

Abstract
Backgroud
Dysregulation of claudin-6 (CLDN6) expression in cancers has been widely documented. However, no study has reported a complete mechanistic understanding of CLDN6 regulation and function in endometrial carcinoma (EC) progression. In the current study, we aimed to assess the expression and biological functions of CLDN6 in EC.
Methods
Firstly, the expression level of CLDN6 in EC was measured based on The Cancer Genome Atlas (TCGA) database. Then, qRT-PCR and western blotting were implemented to detect the expression levels of CLDN6 in 82 pairs of EC tissues and corresponding non-tumor tissues, as well as EC cell line HEC-1B. After knockdown of CLDN6, with the attempt to assess whether CLDN6 reduction had positive effects on the cell proliferation, clone formation, invasion and migration abilities of HLC-1Bs, cell counting kit-8 (CCK-8) assay (24, 48, 72 and 96 hours post-transfection), clone experiment, and invasion and migration assays were conducted. Through western blotting analysis, CLDN6-mediated phosphatidylinositol 3-kinase (PI3K) pathway was evaluated.
Results
Based on the data of TCGA database, clinical patients and cell line HEC-1B, CLDN6 was up-regulated in EC compared with normal. Univariate as well as multivariate COX analysis indicated that CLDN6 expression can act as an independent prognostic factor for overall survival of EC. Further, knockdown of CLDN6 significantly inhibited HEC-1B cell proliferation, suppressed the colony numbers of HEC-1-B cells, and restrained the invasive and migratory ability of HEC-1-B cells. Importantly, through western blot analysis, we found that inhibition of CLDN6 remarkably decreased p-AKT, p-PI3K, and mTOR expression level in EC HEC-1B cell line.
Conclusion
Our data underscore the significance of CLDN6 in EC progression, and CLDN6 is a new candidate oncogene in EC. Our findings propose that targeting CLDN6 might offer future clinical utility in EC.
Keywords: endometrial carcinoma, CLDN6, knockdown, proliferation, PI3K/AKT pathway, prognostic, invasion
Introduction
Endometrial carcinoma (EC) is one of the 3 most common gynecologic cancers, with about 200,000 new diagnoses worldwide annually.1 With the increase in obesity and the reduction in physical activity, the occurrence of EC is rising and exhibits a trend in younger women.2 Currently, surgery, chemotherapy, and radiotherapy are the main therapeutic strategies for EC. Nevertheless, only a minority of EC patients are sensitive to these treatments.3 Moreover, type II EC patients have a poor prognosis with 5-year survival rate <35%.4 Thus, identification of novel therapeutic targets and further elucidation of the molecular mechanisms underlying the tumorigenesis and progression of EC may have a major impact on the health of women.
Claudins (CLDNs) are critical transmembrane proteins in tight junction, which function primarily as a barrier against paracellular transport between epithelial cells and the CLDN family. These proteins play important roles in cellular adhesion, polarity, permeability, and glandular differentiation.5,6 Abnormal expression of CLDNs has been considered as a molecular mechanism of the progression of cancer, for example, breast cancer, prostate cancer, and ovarian cancer.7–10 CLDN6, a member of CLDN family, is located on 16p13.3 and maintains cell-cell junctions in epithelial cell sheets, and the expression of this gene is mainly observed in mouse embryonic stem cells and primitive germ cell tumors.11 CLDN6 has been demonstrated to suppress cancer cell growth and induce cell apoptosis.12,13 In a former study, CLDN6 has been demonstrated to function as a tumor suppressor in breast cancer, inhibiting the breast cancer cell growth, migration, as well as invasion through p38/mitogen-activated protein kinase (MAPK) pathway.14 Moreover, another study has suggested that silencing of CLDN6 enhances migration ability of the human breast epithelium cell line HBL-100.12 Zavala-Zendejas et al15 have implicated that increased expression of CLDN6 can promote the tumorigenic properties of a gastric adenocarcinoma cell line. In addition, CLDN6 is a biomarker for pediatric tumors.16 However, so far, there is no published report on the CLDN6 expression in EC tissues, and the exact mechanism underlying CLDN6 in EC remains unclear.
Therefore, in our study, we designed to investigate the expression of CLDN6 and its clinical-pathological characteristics in EC. As cancer is characterized by increased migratory/invasive capacity, we examined the migratory/invasive ability of HEC-1-B cells with CLDN6 knockdown in the current study. Transwell assay results showed that knockdown of CLDN6 caused significant suppression of migratory/invasive capacity. Our results demonstrated that CLDN6 might serve as a potential therapeutic target for EC.
