

Abstract
Background
Visceral leishmaniasis (VL), caused by Leishmania donovani complex parasites, is a neglected parasitic disease that is generally fatal if untreated. Despite decades of research to develop a sensitive VL diagnostic test, definitive diagnosis of VL still mainly relies on the visualization of the parasite in aspirates from the spleen, liver or bone marrow, an invasive and dangerous process with variable sensitivity. A sensitive assay that can detect Leishmania antigen from blood samples will help confirm cause, cure or recurrence of VL.
Methods
In this study, rabbit polyclonal antibodies were raised against eight recombinant Leishmania proteins that are highly abundant in Leishmania. The antibodies were purified and labeled with biotin for developing a prototype sandwich enzyme-linked immunosorbent assay (ELISA).
Results
The ELISA for the Leishmania 40S ribosomal protein S12 detected target antigen with the highest sensitivity and specificity and could detect 1 pg of purified protein or as few as 60 L. donovani parasites. The 40S ribosomal protein S12 sandwich ELISA could detect the target antigen from Peripheral Blood Mononuclear Cell (PBMC) samples in 68% of VL patients and post-kala-azar dermal leishmaniasis (PKDL) patients, providing an estimation of parasitemia ranging from 15 to 80 amastigotes per ml of blood.
Conclusion
These results indicate that the 40S ribosomal protein S12 sandwich ELISA warrants further tests with more clinical samples of VL patients and other parasitic diseases. It is hopeful that this ELISA could become a useful tool for confirming VL diagnosis, monitoring treatment progress, disease recurrence and possibly detecting asymptomatic Leishmania infections with a high parasite load.
Keywords: Visceral Leishmaniasis, Diagnosis, ELISA, PBMC, 40S ribosomal protein S12
Background
Leishmaniasis is caused by the protozoan Leishmania parasites, which are transmitted by the bite of infected sandflies. Depending on the infecting species, Leishmania infection can cause cutaneous leishmaniasis (CL), mucocutaneous leishmaniasis (MCL) or visceral leishmaniasis (VL). VL, also known as kala-azar, is the most serious form of the disease and is frequently fatal if left untreated [1, 2]. VL is highly endemic in the Indian subcontinent, East Africa and parts of South America. An estimated 50,000 to 90,000 new cases of VL occur worldwide each year (http://www.who.int/news-room/fact-sheets/detail/leishmaniasis). Due to the AIDS epidemic, coinfection with human immunodeficiency virus (HIV) has increased VL cases in some parts of the world [3]. In addition, Leishmania infantum infection causes visceral disease in domestic dogs, which are the major vertebrate reservoirs for transmission to humans in Latin America and Southern Europe [4, 5].
VL is characterized by irregular bouts of fever, weight loss, enlargement of the spleen and liver, and anemia. However, these clinical features are not specific and can be mistaken for other common illnesses associated with fever including malaria. Moreover, infection with Leishmania does not always lead to clinical illness, asymptomatic infections are common, and it is unknown whether these individuals represent a source of transmission [6, 7]. Although there are some drawbacks associated with the current treatments, VL is a life-threatening disease that is curable with proper treatment [7]. Therefore, rapid and accurate diagnosis of visceral Leishmania infection is important for patients to receive prompt treatment, determine cure or an indication of relapse, and thus prevent further transmission of this disease [7].
Currently, diagnosis of VL is made by combining clinical symptoms with parasitological or serological tests. Assays based on detection of parasite-specific antibodies (such as the rK39 test) have proven to be efficient for VL diagnosis. The rK39 immunochromatographic test (ICT) is easy to perform, rapid and inexpensive. However, because the rK39 ICT detects antibodies, it cannot distinguish relapse cases from past infection, or active disease from asymptomatic infection and cannot be used as a test of cure [8–10]. The rK39 ICT is less effective in VL patients co-infected with HIV and is more sensitive for VL diagnosis in Asia than in Africa [8–10], though the new rK28 ICT has improved the detecting sensitivity of VL cases in Africa [11].
Nucleic acid-based diagnostics such as polymerase chain reaction (PCR) are the most sensitive method to detect the presence of parasites in clinical samples, but they are expensive and restricted to referral hospitals and research centers, though this situation could be improved with development of loop-mediated isothermal amplification (LAMP) assays where there has been recent progress [12–17]. Definitive diagnosis of VL still requires microscopic identification of the parasite in internal organs such as in spleen, liver or bone marrow aspirates, an invasive and dangerous process with varied sensitivity (53–99%) [8–10]. Therefore, development of an assay that can sensitively detect Leishmania antigen from blood or urine samples would be helpful for rapid and definitive VL diagnosis, test of cure and relapse [18–24].
Based on the hypothesis that abundant Leishmania proteins could be easier to detect than low abundance proteins, we raised rabbit polyclonal antibodies against eight Leishmania proteins previously reported to be highly abundant in Leishmania [25–27]. With these rabbit antisera, we developed a direct enzyme-linked immunosorbent assay (ELISA), and a sandwich ELISA with purified antibodies labeled with biotin for detection of the Leishmania antigens. The sandwich ELISA against the Leishmania 40S ribosomal protein S12 provided the highest sensitivity and specificity. Importantly, the sandwich ELISA could detect Leishmania 40S ribosomal protein S12 antigen in PBMC lysates prepared from VL patients and post-kala-azar dermal leishmaniasis (PKDL) patients. These results suggest that the 40S ribosomal protein S12 sandwich ELISA could represent a useful test for confirming VL diagnosis and for monitoring treatment progress and relapse.
Methods
Selection of abundant Leishmania proteins
The ten soluble and abundant Leishmania proteins we selected (Table 1) were based on previous Leishmania proteomic analysis, these proteins were present as large and dense spots in two-dimension gel electrophoresis and were identified using mass spectrometry [25, 26]. The abundance of these selected proteins was also confirmed by our own more recent proteomic study on L. donovani [27]. Most of these abundant Leishmania proteins except aldolase (LmjF.36.1260) have low homology to human proteins.
Table 1.
A list of abundant Leishmania proteins selected for production of recombinant proteins in E. coli
| Systematic Name | Simplified Name | Encoded Protein | Molecular weight (kDa) |
|---|---|---|---|
| LmjF.05.0350 | 535a | Trypanothione reductase | 53.1 |
| LmjF.05.0450 | 545 | Kinetochore related protein | 22.3 |
| LmjF.05.0830 | 583 | Methylthioadenosine phosphorylase | 33.4 |
| LmjF.13.0570 | 1357 | 40S ribosomal protein S12 | 15.6 |
| LmjF.24.1500 | 2415 | Translationally controlled tumor protein | 19.4 |
| LmjF.32.0460 | 3246a | Prostaglandin F synthase | 31.8 |
| LmjF.35.0820 | 3582 | Aspartate aminotransferase | 45.9 |
| LmjF.24.2110 | 24,211 | 3-Hydroxy-3-methyl glutaryl-CoA synthase | 55.2 |
| LmjF.36.1260 | 36,126 | Fructose-1,6-bisphosphate aldolase | 40.8 |
| LmjF.36.6760 | 36,676 | ATP synthase delta (OSCP) subunit | 28.9 |
aBecause of low yield, these proteins were not sent for production of antisera
Construction of bacterial expression vectors
For convenience of cloning, we have replaced the sequence (Nde I to Not I) containing the S. Tag and the multiple cloning sites in pET29 bacterial expression vector (Novagen) with following sequence containing a His-Tag sequence and new multiple cloning sites (Hind III, Kpn I, EcoR I, BamH I, Bgl II and Not I): CATATGGCACATCACCACCACCATCACAAGCTTGGTACCGAATTCGGATCCAGATCT GTAGCGGCCGC. We re-named the modified pET29 vector as pET29w. Accordingly, the primers with corresponding restriction enzyme site added at the 5′ end were designed and used to amplify the gene sequences of these abundant Leishmania proteins by PCR using L. donovani genomic DNA as the template. The Leishmania gene PCR products with expected sizes were digested with restriction enzymes and cloned into the corresponding sites of pET29w bacterial expression vector. A list of these primer pairs is shown in Table 2.
