

Abstract
Obesity is associated with cancer risk and its link with liver cancer is particularly strong. Obesity causes non-alcoholic fatty liver disease (NAFLD) that could progress to hepatocellular carcinoma (HCC). Chronic inflammation likely plays a key role. We carried out a bioassay in the high fat diet (HFD) fed C57BL/6J mice to provide insight into the mechanisms of obesity-related HCC by studying γ-OHPdG, a mutagenic DNA adduct derived from lipid peroxidation. In an 80-week bioassay, mice received a low fat diet (LFD), high fat diet (HFD) or HFD with 2% Theaphenon E (HFD+TE). HFD mice developed a 42% incidence of HCC and LFD mice a 16%. Remarkably, TE, a standardized green tea extract formulation, completely blocked HCC in HFD mice with a 0% incidence. γ-OHPdG measured in the hepatic DNA of mice fed HFD and HFD+TE showed its levels increased during early stages of NAFLD in HFD mice and the increases were significantly suppressed by TE, correlating with the tumor data. Whole exome sequencing showed an increased mutation load in the liver tumors of HFD mice with G>A and G>T as the predominant mutations, consistent with the report that γ-OHPdG induces G>A and G>T. Furthermore, the mutation loads were significantly reduced in HFD+TE mice, particularly G>T, the most common mutation in human HCC. These results demonstrate in a relevant model of obesity-induced HCC that γ-OHPdG formation during fatty liver disease may be an initiating event for accumulated mutations that leads to HCC and this process can be effectively inhibited by TE.
Keywords: Hepatocellular carcinoma, lipid peroxidation, DNA adduct, mutation load, high fat diet, green tea, EGCG
Introduction
Hepatocellular carcinoma (HCC) is responsible for nearly 700,000 deaths each year worldwide (1). The risk factors of HCC include aflatoxin exposure, HBV and HCV infection, alcohol consumption and obesity (2, 3). Obesity, which causes non-alcoholic fatty liver disease (NAFLD), is strongly linked to HCC and is the fastest growing risk factor for liver cancer in the US as 40% of this population is obese and 30% suffers from NALFD (4–6). It is pivotal that we understand how obesity promotes HCC by studying the underlying molecular mechanisms.
Obesity is a chronic inflammatory disease that elicits oxidative stress via reactive oxygen species (ROS) and lipid peroxidation (LPO). Several mechanisms have been proposed for obesity-induced hepatocarcinogenesis, including aberrations in the signaling pathways of IL-6, PTEN, NF-κB, Hedgehog, TNF-α and STAT3 (7, 8). However, evidence of oxidative stress-induced DNA damage in this process is scarce. An earlier study in human NAFLD showed that 4-hydroxy-2-nonenal modified protein expression, a marker of LPO, as well as 8-oxo-dG, a bench mark of oxidative DNA damage, were elevated (9). We hypothesize that fatty livers caused by high fat diet (HFD) are challenged with increased LPO and, consequently, raised the levels of γ-OHPdG, a LPO-derived mutagenic DNA adduct, leading to higher mutation loads that set the stage for HCC development and that this process can be prevented by Theaphenon E (TE), a standardized green tea extract formulation,.
The accumulation of fat in the liver, known as steatosis, is a risk factor of NAFLD and non-alcoholic steatohepatitis (NASH) (10). Fatty livers increase inflammation and oxidative DNA damage (11) and may progress to fibrosis and cirrhosis due to liver damage caused by LPO and ROS (12). Cirrhosis, a major risk factor for HCC, develops in nearly 2% of the NAFLD population (13). NAFLD can also progress to HCC without cirrhosis (14).
Lipid metabolism changes, genetic alterations and ROS production are the molecular signatures of fatty liver disease (15, 16). A recent study showed that NAFLD causes selective killing of CD4+T lymphocytes by ROS in mitochondria due to accumulation of linoleic acid that promotes heptocarcinogenesis (17). ROS in hepatocytes can be a source of DNA damages . If the damages persist at the critical genes due to lack of repair, they may lead to mutations that drive cancer development. We focused on γ-OHPdG as a mutagenic bulky adduct derived from LPO (18). γ-OHPdG induces primarily G>T and G> A mutations and it preferentially binds at the mutation hotspots of human p53 gene and it is implicated in human cancers (19–21). However, its mutagenicity in vitro may vary and depends on many factors, for example, sequence context (22). In the present study, we examined in a HFD diet-induced HCC model the relationships of γ-OHPdG and the total mutation loads with HCC as a mechanism of obesity-induced hepatocarinogenesis and studied its prevention by TE.
Methods
Animal bioassay
Two hundred and twenty male C57Bl/6J mice of three weeks of age were purchased from The Jackson Laboratory (Bar Harbor, ME. Stock No. 000664). The mice were housed and maintained under conventional conditions using micro-Isolator system cage top and polycarbonate mouse cage bottom with the controlled room temperature of 68–70 ˚F and 68% humidity on a 12h light cycle. Mice were randomly divided into three groups; 100 mice in HFD group, 60 mice in LFD and 60 in HFD+TE groups. They were fed either a HFD, LFD, or HFD+TE diet starting at four weeks of age. The diet was obtained from Dyets Inc. (Bethleham, PA). The HFD (Cat # 402400) had 60% kcal fat, 20% kcal protein, and 20% kcal carbohydrates. The LFD (#404360) was 12% kcal fat, 29% kcal protein, and 59% kcal carbohydrates. The HFD+TE diet was the same high fat diet above, supplemented with 2% TE (Tokyo, Japan). The 2% TE was chosen based on previous reported bioassays (23). Diet was stored at 4o C and changed weekly. The food consumption was measured weekly and the weekly average consumption was obtained by dividing the number of mice in each cage. Liver tissues from five mice were collected per group at 0, 5, 10, 25, 35, 50, 55, 60, 65 and 70 weeks on the diet and the bioassay was terminated at week 80. Livers were weighed and imaged for gross morphology and sections were formalin fixed for IHC. Food consumption and body weight were recorded weekly. The bioassay was conducted in conformity with PHS policy and approved by the AAALAC accredited Georgetown University institutional review board and institutional animal care and use committee.