Materials and methods
Patient samples
This study was approved by the Research Ethics Committee of Danyang People’s Hospital of Jiangsu Province, China. Specimens were collected after obtaining written informed consents of all patients. The EC tissues and their matched adjacent non-tumorous (ANT) tissues were collected from 82 cases of EC patients and 82 ANT samples through surgical resection at Danyang People’s Hospital of Jiangsu Province between March 2009 and January 2013, and were confirmed by histopathological evaluation. These samples were immediately frozen and stored at −80°C until use. All EC patients received no preoperative treatment, for example, radiotherapy or chemotherapy. The overall survival (OS) was defined as the interval between the surgery and the date of death or the end of follow-up.
CLDN6 expression in The Cancer Genome Atlas (TCGA) database
TCGA covers the clinical data from 552 samples of EC and 35 normal tissue samples. The difference of CLDN6 expression level between normal tissues and EC samples was analyzed using limma (version 3.36.3; http://bioconductor.org/packages/release/bioc/html/limma.html).17 The data were downloaded on November 19, 2017.
Cell line and culture
HEC-1-B cells are EC cells of human epithelial origin. EC cell line (HEC-1-B) and normal endometrial cells (ESC) were obtained from the Shanghai Cell Bank at the Chinese Academy of Sciences (Shanghai, China). The cells were maintained in Roswell Park Memorial Institute (RPMI)-1640 supplemented with 10% FBS (Gibco), 100 U/mL penicillin, and 0.1 mg/mL streptomycin at a condition of a humidified atmosphere of 5% CO2 at 37°C until 75% confluent.
Knockdown of CLDN6 in EC cell line and transfection
The high CLDN6 expression in EC cell line HEC-1-B was used to establish the stable CLDN6 knockdown cell line. Small interfering RNA (siRNA) targeting CLDN6 (si-CLDN6) was applied to suppress the expression of CLDN6 gene in HEC-1-B cell line. A negative control siRNA was commercially generated by transfecting cells with the vector constructed by targeting a sequence that did not yield any appreciable knockdown of the protein production (Shanghai GenePharma Co. Ltd, Shanghai, China). In accordance with the manufacturer’s instruction, siRNA (sense [5′-AAGAUUUGCAGACCAGUAGAGGCCA-3′] and antisense [5′-UGGCCUCUACUGGUCUGCAAAUCU U-3′]) were transfected by using Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA). Interference efficiency was determined by qRT-PCR and western blotting.
RNA extraction and qRT-PCR
Total RNA of the tissue samples and cell line were extracted using the Trizol reagent (Thermo Fisher Scientific, Waltham, MA, USA) based on the manufacturer’s instructions. Total RNA was reverse transcribed into cDNA using a Reverse Transcription Kit (Takara Biotechnology Co., Ltd., Dalian, China) on the basis of the manufacturer’s instructions. The CLDN6 expression was measured by qRT-PCR using glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as an internal control, and the data collection was implemented on the Bio-Rad System (Bio-Rad Laboratories Inc., Hercules, CA, USA) based on the manufacturer’s instructions. The primer sequences were: CLDN6, forward: 5′-TGTTCGGCTTGCTGGTCTAC-3′, reverse: 5′-CGGGGATTAGCGTCAGGAC-3′; GAPDH, forward: 5′-GGAGCGAGATCCCTCCAAAAT-3′, reverse: 5′-GGCTGTTGTCATACTTCTCATGG-3′. Ct method was applied to quantify the transcripts. The average value in each triplicate was used to calculate the relative amount of CLDN6 using 2−ΔΔCt method relative to GAPDH. The PCR conditions were set as 95°C for 5 minutes, followed by 40 cycles of 95°C for 30 seconds, 60°C for 45 seconds, and 72°C for 30 minutes.
Cell counting kit-8 assay and colony-formation assay
In the present study, cell counting kit-8 (CCK-8) assay was used to evaluate the cell growth at 24, 48, 72, and 96 hours after CLDN6 knockdown according to the previously described report.18 Specifically, HEC-1-B cells, with and without transfection, were incubated in 96-well plates in 100 µL of RPMI-1640 containing 10% FBS and cultured at 37°C for 4 days. Cell viability was documented every 24 hours. Then, CCK-8 dye as well as culture medium (at a ratio of 1:10) were added to each well and incubated for 1.5 hour at 37°C at 5% CO2. After that, we read the absorbance by means of a microplate reader at 450 nm. The OD450 values were proportional to the total number of live cells. This proliferation experiment was repeated at least 3 times.
For the colonies’ formation, si-CLDN6 and si-control cells were placed into fresh 6-well plates and cultured in media supplementing with 10% FBS, replacing the cell medium every 4 days. Following 2 weeks, the colonies were fixed with 4% paraformaldehyde and stained using 0.1% crystal violet (Sigma-Aldrich Co., St Louis, MO, USA). The visible colonies containing >50 cells were manually calculated. Each assay was duplicated at least 3 times independently.
