

Abstract
Profiling using high-throughput MS has discovered an overwhelming number of novel protein phosphorylation sites (“phosphosites”). However, the functional relevance of these sites is not always clear. In light of recent studies on the evolutionary mechanism of phosphorylation, we have developed CPhos, a Java program that can assess the conservation of phosphosites among species using an information theory-based approach. The degree of conservation established using CPhos can be used to assess the functional significance of phosphosites. CPhos has a user friendly graphical user interface and is available both as a web service and as a standalone Java application to assist phosphoproteomic researchers in analyzing and prioritizing lists of phosphosites for further experimental validation. CPhos can be accessed or downloaded at http://helixweb.nih.gov/CPhos/.
Keywords: Bioinformatics, Conservation, Functional significance, Information theory, Phosphorylation sites
Studies of protein post-translational modifications, especially phosphorylation, have been greatly aided by recent advancements in enrichment techniques and high-throughput MS. These methods have enabled the identification of a remarkable number of previously unreported phosphosites that may serve regulatory roles in cellular processes [1, 2]. In fact, databases such as Phospho. ELM and PhosphoSitePlus have over 42 000 and 170 000 unique phosphosites recorded to date, respectively [3,4]. However, there has been speculation that a large number of the reported modification sites are not functionally significant [5–7].
There are a number of tools freely available for predicting phosphorylation sites using a wide array of approaches, including decision trees, position-specific scoring matrices, artificial neural networks, hidden Markov models, and support vector machines. Tools such as NetPhos [8], DISPHOS [9], and scan-x [10] focus on general prediction of phosphorylatable serine, threonine, or tyrosine. Although this provides valuable insight to which sites can be potentially phosphorylated, the functional significance of those sites is not addressed. Another group of predictors focuses on kinase-specific phosphorylation sites. This group includes Scan-site [11], NetPhosK [12], Kinase-Phos [13], NetPhorest [14], GPS[15],PSSP[16],and PredPhospho[17] among others [18]. These tools provide greater functional information on kinase-substrate relationships. However, the limited knowledge on phosphorylation site preferences of kinases restricts predictions made by these tools to only the well-characterized kinase families.
One alternative way to potentially address the functional relevance of a discovered or predicted site is to assess the evolutionary conservation of these sites across species. Several studies have examined the evolution of phosphorylation, supporting the notion that conserved phosphosites among different species are more likely to be functional than nonconserved phosphosites in both ordered and disordered regions [19–21]. In light of these studies, one approach utilizes the conserved domain database to map phosphosites onto conserved domains across three species [22]. However, the limited size of the database and species greatly restricts the coverage for analyses. In addition, this and other conservation-based approaches [23–25] have yet to capture specifically the evolution of phosphorylation events, which recently Ferrell et al.[26]showed through comparative genomics that some well conserved activating phosphorylation sites appear to have evolved from acidic Asp/Glu residues.
In this paper, we propose to address the functional relevance problem by the use of an information theory-based approach to capture the conservation and most importantly the Asp/Glu to pSer/pThr substitutions observed in the evolution of some phosphosites as a way to distinguish potentially functional from potentially nonfunctional phosphosites. More specifically, we utilized Shannon’s information theory to assess the information content (IC) in the aligned phosphosequences [27] and to generate normalized weighted conservation scores for the phosphosite and the flanking regions. In the set of aligned sequences, the entropy, or level of uncertainty, for the position can be represented as
where Pa,i represents the probability of amino acid a at a position i. Therefore, the maximum value for Shannon’s entropy is log2|Σ|, where is the alphabet size of the group (e.g. for a nondegenerate analysis with 20 amino acids, log2|20| = 4.32 bits). The IC is considered the opposite of entropy and is defined as
Thus, IC measures the amount of “information” in a multiple alignment with the highest IC corresponding to high conservation. We next wanted to utilize this IC calculation to capture the functional relevance by taking into account the Asp/Glu to pSer/pThr substitutions observed in the evolution of some activating phosphosites [26]. As such, for each position in the alignment with identified phosphorylation, the algorithm calculates a normalized sum of three individually calculated IC scores. The first IC score is calculated assuming no degeneracy in the amino acids. To account for substitutions of amino acids with similar chemical properties, a second IC score is calculated using the following degenerate groupings: {KRH, DE, ST, NQ, FWY, ILMVA, G, P, C}. To also account for the Asp/Glu to pSer/pThr substitutions, a third IC score is calculated with the degenerate grouping: {KRH, STDE, NQ, FWY, ILMVA, G, P, C}. Therefore, the final normalized conservation score is calculated as follows:
Where Φ = {nondegenerate, degenerate [KRH, DE, ST, NQ, FWY, ILMVA, G, P, C], degenerate [KRH, STDE, NQ, FWY, ILMVA, G, P, C]} ᴧ = {20, 9, 8}, and ᴧ corresponds to the number of sets for each grouping.