Tumors and H & E and immunohistochemistry (IHC)
At each sacrifice during the bioassay, Livers were examined for fatty change, fibrosis and formation of tumors. When tumors were visible they were counted, measured, and those over two mm were collected, formalin fixed and FFPE blocks were made to prepare slides for histological analysis and confirmation. Tissues were fixed for at least 24 hours in 10% neutral buffered formalin, dehydrated through a graded series of alcohols, cleared in xylenes, infiltrated with paraffin wax and embedded in wax. Tissues were cut in five micron sections and placed onto Superfrost Plus charged slides (Fisher). For pathology evaluation, routine hematoxylin and Eosin (Suripath) staining was performed on a Leica Autostainer XL and Masson’s trichrome staining performed as per standard protocols. For IHC, heat induced epitope retrieval (HIER) was performed by immersing the tissue sections at 98oC for 20 minutes in Diva Decloaker (Biocare). Immunohistochemical staining was performed using a horseradish peroxidase labeled polymer (Dako/Agilent K4003) according to manufacturer’s instructions. Briefly, slides were treated with 3% hydrogen peroxide and 10% normal goat serum for 10 minutes each, and exposed to primary antibodies for Ki67 (Biocare, cat. CRM325, 1:50 dilution in VanGough diluent) overnight at 4oC. Slides were exposed to anti-rabbit HRP labeled polymer for 30 minutes. and DAB chromagen (Dako/Agilent) for five minutes. Slides were counterstained with Hematoxylin (Fisher, Harris Modified Hematoxylin), blued in 1% ammonium hydroxide, dehydrated, and mounted with Acrymount. All washes were performed with Tris Buffered Saline with 0.5% Tween. Consecutive sections with the primary antibody omitted were used as negative controls. The H&E slides for liver pathology were evaluated by a board certified and practicing pathologist. Masson’s Trichrome staining was scored using the scale: 0 –Normal, 1 –Expanded, 2 –Bridging, 3 -Focal Nodule, 4 -Diffuse (Cirrhosis). The diagnosis of HCC was determined based on the presence of standard architectural and/or cytological atypia including increased thickness of hepatic cords, nuclear enlargement, prominent nucleoli and multi nucleation and the presence of cytoplasmic globules and abundant Mallory’s hyalin, particularly in comparison with adjacent non-neoplastic hepatic parenchyma.
Liver Function and Injury
The levels of ALT and AST were assessed using Activity Assay Kits (Sigma-Aldrich #MAK052 and MAK055, respectively) according to manufacturer’s protocol. ALT and AST were detected in the serum of the HFD, LFD and HFD+TE mice collected and frozen at each time point.
Quantification of γ-OHPdG in mouse livers
LC-MS/MS ---DNA samples from the flash frozen mouse liver samples were isolated using QIAGEN Blood and Cell Culture Maxi Kit (#13362) following the protocol for tissue extraction as recommended by the manufacturer. DNA concentration was determined by using a NanoDrop-1000 spectrophotometer (O.D. ratios at 260/280nm are 1.8 of higher). Whole blood was sent for whole exome sequencing (Otogenetics, Atlanta, GA, USA) as baseline. γ-OHPdG standards and stable-isotope labelled internal standards were prepared as described in a previous publication (24). Enzymatic hydrolysis of DNA samples was performed using at least 0.2 mg DNA and a portion of each sample was used following hydrolysis for dG analysis as described previously (24). The levels of γ-OHPdG in each DNA sample were expressed as a ratio of γ-OHPdG to dG x 109.
IHC---Livers of five mice from each time point (weeks 5, 25 and 50) were processed for staining using a monoclonal antibody (A2) developed in our lab against γ-OHPdG (25). IHC was performed as described for Ki67 with the following modifications: deparaffinization of slides in Xylenes was extended by 30 additional minutes, antigen retrieval was performed for 40 minutes in Citrate buffer pH 6.0, and primary antibody was applied for 1 hour at room temperature and applied prior to hydrogen peroxide treatment. Eight to 23 images were randomly selected from each slide. The adduct levels were quantified based on distribution (scale 0 to 3, respectively, representing 0, 30, 60 and 90% of positively stained cells) and intensity (from a scale of 0 to 3 and score 3 is the highest) in each image.
Whole Exome Sequencing
Whole exome sequencing from mouse genomic DNA (gDNA) was performed by Otogenetics (Atlanta, GA, USA) using tumor and non-tumor liver tissues. Sequencing libraries were prepared using the Agilent SureSelect Target Enrichment System. Paired-end sequencing was performed on Illumina HiSeq2500 platform (PE 100–125, 50X). The liver samples that were analyzed included four mice from the HFD group without tumor at the final collection time point and nine tumors from mice from the HFD group (6 from week 80, 2 week 70, 1 week 60). In addition, two blood samples used as background controls from the final time point (one each from HFD and HFD+TE) were sequenced. FASTQ sequencing output was aligned to mouse genome GRCm38/mm10 using bwa-mem , followed by de-duplication using Picard and indel realignment using GATK (Broad Institute) . Base quality scores were recalibrated (BQSR GATK module) using MGP V5 from the Sanger Mouse Genomes Project to facilitate sensitive variant detection . Variants were called and filtered using LoFreq . P-value cutoffs for detection were LoFreq recommended defaults (FDR <= 0.05). VCF files were annotated in R using the VariantAnnotation package . Variants detected by LoFreq, at any allele frequency, were included in quantification of the total number of separate mutations, with context sequence provided by GRCm38. Plotting and statistics were produced using custom R code, available on request. VCF files are available from the European Variation Archive (https://www.ebi.ac.uk/eva/, Accession: PRJEB24306). Variant effect prediction was created using the VEP (v86) tool in conjunction with dbSNP (v146) and GENCODE M11 for mouse genome GRCm38.p4 (Table S4)
Statistical Analysis Methods
To compare outcomes among LFD, HFD and HFD+TE groups, Student’s test, Chi-square test and Fisher’s exact test (for categorical variables) were used. Because the sample size in each group at each time-point is small, the Wilcoxon rank sum test was used to compare the levels of γ-OHPdG obtained from LC-MS/MS and the regression analysis was employed to determine the levels changes over time in each group. Student’s t-test was used to compare γ-OHPdG values in HFD vs. HFD+TE mouse livers obtained from IHC. To determine the significance between LFD, HFD and HFD+TE samples for liver to body weight ratio, ALT and AST, the student’s t-test was performed. To calculate the difference in tumor number and size between LFD, HFD and HFD+TE tumor samples, chi-square and Wilcoxon rank sum test are used. A p-value of <.05 was used for a statistical significance.
Results
In an 80-week tumor bioassay five mice from each group were sacrificed at 11 sequential intervals that represent the progression of NAFLD to HCC (Fig. 1A). After normalization, no significant difference in food consumption between HFD, LFD and HFD+TE mice was observed (Fig. S1). HFD+TE group showed the least weight gains, despite consuming a similar amount or more food than the other groups (Fig. 1B). Throughout the bioassay, HFD+TE mice appeared healthy and lean without overt adverse effects. Fig. 1C shows the representative mice at termination. HFD+TE mice generally had a significantly lower liver to body weight ratio than HFD and LFD mice (Fig. 1D).