Table 2.
A list of primers used to amplify genes encoding abundant Leishmania proteins
| Systematic Name | Primer pairs used for PCR | Gene size (bp) |
|---|---|---|
| LmjF.05.0350 | 5’ cccaagcttaccATGTCCCGCGCGTACGACCTCG | 1476 |
| 5’ ggaagatctgtGAGGTTGCTGCTCAGCTTTTCG | ||
| LmjF.05.0450 | 5’ cccaagcttaccATGGCTGACGAAGGCGCTATAGA | 591 |
| 5’ ggaagatctgtCTTCTTCGTCGTGGCCTTCACAG | ||
| LmjF.05.0830 | 5’ cccaagcttaccATGTACGGCAACCCGCACAAGGA | 921 |
| 5’ ggaagatctgtCGGGGCGAACTGCGGGTACTTGC | ||
| LmjF.13.0570 | 5’ cggggtaccATGGCTGAGGAAACCGTCCGTGTTG | 426 |
| 5’ cgcggatccgtGTGCAGCTGAGACAGCAGGTAGTCC | ||
| LmjF.24.1500 | 5’ cggggtaccATGAAGATCTTCAAGGACGTGCTG | 513 |
| 5’ cgcggatccgtGACGCGCTCGCCCTTCAAGCCATC | ||
| LmjF.32.0460 | 5’ cccaagcttaccATGGCTGTTAAGTGCACGCACG | 840 |
| 5’ ggaagatctgtCTTGCGCTCGGTTGGGAAGAAGG | ||
| LmjF.35.0820 | 5’ cccaagcttaccATGTCCACGCAGGCGGCCATG | 1239 |
| 5’ ggaagatctgtCTCACGATTCACATTGCGCACA | ||
| LmjF.24.2110 | 5’ cggggtaccATGATGCGCAACACCTGTCTTAG | 1506 |
| 5’ cgcggatccgtCTGGATGTAGCGGTAGTACTCA | ||
| LmjF.36.1260 | 5’ cccaagcttaccATGTCGCGTGTGACGATCTTTCAG | 1116 |
| 5’ cgcggatccgtATAGATGTTGCCTTTGACGTACAG | ||
| LmjF.36.6760 | 5’ cggggtaccATGTTCCGCCGTCTCTCCGTG | 777 |
| 5’ cgcggatccgtAACACCGCTCTTGAGCTCCTCG |
Expression and purification of recombinant Leishmania proteins in E. coli BL21 or Rosetta cells
We isolated the recombinant Leishmania proteins from bacteria by following the manufacturer’s protocol (Novagen pET system manual). Some of these recombinant proteins formed Inclusion Bodies after Isopropyl β-D-1-thiogalactopyranoside (IPTG) induction of the culture and were first urea-solubilized before using Ni-NTA Agarose resin (Qiagen) to purify. Depending on the protein, we obtained 2–10 mg of each recombinant protein from 1 L shake cultures for polyclonal antibody production.
Production of rabbit polyclonal antibodies
The rabbit polyclonal antibodies (antisera) against these abundant Leishmania proteins were produced by Syd Labs, Inc. (Natick, MA, USA). The detailed procedures can be found in the website of Syd Labs, Inc. (https://www.sydlabs.com). One or two rabbits were immunized for each Leishmania recombinant protein with Freund’s adjuvant (SIGMA, USA). About 0.5 ml of pre-immune rabbit serum and 40 to 80 ml of antiserum with ELISA titers > 1:20,000 were obtained within 4 months for each antigen. The antisera were kept at 4 °C for current use and in a − 20 °C freezer for longer storage.
Leishmania strains and macrophage infection
The L. donovani 1S/Cl2D and Leishmania major Friedlin V9 strains used in this study were routinely cultured at 27 °C in M199 medium (Cat # M0393, SIGMA) supplemented with 10% heat-inactivated fetal bovine serum, 40 mM HEPES (pH 7.4), 0.1 mM adenine, 5 mg l− 1hemin, 1 mg l− 1 biotin, 1 mg l− 1 biopterine, 50 U ml− 1 penicillin and 50 μg ml− 1 streptomycin. Cultures were passaged to fresh medium in a 20-fold dilution once a week. Infection of cultured macrophages was performed as described [28].
Western blot analysis of abundant Leishmania proteins
Whole cell lysates were prepared in sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer from L. major promastigotes, L. donovani promastigotes, L. donovani axenic amastigotes, and human H1299 epithelial cells. After boiling in water for 3 min and centrifuging at 15,000 RPM (Revolutions per minute) for 10 min, the cell lysate supernatants were separated in SDS-PAGE gel and subjected to Western blot analysis [29]. Rabbit antiserum was diluted 1:1000 for Western blot analysis to detect target proteins in Leishmania cell lysates (4 × 107 Leishmania cells per lane).
Isolation of peripheral blood mononuclear cells (PBMCs) from clinical samples
The diagnosis for most of the VL patients at the Rejandra Memorial Research Institute of Medical Sciences (RMRIMS), Patna, Bihar was confirmed by spleen or bone marrow biopsies. PKDL cases at RMRIMS were confirmed by skin smear microscopy for LD bodies (Amastigotes). The healthy controls were all rK39-negative from the out-patient department for regular check-up. Total 14 VL patients, 5 likely VL cases which had typical VL symptoms and were rK39-positive but biopsy-negative, 3 PKDL patients and 12 healthy controls were recruited. The 14 VL cases included 10 males with age range from 3 to 52 years old and 4 females with age range 18 to 35; the 5 likely VL cases were 3 males with age range 14 to 45 and 2 females with age 40 and 45; the 3 PKDL patients included 1 male aged 30, and 2 females aged 18 and 40; the 12 healthy controls were 7 males and 5 females with age range from 24 to 34. Blood (5 ml per patient) drawn from patients before treatment were utilized for preparation of PBMCs with Ficoll-density separation [30]. PBMC lysates were prepared by lysing cells in 1% NP-40 in PBS with proteinase inhibitors (cOmplete Cocktail Tablets, SIGMA) at the concentration of 1 × 107 cells/ml. After incubation on ice for 30 min with occasional mixing, the lysates were centrifuged at 4 °C at high speed (15,000 RPM) for 15 min, and the supernatant was kept and stored at -20 °C until use.
Direct ELISA
L. donovani promastigote lysate was prepared by lysing cells in 1% NP-40 in PBS (Phosphate-buffered saline) with proteinase inhibitors at the concentration of 4 × 108 cells/ml. After centrifugation at 4 °C at high speed for 15 min, the supernatant was stored at -20 °C until use.