Next, we wanted to apply this same calculation for regions flanking each phosphorylation site. The algorithm calculates this with two IC scores for each position (with degeneracy and without degeneracy) and normalizes the sum of all the six positions upstream and downstream of the phosphosites.
where Γ = {± six neighboring residues excluding phosphor-site} ψ= {nondegenerate, degenerate[KRH, DE, ST, NQ, FWY, ILMVA, G, P, C]} Ω= {20, 9}
We implemented the algorithm and a series of upstream data preprocessing steps (Fig. 1) in Java with a graphical user interface (Fig. 2). The program accepts input as a list of phosphopeptides (phosphosites annotated with the symbol * after the residue). This is merged with the RefSeq proteome database to derive the parent protein for each peptide and the corresponding phosphosite residue number.
Figure 1.
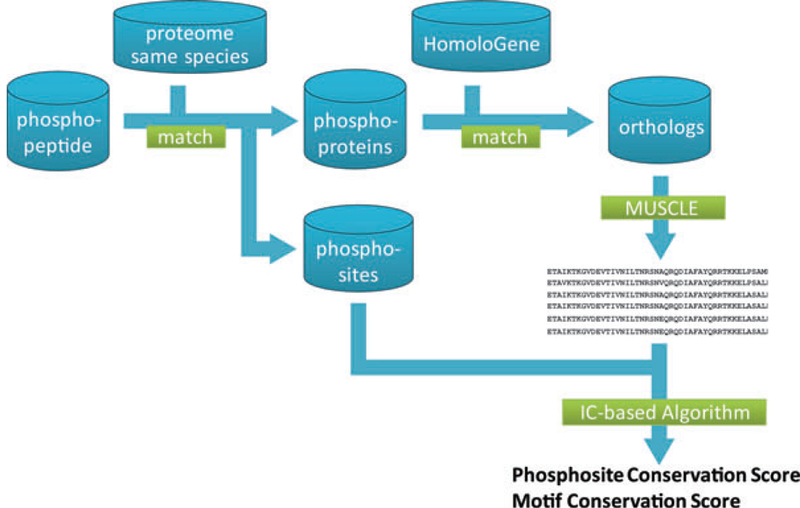
Program workflow of the conservation scores calculation.
Figure 2.
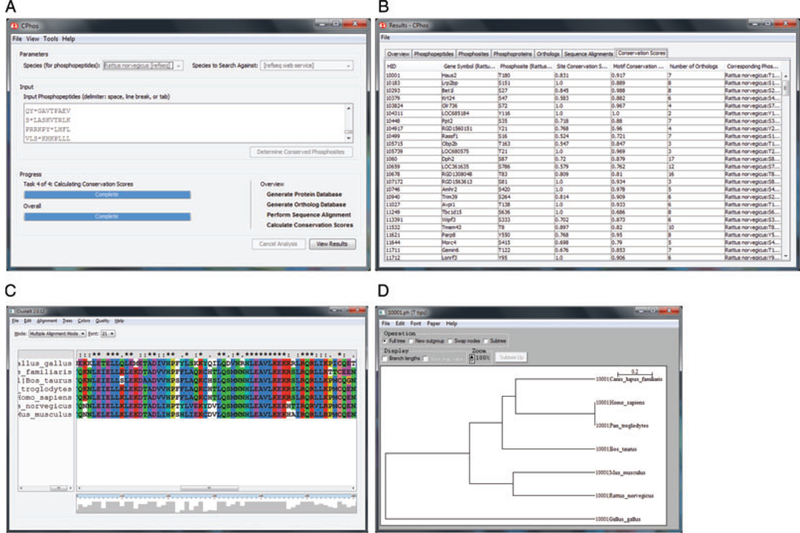
CPhos graphical user interface. (A) Main window for user input. The user can specify the species of the input phosphopeptides and species to derive the orthologs for analysis. Additional settings are available in a separate preferences window. (B) Results window showing a summary page, list of phosphopeptides, phosphosites, phosphoproteins, orthologs, sequence alignments, and conservation scores. Results can be exported as .CSV files. (C) Sequence alignment can be visualized using ClustalX. (D) Phylogenetic tree of the species of the orthologs can be visualized using NJplot.