Figure 1.
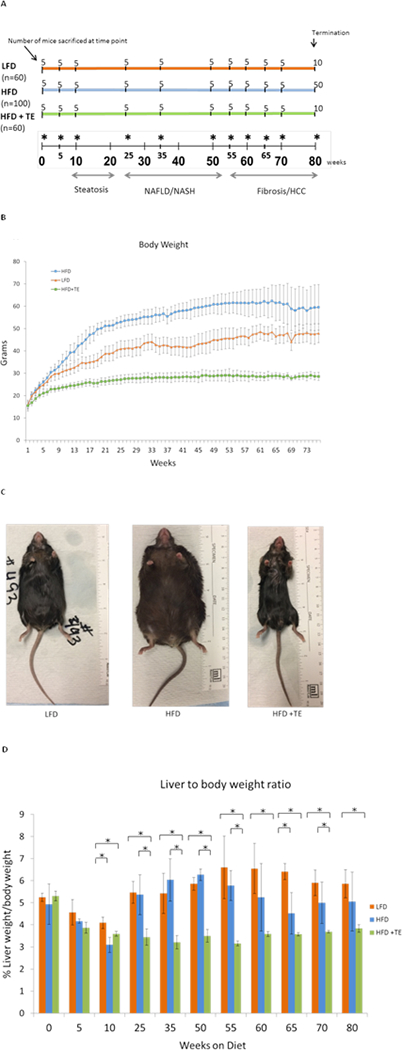
(A) The number of mice collected at each time point across the duration of the tumor bioassay is shown (* indicates the collection time point). The time points indicate the number of weeks the mice were on each specific diet, starting at 0 weeks (4 weeks of age) and “n” is total number of mice in each group at the beginning of the bioassay. The disease stages based pathological examination are indicated progressively from steatosis to NAFLD/NASH to HCC. (B) Body weight curves of mice from the three groups. The mice were weighed weekly and the average weight of the animals in each group high fat diet (HFD), low fat diet (LFD) and high fat diet+2% TE (HFD+TE) was used. (C) Representative mouse from each diet group at the termination of 80 weeks. (D) The liver to body weight ratios (%) of the mice collected at each time point. Error bars represent standard deviation from five mice of each group of the designated time point. * indicates p<0.05 (Student’s t-test).
The livers of LFD and HFD mice showed progressive tissue expansion and discoloration due to lipid accumulation (Fig. 2A). In contrast, livers from HFD+TE mice retained a healthy red color and shiny smooth texture. HFD mice developed steatotic livers at earlier stages. Increasing lipid droplet formation, including macrovesicular and microvesicular steatosis, were seen in LFD and HFD groups, whereas HFD+TE mice showed little or no sign of lipid accumulation (Fig. 2B). Additionally, features of NASH, including ballooning of hepatocytes, increased portal and lobular inflammatory infiltrate, neutrophils surrounding lipid-laden hepatocytes and scattered Mallory’s hyaline were observed in HFD and LFD mice starting at Weeks 25 and 50, respectively (Fig. 2B, inlets). One out of five mice developed NASH as early as 25 weeks in HFD group and NASH became more frequent at the later weeks (Fig. 2B). Masson’s trichrome staining showed mild fibrosis in some mice at 65 and 80 weeks after feeding LFD and HFD (Fig. 2C). Neither NASH nor fibrosis was found in HFD+TE mice (Fig. 2B and Fig. 2C). No cirrhosis was observed in HFD and LFD mice. The occasional fibrosis and the lack of cirrhosis in HFD-fed mice has been previously reported (26). The ALT increased after 35 weeks in HFD and LFD mice, however, ALT remained low in HFD+TE mice during the bioassay (Fig. 3A). Similarly, the AST activity was also reduced in HFD+TE mice, specifically at the 35 and 65 weeks (Fig. 3B), although the changes were not as dramatic. Ki67 staining showed increased hepatic cell proliferation during NAFLD in HFD mice compared to HFD+TE mice (Fig.S2). These results showed that mice fed HFD+TE had reduced liver toxicity and fatty liver diseases compared to HFD and LFD mice.
Figure 2. Comparison of liver morphology and histology among the groups.
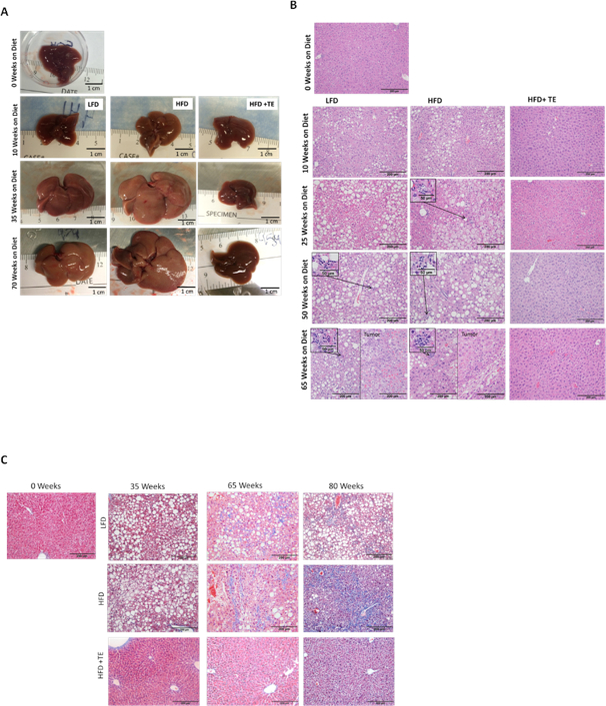
(A) The gross morphology of representative non-tumor livers from 0, 10, 35 and 70 weeks on the diet. (B) H&E staining of livers at 0, 10, 35, 50 and 70 weeks on the diet (20X, scale bar indicates 200 μm). NASH found in livers of mice from 25 weeks and 50 weeks from HFD and LFD group, respectively, is shown in the Inlets. (C) Masson’s trichrome staining of representative sections at 0, 35, 65 and 80 weeks on diet. Blue staining indicates locations of expanded fibrotic tissue. Scale bar indicates 200 μm.
Figure 3.