Wound-healing assay
Cells were cultured to confluency and wounds were created by dragging a 10-µL pipette tip through the monolayer. The cells were allowed to migrate for 24 hours. The wound images were monitored using microscopy (Olympus, Tokyo, Japan) at 0 and 24 hours after wounding. The widths of the wounded areas were measured at 0 (W0) and 24 hours (W24). The relative migration distance was calculated as (W0–W24)/W0×100%. A minimum of 5 random areas were recorded.
Cell invasion and migration assays
In order to reveal the effects of CLDN6 on cell migration and invasion, trans-well experiments were implemented using 6.5 mm transwell chambers with 8.0 µm pore-size polycarbonate membranes with or without Matrigel (BD Biosciences). In the invasion experiment, the upper chambers were first coated with 100 µL of Matrigel. In both invasion and migration assays, cells were suspended in serum-free medium at a 1:6 dilution (BD Biosciences) and incubated at 37°C for 4 hours. Subsequently, cells were loaded onto the top chamber of the transwell at a density of 5×105 cells/mL (200 µL/chamber). The lower chambers were filled with 500 µL complete media containing 10% FBS. After overnight incubation at 37°C in an air/5% CO2 atmosphere, non-invasive cells were removed from the top chambers with cotton swabs. The remaining cells that were attached to the underside of the membranes were fixed using 4% paraformaldehyde and stained with 0.1% crystal violet for 20 minutes. Next, the cells were counted in 5 randomly selected fields of view using a microscope (Nikon ECLIPSE 80i system), and the average value was recorded for every field. Each experiment was conducted at least 3 times.
Western blotting analysis
Total proteins were extracted from the HEC-1-B cells after complete cell lysis and separated by SDS-PAGE. Our interested proteins were determined based on corresponding specific antibodies after transferring to polyvinylidene difluoride membranes. The following antibodies were used for analysis: anti-AKT at 1:1,000 dilution, anti-p-AKT (1:1,000), anti-mTOR (1:1,000), anti-PI3K (1:1,000), anti-p-PI3K (1:1,000), anti-CLDN6 (1:1,000), and anti-GAPDH (1:5,000, GAPDH was used as an internal reference). Then, the membranes were washed 3 times with Tris-buffered saline with Tween 20 at room temperature and incubated using a horseradish peroxidase-conjugated anti-rabbit IgG as the secondary antibody (Santa Cruz Biotechnology Inc.) for 2 hours at 1:5,000 dilution. An enhanced chemiluminescence (ECL) western blot kit was used to detect all bands by means of ECL system (GE Healthcare Life Sciences). Finally, protein bands were quantified using Bio-Rad Quantity One 1-D software.
Statistical analyses
In the current work, we used SPSS 22.0 software to perform all statistical analyses. Each assay was implemented at least 3 times. The data were presented as the mean ± SD, and the comparison between 2 groups was performed using a chi-squared test, and multigroups were compared using 1-way ANOVA test. The survival curve was visualized using Kaplan–Meier approach and log-rank test. Cox proportional hazards model was applied to analyze the prognostic factors. Each variable having statistical significance within the univariate analysis was put into multivariate analysis models for evaluating the independent prognostic values on EC. P<0.05 was considered to be statistically significant.
Results
Expression of CLDN6 in EC tissues and cell line
To begin with, the CLDN6 expression level in EC samples (EC samples =552; normal samples =35) was analyzed on the basis of the RNA-Seq data obtained from TCGA database. Figure 1A shows that CLDN6 expression level was overexpressed in EC tissues (P=8.29E−15). Then, the relative expression level of CLDN6 in EC tissues (n=82) compared with corresponding ANT tissues (n=82) was examined by means of RT-PCR and normalized to GAPDH. As shown in Figure 1B, the CLDN6 level was remarkably upregulated in EC tissues compared with corresponding ANT tissues (P<0.001). These indicated that abnormal CLDN6 expression might be associated with EC pathogenesis. Subsequently, the expression of CLDN6 in the EC cell line HEC-1-B was evaluated using qRT-PCR. The PCR results demonstrated that HEC-1-B cells showed a higher expression of CLDN6 compared with the normal human ESC (P<0.001, Figure 1C).
Figure 1.

CLDN6 expression level in EC was significantly upregulated.
Notes: (A) CLDN6 expression level of patients with EC (n=552) and controls (n=35) from the TCGA database was analyzed. (B) Relative expression levels of CLDN6 in EC tissues (n=82) relative to corresponding ANT tissues (n=82). CLDN6 expression was examined using PCR and normalized to GAPDH. (C) Relative expression levels of CLDN6 in EC cell line HEC-1-B relative to corresponding control cell. CLDN6 expression was examined using qRT-PCR and normalized to GAPDH. **P<0.001.