By default, CPhos includes three proteome databases (rat, mouse, and human). However, users can manually download additional proteome databases to analyze peptides derived from other species. In case of multiple protein matches, the peptide is omitted from further analysis. The list of phosphoproteins is merged with the HomoloGene (NCBI) and RefSeq proteome database to obtain the orthologous sequences for each protein. Due to the limited size of paralogs in the current HomoloGene database, only orthologs are analyzed. The user has the option to obtain the sequences for all orthologs or limit the orthologs to specific groups (e.g. mammalian).
The maximum number of orthologs is limited by what is available in the HomoloGene database. The default option is set to all orthologs to achieve the maximal sampling and to more likely capture potential phosphorylation evolutionary events. The sequences are aligned using the first two iterations of MUSCLE algorithm (v3.8.31) [28]. Our tests suggest that additional iterative stages provide minimal improvement on the sequence alignment quality while at the expense of performance. The final results after phosphosite and motif conservation score calculations are displayed in a separate output window and can also be exported as .CSV files. In addition, the program includes visualization tools ClustalX (v2.0.12) for the sequence alignment and NJplot (v2.3) for the neighbor-joining phylogenetic tree results.
The performance evaluation of CPhos was carried out on a server with an Intel(R) Xeon(R) CPU E5620 with clock speed of 2.40GHz, 12288 KB cache, and 32GB RAM. The server had Ubuntu SMP (2.6.32–31-generic) operating system and algorithm has been implemented in Java(TM) SE Runtime Environment (build 1.6.0 20). Using the machine and operating system, the implemented program CPhos was able to perform calculations for 64 000 randomly drawn phosphopeptides from the rat proteome in about 6 h (Supplementary Information Table S1 and Fig. 3A).
Figure 3.
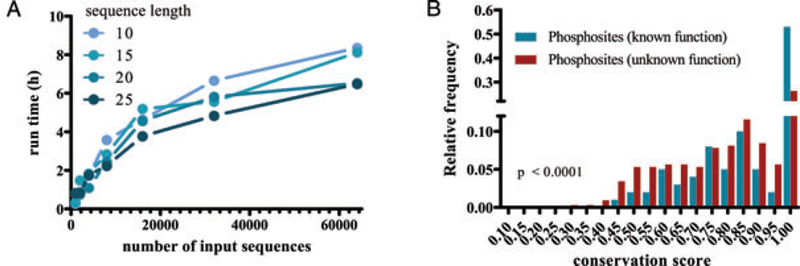
Validation of CPhos. (A) Run time analysis of CPhos on a set of randomly drawn phosphopeptides from the rat proteome using a Linux server. (B) Normalized histogram of phosphosite conservation scores on a dataset containing phosphosites from human proteome with known and unknown [10]. P value was determined using Mann–Whitney test
For improved availability and compatibility, we have also implemented CPhos using Java servlet and deployed on a web server with Apache Tomcat 7.0.12. The web service is hosted by the National Institute of Health (NIH) Helix and Biowulf cluster systems (http://helixweb.nih.gov/).
