previous studies have demonstrated that AcSDKP accumulates in NKO mice.23 Whether the two-fold increase of urinary AcSDKP observed in NKO mice is enough to reach a therapeutic level is unknown. However, previous studies have demonstrated that a two- to three-fold increase in urine AcSDKP is usually associated with beneficial effects in several models of renal and cardiovascular disease.23,26,57 Further evidence of the therapeutic role of AcSDKP in our model of diabetic nephropathy was obtained after treating diabetic NKO mice with S17092. Here, urinary AcSDKP levels decreased and sodium handling and renal ENaC levels were indistinguishable from those observed in diabetic WT mice (Figures 7 and 8). Although we provide substantial evidence indicating that the protective phenotype of diabetic NKO mice is mediated through an accumulation of AcSDKP, other ACE N-domain substrates or products, not evaluated in this work, might still be involved in these effects.
Our studies suggest that AcSDKP, either directly or indirectly, regulates ENaC activity. Several reports have shown that ENaC activation involves a proteolytic cleavage of the extracellular domain of the α and γ subunits by serine proteases, such as prostasin and furin.44,58 Indeed, the presence of these proteases in the urine is often associated with proteolytic activation of ENaC in several pathologic conditions.59,60 Research demonstrated that prostasin reaches the urine through an aberrant glomerular filtration, whereas furin, an intracellular convertase-type protease, is probably released from injured tubular epithelial cells rather than glomerular leakage.60 In diabetic NKO mice, we found less mesangial expansion, less reduction of glomerular CD31 expression, lower tubular fibrosis, and lower levels of the profibrotic cytokine TGF-β compared with diabetic WT mice (Figures 5 and 6). This protective phenotype was associated with decreased levels of prostasin and furin in the urine. Thus, AcSDKP may indirectly regulate ENaC by preserving the kidney ultrastructure of diabetic NKO and decreasing the leakage of serine proteases to the distal portion of the nephron.
Another potential role of AcSDKP in sodium handling might be mediated through the modulation of renal inflammation. Several studies have underscored a critical role of inflammation in the pathogenesis of diabetic kidney disease and sodium retention. This inflammatory response injures the renal parenchyma and promotes defective sodium handling that predisposes to hypertension.61 ENaC has been described as a potential mediator of inflammation-induced sodium retention. Research has shown that TNF-α activates ENaC during diabetic nephropathy.47 On the other hand, AcSDKP exhibits renal antifibrotic and anti-inflammatory effects in various experimental models of kidney disease, including diabetic nephropathy.62 Many of the beneficial effects of AcSDKP are mediated through the inhibition of TGF-β–related intracellular signaling pathways such as the transcription factors Smads.63 In addition, AcSDKP inhibits the key proinflammatory transcriptional factor, NFκB64; reduces the mRNA levels of TNF-α and IL-1β65; and exerts a renal protective effect in Dahl salt-sensitive rats.66 Thus, the increased sodium excretion observed in diabetic NKO mice might be mediated through the anti-inflammatory effects of AcSDKP. For example, renal TNF-α was decreased in diabetic NKO mice (Figure 4) and restored after S17092 treatment (Figure 8), suggesting that the lower ENaC activity of diabetic NKO mice might be a consequence of AcSDKP-mediated downregulation of TNF-α expression. Research demonstrated that IL-6 is also capable of stimulating ENaC expression and activity in mouse cortical collecting duct cells.67 However, our ELISA studies show that IL-6 was similar in all diabetic groups, discarding a potential role of this cytokine in controlling ENaC expression in this model of diabetes. The regulation of ENaC by IL-1β has also been broadly investigated in the last decade. Although most reports indicate that IL-1β downregulates ENaC expression in vitro,68,69 the in vivo effect might be influenced by the underlying pathologic condition. For example, it is possible that IL-1β indirectly controls ENaC expression/activity through the induction of TNF-α by renal inflammatory cells. In fact, recent reports demonstrate that IL-1β recruits and activates macrophages to produce TNF-α.70,71 This hypothesis agrees with our observation that diabetic WT and CKO mice display significant renal macrophage infiltration compared with diabetic NKO mice (Figure 5). Further evidence of the anti-inflammatory effect of AcSDKP was obtained from our in vitro experiments. Previous reports showed that renal epithelial cells produce IL-1β in response to high glucose.72 Here, we observed that high-glucose–induced IL-1β production was dramatically reduced in epithelial cells from NKO mice, a process mediated through AcSDKP accumulation (Figure 9). We also observed that IL-1β is the main cytokine released by tubular cells exposed to high glucose. However, immunohistochemical analysis of kidney samples revealed that, after 6 months of diabetes, other cytokines besides IL-1β are found in renal tubules, suggesting that epithelial cells are also an important source of these inflammatory cytokines during diabetic kidney disease. Thus, IL-1β might be an early cytokine produced in response to high glucose that triggers the production of other inflammatory cytokines either by tubular epithelium or inflammatory cells. The role of AcSDKP in ameliorating diabetic nephropathy might involve inhibition of the initial IL-1β boost.