Abbreviations: UCEC, Uterine Corpus Endometrial Carcinoma; ANT, adjacent non-tumorous; CLDN6, claudin-6; EC, endometrial carcinoma; ESC, endometrial cells; TCGA, The Cancer Genome Atlas.
Relationship of CLDN6 expression level with the clinicopathological factors in EC patients
In an attempt to reveal the clinical relevance of CLDN6 expression in EC patients, we conducted the correlation analysis between CLDN6 expression and clinicopathological factors, for instance, age, histological type, clinical stage, and grade. The relationship of CLDN6 expression level with the clinicopathological factors in EC is shown in Table 1. From this table, we observed that the expression level of CLDN6 was significantly correlated to age (P=0.002), histological type (P≤0.0001), clinical stage (P=0.002), and grade (P≤0.0001).
Table 1.
Clinical association between CLDN6 expression and clinicopathological variables in EC patients
| Characteristics | Expression of CLDN6
|
P-value | |
|---|---|---|---|
| Low | High | ||
|
| |||
| Age (years) | 0.002* | ||
| <60 | 16 | 4 | |
| ≥60 | 25 | 37 | |
| Histological type | ≤0.0001* | ||
| Endometrioid endometrial adenocarcinoma | 34 | 5 | |
| Mixed serous and endometrioid | 3 | 3 | |
| Serous endometrial adenocarcinoma | 4 | 33 | |
| Clinical stage | 0.002* | ||
| I+II | 29 | 15 | |
| III+IV | 12 | 26 | |
| Grade | ≤0.0001* | ||
| G1+G2 | 22 | 1 | |
| G3+G4 | 19 | 40 | |
Notes: The chi-squared test was used to evaluate the correlation between gene expression and clinical features. The expression of CLDN6 gene is associated with age, histological type, clinical stage, and grade in patients with EC.
P, statistically significant.
Abbreviations: CLDN6, claudin-6; EC, endometrial carcinoma.
High level of CLDN6 is predictive of worse prognosis of EC patients
The relationship between the CLDN6 expression and the survival time of EC patients was further analyzed using Kaplan–Meier analysis and log-rank test. We found that EC patients with high CLDN6 expression exhibited a worse prognosis, relative to those with low level of CLDN6 (Figure 2, P=0.005). Next, in order to further prove the prognostic role of CLDN6 in EC patients, univariate and multivariate COX proportional hazards analyses were conducted to analyze the independent prognostic factors for survival in EC patients. Univariate analysis demonstrated that the histological type (P≤0.0001), clinical stage (P=0.0003), and CLDN6 expression (P=0.008) were significantly related to the OS of EC patients, as shown in Table 2. However, multivariate analysis suggested that histological type was an independent prognostic factor for the OS of EC patients (Table 2, P=0.016). Accordingly, these results indicated that upregulated expression of CLDN6 might predicate a poor prognosis of patients with EC.
Figure 2.
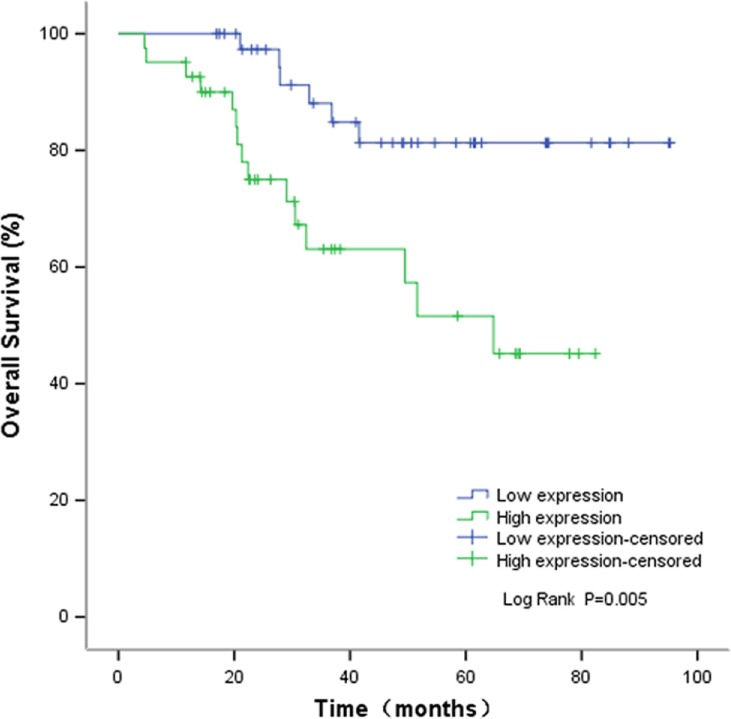
Kaplan–Meier OS curves based on CLDN6 expression level.