The ELISA plate (Immulon cat#: 62402–972 from VWR) was coated with 50 μl (per well) cell lysate diluted in carbonate/bicarbonate buffer (1.89 g NaHCO3 and 0.954 g Na2CO3 in 500 ml H2O, pH 9.6) and incubated at 4 °C in a moist chamber overnight. The plate was washed 2 times with 200 μl wash buffer (0.05% Tween 20 in PBS) then blocked with 200 μl/well blocking buffer (5% nonfat dry milk in 0.1% PBS/T) for 1 h at 37 °C (covered with an adhesive plastic). The plate was washed 2 times with wash buffer, and 100 μl rabbit antiserum in 1:2000 dilution in blocking buffer was added to each well for 1.5 h at 37 °C or overnight at 4 °C. The plate was washed 3 times with wash buffer and 100 μl of horseradish peroxidase (HRP)-linked anti-rabbit antibody (ECL NA934V 1:5000 in blocking buffer) was added into each well for 1 h at 37 °C. The plate was washed 4 times with wash buffer followed by adding 50 μl/well of the HRP substrate TMB (eBioscience). After 10 min incubation at room temperature, the reactions were stopped with 25 μl of 1 N H2SO4. The color change in wells was read at 450 nm absorbance.
Purification and labelling of rabbit anti-Leishmania protein antibodies
To set up the sandwich ELISA, Immunoglobulin G (IgG) was first purified from the rabbit antisera with the Melon Gel IgG Purification System (Cat # 45206, Thermo Fisher Scientific), which purifies antibodies from serum by removing non-relevant proteins. After dilution with the Melon gel purification buffer (1:10) and passing through the Melon gel purification column, 100 μl of serum generated 1000 μl of purified IgG. One-half of the purified IgGs were biotin labeled using the Thermo Scientific EZ-Link Sulfo-NHS-SS-Biotin (sulfosuccinimidyl-2-[biotinamido]ethyl-1,3-dithiopropionate, Cat # 21331). 7 μl Sulfo-NHS-SS-Biotin (6 mg/ml, freshly prepared) was added into 500 μl purified rabbit IgG and labeled at 4o C overnight. Free Sulfo-NHS-SS-Biotin was removed from the labeling reaction by dialysis in PBS.
Sandwich ELISA
A 96-well ELISA plate was coated with 100 μl/well of capture antibody in carbonate/bicarbonate coating buffer (i.e. 0.5 μl capture ab + 100 μl coating buffer; 1:200 dilution). The plate was sealed and incubated overnight at 4 °C in a moist chamber. Wells were aspirated and washed 5 times with 250 μl/well wash buffer (0.05%Tween 20 in PBS) allowing time for soaking (~ 1 min) during each wash step. Absorbent paper was used to remove any residual buffer. Wells were blocked with 200 μl/well of 2% bovine serum albumin (BSA) in PBS, incubated at 37 °C for 2 h, and aspirated and washed 2 times. A 2-fold serial dilution of the standards (L. donovani promastigote lysate alone or mixed with the healthy control PBMC lysate) was performed with assay buffer (1% BSA in wash buffer) with a final volume of 100 μl per well to generate the standard curve. PBMC lysates of clinical samples (100-250 μl) prepared as above were added into each well. The plate was covered, incubated overnight at 4 °C, aspirated free of buffer, and washed 5 times. Detection antibody (100 μl/well) diluted in assay buffer (i.e., 0.25 μl Detection ab-Biotin + 100 μl Assay buffer; 1:400 dilution) was added per well, and the plate was sealed and incubated at 37 °C for 2 h. Wells were aspirated and washed 5 times, and 100 μl/well of Avidin-HRP (eBioscience) diluted in assay buffer (1:250 dilution) was added and incubated at room temperature for 30 min to 1 h. Wells were aspirated and washed 10 to 14 times with the wells soaking for 2 min for each wash. Substrate solution (TMB; 100 μl/well) was added, and plates incubated at room temperature for 15 min. The reactions were stopped by adding 50 μl/well of Stop Solution (2 N H2SO4), and absorbance was read at 450 nm.
Generation of monoclonal antibodies against peptides of L. donovani 40S ribosomal protein S12
Monoclonal antibodies against L. donovani 40S ribosomal protein S12 peptides were generated by Abmart (Shanghai) Co. Ltd. However, none of these anti-peptide monoclonal antibodies could recognize the native L. donovani 40S ribosomal protein S12 (See discussion). The twelve peptide sequences used to generate the monoclonal antibodies are as follows: NVVVDVAPES,EETVRVEVPA,EVPAVEENVV,VAGEVTKTLK,QKALEANGLV,FGERTKALDY,ESLEDAVRIV,NVEEREKLAQ,TALAKQGNID,VRGLSEVART,QWAGLVRRDV,EDEEYKKLVT.
Results
Expression and purification of recombinant Leishmania proteins in E. coli
We hypothesized that abundant Leishmania proteins would be more easily detected from clinical samples than low abundance proteins. Therefore, we selected ten soluble and abundant Leishmania proteins that were previously identified by proteomic analysis using two-dimension gel electrophoresis and mass spectrometry [25, 26] for production as recombinant proteins in E. coli (Table 1). The coding sequences of these abundant Leishmania proteins were amplified by PCR from L. donovani genomic DNA and cloned into the pET29w expression vector, placing the His-tag at the N-terminus of the expressed recombinant proteins. The His-tagged proteins were purified from E. coli lysates on nickel-charged resin columns and the purity was verified by SDS-PAGE (Fig. 1). Eight of the recombinant proteins that met the required yield and purity were used to immunize rabbits to produce polyclonal antisera.
Fig. 1.

Production and purification of the recombinant abundant Leishmania proteins. The eight abundant Leishmania protein genes were PCR amplified from L. donovani and cloned into the pET29w expression vector. Recombinant proteins containing an N-terminal His-tag were expressed in E. coli BL21 and purified with Ni-NTA agarose. Purity was evaluated by SDS-PAGE and Coomassie blue staining. These recombinant Leishmania proteins were sent out for rabbit polyclonal antibody production
Western blot analysis of antisera against Leishmania and human cells
To determine whether the resulting antisera could specifically recognize the Leishmania proteins, whole cell lysates prepared from L. major promastigotes, L. donovani promastigotes, L. donovani axenic amastigotes, and human H1299 epithelial cells were separated in SDS-PAGE gel and subjected to Western blot analysis. As shown in Fig. 2, all the rabbit antisera could detect the corresponding Leishmania proteins of the correct size. Importantly, Leishmania proteins were equally well expressed in L. major promastigotes, L. donovani promastigotes, and L. donovani axenic amastigotes, though some had one or multiple detectable non-specific bands depending on the antisera. These additional protein bands could be the partially degraded protein products (smaller bands for 545, 2415 and 36,676), products with post translation modifications or oligomers (larger bands for 1357 and 3582 proteins), or cross reaction with other proteins (the multiple additional bands seen for 583, 36,126 and 24,211). Except for anti-3582 serum, these antisera had no or low cross reactivity to human proteins in H1299 cell lysate.
Fig. 2.