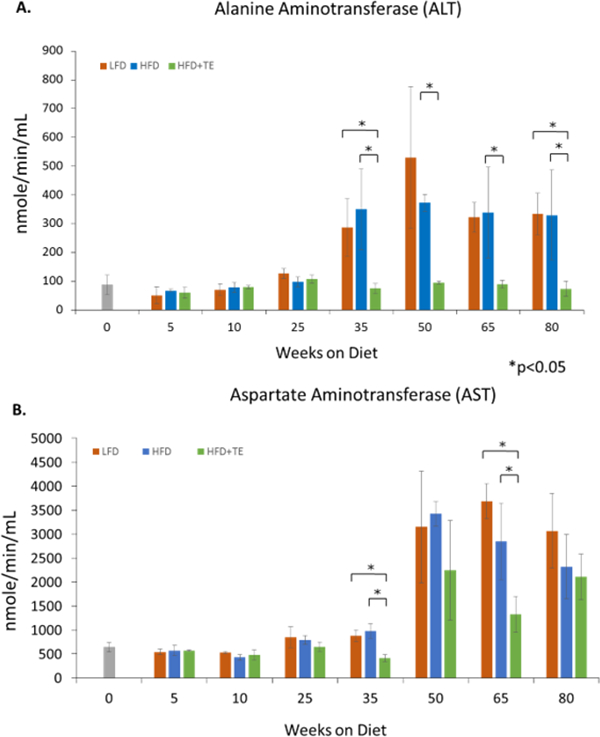
(A) Alanine aminotransferase (ALT) levels were significantly higher in HFD fed mice than the HFD+TE at all time points after 35 weeks and also in the LFD group as compared to the HFD+TE groups at 35 and 80 weeks. (* p<0.05, Student’s t test). (B) Aspartate aminotransferase (AST) levels were higher in the LFD and HFD group than the HFD+TE group at 35 and 65 weeks. Error bars represent standard deviation.
HCC was verified by H & E (Fig. 2B). Liver tumors became palpable at Week 55 in HFD and LFD groups (Fig. 4A, Table S1). At termination, the total tumor numbers between LFD and HFD groups showed no significant difference (p=0.9076), however, the tumor size were significantly lower in LFD than HFD mice (p=0.0287) (Fig. 4B, C). The overall tumor incidence was 42% in HFD, 16% in LFD and 0% in HFD+TE mice (Fig. 4D, Table S1 and S2) and there is a significant difference among the three groups (p<0.0001). Remarkably, not a single tumor was found in HFD+TE mice throughout the bioassay. TE diet also prevented NASH and fibrosis in HFD mice (Table S3). At termination, the mice fed HFD had a significantly high mortality rate at 22% compared to 5% in the LFD and 3% in HFD+TE, based on the assumption that all mice sacrificed at intervals survive at the end of the bioassay (Fig. 4E, Tables S1 and S2).
Figure 4. Tumor formation in mice fed HFD, LFD and HFD+TE.
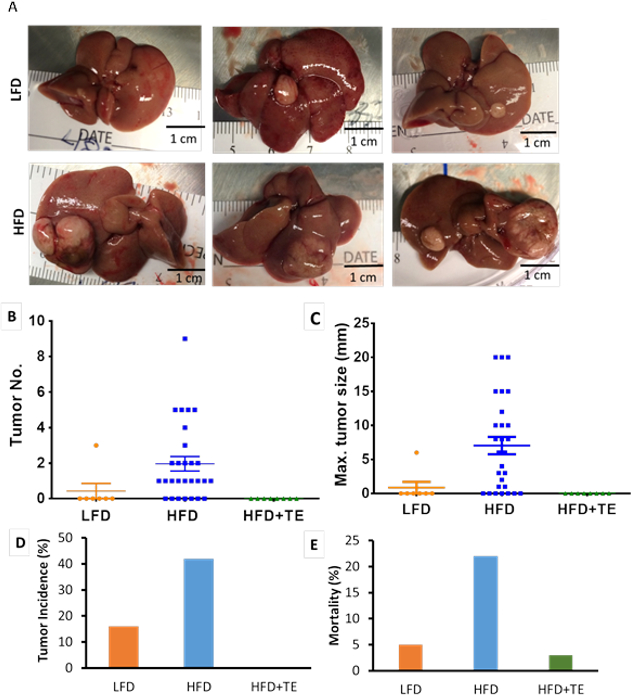
(A) Tumors found in LFD mice (55, 65 and 70 weeks left to right) and HFD mice (60, 70 and 80 weeks left to right). No tumors found in HFD+TE mice. (B) The overall tumor multiplicity is significantly higher in the HFD compared to LFD and high fat diet+2% TE (HFD+TE). No significant difference between LFD and HFD groups in tumor numbers (p=0.9076). (C) The overall tumor size is significantly higher in HFD compared to LFD (p=0.0287 based on Chi-square test) and HFD+TE mice. Error bars indicate standard error of difference. (D) The overall tumor incidence in LFD, HFD and HFD+TE groups. The Chi-square test shows that there is significantly different for tumor incidence among the three groups (p<0.0001). The Fisher’s exact test shows that there are significant different of tumor incidence not only between LFD or HFD and HFD+TE groups (p=0.0012 and <0.0001, respectively), but also between LFD and HFD groups (p=0.0011). (E) The mortality rate during the entire bioassay in LFD, HFD and HFD+TE groups. Significant differences among three groups (p=0.0003) by chi-square test.
To study γ-OHPdG as an initiating event in HCC, we first measured its levels in hepatic DNA from five mice at 0, 10, 25, 35 and 50 weeks between HFD and HFD+TE groups using a LC-MS/MS method (24). Although an unexpected larger variability than other models we have studied was found (27), an increasing trend of γ-OHPdG levels was noted in HFD group during the steatosis and NALFD stages (25 to 35 weeks), followed by a decline at Week 50 (Fig. 5A). The Wilcoxon rank sum test revealed a trend of lower adduct levels in HFD+TE mice compared to HFD mice. Due to the small animal number a significant difference was detected only at Week 50 with its levels reducing from 148 in HFD to 50 adducts/dGx109 in HFD+TE groups (p<0.03). To verify, we further quantified γ-OHPdG by IHC method, based on distribution (percentage of positively stained cells) and intensity (score 0 to 3), using a monocloncal antibody previously developed in our lab (25). The results corroborate the data from LC-MS/MS, showing a small, but significant, increase of adduct in HFD mice from 5 to 25 weeks (p=0.0478 distribution), followed by a decline at Week 50 (p=0.019 distribution and p= 0.0095 intensity). While TE treatment showed no difference at Week 5, it significantly suppressed γ-OHPdG levels in HFD group at Week 25 (p=0.03 and p=0.0002, respectively) and Week 50 (p=0.0016 and p=0.0006, respectively) (Fig. 5B). The representative IHC images are shown in Figure 5C.
Figure 5.