Note: The relationship between the CLDN6 expression and the survival time of EC patients was analyzed using Kaplan–Meier analysis and log-rank test.
Abbreviations: CLDN6, claudin-6; EC, endometrial carcinoma; OS, overall survival.
Table 2.
Univariate and multivariate analysis of clinical prognostic factors of EC
| Variables | Univariate analysis
|
Multivariate analysis
|
||||
|---|---|---|---|---|---|---|
| P-value | HR | 95% CI | P-value | HR | 95% CI | |
|
| ||||||
| CLDN6 expression (high/low) | 0.008* | 3.629 | 1.402–9.398 | 0.861 | 0.902 | 0.285–2.858 |
| Histological type | ≤0.0001* | 3.038 | 1.657–5.568 | 0.016* | 2.624 | 1.194–5.764 |
| Clinical stage (I–IV) | 0.003* | 4.319 | 1.665–11.202 | 0.226 | 1.921 | 0.668–5.526 |
Note:
P-value is statistically significant.
Abbreviations: CLDN6, claudin-6; EC, endometrial carcinoma.
Knockdown of CLDN6 inhibits cell proliferation and growth of HEC-1-B cells after transfection
To investigate the role of CLDN6 in EC cells, the CLDN6-specific si-CLDN6 was designed and transfected into EC cells to further determine its effect on the cell growth of EC cell in vitro. The expression level of CLDN6 was detected by qRT-PCR and western blotting 72 hours after transfection (Figure 3). As listed in Figure 3, si-CLDN6-transfected cells displayed a significant reduction in both mRNA and protein level of CLDN6 compared with the control group in HEC-1-B cells (P<0.001 in the results in qRT-PCR and western blotting).
Figure 3.

Knockdown efficiency was determined by qRT-PCR (A) and western blotting (B and C) in EC cells.
Notes: **P<0.001. Data represent the mean ± SD.
Abbreviations: CLDN6, claudin-6; EC, endometrial carcinoma.
CCK-8 assay results revealed that the CLDN6 knockdown obviously suppressed the proliferation rate of HEC-1-B cells (Figure 4) at 48 (P<0.05), 72 (P<0.001), and 96 hours (P<0.001). Consistently, a colony-formation assay implicated that CLDN6 knockdown significantly inhibited the colony numbers of HEC-1-B cells (P<0.001, Figure 5).
Figure 4.
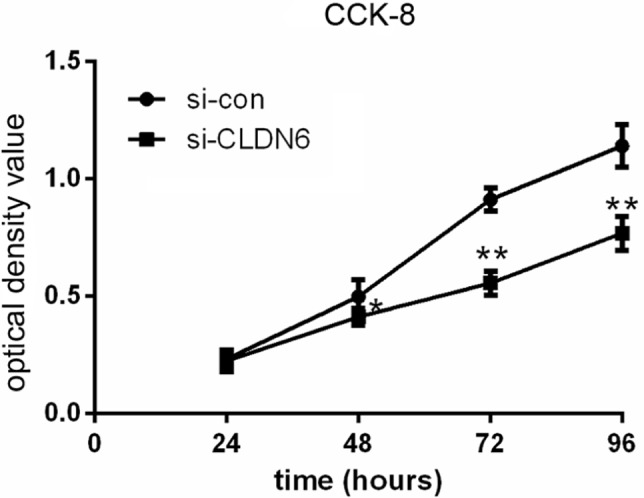
Knockdown CLDN6 in EC cells significantly reduced the proliferative abilities, as determined by CCK-8 assay.
Note: *P<0.05, **P<0.001.
Abbreviations: CCK-8, cell counting kit-8; CLDN6, claudin-6; EC, endometrial carcinoma.
Figure 5.

Colony-formation assays suggested that knockdown of CLDN6 significantly reduced the colony-forming ability of EC cells.
Notes: (A) Colonies in the two groups were showed under a microscope. (B) Bar chart showed the number of colonies. **P<0.001.
Abbreviations: CLDN6, claudin-6; EC, endometrial carcinoma.
Inhibition of migratory/invasive capacity by CLDN6 knockdown
Transwell assays were carried out to assess the potential effects of CLDN6 knockdown on the migratory and invasive abilities. As shown in Figure 6, invasive and migratory cell proportion in HEC-1-B cells with transfection was significantly lower than those in un-transfected HEC-1-B cells (P<0.001), suggesting a suppression of invasive and migratory ability of HEC-1-B cells after CLDN6 was knocked down. After 24 hours of wound generation in the cell monolayer, the wound of the CLDN6 knockdown cells was wider than that of the control cells.