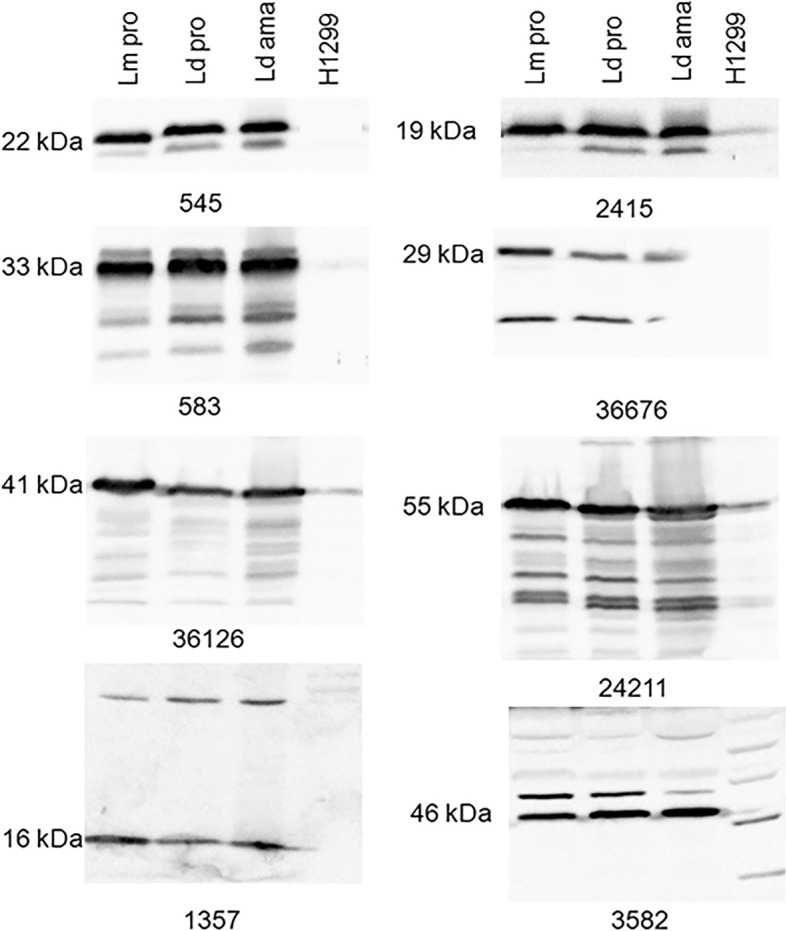
Western blot analysis show all eight rabbit antisera generated in this study specifically detected the corresponding Leishmania proteins. The lysate of 4 × 107 Leishmania cells or 1.5 × 105 Human H1299 cells was loaded into each lane and separated on SDS-PAGE. The specific Leishmania proteins were detected with rabbit antisera in 1:1000 dilution and goat anti-rabbit IgG labeled with horseradish peroxidase. Lm pro: L. major promastigotes; Ld pro: L. donovani promastigotes; Ld ama: L. donovani axenic amastigotes; H1299: Human H1299 cell line. Note: similar amounts of these abundant Leishmania proteins were detected in L. major, L. donovani promastigotes, and L. donovani axenic amastigotes, while these sera exhibited variable cross reactivities to human proteins in the H1299 cell lysate
ELISA analysis of antisera against L. donovani proteins
To establish the sensitivity of these rabbit antisera in detecting L. donovani antigens in an ELISA format, microtiter plates were coated with L. donovani promastigote lysates (ranging from 10 to 1 million promastigotes per well). Rabbit antisera were diluted 1: 2000 and added to the plate followed by the HRP-linked anti-rabbit antibody and the HRP substrate TMB, and the color change at 450 nm absorbance were measured. As shown in Fig. 3a, six of the eight antisera could detect the corresponding antigen from the L. donovani cell lysate in this assay (36,676 and 3582 were the exceptions). Antiserum for 1357 (40S ribosomal protein S12) showed the highest sensitivity with a detection limit of approximately 500 L. donovani promastigotes per well.
Fig. 3.
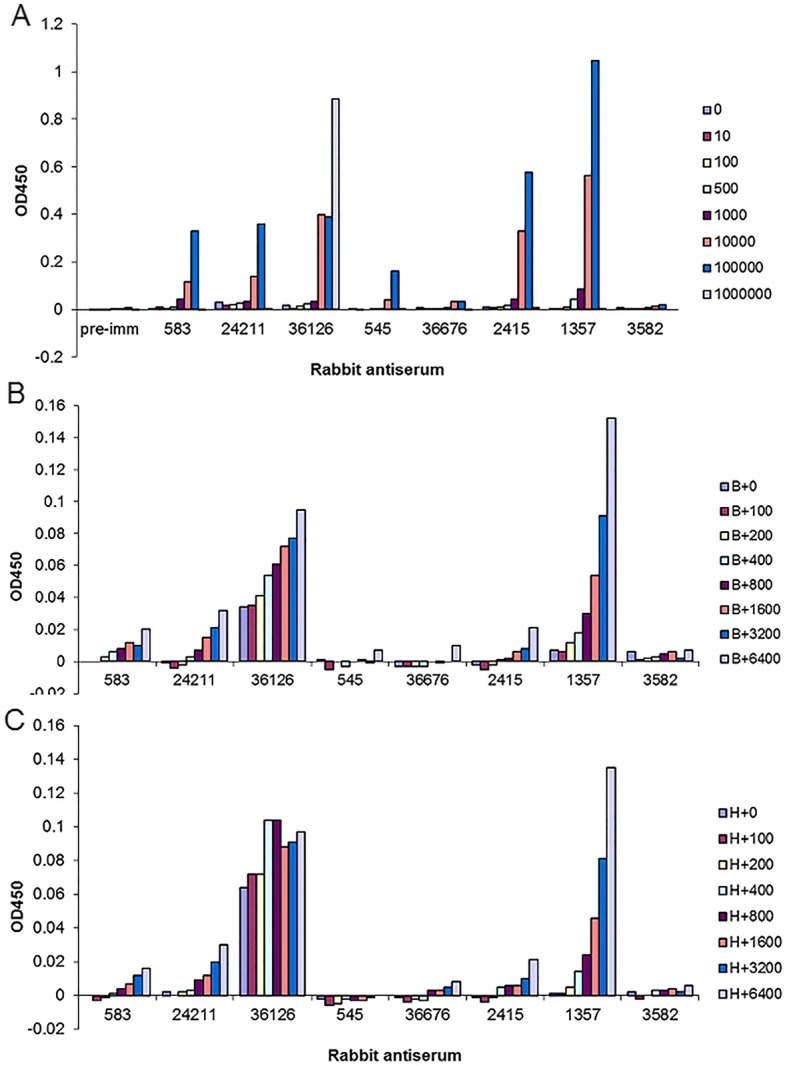
The rabbit serum against Leishmania 40S ribosomal protein S12 antigen (1357) displayed the highest sensitivity and specificity in a direct ELISA. To determine whether these rabbit antisera can detect the corresponding antigen in L. donovani cell lysate in traditional ELISA, ELISA plates were directly coated with L. donovani promastigote lysate ranging from 10 to 1 million promastigotes per well as indicated in a. Each rabbit antiserum in 1:2000 dilution was added to the plate, followed by adding the HRP-linked anti-rabbit antibody and HRP substrate TMB. The color change proportional to the level of target antigen detected was measured at A450nm. b and c To determine whether these antisera are able to detect Leishmania antigen in L. donovani-infected macrophages, the lysate of L. donovani-infected B10R-mouse macrophages were mixed with non-infected B10R macrophage lysate (b) or human H1299 cell lysate (c) in different ratios (denoted as B or H in the figure legends, respectively) and coated on the plate so that each well contained the same amount of cell lysate from a total 3000 (infected plus non-infected) mammalian cells where the total number of amastigotes ranged from 100 to 6400 per well, as indicated; the plate was then hybridized with these rabbit antisera as a
To determine whether these antisera could detect Leishmania antigen in L. donovani-infected macrophages, mouse macrophage B10R cells were infected with L. donovani. The lysates of L. donovani-infected macrophages were mixed with non-infected macrophage lysates in different ratios and coated on the 96-well plate so that each well contained the same amount of cell lysate from 3000 macrophages (infected plus non-infected) with amastigote numbers ranging from 100 to 6400 per well. As shown in Fig. 3b, the anti-1357 antiserum could detect down to approximately 200 amastigotes under these conditions and exhibited a L. donovani amastigote, dose-dependent increase in the signal. Similarly, signal was detected with human H1299 cell lysates mixed with L. donovani infected macrophages down to ~ 200 amastigotes (Fig. 3c). In contrast, although relatively strong signals were detected for the antiserum to Leishmania aldolase (36126), a significant background was also observed for this anti-36,126 serum (Fig. 3b and c), likely due to the high sequence identity for the Leishmania and human aldolase proteins. Taken together, out of the eight antisera, only the anti-1357 serum to the 40S ribosomal protein S12 gave applicable results and could detect approximately 200 amastigotes per 3000 macrophage lysates per well.