In conclusion, this study shows that diabetic mice lacking a functional ACE N-domain display improved sodium excretion in response to a saline challenge, less activity of the distal sodium channel ENaC, lower renal inflammation, and ameliorated renal injury compared with diabetic WT or CKO mice. We also provide evidence suggesting that AcSDKP accumulation in the kidney mediates the protective phenotype of NKO mice. These beneficial effects were not associated with changes in intrarenal angiotensin II levels. To our knowledge, this is the first report describing AcSDKP as a regulator of sodium handling in the distal portion of the nephron.
Disclosures
None.
Supplementary Material
Acknowledgments
The authors thank F. Chung for his assistance in immunohistochemistry studies and B. Taylor for his assistance in preparing this manuscript.
This study was supported by a grant from The International Research Fund for Subsidy of Kyushu University School of Medicine Alumni and Senshin Medical Research Foundation to M.E.; Western University of Health Sciences internal funds to S.F.; National Institutes of Health (NIH) 5R01DK083785-08 to A.A.M.; NIH R03DK101592 to R.A.G.-V.; NIH R01HL110353, R21AI114965, P01HL129941, and American Heart Association (AHA) Grant-in-Aid 17GRNT33661206 to K.E.B.; and Cedars-Sinai Internal Funds, AHA Scientist Development Grant 16SDG30130015, and Diabetic Research Center Pilot and Feasibility P30DK063491 to J.F.G..
M.E., K.E.B., and J.F.G. conceived and designed research; M.E., E.A.B., L.C.V., Z.K., D.Y.C., J.E.T., and J.F.G. performed experiments; M.E., L.C.V., S.F., A.A.M., J.E.T., R.A.G.-V., K.E.B., and J.F.G. analyzed data; M.E., E.A.B., L.C.V., A.A.M., K.E.B., and J.F.G. interpreted results of experiments; M.E. and J.F.G. prepared figures; J.F.G. drafted the manuscript; M.E., L.C.V., S.F., A.A.M., R.A.G.-V., K.E.B., and J.F.G. edited and revised the manuscript; all authors approved the final version of the manuscript.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2018030323/-/DCSupplemental.
References
- 1.Fernandez-Fernandez B, Ortiz A, Gomez-Guerrero C, Egido J: Therapeutic approaches to diabetic nephropathy--beyond the RAS. Nat Rev Nephrol 10: 325–346, 2014 [DOI] [PubMed] [Google Scholar]
- 2.Duran-Salgado MB, Rubio-Guerra AF: Diabetic nephropathy and inflammation. World J Diabetes 5: 393–398, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McMaster WG, Kirabo A, Madhur MS, Harrison DG: Inflammation, immunity, and hypertensive end-organ damage. Circ Res 116: 1022–1033, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rodríguez-Iturbe B, Franco M, Tapia E, Quiroz Y, Johnson RJ: Renal inflammation, autoimmunity and salt-sensitive hypertension. Clin Exp Pharmacol Physiol 39: 96–103, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lanaspa MA, Ishimoto T, Cicerchi C, Tamura Y, Roncal-Jimenez CA, Chen W, et al.: Endogenous fructose production and fructokinase activation mediate renal injury in diabetic nephropathy. J Am Soc Nephrol 25: 2526–2538, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Buren PN, Toto R: Hypertension in diabetic nephropathy: Epidemiology, mechanisms, and management. Adv Chronic Kidney Dis 18: 28–41, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Châtel R, Weidmann P, Flammer J, Ziegler WH, Beretta-Piccoli C, Vetter W, et al.: Sodium, renin, aldosterone, catecholamines, and blood pressure in diabetes mellitus. Kidney Int 12: 412–421, 1977 [DOI] [PubMed] [Google Scholar]