Figure 6.

Migratory/invasive capacity was measured using wound-healing and transwell assay.
Notes: (A) Microscopic images of HEC-1-B cells were assessed by wound-healing assay in two groups. (B) Microscopic images of migratory and invasive cell passing though the microwells of the transwell chamber were detected by transwell assay. (C) The number of migratory and invasive cells was counted. **P<0.01 vs si-con group. Data were presented as the mean ± SD of 3 independent experiments.
Abbreviation: CLDN6, claudin-6.
CLDN6 knockdown suppresses the activation of PI3K signaling of HEC-1-B cells
To further explore whether the abnormal CLDN6 expression mediates the PI3K signaling, a series of biomarkers including AKT, PI3K, and p70S6K were examined based on western blotting analysis. As shown in Figure 7, CLDN6 knockdown remarkably decreased the p-AKT, p-PI3K, and mTOR in HEC-1-B cells (P<0.001).
Figure 7.

Effect of CLDN6 knockdown on activation of PI3K pathway in HEC-1B cells.
Notes: (A) The expressions of PI3K, AKT, and mTOR in EC cells 72 h after transfection examined using western blotting analysis. (B) Bars present means ± SD. The content of protein was normalized with the expression of GAPDH. **P<0.001 compared with the respective non-transfected group.
Abbreviations: CLDN6, claudin-6; EC, endometrial carcinoma.
Discussion
One of the first steps in cancer progression and metastasis is loss of cell-to-cell adhesion.19 As we all know, CLDNs are important for tight junction formation, but the changes in their expression or localization often result in the changes of the tight junction structure and function, which exert key functions in the transform in motility, cancer progression, invasion, and metastasis.20 CLDN6 was overexpressed in multiple-tumor tissues (such as gastric cancer, esophageal squamous cell carcinoma, and lung cancer) and its expression was correlated with tumor invasiveness and metastasis, and thus it might be a prognostic factor of poor outcome.21–23 However, whether CLDN6 regulated the progression of EC is still unknown. To the best of our knowledge, this is the first report in which we assess the prognostic impact of CLDN6 on EC. In our study, CLDN6 knockdown decreased cell proliferation and migration, accompanied by the suppression of PI3K/AKT/mTOR pathway in HEC-1-B cells.
In our study, by using CLDN6 knockdown of HEC-1-B cells as a model, we found the significantly lower rates of cell proliferation in CLDN6 knockdown cells compared with control cells. Additionally, the knockdown of CLDN6 was observed to result in a decreased migratory and invasive ability in HEC-1-B cells. Our results were in line with previous studies, indicating an oncogenic role of CLDN6.24–26 However, the exact mechanisms remain poor. Significantly, a former report has demonstrated that enhanced proliferation and migration of tumor cells may be caused by the altered change in ionic microenvironments in the lateral membranes.27 Moreover, epithelial to mesenchymal transition (EMT) is one of the mechanisms of tumor migration and invasion.6–8 CLDN6 may suppress migration and invasion of cancer cells via EMT inhibition.26 However, the mechanisms that could explain how CLDN6 regulate cell proliferation rate, migration, and invasiveness still remain to be revealed.
Activation of PI3K/AKT/mTOR signaling pathway plays a critical role in the initiation and progression of many cancers, for example, colon, bladder, and ovarian cancers.28–30 Activated PI3K recruits AKT to the plasma membrane, inducing its activation, which in turn phosphorylates various proteins, for instance, mTOR, caspase-3, and so on.31,32 Gille et al33 have demonstrated that the stimulation of mTOR leads to the activation of PI3K in endothelial cells, increases cell migration, and induces angiogenesis. Trisciuoglio et al34 also have suggested that PI3K-dependent pathway participates in the angiogenesis. Significantly, the formation of new vessels has been proven to cause EC progression.35 In our study, a decreased expression of PI3K/AKT/mTOR signaling pathway in CLDN6 knockdown cells was also found, suggesting that PI3K/AKT/mTOR might be an important pathway involved in altering the phenotype of the cells following CLDN6 knockdown. Of note, our results are consistent with a previous study that reported that PI3K/AKT/mTOR signaling pathway is activated in EC pathogenesis.36 Significantly, Zhang et al37 have demonstrated that miRNA-101 decreased HEC-1-A cell proliferation and invasion through attenuating the activity of the pathway of PI3K/AKT/mTOR. Thus, we infer that CLDN6 inhibition might, to a certain degree, regulate the suppression of PI3K/AKT/mTOR signaling pathway in EC, thereby inhibiting tumor initiation and progression.