The weak-reacting signals for antigens 36,676, 3582 and 545 could have been due either to poor binding of these Leishmania proteins to the plate or poor recognition of these non-denatured or partially denatured proteins. All the antisera except 36,676 and 3582 showed good increases in signal intensity as the number of L. donovani promastigotes increased in the lysates, at least up to 100,000 promastigotes (Fig. 3a). However, only 36,126 antiserum produced a signal using 1 million L. donovani cells (see below).
It is noteworthy that a Leishmania sandwich ELISA developed by Ferrua et al. could detect circular Leishmania antigen in sera of L. infantum infected visceral leishmaniasis patients with sensitivity like that shown in Fig. 3 [18]. Since the maximum binding capacity of a 96-well ELISA plate well is about 1 μg protein, it is necessary to dilute the Leishmania lysate to a concentration of 20 μg/ ml (1 μg/50 μl/ well) in coating buffer. If the protein concentration is higher than 1 μg/well, then all the binding sites will be saturated and other proteins could compete with the target antigen for binding sites. This could result in loss of signal, consistent with our observation (Fig. 3a) at lysates from one million L. donovani promastigotes containing about 4 μg protein which could compete with target antigen binding [18].
Sandwich ELISA for Leishmania 40S ribosomal protein S12 antigen
Since the binding capacity is limited in the direct ELISA, it could be difficult to detect the antigen if present at low levels or if they fail to adhere to the plastic. In this case, a sandwich (or capture) ELISA may be more sensitive since more of the antigen can be immobilized on the plate.
In this study the rabbit polyclonal antibodies were used as both the capture antibody and the detection antibody in the sandwich ELISA. Total IgG were purified from each of the rabbit antisera, unlabeled purified IgGs (1:2000 dilution) were coated on the ELISA plate as protein-capture antibodies, and one-half of the purified IgG was biotinylated as detection antibody. After blocking and washing, the L. donovani promastigote lysate ranging from 100 to 1 million cells was added into each well of the ELISA plate. The captured Leishmania antigen was detected with the biotin-labelled IgGs, avidin-HRP, and TMB substrate. As shown in the sandwich ELISA of Fig. 4, only the 40S ribosomal S12 antigen (1357) displayed a dose-dependent signal that became saturated beyond an input of 100,000 promastigotes.
Fig. 4.
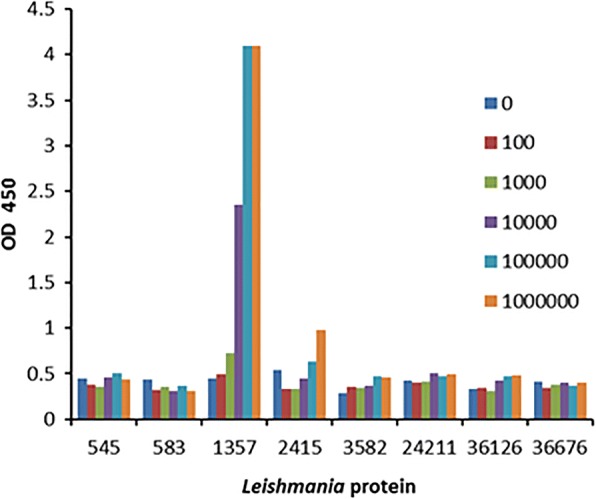
Leishmania 40S ribosomal protein S12 antigen (1357) sandwich ELISA had the highest sensitivity and strongest signal among the sandwich ELISAs prepared from the eight rabbit antisera. Unlabeled capture IgG and biotinylated detection IgG were prepared from each of the rabbit sera and tested in sandwich ELISAs. The capture IgG was used at a 1:2000 dilution. Whole cell lysates of 100 to 1 million L. donovani promastigotes were added to the wells of the 96-well ELISA plate. Captured antigen was detected with the homologous biotinylated IgG, Avidin-HRP, and TMB substrate, and absorbance measured at 450 nm
The 40S ribosomal protein S12 (1357) sandwich ELISA was optimized by further dialysis to remove any free biotin conjugates, and various dilutions of capture antibody and detection antibody were tested to improve the signal to noise ratio. After optimization it was possible to detect as little as 1 pg of purified recombinant 40S ribosomal protein S12 (Fig. 5a) and as few as 60 L. donovani parasites either tested alone or mixed with 500,000 human PBMCs from healthy donors (Fig. 5b), which represents PBMCs from about 0.75 ml of blood.
Fig. 5.