We validated CPhos using a published dataset containing approximately 500 phosphosites from human proteome, with a portion annotated with known function and the rest from large-scale MS studies with unknown function [20]. We posit that the phosphosites with unknown function are more likely to contain a mixture of true functional and nonfunctional sites and have a smaller number of high scoring sites compared to phosphosites with known function. This was indeed what we observed (Supplementary Information Table S2 and Fig. 3B). Interestingly, a portion of phosphosites with unknown function also had high conservation scores. These may represent potentially functional sites that can be further validated with different experimental approaches. For example, both CRYBA1 T127 and RANBP9 S483 have a phosphosite conservation score of 1, which always correspond to a perfect identity among all the sites examined and suggests potential functional roles (Supplementary Information Figs. S1 and S2). However, the motif conservation score for the same sites for RANBP9 only scored 0.75 compared to that for CRYBA1, which scored 0.964. This provides additional information that CRYBA1 T127 may be conserved as a result of its position within a functional domain while RANBP9 S483 may be a specific functional conservation for interaction with kinase(s). T127 of CRYBA1 is in fact found within the beta/gamma crystalline “Greek key” 3 domain. High- and low-scoring phosphosites can also occur on the same protein and facilitates prioritization for experimental validation given a protein of interest. For example, PHF16 is a protein in the histone acetyltransferase complex thought to be involved in transcriptional regulation. However, the detailed function of this protein and the role of many novel post-transcriptional modifications remain unexplored. PHF16 S85 has a lower score (0.606) compared to that for S715 (0.887) (Supplementary Information Figs. S3 and S4). PHF16 S715 is a much more conserved site and has glutamic acid in a lower species (Caenorhabditis elegans) before evolving to serine. This suggests that site S715 may be a functionally significant site and merit further investigation (e.g. mutagenesis with subsequent in vitro and/or in vivo phenotype characterization).
It is of note that the limited number of orthologs in the current version of HomoloGene introduces an underestimation of entropy. Rudimentary correction techniques such as Miller–Madow correction are inaccurate under the regime of small observation and large alphabet sizes. However, as illustrated in the examples above, the scores from CPhos present a preliminary first step in analyzing phosphosites for potential functional significance. Future work on CPhos will aim to incorporate additional algorithmic improvements such as small sample and background frequency corrections and to account for nonpositional conserved phosphosites.
In conclusion, we have developed an information theory-based algorithm implemented in Java with a user-friendly graphical user interface to facilitate the prioritization of phosphosites for functional experimental validation. CPhos is freely available both as a standalone software and as a web service (http://helixweb.nih.gov/CPhos/). We believe that the implemented tool will prove useful to the computational, mass spectrometry and proteomics communities alike.
Supplementary Material
Acknowledgments
We thank members of the Knepper lab and Kelly Brock for discussions and suggestions. This work was supported by the Intramural Budget of the NHLBI (NHLBI Project no. Z01-HL001285). Boyang Zhao was a student intern from the University of Michigan in the National Institutes of Health Biomedical Engineering Summer Internship Program, funded by the National Institute of Biomedical Imaging and Bioengineering.