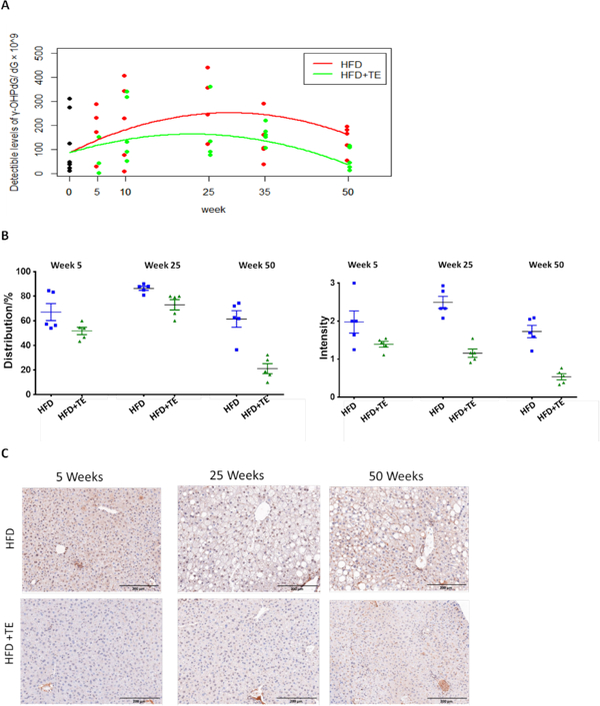
(A) γ-OHPdG levels in hepatic DNA measured by LC-MS/MS in mice fed HFD and HFD+TE for 0, 5, 10, 25, 35 and 50 weeks. The Wilcoxon rank sum test shows that the detectible levels of γ-OHPdG in HFD+TE group are significantly lower than that in HFD group (p=0.03) at week 50. (B) Quantification of γ-OHPdG by IHC staining in mouse livers from HFD vs. HFD+TE groups based on distribution (% of positively stained cells) and intensity (0 to 3) at week 5 (p=0.09 and p=0.12, respectively), week 25 (p=0.03 and p=0.0002, respectively) and week 50 (p=0.0016 and p=0.0006, respectively). In HFD mice, an increase of adduct levels from 5 to 25 weeks (p=0.0478 distribution and p=0.1699 intensity), followed by a decline at Week 50 (p=0.0190 and p= 0.0095, respectively). In HFD+TE mice, an increase from 5 to 25 weeks (p=0.0041 distribution), followed by decrease at Week 50 (p<0.0001 and p=0.0023, respectively) (C) Representative IHC staining of livers obtained from mice fed HFD vs. HFD+TE at weeks 5, 25 and 50.
To examine the relationships of γ-OHPdG with somatic mutations, whole exome sequencing was performed using livers from 13 mice in HFD (9 tumors and 4 normal) and 4 in HFD+TE group (all normal). The overall mutation loads were higher in the tumors from HFD mice than the normal tissues from HFD mice and HFD+TE group (p=0.0108 and p=0.0056, respectively) (Fig. 6A and B). G>A and G>T were over-represented in HCC of HFD mice. When the proportion of specific somatic mutations was compared, G>T was the only mutation significantly decreased in HFD+TE mice (p=0.0336) (Fig. 6A and C). Examination of the average mutations in each sample based on sequence context shows, again, the mutations in each sample were reduced by TE, and G>T mutations are evidently reduced(Fig. S3). However, there is no indication of sequence–dependence in all mutations. In human HCC, base substitution mutations in β-catenin and p53 genes are common (28, 29). However, no such mutations were detected in the mouse liver tumors. We did identify mutations at codon 61 in Hras, a mutation hotspot of certain human cancers (30), in the liver tumors obtained from five out of nine mice, among them three are G>T mutation.
Figure 6.
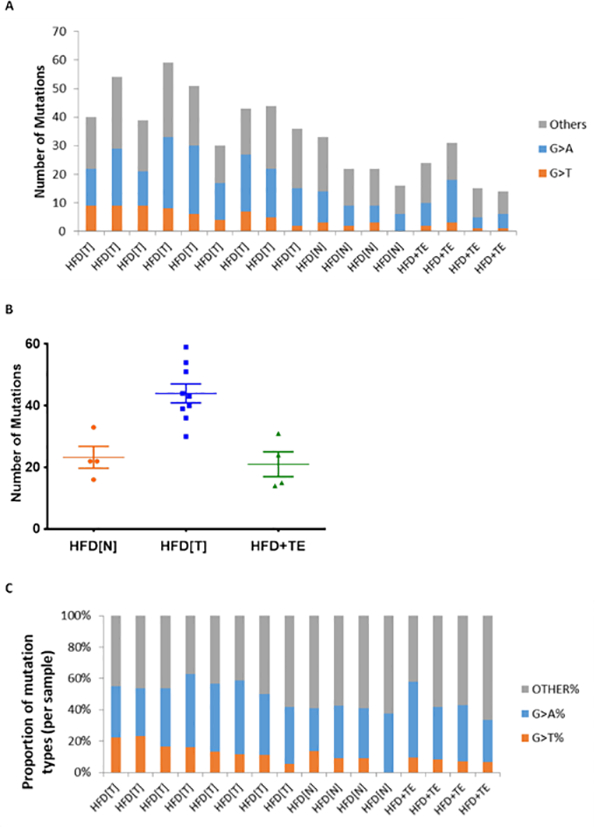
(A) Number and types of base substitution mutation of liver tumors (T) from nine mice and non-tumor liver tissue (N) from four mice on HFD and normal liver tissues from four mice on HFD+TE. (B) Dot plot of mutations in the liver tissue of each mouse from HFD (N), HFD(T) and HFD+TE (p=0.0108 and p=0.0056 for HFD (T) vs. HFD (N) and HFD (T) vs. HFD+TE normal, respectively, based on Wilcoxon rank sum test). (C) Proportions of mutation types in livers of HFD (T) vs. HFD (N) vs. HFD+TE. A significant decrease in G>T was found in HFD+TE group as compared to HFD (T) (p=0.0336), but not HFD (T) vs. HFD (N) (p=0.1047) and HFD (N) vs. HFD+TE (p=0.6631), based on Wilcoxon rank sum test. No difference was seen with G>A among the three groups.