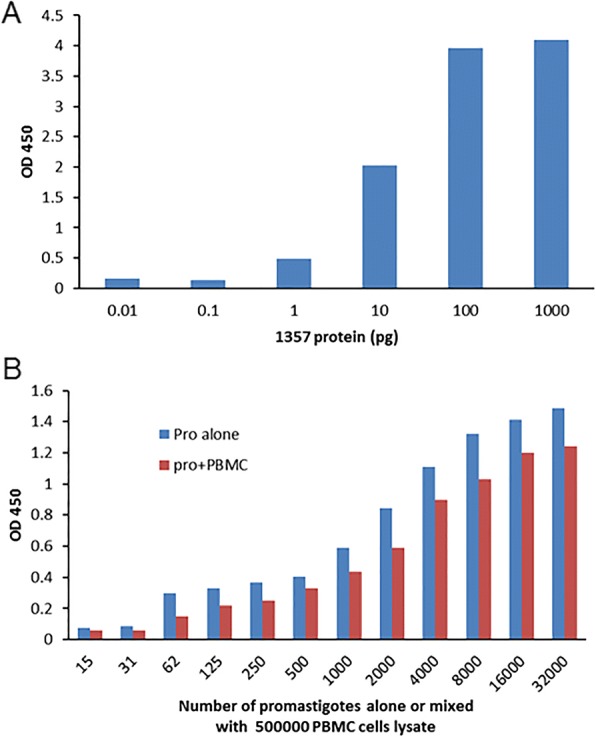
Leishmania 40S ribosomal protein S12 sandwich ELISA displayed high sensitivity and specificity. This ELISA assay can detect as low as 1 pg of purified recombinant 40S ribosomal protein S12 (a), the lysate of 60 Leishmania donovani parasites alone or mixed with 500,000 PBMCs prepared from healthy controls (b), which equals about 60 Leishmania amastigotes in 750 μl blood. Note: the detecting signal intensities were slightly lower in spiked PBMC lysate due to the matrix effect
Analysis of clinical samples with the Leishmania 40S ribosomal protein S12 (1357) sandwich ELISA
To determine whether this sandwich ELISA could detect the Leishmania 40S ribosomal protein S12 antigen from the clinical samples, PBMCs were isolated from 12 healthy controls, 14 VL patients, 3 post-kala-azar dermal leishmaniasis (PKDL) patients, and 5 likely VL cases before treatment at the Rejandra Memorial Research Institute of Medical Sciences (RMRIMS), Patna, Bihar, India. Leishmania donovani (LD) bodies (amastigotes) in the spleen or bone marrow biopsies were positive for the 14 confirmed VL patients, and negative for the 5 likely VL cases that were, however, positive on rK39 Rapid strip test and displayed clinical symptoms of VL including prolonged fever and enlarged spleen (Fig. 6b). All three PKDL cases were confirmed by skin smear microscopy for LD bodies and the 12 healthy controls were from the out-patient department and rK39 negative.
Fig. 6.
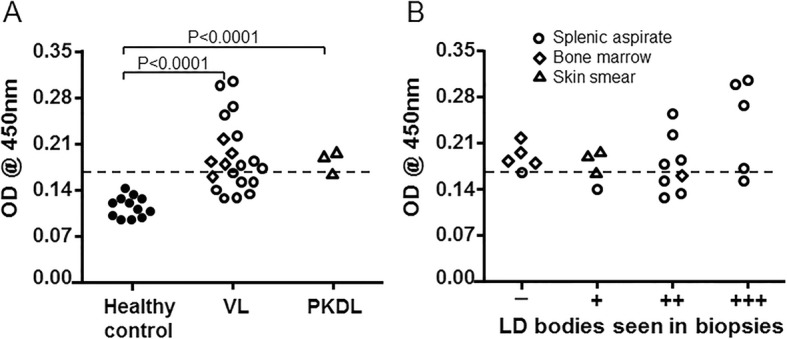
Leishmania 40S ribosomal protein S12 sandwich ELISA was able to detect the target antigen specifically in the clinical samples. a This novel ELISA detected Leishmania 40S ribosomal protein S12 antigen in lysates of PBMCs isolated from Visceral Leishmaniasis (VL, including 5 likely VL cases) and post-kala-azar dermal leishmaniasis (PKDL) patients before treatment. The A450 value differences are statistically significant (Student’s t-test); P < 0.0001 between the healthy controls and VL or PKDL patients. The dashed line is the standard cut-off value of the mean of healthy control +3SD (standard deviation). b This ELISA could detect the target antigen in lysates of PBMCs isolated from patients who were LD-body (Amastigote) negative in bone marrow or splenic biopsies, and the absorbance values for the target antigen were roughly correlated to the number of LD bodies seen in the spleen biopsies of VL patients. Open circles represent VL and likely VL cases examined by splenic aspirate; Open diamond, VL and likely VL case examined by bone marrow biopsy; Open triangle, PKDL patient confirmed by skin smear microscopy; Filled circle, healthy control
As shown in Fig. 6a, the A450 value differences were statistically significant (P < 0.0001) between these VL patients (median value, 0.191) and healthy controls (median value, 0.117) or between PKDL patients (median value, 0.185) and healthy controls. It is also noteworthy that while the ELISA values correlate roughly with the score of parasite levels from splenic biopsies (Fig. 6b), this ELISA was also capable of detecting the target 40S ribosomal protein S12 antigen in the PBMC samples of the 5 likely VL cases who were LD-body negative (Fig. 6b). If using the mean of the healthy controls +3SD (standard deviation) as the standard cut-off value (dashed line), 68% (15/22) of the rK39-positive cases (14 VL patients plus 5 likely VL cases) and 3 PKDL patients can be identified as Leishmania 40S ribosomal protein S12 antigen positive. Considering the sensitivity of this ELISA (Fig. 5b), this indicates that these VL patients could have approximately 15–80 amastigotes per ml of their blood.
Discussion
An ELISA was developed to detect Leishmania proteins based on the assumption that abundant proteins could be more easily detected than proteins of low abundance in clinical samples. Antibodies raised against the 40S ribosomal protein S12 antigen (1357) outperformed all the other antisera tested and could detect the presence of L. donovani in PBMCs from VL patients. Although all the generated rabbit antisera could bind specifically to the corresponding Leishmania proteins in denatured form by Western blot analysis, antiserum to the 40S ribosomal S12 antigen was far superior to the other generated antibodies at recognizing their corresponding native Leishmania proteins in the ELISA. One explanation for this is that these E. coli expressed recombinant proteins were in a different, possibly denatured form that differs from that of the native Leishmania proteins. Moreover, we have recently found that antisera raised against some additional abundant Leishmania proteins (LdBPK_140910.1; LdBPK_250740.1; LdBPK_363750.1; LdBPK_321910.1 and LdBPK_180690.1) also failed to recognize the native Leishmania proteins despite working in Western blot analysis (data not shown). Therefore, future attempts should consider different methods for production and purification of recombinant proteins for generation of antibodies. Different adjuvants could also be considered to produce antibodies that recognize the native Leishmania proteins. Another relevant observation from this study is that we synthesized twelve peptide sequences (ten amino acid each) of the 40S ribosomal S12 antigen to generate anti-peptide monoclonal antibodies. However, possibly due to the native protein folding which may hide these epitopes, none of these anti-peptide monoclonal antibodies recognized the native 40S ribosomal protein S12 despite binding to the cognate peptides (See these peptides sequences in Methods).
The 40S ribosomal protein S12 antigen (1357) antiserum could be used to develop a sandwich ELISA that could detect as low as 1 pg purified recombinant 40S ribosomal protein S12 and approximately 60 Leishmania donovani parasites. Notably, this ELISA was able to detect the Leishmania 40S ribosomal protein S12 antigen in 68% PBMC samples of VL and PKDL patients including the likely VL cases that were LD-body negative in splenic or bone marrow biopsies, but rK39 positive with clinical symptoms. This ELISA also provided a quantitative estimation of the parasitemia for these positive cases. This suggests that this ELISA could be further developed to detect parasitemia for test of cure and relapse and could also be helpful to confirm VL diagnosis of equivocal cases.