Abbreviation:
- IC
information content
Footnotes
References
- [1].Schmelzle K,White FM,Phosphoproteomic approaches to elucidate cellular signaling networks. Curr. Opin. Biotechnol 2006, 17, 406–414. [DOI] [PubMed] [Google Scholar]
- [2].Zhao B, Knepper MA, Chou C-L, Pisitkun T, Large-scale phosphotyrosine proteomic profiling of rat renal collecting duct epithelium reveals predominance of proteins involved in cell polarity determination. Am. J. Physiol. Cell. Physiol 2012, 302, C27–C45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hornbeck PV, Chabra I, Kornhauser JM, Skrzypek E, Zhang B, PhosphoSite: a bioinformatics resource dedicated to physiological protein phosphorylation. Proteomics 2004, 4, 1551–1561. [DOI] [PubMed] [Google Scholar]
- [4].Dinkel H,Chica C,Via A,Gould CM et al. ,Phospho.ELM: a database of phosphorylation sites–update 2011. Nucleic Acids Res 2011, 39, D261–D267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lienhard GE, Non-functional phosphorylations? Trends Biochem. Sci 2008, 33, 351–352. [DOI] [PubMed] [Google Scholar]
- [6].Levy ED, Landry CR, Michnick SW, Signaling through cooperation. Science 2010, 328, 983–984. [DOI] [PubMed] [Google Scholar]
- [7].Beltrao P, Albanese V, Kenner LR, Swaney DL et al. ,` Systematic functional prioritization of protein posttranslational modifications. Cell 2012, 150, 413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Blom N, Gammeltoft S, Brunak S, Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol 1999, 294, 1351–1362. [DOI] [PubMed] [Google Scholar]
- [9].Iakoucheva LM, Radivojac P, Brown CJ, O’Connor TR et al. , The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res 2004, 32, 1037–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Schwartz D, Chou MF, Church GM, Predicting protein post-translational modifications using meta-analysis of proteome scale data sets. Mol. Cell. Proteomics 2009, 8, 365– 379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Obenauer JC, Cantley LC, Yaffe MB, Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res 2003,31,3635– 3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Blom N, Sicheritz-Ponten T, Gupta R, Gammeltoft S,´ Brunak, S., Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 2004, 4, 1633–1649. [DOI] [PubMed] [Google Scholar]
- [13].Wong Y-H, Lee T-Y, Liang H-K, Huang C-M et al. , Kinase-Phos 2.0: a web server for identifying protein kinase-specific phosphorylation sites based on sequences and coupling patterns. Nucleic Acids Res 2007, 35, W588–W594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Miller ML, Jensen LJ, Diella F, Jørgensen C et al. , Linear motif atlas for phosphorylation-dependent signaling. Sci. Signal 2008, 1, ra2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Xue Y,Ren J,Gao X,Jin C et al. ,GPS2.0,a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol. Cell. Proteomics 2008, 7, 1598–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Xue Y, Li A, Wang L, Feng H, Yao X, PPSP: prediction of PK-specific phosphorylation site with Bayesian decision theory. BMC Bioinformatics 2006, 7, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kim JH, Lee J, Oh B, Kimm K, Koh I, Prediction of phosphorylation sites using SVMs. Bioinformatics 2004, 20, 3179–3184. [DOI] [PubMed] [Google Scholar]
- [18].Trost B, Kusalik A, Computational prediction of eukaryotic phosphorylation sites. Bioinformatics 2011, 27, 2927– 2935. [DOI] [PubMed] [Google Scholar]
- [19].Ba ANN, Moses AM, Evolution of characterized phosphorylation sites in budding yeast. Mol. Biol. Evol 2010, 27, 2027–2037. [DOI] [PubMed] [Google Scholar]
- [20].Landry CR, Levy ED, Michnick SW, Weak functional constraints on phosphoproteomes. Trends Genet 2009, 25, 193–197. [DOI] [PubMed] [Google Scholar]
- [21].Malik R, Nigg EA, Korner R, Comparative conservation¨ analysis of the human mitotic phosphoproteome. Bioinformatics 2008, 24, 1426–1432. [DOI] [PubMed] [Google Scholar]
- [22].Sridhara V, Marchler-Bauer A, Bryant SH, Geer LY, Automatic annotation of experimentally derived, evolutionarily conserved post-translational modifications onto multiple genomes. Database 2011, 2011, bar019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lai ACW, Nguyen Ba AN, Moses AM, Predicting kinase substrates using conservation of local motif density. Bioinformatics 2012, 28, 962–969. [DOI] [PubMed] [Google Scholar]
- [24].Lam HYK, Kim PM, Mok J, Tonikian R et al. , MOTIPS: automated motif analysis for predicting targets of modular protein domains. BMC Bioinformatics 2010, 11, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Pei J, Grishin NV, AL2CO: calculation of positional conservation in a protein sequence alignment. Bioinformatics 2001, 17, 700–712. [DOI] [PubMed] [Google Scholar]
- [26].Pearlman SM, Serber Z, Ferrell JE, A mechanism for the evolution of phosphorylation sites. Cell 2011, 147, 934–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Shannon CE, A mathematical theory of communication. Bell Syst. Tech. J 1948, 27, 379–423, 623–656. [Google Scholar]
- [28].Edgar RC, MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 2004, 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.