However, we found that by multivariate analysis, CLDN6 expression does not significantly affect the OS. It is possible that the adjusted OS curve might not show any difference based on CLDN6 expression. Therefore, it is necessary to increase the sample size and samples of different pathological stages and grades for further study. Moreover, the information in many materials in TCGA is incomplete, which has an impact on relevant statistical analysis. We are now increasing the collection of our own clinical cases and hope that this issue can be better explained in the future.
Taken together, our study has implicated that upregulation of CLDN6 expression results in a relatively malignant phenotype mediated through PI3K/AKT/mTOR signaling pathway in the EC cell line HEC-1-B, further supporting the hypothesis that CLDN6 is likely to be a promoter in EC. Increased expression of CLDN6 might contribute to the malignant progression of EC.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Jemal A, Murray T, Ward E, Samuels A. Erratum: Cancer statistics, 2005 (Ca – A Cancer Journal for Clinicians (January/February 2005) 55 (10–30)) CA Cancer J Clin. 2005;55(4):259. doi: 10.3322/canjclin.55.1.10. [DOI] [PubMed] [Google Scholar]
- 2.Schouten LJ, Goldbohm RA, van den Brandt PA, Anthropometry PVDB. Anthropometry, physical activity, and endometrial cancer risk: results from the Netherlands Cohort Study. J Natl Cancer Inst. 2004;96(21):1635–1638. doi: 10.1093/jnci/djh291. [DOI] [PubMed] [Google Scholar]
- 3.Humber C, Tierney J, Symonds P, et al. Chemotherapy for advanced, recurrent or metastatic endometrial carcinoma. Cochrane Database Syst Rev. 2005;5(3):CD003915. doi: 10.1002/14651858.CD003915.pub3. [DOI] [PubMed] [Google Scholar]
- 4.Alvarez T, Miller E, Duska L, Oliva E. Molecular profile of grade 3 endometrioid endometrial carcinoma: is it a type I or type II endometrial carcinoma? Am J Surg Pathol. 2012;36(5):753. doi: 10.1097/PAS.0b013e318247b7bb. [DOI] [PubMed] [Google Scholar]
- 5.Lu S, Singh K, Mangray S, et al. Claudin expression in high-grade invasive ductal carcinoma of the breast: correlation with the molecular subtype. Mod Pathol. 2013;26(4):485–495. doi: 10.1038/modpathol.2012.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Günzel D, Yu AS, Asl Y. Claudins and the modulation of tight junction permeability. Physiol Rev. 2013;93(2):525–569. doi: 10.1152/physrev.00019.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oshima T, Miwa H. Gastrointestinal mucosal barrier function and diseases. J Gastroenterol. 2016;51(8):768–778. doi: 10.1007/s00535-016-1207-z. [DOI] [PubMed] [Google Scholar]
- 8.Morohashi S, Kusumi T, Sato F, et al. Decreased expression of claudin-1 correlates with recurrence status in breast cancer. Int J Mol Med. 2007;20(2):139. [PubMed] [Google Scholar]
- 9.Maeda T, Murata M, Chiba H, et al. Claudin-4-targeted therapy using Clostridium perfringens enterotoxin for prostate cancer. Prostate. 2012;72(4):351–360. doi: 10.1002/pros.21436. [DOI] [PubMed] [Google Scholar]
- 10.Seo HW, Rengaraj D, Choi JW, et al. Claudin 10 is a glandular epithelial marker in the chicken model as human epithelial ovarian cancer. Int J Gynecol Cancer. 2010;20(9):1465. [PubMed] [Google Scholar]
- 11.Rendon-Huerta EP, Torres-Martínez AC, Montaño L. CLDN6 (claudin 6) Atlas Genet Cytogenet Oncol Haematol. 2013;17(6):396–399. [Google Scholar]
- 12.Ren Y, Wu Q, Liu Y, Xu X, Quan C. Gene silencing of claudin-6 enhances cell proliferation and migration accompanied with increased MMP-2 activity via p38 MAPK signaling pathway in human breast epithelium cell line HBL-100. Mol Med Rep. 2013;8(5):1505–1510. doi: 10.3892/mmr.2013.1675. [DOI] [PubMed] [Google Scholar]
- 13.Xu X, Jin H, Liu Y, et al. The expression patterns and correlations of claudin-6, methy-CpG binding protein 2, DNA methyltransferase 1, histone deacetylase 1, acetyl-histone H3 and acetyl-histone H4 and their clinicopathological significance in breast invasive ductal carcinomas. Diagn Pathol. 2012;7(1):33. doi: 10.1186/1746-1596-7-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu Q, Liu X, Liu YF, et al. Inhibition of p38 activity reverses claudin-6 induced cell apoptosis, invasion, and migration. Chin Med J (Engl) 2013;126(18):3539–3544. [PubMed] [Google Scholar]