While this project was in progress, three Leishmania antigen detection tests were reported including a triple Leishmania protein detection ELISA (DetectoGen Inc., USA), a Leishmania antigen ELISA (Kalon Biologicals Ltd., UK) and a Leishmania Antigen Detect™ ELISA (Infectious Disease Research Institute, USA) [20–23]. All these tests detect Leishmania antigens with slight differences in sensitivity from urine samples of VL patients, and all could potentially be used to monitor treatment progress. The Leishmania antigen ELISA and Leishmania Antigen Detect™ ELISA were developed using polyclonal antibodies raised against the whole Leishmania donovani cell lysate [23]. The DetectoGen ELISA was developed by raising polyclonal antibodies against three Leishmania proteins identified in VL patients’ urine samples [20]. Interestingly, like the 40S ribosomal protein S12 (16 kDa), all these three target proteins used in the DetectoGen Inc., capture ELISA (iron superoxide dismutase, 22 kDa; tryparedoxin, 17 kDa; and nuclear transport factor 2, 14 kDa) are also small and abundant Leishmania proteins. The 40S ribosomal protein S12 ELISA being reported in our study has similar sensitivity to the triple protein ELISA (DetectoGen), and is approximately two times more sensitive than the whole cell lysate (WCL) antigen ELISAs. The DetectoGen ELISA has a detection limit ranging from 4 to 100 pg/ml of the target antigens [20–22]. The Detect™ ELISA detection limit is 4 ng/ml, equivalent to about 100 Leishmania parasites per well [23]. We have yet to test whether the 40S ribosomal protein S12 antigen is present in VL patient urine but, if it is, it would be worth determining whether the 40S ribosomal protein S12-IgG complements those in the DetectoGen test, which is also based on anti-recombinant protein antibodies.
Compared with capture ELISAs based on individual antigens, the advantages of WCL capture ELISAs are (1) the ease of preparing the WCL antigens and (2) the greater range of Leishmania proteins (native or denatured) present in various clinical samples that could be recognized by the polyclonal antibodies, potentially resulting in a more sensitive test. In fact, the WCL ELISA may detect predominantly abundant Leishmania proteins that are also present in the urine samples. Based on its genome sequence, Leishmania has approximately 8000 potential proteins, and many of these proteins could significantly increase the likelihood of cross reactions to human proteins, which would increase the background signal. Therefore, it would be useful to compare all the available Leishmania antigen detection ELISAs side-by-side with various clinical samples including urine, blood and PBMCs. With this information, it may be possible to further improve the sensitivity and specificity for Leishmania antigen detection.
Conclusions
Progress is described in the development of a sandwich ELISA for detecting Leishmania 40S ribosomal protein S12 in PBMCs of VL and PKDL patients. Though more clinical samples of VL patients and other parasitic diseases such as malaria and trypanosomiasis should be tested to verify its sensitivity and specificity, such a test would be useful for confirming VL diagnosis, relapse cases and monitoring treatment progress. With some refinement and/or in combination with other recently described Leishmania antigen-detecting reagents [20], more sensitive Leishmania antigen detection tests could be developed.
Acknowledgements
We would like to thank all the VL and PKDL patients and Healthy donors for providing blood samples for this study.
Funding
This work was supported by grants from the Bill and Melinda Gates Foundation (OPP49932 and OPP1084251) to SGR and the Canadian Institutes of Health Research to GM. These granting agencies were not involved in data analysis or interpretation.
Availability of data and materials
The datasets and materials used and/or analyzed during the current study will be available from the corresponding author on reasonable request.
Abbreviations
- BSA
Bovine serum albumin
- ELISA
Enzyme linked immunosorbent assay
- IgG
Immunoglobulin G
- IPTG
Isopropyl β-D-1-thiogalactopyranoside
- PBMC
Peripheral blood mononuclear cell
- PBS
Phosphate-buffered saline
- PCR
Polymerase chain reaction
- PKDL
Post-kala-azar dermal leishmaniasis
- rK39 ICT
rK39 immunochromatographic test
- SDS-PAGE
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
- VL
Visceral leishmaniasis
Authors’ contributions
Conceived and designed the experiments: GM, WWZ. Performed the experiments: WWZ, AKG, RM, JW, AP, PL. Analyzed the data: WWZ, AKG and GM. Provided research reagents: MN, PD, SGR and GM. Supervised the research: GM, PD, SGR. Wrote the paper: WWZ, RFH and GM. All authors read and approved the final manuscript.
Ethics approval and consent to participate
This study was approved by the Institutional Review Boards and Human Ethical Committee of the Rajendra Memorial Research Institute of Medical Sciences (Patna, India) under the project head of “Grand Challenges Canada”. Human PBMCs from whole blood were isolated from healthy and VL/PKDL volunteers after written, informed consent as per standard guidelines.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Wen-Wei Zhang, Email: wenwei.zhang@mcgill.ca.
Ayan Kumar Ghosh, Email: akg.rkmvp@gmail.com.
Raodoh Mohamath, Email: Raodoh.Mohamath@idri.org.
Jacqueline Whittle, Email: jacqueline.whittle@idri.org.
Alessandro Picone, Email: alessandro.picone@idri.org.
Patrick Lypaczewski, Email: patrick.lypaczewski@mail.mcgill.ca.
Momar Ndao, Email: momar.ndao@mcgill.ca.
Randall F Howard, Email: Randall.Howard@idri.org.
Pradeep Das, Email: drpradeep.das@gmail.com.
Steven G Reed, Email: Steven.Reed@idri.org.
Greg Matlashewski, Phone: 514-398-7479, Email: greg.matlashewski@mcgill.ca.