- 8.Zaika O, Mamenko M, Staruschenko A, Pochynyuk O: Direct activation of ENaC by angiotensin II: Recent advances and new insights. Curr Hypertens Rep 15: 17–24, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.San-Cristobal P, Pacheco-Alvarez D, Richardson C, Ring AM, Vazquez N, Rafiqi FH, et al.: Angiotensin II signaling increases activity of the renal Na-Cl cotransporter through a WNK4-SPAK-dependent pathway. Proc Natl Acad Sci USA 106: 4384–4389, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nguyen MT, Lee DH, Delpire E, McDonough AA: Differential regulation of Na+ transporters along nephron during ANG II-dependent hypertension: Distal stimulation counteracted by proximal inhibition. Am J Physiol Renal Physiol 305: F510–F519, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Price DA, Porter LE, Gordon M, Fisher ND, De’Oliveira JM, Laffel LM, et al.: The paradox of the low-renin state in diabetic nephropathy. J Am Soc Nephrol 10: 2382–2391, 1999 [DOI] [PubMed] [Google Scholar]
- 12.Christlieb AR, Kaldany A, D’Elia JA: Plasma renin activity and hypertension in diabetes mellitus. Diabetes 25: 969–974, 1976 [DOI] [PubMed] [Google Scholar]
- 13.Zimpelmann J, Kumar D, Levine DZ, Wehbi G, Imig JD, Navar LG, et al.: Early diabetes mellitus stimulates proximal tubule renin mRNA expression in the rat. Kidney Int 58: 2320–2330, 2000 [DOI] [PubMed] [Google Scholar]
- 14.Park S, Bivona BJ, Kobori H, Seth DM, Chappell MC, Lazartigues E, et al.: Major role for ACE-independent intrarenal ANG II formation in type II diabetes. Am J Physiol Renal Physiol 298: F37–F48, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harrison-Bernard LM, Imig JD, Carmines PK: Renal AT1 receptor protein expression during the early stage of diabetes mellitus. Int J Exp Diabetes Res 3: 97–108, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Campbell DJ, Kelly DJ, Wilkinson-Berka JL, Cooper ME, Skinner SL: Increased bradykinin and “normal” angiotensin peptide levels in diabetic Sprague-Dawley and transgenic (mRen-2)27 rats. Kidney Int 56: 211–221, 1999 [DOI] [PubMed] [Google Scholar]
- 17.Anderson S: Physiologic actions and molecular expression of the renin-angiotensin system in the diabetic rat. Miner Electrolyte Metab 24: 406–411, 1998 [DOI] [PubMed] [Google Scholar]
- 18.Vora JP, Oyama TT, Thompson MM, Anderson S: Interactions of the kallikrein-kinin and renin-angiotensin systems in experimental diabetes. Diabetes 46: 107–112, 1997 [DOI] [PubMed] [Google Scholar]
- 19.Eriguchi M, Lin M, Yamashita M, Zhao TV, Khan Z, Bernstein EA, et al. : Renal tubular ACE-mediated tubular injury is the major contributor to microalbuminuria in early diabetic nephropathy. Am J Physiol Renal Physiol 314: F531–F542, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen XZ, Li P, Weiss D, Fuchs S, Xiao HD, Adams JA, et al.: Mice with enhanced macrophage angiotensin-converting enzyme are resistant to melanoma. Am J Pathol 170: 2122–2134, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khan Z, Shen XZ, Bernstein EA, Giani JF, Eriguchi M, Zhao TV, et al.: Angiotensin-converting enzyme enhances the oxidative response and bactericidal activity of neutrophils. Blood 130: 328–339, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bernstein KE, Ong FS, Blackwell WL, Shah KH, Giani JF, Gonzalez-Villalobos RA, et al.: A modern understanding of the traditional and nontraditional biological functions of angiotensin-converting enzyme. Pharmacol Rev 65: 1–46, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li P, Xiao HD, Xu J, Ong FS, Kwon M, Roman J, et al.: Angiotensin-converting enzyme N-terminal inactivation alleviates bleomycin-induced lung injury. Am J Pathol 177: 1113–1121, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumar N, Nakagawa P, Janic B, Romero CA, Worou ME, Monu SR, et al.: The anti-inflammatory peptide Ac-SDKP is released from thymosin-β4 by renal meprin-α and prolyl oligopeptidase. Am J Physiol Renal Physiol 310: F1026–F1034, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharma U, Rhaleb NE, Pokharel S, Harding P, Rasoul S, Peng H, et al.: Novel anti-inflammatory mechanisms of N-Acetyl-Ser-Asp-Lys-Pro in hypertension-induced target organ damage. Am J Physiol Heart Circ Physiol 294: H1226–H1232, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shibuya K, Kanasaki K, Isono M, Sato H, Omata M, Sugimoto T, et al.: N-acetyl-seryl-aspartyl-lysyl-proline prevents renal insufficiency and mesangial matrix expansion in diabetic db/db mice. Diabetes 54: 838–845, 2005 [DOI] [PubMed] [Google Scholar]