Discussion
As a disease of chronic inflammation, ROS generation and the related mechanisms are likely to be important in obesity-induced hepatocarcinogenesis. Inflammation-associated signaling pathways have been studied, however, the role of oxidative DNA damages in this process is poorly understood. As fatty livers and inflammation are crucial to elicit oxidative DNA damage, we chose to focus on γ-OHPdG, a well-studied LPO-derived mutagenic adduct. We recently reported that hepatic γ-OHPdG levels not only correlate well with HCC in Xpa −/− and DEN exposed mice, it also serves as a biomarker for HCC recurrence in patients after resection (27). We showed that C57BL/6J mice predisposed to obesity and HCC by HFD can be effectively prevented by TE. This model is more relevant than the genetically modified, the methionine- and choline-deficient diet and chemical–induced HCC models (31, 32). TE treatment significantly lowered adduct levels in mice fed HFD (Fig. 5A and B), correlating well with its inhibition of HCC. These results support the paradigm that higher levels of mutagenic adduct lead to higher mutation loads that increase the risk for HCC. This idea is reinforced by our data that hepatic cell proliferation was increased in NAFLD of HFD mice (Fig. S2) (33). Conversely, the diminished steatotic tissues with lowered adduct levels by TE in HFD mice, combining with its effect to decrease hepatic cell proliferation during NAFLD (Fig. S2), reduces overall mutation loads and, consequently, lowers HCC incidence.
Two independent methods, LC-tandem MS and IHC, were used to determine the hepatic γ-OHPdG levels. Both confirm the conclusions that TE suppresses the hepatic adduct levels caused by HFD during the stages of NAFLD. Studies have shown that γ-OHPdG induces predominantly G>T and G>A mutations (19, 20). These mutations also occur frequently in the liver tumors of HFD mice (Fig. 6). The G>T mutations, not commonly associated with other human solid cancers except for lung cancer, are over-represented in human HCC (29). The decreased γ-OHPdG levels by TE is reflected in the lower number of total mutational loads in liver tissues of these mice, particularly G>T mutations. The fact that G>T was the only mutation detected at a significantly lower frequency in liver tissues of mice treated with TE, albeit the sample size is small, implicates its role in HCC. Collectively, these results lend support to the role of γ-OHPdG in hepatocarcinogenesis in this model. It should be mentioned, however, this is only circumstantial evidence that further investigations are needed to better understand the mechanisms. γ-OHPdG is not the only DNA adduct derived from LPO and, more broadly, from oxidative stress. The fact that TE completely blocked HCC in HFD mice, yet only partially blocked hepatic γ-OHPdG formation, may be explained by the assumption that a critical or threshold adduct level exists for HCC initiation or that other mechanisms may be involved, such as the loss of CD4+T cells (17), and γ-OHPdG is only partially responsible for HCC development .
Studies have shown that G>T transversion is over-represented somatic mutations in HCC from patients in France that are developed from non-cirrhotic livers, implicating the exposure of DNA-damaging agents in hepatocarcinogensis which is independent of cirrhotic status (29). A widely recognized liver carcinogen aflatoxin B1 is an agent that induces G>T mutations (34). However, it is unlikely that this population is exposed to aflatoxin B1. As the causative agents remain elusive, our results may shed some light on the etiological agent. Whole genome sequencing of human HCC has identified missense mutations in the β-catenin (33%) and p53 (21%) genes are the most prevalent (28, 29). The more common G > T transversions in human HCC are found in the non-transcribed (NT) strand than in the transcribed (T) strand, indicating that a substantial number of the mutations in HCC are induced by bulky DNA damage (29, 35). However, none of the nine mouse liver tumors examined showed mutations in p53 and β-catenin. Similar observations were made in liver tumors from Xpa−/− mice (27). This is somewhat unexpected because γ-OHPdG has been shown to preferentially form at mutational hotspots in human p53 (21). A larger sample size may be needed to ascertain the mutation frequencies in these genes or a different set of genes are involved in HCC in these mice. Regardless, our sequencing data revealed variants of other genes that may be informative (Table S4). For example, five out of the nine liver tumors from the HFD group showed mutations in codon 61 of HRas and three out of the five samples are G>T (Q61K) mutation. The mutation at codon 61 in Hras was reported as a hotspot in certain human cancers, although its relationship with HCC has not been established (30). A recent study identified HRas mutation at codon 61 in HCC in obese mice exposed to DEN (36). Our study is the first to demonstrate HRas mutation at codon 61 of liver tumors in a diet-induced obesity model, suggesting its potential role in hepatocarcinogenesis associated with NAFLD caused by HFD.
Chemopreventive agents, such as metformin and acyclic retinoid, for HFD-induced HCC have been studied (37, 38). Phytochemicals are a rich source of potential cancer chemotherapeutics and chemopreventive agents. EGCG, a major catechin in green tea, reduces body weight gains and lipid accumulation in livers of HFD-fed mice (39). Antioxdiant catechins in green tea are known to nullify the adverse effects of LPO (40). The cancer chemoprevention of green tea and its catechins has been studied in chemically-induced hepatocarcinogenesis . The total protection against HCC in mice receiving HFD by TE is remarkable. TE constitutes approximately 70% EGCG which has been shown to suppress NASH in obese rats (41). TE also reduced body weight gains caused by HFD, possibly related to green tea’s effects on thermogenesis and fat oxidation (42). In this context, a recent study reported that caloric restriction diet inhibits DEN-induced liver tumorigenesis in mice which may involve multifaceted mechanisms (43). The antioxidant effects of catechins may be due in part to increased activities of catalase, superoxide dismutase and glutathione peroxidase (44). Other possible mechanisms include altered DNA methylation patterns and nuclear factor erythroid 2-related factor 2 (Nrf2) pathway (45). Green tea has also been shown to reduce ROS production and inhibit downstream cell proliferation pathways (46).
The absence of cirrhosis in this model is not unexpected. Several HFD models show no indication of fibrosis and the progressive fibrosis only occurred in mice fed HFD supplemented with high fructose (26, 47, 48). It appeared that NAFLD is sufficient for HCC dcelopment in the HFD model as cirrhosis is not always necessary for HCC in humans and mice (49). While the model has limitations, the results suggest that targeting NAFLD in obesity by TE may be a viable approach to prevent HCC. A prerequisite for HCC prevention strategy is to identify individuals at high risks. We found elevated hepatic γ-OHPdG levels in patients of early NAFLD followed by a decline during fibrosis and cirrhosis, reminiscent of the present study (50). In a recent study we also reported that γ-OHPdG predicts recurrence in HCC patients (27). These results support γ-OHPdG is a useful early biomarker of HCC and its application in identifying individuals with increased HCC risk warrants further investigation.