References
- 1.Alvar J, Vélez ID, Bern C, Herrero M, Desjeux P, Cano J, Jannin J, den Boer M. Leishmaniasis worldwide and global estimates of its incidence. PLoS One. 2012;7:e35671. doi: 10.1371/journal.pone.0035671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ready PD. Epidemiology of visceral leishmaniasis. Clin Epidemiol. 2014;3(6):147–154. doi: 10.2147/CLEP.S44267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alvar J, Aparicio P, Aseffa A, Den Boer M, Cañavate C, Dedet JP, et al. The relationship between leishmaniasis and AIDS: the second 10 years. Clin Microbiol Rev. 2008;21:334–359. doi: 10.1128/CMR.00061-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belazzoug S. Leishmaniasis in Mediterranean countries. Vet Parasitol. 1992;44:15–19. doi: 10.1016/0304-4017(92)90139-Z. [DOI] [PubMed] [Google Scholar]
- 5.Quinnell RJ, Courtenay O. Transmission, reservoir hosts and control of zoonotic visceral leishmaniasis. Parasitology. 2009;136:1915–1934. doi: 10.1017/S0031182009991156. [DOI] [PubMed] [Google Scholar]
- 6.Banu SS, Meyer W, Ahmed BN, Kim R, Lee R. Detection of Leishmania donovani in peripheral blood of asymptomatic individuals in contact with patients with visceral leishmaniasis. Trans R Soc Trop Med Hyg. 2016;110(5):286–293. doi: 10.1093/trstmh/trw027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singh OP, Hasker E, Boelaert M, Sundar S. Elimination of visceral leishmaniasis on the Indian subcontinent. Lancet Infect Dis. 2016;16(12):e304–e309. doi: 10.1016/S1473-3099(16)30140-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sundar S, Rai M. Laboratory diagnosis of visceral leishmaniasis. Clin Diagn Lab Immunol. 2002;9:951–958. doi: 10.1128/CDLI.9.5.951-958.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Srivastava P, Dayama A, Mehrotra S, Sundar S. Diagnosis of visceral leishmaniasis. Trans R Soc Trop Med Hyg. 2011;105:1–6. doi: 10.1016/j.trstmh.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sakkas H, Gartzonika C, Levidiotou S. Laboratory diagnosis of human visceral leishmaniasis. J Vector Borne Dis. 2016;53(1):8–16. [PubMed] [Google Scholar]
- 11.Pattabhi Sowmya, Whittle Jacqueline, Mohamath Raodoh, El-Safi Sayda, Moulton Garner G., Guderian Jeffrey A., Colombara Danny, Abdoon Asem O., Mukhtar Maowia M., Mondal Dinesh, Esfandiari Javan, Kumar Shailendra, Chun Peter, Reed Steven G., Bhatia Ajay. Design, Development and Evaluation of rK28-Based Point-of-Care Tests for Improving Rapid Diagnosis of Visceral Leishmaniasis. PLoS Neglected Tropical Diseases. 2010;4(9):e822. doi: 10.1371/journal.pntd.0000822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khan MG, Bhaskar KR, Salam MA, Akther T, Pluschke G, Mondal D. Diagnostic accuracy of loop-mediated isothermal amplification (LAMP) for detection of Leishmania DNA in buffy coat from visceral leishmaniasis patients. Parasit Vectors. 2012;3(5):280. doi: 10.1186/1756-3305-5-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khan MG, Bhaskar KR, Kikuchi M, Salam MA, Akther T, Haque R, et al. Comparison of PCR-based diagnoses for visceral leishmaniasis in Bangladesh. Parasitol Int. 2014;63(2):327–331. doi: 10.1016/j.parint.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 14.Abbasi I, Kirstein OD, Hailu A, Warburg A. Optimization of loop-mediated isothermal amplification (LAMP) assays for the detection of Leishmania DNA in human blood samples. Acta Trop. 2016;162:20–26. doi: 10.1016/j.actatropica.2016.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Verma S, Singh R, Sharma V, Bumb RA, Negi NS, Ramesh V, et al. Development of a rapid loop-mediated isothermal amplification assay for diagnosis and assessment of cure of Leishmania infection. BMC Infect Dis. 2017;17(1):223. doi: 10.1186/s12879-017-2318-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mukhtar M, Ali SS, Boshara SA, Albertini A, Monnerat S, Bessell P, et al. Sensitive and less invasive confirmatory diagnosis of visceral leishmaniasis in Sudan using loop-mediated isothermal amplification (LAMP) PLoS Negl Trop Dis. 2018;12(2):e0006264. doi: 10.1371/journal.pntd.0006264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adams ER, Schoone G, Versteeg I, Gomez MA, Diro E, Mori Y, et al. Development and evaluation of a novel LAMP assay for the diagnosis of Cutaneous and Visceral Leishmaniasis. J Clin Microbiol. 2018; pii: doi: 10.1128/JCM.00386-18. [DOI] [PMC free article] [PubMed]
- 18.Ferrua B, Rol N, Michel G, Marty P. Antigenemia in patients with mediterranean visceral leishmaniasis. J Clin Microbiol. 2009;47(11):3760–3762. doi: 10.1128/JCM.00649-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salam MA, Khan MG, Bhaskar KR, Afrad MH, Huda MM, Mondal D. Peripheral blood buffy coat smear: a promising tool for diagnosis of visceral leishmaniasis. J Clin Microbiol. 2012;50(3):837–840. doi: 10.1128/JCM.05067-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abeijon C, Kashino SS, Silva FO, Costa DL, Fujiwara RT, Costa CH, Campos-Neto A. Identification and diagnostic utility of Leishmania infantum proteins found in urine samples from patients with visceral leishmaniasis. Clin Vaccine Immunol. 2012;19(6):935–943. doi: 10.1128/CVI.00125-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abeijon C, Campos-Neto A. Potential non-invasive urinebased antigen (protein) detection assay to diagnose active visceral leishmaniasis. PLoS Negl Trop Dis. 2013;7:e2161. doi: 10.1371/journal.pntd.0002161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abeijon C, Singh OP, Chakravarty J, Sundar S, Campos-Neto A. Novel antigen detection assay to monitor therapeutic efficacy of visceral Leishmaniasis. Am J Trop Med Hyg. 2016;95(4):800–802. doi: 10.4269/ajtmh.16-0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vallur AC, Tutterrow YL, Mohamath R, Pattabhi S, Hailu A, Abdoun AO, et al. Development and comparative evaluation of two antigen detection tests for visceral Leishmaniasis. BMC Infect Dis. 2015;15:384. doi: 10.1186/s12879-015-1125-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diro Ermias, Yansouni Cedric P., Takele Yegnasew, Mengesha Bewketu, Lynen Lutgarde, Hailu Asrat, van Griensven Johan, Boelaert Marleen, Büscher Philippe. Diagnosis of Visceral Leishmaniasis Using Peripheral Blood Microscopy in Ethiopia: A Prospective Phase-III Study of the Diagnostic Performance of Different Concentration Techniques Compared to Tissue Aspiration. The American Journal of Tropical Medicine and Hygiene. 2016;96(1):190–196. doi: 10.4269/ajtmh.16-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Drummelsmith J, Brochu V, Girard I, Messier N, Ouellette M. Proteome mapping of the protozoan parasite Leishmania and application to the study of drug targets and resistance mechanisms. Mol Cell Proteomics. 2003;2(3):146–155. doi: 10.1074/mcp.M200085-MCP200. [DOI] [PubMed] [Google Scholar]
- 26.Bente M, Harder S, Wiesgigl M, Heukeshoven J, Gelhaus C, Krause E, et al. Developmentally induced changes of the proteome in the protozoan parasite Leishmania donovani. Proteomics. 2003;3(9):1811–1829. doi: 10.1002/pmic.200300462. [DOI] [PubMed] [Google Scholar]
- 27.McCall LI, Zhang WW, Dejgaard K, Atayde VD, Mazur A, Ranasinghe S, et al. Adaptation of Leishmania donovani to cutaneous and visceral environments: in vivo selection and proteomic analysis. J Proteome Res. 2015;14(2):1033–1059. doi: 10.1021/pr5010604. [DOI] [PubMed] [Google Scholar]
- 28.Buates S, Matlashewski G. General suppression of macrophage gene expression during Leishmania donovani infection. J Immunol. 2001;166(5):3416–3422. doi: 10.4049/jimmunol.166.5.3416. [DOI] [PubMed] [Google Scholar]
- 29.Zhang WW, Ramasamy G, McCall LI, Haydock A, Ranasinghe S, Abeygunasekara P, et al. Genetic analysis of Leishmania donovani tropism using a naturally attenuated cutaneous strain. PLoS Pathog. 2014;10(7):e1004244. doi: 10.1371/journal.ppat.1004244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fuss IJ, Kanof ME, Smith PD, Zola H. Isolation of whole mononuclear cells from peripheral blood and cord blood. Curr Protoc Immunol. 2009; Chapter 7: Unit7.1. doi: 10.1002/0471142735.im0701s85. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets and materials used and/or analyzed during the current study will be available from the corresponding author on reasonable request.