- 27.Castoldi G, di Gioia CR, Bombardi C, Preziuso C, Leopizzi M, Maestroni S, et al.: Renal antifibrotic effect of N-acetyl-seryl-aspartyl-lysyl-proline in diabetic rats. Am J Nephrol 37: 65–73, 2013 [DOI] [PubMed] [Google Scholar]
- 28.Nagai T, Kanasaki M, Srivastava SP, Nakamura Y, Ishigaki Y, Kitada M, et al.: N-acetyl-seryl-aspartyl-lysyl-proline inhibits diabetes-associated kidney fibrosis and endothelial-mesenchymal transition. BioMed Res Int 2014: 696475, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nitta K, Shi S, Nagai T, Kanasaki M, Kitada M, Srivastava SP, et al.: Oral administration of N-acetyl-seryl-aspartyl-lysyl-proline ameliorates kidney disease in both type 1 and type 2 diabetic mice via a therapeutic regimen. BioMed Res Int 2016: 9172157, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kumar N, Liao TD, Romero CA, Maheshwari M, Peterson EL, Carretero OA: Thymosin β4 deficiency exacerbates renal and cardiac injury in angiotensin-II-induced hypertension. Hypertension 71: 1133–1142, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fuchs S, Xiao HD, Cole JM, Adams JW, Frenzel K, Michaud A, et al.: Role of the N-terminal catalytic domain of angiotensin-converting enzyme investigated by targeted inactivation in mice. J Biol Chem 279: 15946–15953, 2004 [DOI] [PubMed] [Google Scholar]
- 32.Fuchs S, Xiao HD, Hubert C, Michaud A, Campbell DJ, Adams JW, et al.: Angiotensin-converting enzyme C-terminal catalytic domain is the main site of angiotensin I cleavage in vivo. Hypertension 51: 267–274, 2008 [DOI] [PubMed] [Google Scholar]
- 33.Tikellis C, Brown R, Head GA, Cooper ME, Thomas MC: Angiotensin-converting enzyme 2 mediates hyperfiltration associated with diabetes. Am J Physiol Renal Physiol 306: F773–F780, 2014 [DOI] [PubMed] [Google Scholar]
- 34.Cavasin MA, Rhaleb NE, Yang XP, Carretero OA: Prolyl oligopeptidase is involved in release of the antifibrotic peptide Ac-SDKP. Hypertension 43: 1140–1145, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Veiras LC, Girardi ACC, Curry J, Pei L, Ralph DL, Tran A, et al.: Sexual dimorphic pattern of renal transporters and electrolyte homeostasis. J Am Soc Nephrol 28: 3504–3517, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Giani JF, Eriguchi M, Bernstein EA, Katsumata M, Shen XZ, Li L, et al.: Renal tubular angiotensin converting enzyme is responsible for nitro-L-arginine methyl ester (L-NAME)-induced salt sensitivity. Kidney Int 91: 856–867, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Giani JF, Janjulia T, Kamat N, Seth DM, Blackwell WL, Shah KH, et al.: Renal angiotensin-converting enzyme is essential for the hypertension induced by nitric oxide synthesis inhibition. J Am Soc Nephrol 25: 2752–2763, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schreiber A, Shulhevich Y, Geraci S, Hesser J, Stsepankou D, Neudecker S, et al.: Transcutaneous measurement of renal function in conscious mice. Am J Physiol Renal Physiol 303: F783–F788, 2012 [DOI] [PubMed] [Google Scholar]
- 39.Giani JF, Bernstein KE, Janjulia T, Han J, Toblli JE, Shen XZ, et al.: Salt sensitivity in response to renal injury requires renal angiotensin-converting enzyme. Hypertension 66: 534–542, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Todkar A, Picard N, Loffing-Cueni D, Sorensen MV, Mihailova M, Nesterov V, et al.: Mechanisms of renal control of potassium homeostasis in complete aldosterone deficiency. J Am Soc Nephrol 26: 425–438, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Giani JF, Muñoz MC, Pons RA, Cao G, Toblli JE, Turyn D, et al.: Angiotensin-(1-7) reduces proteinuria and diminishes structural damage in renal tissue of stroke-prone spontaneously hypertensive rats. Am J Physiol Renal Physiol 300: F272–F282, 2011 [DOI] [PubMed] [Google Scholar]