Because of its increasing prevalence, obesity and NAFLD is believed to be a more significant risk for HCC than diabetes and viral infection combined. An effective and practical preventative strategy based on a better understanding of its mechanism is urgently needed. Dietary-related antioxidants derived from natural sources offer several advantages as cancer preventive agents because they are relatively low cost, readily available and, in general, of little toxicity. Our findings that TE, that is well tolerated in hepatitis patients (51), effectively prevents NAFLD, reduces γ-OHPdG and total mutational loads and, subsequently, blocks HCC development in HFD-fed mice raise the possibility of its clinical application in preventing HCC associated with obesity.
Supplementary Material
Acknowledgements
We thank Dr. Bin Gao from NIAAA and Dr. Moon-Shong Tang from NYU School of Medicine for the discussions and Dr. Yukihiko Hara for providing Theaphenon E. We thank the technical assistance of the Animal, Proteomic, Histopathology, Cytometry and Microscopy and Imaging Shared Resources of the Lombardi Comprehensive Cancer Center at Georgetown University Medical Center. This work is supported by NIH/NCI/CCSG grant P30-CA051008 and NIH/NCI 5R01CA190678.
Financial support: This work was supported by the NCI grant RO1-CA-134892
Footnotes
Conflict of interest: The authors listed have no affiliations with or involvement in any organization or entity with any financial interest or non-financial interest in the subject matter or materials discussed in this manuscript.
REFERENCES
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin 2015. [DOI] [PubMed] [Google Scholar]
- 2.Alzahrani B, Iseli TJ, Hebbard LW. Non-viral causes of liver cancer: Does obesity led inflammation play a role? Cancer Letters 2014;345:223–229. [DOI] [PubMed] [Google Scholar]
- 3.Moudgil V, Redhu D, Dhanda S, Singh J. A review of molecular mechanisms in the development of hepatocellular carcinoma by aflatoxin and hepatitis B and C viruses. J Environ Pathol Toxicol Oncol 2013;32:165–175. [DOI] [PubMed] [Google Scholar]
- 4.Baffy G, Brunt EM, Caldwell SH. Hepatocellular carcinoma in non-alcoholic fatty liver disease: an emerging menace. J Hepatol 2012;56:1384–1391. [DOI] [PubMed] [Google Scholar]
- 5.Saran U, Humar B, Kolly P, Dufour JF. Hepatocellular carcinoma and lifestyles. J Hepatol 2016;64:203–214. [DOI] [PubMed] [Google Scholar]
- 6.Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med 2003;348:1625–1638. [DOI] [PubMed] [Google Scholar]
- 7.Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, Osterreicher CH, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010;140:197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bohinc BN, Diehl AM. Mechanisms of disease progression in NASH: new paradigms. Clin Liver Dis 2012;16:549–565. [DOI] [PubMed] [Google Scholar]
- 9.Seki S, Kitada T, Yamada T, Sakaguchi H, Nakatani K, Wakasa K. In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. J Hepatol 2002;37:56–62. [DOI] [PubMed] [Google Scholar]
- 10.Neuschwander-Tetri BA, Caldwell SH. Nonalcoholic steatohepatitis: summary of an AASLD Single Topic Conference. Hepatology 2003;37:1202–1219. [DOI] [PubMed] [Google Scholar]
- 11.Podrini C, Borghesan M, Greco A, Pazienza V, Mazzoccoli G, Vinciguerra M. Redox homeostasis and epigenetics in non-alcoholic fatty liver disease (NAFLD). Curr Pharm Des 2013;19:2737–2746. [DOI] [PubMed] [Google Scholar]
- 12.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest 2005;115:209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Michelotti GA, Machado MV, Diehl AM. NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol 2013;10:656–665. [DOI] [PubMed] [Google Scholar]
- 14.Dongiovanni P, Romeo S, Valenti L. Hepatocellular carcinoma in nonalcoholic fatty liver: role of environmental and genetic factors. World J Gastroenterol 2014;20:12945–12955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chavez-Tapia NC, Rosso N, Tiribelli C. In vitro models for the study of non-alcoholic fatty liver disease. Curr Med Chem 2011;18:1079–1084. [DOI] [PubMed] [Google Scholar]
- 16.DeAngelis RA, Markiewski MM, Taub R, Lambris JD. A high-fat diet impairs liver regeneration in C57BL/6 mice through overexpression of the NF-kappaB inhibitor, IkappaBalpha. Hepatology 2005;42:1148–1157. [DOI] [PubMed] [Google Scholar]
- 17.Ma C, Kesarwala AH, Eggert T, Medina-Echeverz J, Kleiner DE, Jin P, Stroncek DF, et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature 2016;531:253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nath RG, Chung FL. Detection of exocyclic 1,N2-propanodeoxyguanosine adducts as common DNA lesions in rodents and humans. Proc Natl Acad Sci U S A 1994;91:7491–7495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang IY, Chan G, Miller H, Huang Y, Torres MC, Johnson F, Moriya M. Mutagenesis by acrolein-derived propanodeoxyguanosine adducts in human cells. Biochemistry 2002;41:13826–13832. [DOI] [PubMed] [Google Scholar]
- 20.Minko IG, Washington MT, Kanuri M, Prakash L, Prakash S, Lloyd RS. Translesion synthesis past acrolein-derived DNA adduct, gamma -hydroxypropanodeoxyguanosine, by yeast and human DNA polymerase eta. J Biol Chem 2003;278:784–790. [DOI] [PubMed] [Google Scholar]
- 21.Feng Z, Hu W, Hu Y, Tang MS. Acrolein is a major cigarette-related lung cancer agent: Preferential binding at p53 mutational hotspots and inhibition of DNA repair. Proc Natl Acad Sci U S A 2006;103:15404–15409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Minko IG, Kozekov ID, Harris TM, Rizzo CJ, Lloyd RS, Stone MP. Chemistry and biology of DNA containing 1,N(2)-deoxyguanosine adducts of the alpha,beta-unsaturated aldehydes acrolein, crotonaldehyde, and 4-hydroxynonenal. Chem Res Toxicol 2009;22:759–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Emami A, Dyba M, Cheema AK, Pan J, Nath RG, Chung FL. Detection of the acrolein-derived cyclic DNA adduct by a quantitative 32P-postlabeling/solid-phase extraction/HPLC method: blocking its artifact formation with glutathione. Anal Biochem 2008;374:163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fu Y, Nath RG, Dyba M, Cruz IM, Pondicherry SR, Fernandez A, Schultz CL, et al. In vivo detection of a novel endogenous etheno-DNA adduct derived from arachidonic acid and the effects of antioxidants on its formation. Free