- 42.Terryn S, Jouret F, Vandenabeele F, Smolders I, Moreels M, Devuyst O, et al.: A primary culture of mouse proximal tubular cells, established on collagen-coated membranes. Am J Physiol Renal Physiol 293: F476–F485, 2007 [DOI] [PubMed] [Google Scholar]
- 43.Weinstein SW, Szyjewicz J: Single-nephron function and renal oxygen consumption during rapid volume expansion. Am J Physiol 231: 1166–1172, 1976 [DOI] [PubMed] [Google Scholar]
- 44.Kleyman TR, Carattino MD, Hughey RP: ENaC at the cutting edge: Regulation of epithelial sodium channels by proteases. J Biol Chem 284: 20447–20451, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brenner BM, Lawler EV, Mackenzie HS: The hyperfiltration theory: A paradigm shift in nephrology. Kidney Int 49: 1774–1777, 1996 [DOI] [PubMed] [Google Scholar]
- 46.Song J, Knepper MA, Verbalis JG, Ecelbarger CA: Increased renal ENaC subunit and sodium transporter abundances in streptozotocin-induced type 1 diabetes. Am J Physiol Renal Physiol 285: F1125–F1137, 2003 [DOI] [PubMed] [Google Scholar]
- 47.DiPetrillo K, Coutermarsh B, Soucy N, Hwa J, Gesek F: Tumor necrosis factor induces sodium retention in diabetic rats through sequential effects on distal tubule cells. Kidney Int 65: 1676–1683, 2004 [DOI] [PubMed] [Google Scholar]
- 48.Weidmann P, Ferrari P: Central role of sodium in hypertension in diabetic subjects. Diabetes Care 14: 220–232, 1991 [DOI] [PubMed] [Google Scholar]
- 49.Navar LG, Prieto MC, Satou R, Kobori H: Intrarenal angiotensin II and its contribution to the genesis of chronic hypertension. Curr Opin Pharmacol 11: 180–186, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kobori H, Nangaku M, Navar LG, Nishiyama A: The intrarenal renin-angiotensin system: From physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev 59: 251–287, 2007 [DOI] [PubMed] [Google Scholar]
- 51.Carey RM, Siragy HM: The intrarenal renin-angiotensin system and diabetic nephropathy. Trends Endocrinol Metab 14: 274–281, 2003 [DOI] [PubMed] [Google Scholar]
- 52.Soler MJ, Wysocki J, Ye M, Lloveras J, Kanwar Y, Batlle D: ACE2 inhibition worsens glomerular injury in association with increased ACE expression in streptozotocin-induced diabetic mice. Kidney Int 72: 614–623, 2007 [DOI] [PubMed] [Google Scholar]
- 53.Wysocki J, Ye M, Soler MJ, Gurley SB, Xiao HD, Bernstein KE, et al.: ACE and ACE2 activity in diabetic mice. Diabetes 55: 2132–2139, 2006 [DOI] [PubMed] [Google Scholar]
- 54.Redelinghuys P, Nchinda AT, Sturrock ED: Development of domain-selective angiotensin I-converting enzyme inhibitors. Ann N Y Acad Sci 1056: 160–175, 2005 [DOI] [PubMed] [Google Scholar]
- 55.Kota SK, Meher LK, Jammula S, Kota SK, Modi KD: ACE inhibitors or ARBs for diabetic nephropathy: The unrelenting debate. Diabetes Metab Syndr 6: 215–217, 2012 [DOI] [PubMed] [Google Scholar]
- 56.Srivastava SP, Shi S, Kanasaki M, Nagai T, Kitada M, He J, et al.: Effect of antifibrotic microRNAs crosstalk on the action of N-acetyl-seryl-aspartyl-lysyl-proline in diabetes-related kidney fibrosis. Sci Rep 6: 29884, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peng H, Carretero OA, Brigstock DR, Oja-Tebbe N, Rhaleb NE: Ac-SDKP reverses cardiac fibrosis in rats with renovascular hypertension. Hypertension 42: 1164–1170, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Buhl KB, Oxlund CS, Friis UG, Svenningsen P, Bistrup C, Jacobsen IA, et al. : Plasmin in urine from patients with type 2 diabetes and treatment-resistant hypertension activates ENaC in vitro. J Hypertens 32: 1672–1677; discussion 1677, 2014 [DOI] [PubMed] [Google Scholar]
- 59.Andersen H, Friis UG, Hansen PB, Svenningsen P, Henriksen JE, Jensen BL: Diabetic nephropathy is associated with increased urine excretion of proteases plasmin, prostasin and urokinase and activation of amiloride-sensitive current in collecting duct cells. Nephrol Dial Transplant 30: 781–789, 2015 [DOI] [PubMed] [Google Scholar]
- 60.Zheng H, Liu X, Sharma NM, Li Y, Pliquett RU, Patel KP: Urinary proteolytic activation of renal epithelial Na+ channels in chronic heart failure. Hypertension 67: 197–205, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rodriguez-Iturbe B, Franco M, Johnson RJ: Impaired pressure natriuresis is associated with interstitial inflammation in salt-sensitive hypertension. Curr Opin Nephrol Hypertens 22: 37–44, 2013 [DOI] [PubMed] [Google Scholar]
- 62.Kanasaki K, Nagai T, Nitta K, Kitada M, Koya D: N-acetyl-seryl-aspartyl-lysyl-proline: A valuable endogenous anti-fibrotic peptide for combating kidney fibrosis in diabetes. Front Pharmacol 5: 70, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kanasaki K, Koya D, Sugimoto T, Isono M, Kashiwagi A, Haneda M: N-Acetyl-seryl-aspartyl-lysyl-proline inhibits TGF-beta-mediated plasminogen activator inhibitor-1 expression via inhibition of Smad pathway in human mesangial cells. J Am Soc Nephrol 14: 863–872, 2003 [DOI] [PubMed] [Google Scholar]
- 64.Zhu L, Yang XP, Janic B, Rhaleb NE, Harding P, Nakagawa P, et al.: Ac-SDKP suppresses TNF-α-induced ICAM-1 expression in endothelial cells via inhibition of IκB kinase and NF-κB activation. Am J Physiol Heart Circ Physiol 310: H1176–H1183, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Omata M, Taniguchi H, Koya D, Kanasaki K, Sho R, Kato Y, et al.: N-acetyl-seryl-aspartyl-lysyl-proline ameliorates the progression of renal dysfunction and fibrosis in WKY rats with established anti-glomerular basement membrane nephritis. J Am Soc Nephrol 17: 674–685, 2006 [DOI] [PubMed] [Google Scholar]
- 66.Worou ME, Liao TD, D’Ambrosio M, Nakagawa P, Janic B, Peterson EL, et al.: Renal protective effect of N-acetyl-seryl-aspartyl-lysyl-proline in dahl salt-sensitive rats. Hypertension 66: 816–822, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li K, Guo D, Zhu H, Hering-Smith KS, Hamm LL, Ouyang J, et al.: Interleukin-6 stimulates epithelial sodium channels in mouse cortical collecting duct cells. Am J Physiol Regul Integr Comp Physiol 299: R590–R595, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Roux J, Kawakatsu H, Gartland B, Pespeni M, Sheppard D, Matthay MA, et al.: Interleukin-1beta decreases expression of the epithelial sodium channel alpha-subunit in alveolar epithelial cells via a p38 MAPK-dependent signaling pathway. J Biol Chem 280: 18579–18589, 2005 [DOI] [PubMed] [Google Scholar]
- 69.Choi JY, Choi YS, Kim SJ, Son EJ, Choi HS, Yoon JH: Interleukin-1beta suppresses epithelial sodium channel beta-subunit expression and ENaC-dependent fluid absorption in human middle ear epithelial cells. Eur J Pharmacol 567: 19–25, 2007 [DOI] [PubMed] [Google Scholar]
- 70.Rider P, Carmi Y, Guttman O, Braiman A, Cohen I, Voronov E, et al.: IL-1α and IL-1β recruit different myeloid cells and promote different stages of sterile inflammation. J Immunol 187: 4835–4843, 2011 [DOI] [PubMed] [Google Scholar]
- 71.Jayaraman P, Sada-Ovalle I, Nishimura T, Anderson AC, Kuchroo VK, Remold HG, et al.: IL-1β promotes antimicrobial immunity in macrophages by regulating TNFR signaling and caspase-3 activation. J Immunol 190: 4196–4204, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen K, Zhang J, Zhang W, Zhang J, Yang J, Li K, et al.: ATP-P2X4 signaling mediates NLRP3 inflammasome activation: A novel pathway of diabetic nephropathy. Int J Biochem Cell Biol 45: 932–943, 2013 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.