Radic Biol Med 2014;73:12–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pan J, Awoyemi B, Xuan Z, Vohra P, Wang HT, Dyba M, Greenspan E, et al. Detection of acrolein-derived cyclic DNA adducts in human cells by monoclonal antibodies. Chem Res Toxicol 2012;25:2788–2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hill-Baskin AE, Markiewski MM, Buchner DA, Shao H, DeSantis D, Hsiao G, Subramaniam S, et al. Diet-induced hepatocellular carcinoma in genetically predisposed mice. Hum Mol Genet 2009;18:2975–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fu Y, Silverstein S, McCutcheon JN, Dyba M, Nath RG, Aggarwal M, Coia H, et al. An endogenous DNA adduct as a prognostic biomarker for hepatocarcinogenesis and its prevention by Theaphenon E in mice. Hepatology 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tornesello ML, Buonaguro L, Tatangelo F, Botti G, Izzo F, Buonaguro FM. Mutations in TP53, CTNNB1 and PIK3CA genes in hepatocellular carcinoma associated with hepatitis B and hepatitis C virus infections. Genomics 2013;102:74–83. [DOI] [PubMed] [Google Scholar]
- 29.Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, Calderaro J, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet 2012;44:694–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res 2012;72:2457–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rinella ME, Elias MS, Smolak RR, Fu T, Borensztajn J, Green RM. Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. J Lipid Res 2008;49:1068–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Verna L, Whysner J, Williams GM. N-nitrosodiethylamine mechanistic data and risk assessment: bioactivation, DNA-adduct formation, mutagenicity, and tumor initiation. Pharmacol Ther 1996;71:57–81. [DOI] [PubMed] [Google Scholar]
- 33.Vansaun MN, Mendonsa AM, Lee Gorden D. Hepatocellular proliferation correlates with inflammatory cell and cytokine changes in a murine model of nonalchoholic fatty liver disease. PLoS One 2013;8:e73054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bailey EA, Iyer RS, Stone MP, Harris TM, Essigmann JM. Mutational properties of the primary aflatoxin B1-DNA adduct. Proc Natl Acad Sci U S A 1996;93:1535–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu X, Gu J, Patt Y, Hassan M, Spitz MR, Beasley RP, Hwang LY. Mutagen sensitivity as a susceptibility marker for human hepatocellular carcinoma. Cancer Epidemiol Biomarkers Prev 1998;7:567–570. [PubMed] [Google Scholar]
- 36.Shen J, Tsoi H, Liang Q, Chu ES, Liu D, Yu AC, Chan TF, et al. Oncogenic mutations and dysregulated pathways in obesity-associated hepatocellular carcinoma. Oncogene 2016;35:6271–6280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bhalla K, Hwang BJ, Dewi RE, Twaddel W, Goloubeva OG, Wong KK, Saxena NK, et al. Metformin prevents liver tumorigenesis by inhibiting pathways driving hepatic lipogenesis. Cancer Prev Res (Phila) 2012;5:544–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shimizu M, Sakai H, Shirakami Y, Iwasa J, Yasuda Y, Kubota M, Takai K, et al. Acyclic retinoid inhibits diethylnitrosamine-induced liver tumorigenesis in obese and diabetic C57BLKS/J- +(db)/+Lepr(db) mice. Cancer Prev Res (Phila) 2011;4:128–136. [DOI] [PubMed] [Google Scholar]
- 39.Bose M, Lambert JD, Ju J, Reuhl KR, Shapses SA, Yang CS. The major green tea polyphenol, (−)-epigallocatechin-3-gallate, inhibits obesity, metabolic syndrome, and fatty liver disease in high-fat-fed mice. J Nutr 2008;138:1677–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao BL, Li XJ, He RG, Cheng SJ, Xin WJ. Scavenging effect of extracts of green tea and natural antioxidants on active oxygen radicals. Cell Biophys 1989;14:175–185. [DOI] [PubMed] [Google Scholar]
- 41.Kochi T, Shimizu M, Terakura D, Baba A, Ohno T, Kubota M, Shirakami Y, et al. Non-alcoholic steatohepatitis and preneoplastic lesions develop in the liver of obese and hypertensive rats: suppressing effects of EGCG on the development of liver lesions. Cancer Lett 2014;342:60–69. [DOI] [PubMed] [Google Scholar]
- 42.Westerterp-Plantenga MS. Green tea catechins, caffeine and body-weight regulation. Physiol Behav 2010;100:42–46. [DOI] [PubMed] [Google Scholar]
- 43.Ploeger JM, Manivel JC, Boatner LN, Mashek DG. Caloric Restriction Prevents Carcinogen-Initiated Liver Tumorigenesis in Mice. Cancer Prev Res (Phila) 2017;10:660–670. [DOI] [PubMed] [Google Scholar]
- 44.Lee SF, Liang YC, Lin JK. Inhibition of 1,2,4-benzenetriol-generated active oxygen species and induction of phase II enzymes by green tea polyphenols. Chem Biol Interact 1995;98:283–301. [DOI] [PubMed] [Google Scholar]
- 45.Li W, Guo Y, Zhang C, Wu R, Yang AY, Gaspar J, Kong AN. Dietary Phytochemicals and Cancer Chemoprevention: A Perspective on Oxidative Stress, Inflammation, and Epigenetics. Chem Res Toxicol 2016;29:2071–2095. [DOI] [PubMed] [Google Scholar]
- 46.Yang CS, Wang X, Lu G, Picinich SC. Cancer prevention by tea: animal studies, molecular mechanisms and human relevance. Nat Rev Cancer 2009;9:429–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kohli R, Kirby M, Xanthakos SA, Softic S, Feldstein AE, Saxena V, Tang PH, et al. High-fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology 2010;52:934–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Asgharpour A, Cazanave SC, Pacana T, Seneshaw M, Vincent R, Banini BA, Kumar DP, et al. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J Hepatol 2016;65:579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paradis V, Zalinski S, Chelbi E, Guedj N, Degos F, Vilgrain V, Bedossa P, et al. Hepatocellular carcinomas in patients with metabolic syndrome often develop without significant liver fibrosis: a pathological analysis. Hepatology 2009;49:851–859. [DOI] [PubMed] [Google Scholar]
- 50.Coia H, Ma N, He AR, Kallakury B, Berry DL, Permaul E, Makambi KH, et al. Detection of a lipid peroxidation-induced DNA adduct across liver disease stages. Hepatobiliary Surgery and Nutrition 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Halegoua-De Marzio D, Kraft WK, Daskalakis C, Ying X, Hawke RL, Navarro VJ. Limited Sampling Estimates of Epigallocatechin Gallate Exposures in Cirrhotic and Noncirrhotic Patients With Hepatitis C After Single Oral Doses of Green Tea Extract. Clinical Therapeutics 2012;34:2279–2285.e2271. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.