- 15.Zavala-Zendejas VE, Torres-Martinez AC, Salas-Morales B, Fortoul TI, Montaño LF, Rendon-Huerta EP. Claudin-6, 7, or 9 overexpression in the human gastric adenocarcinoma cell line AGS increases its invasiveness, migration, and proliferation rate. Cancer Invest. 2011;29(1):1–11. doi: 10.3109/07357907.2010.512594. [DOI] [PubMed] [Google Scholar]
- 16.Sullivan LM, Yankovich T, Le P, et al. Claudin-6 is a nonspecific marker for malignant rhabdoid and other pediatric tumors. Am J Surg Pathol. 2012;36(1):73–80. doi: 10.1097/PAS.0b013e31822cfa7e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ritchie ME, Phipson B, Wu D, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang X, Qi H, Wang S, et al. Cellular responses of aniline oligomers: a preliminary study. Toxicol Res. 2012;1(3):201–205. [Google Scholar]
- 19.Jordan NV, Johnson GL, Abell AN. Tracking the intermediate stages of epithelial-mesenchymal transition in epithelial stem cells and cancer. Cell Cycle. 2011;10(17):2865–2873. doi: 10.4161/cc.10.17.17188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martin TA, Jiang WG. Loss of tight junction barrier function and its role in cancer metastasis. Biochim Biophys Acta. 2009;1788(4):872–891. doi: 10.1016/j.bbamem.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 21.Zavala-Zendejas VE, Torres-Martinez AC, Salas-Morales B, Fortoul TI, Montaño LF, Rendon-Huerta EP. Claudin-6, 7, or 9 overexpression in the human gastric adenocarcinoma cell line AGS increases its invasiveness, migration, and proliferation rate. Cancer Invest. 2011;29(1):1–11. doi: 10.3109/07357907.2010.512594. [DOI] [PubMed] [Google Scholar]
- 22.Tsunoda S, Smith E, de Young NJ, et al. Methylation of CLDN6, FBN2, RBP1, RBP4, TFPI2, and TMEFF2 in esophageal squamous cell carcinoma. Oncol Rep. 2009;21(4):1067. doi: 10.3892/or_00000325. [DOI] [PubMed] [Google Scholar]
- 23.Micke P, Mattsson JS, Edlund K, et al. Aberrantly activated claudin 6 and 18.2 as potential therapy targets in non-small-cell lung cancer. Int J Cancer. 2014;135(9):2206–2214. doi: 10.1002/ijc.28857. [DOI] [PubMed] [Google Scholar]
- 24.Liu Y, Jin X, Li Y, et al. DNA methylation of claudin-6 promotes breast cancer cell migration and invasion by recruiting MeCP2 and deacetylating H3Ac and H4Ac. J Exp Clin Cancer Res. 2016;35(1):120. doi: 10.1186/s13046-016-0396-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Birks DK, Kleinschmidt-Demasters BK, Donson AM, et al. Claudin 6 is a positive marker for atypical teratoid/rhabdoid tumors. Brain Pathol. 2010;20(1):140–150. doi: 10.1111/j.1750-3639.2008.00255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu Y, Wang L, Li H, et al. SMAD2 Inactivation Inhibits CLDN6 Methylation to Suppress Migration and Invasion of Breast Cancer Cells. Int J Mol Sci. 2017;18(9):1863. doi: 10.3390/ijms18091863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsukita S, Yamazaki Y, Katsuno T, Tamura A, Tsukita S. Tight junction-based epithelial microenvironment and cell proliferation. Oncogene. 2008;27(55):6930–6938. doi: 10.1038/onc.2008.344. [DOI] [PubMed] [Google Scholar]
- 28.Houédé N, Pourquier P. Targeting the genetic alterations of the PI3K-AKT-mTOR pathway: its potential use in the treatment of bladder cancers. Pharmacol Ther. 2015;145(33):1–18. doi: 10.1016/j.pharmthera.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 29.Zhu L, Derijard B, Chakrabandhu K, Wang BS, Chen HZ, Hueber AO. Synergism of PI3K/Akt inhibition and Fas activation on colon cancer cell death. Cancer Lett. 2014;354(2):355–364. doi: 10.1016/j.canlet.2014.08.038. [DOI] [PubMed] [Google Scholar]
- 30.Denoyelle C, Lambert B, Meryet-Figuière M, et al. miR-491-5p-induced apoptosis in ovarian carcinoma depends on the direct inhibition of both BCL-XL and EGFR leading to BIM activation. Cell Death Dis. 2014;5(10):e1445. doi: 10.1038/cddis.2014.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hy Y, Kim SO, Jin C, et al. β-lapachone-Induced Apoptosis of Human Gastric Carcinoma AGS Cells Is Caspase-Dependent and Regulated by the PI3K/Akt Pathway. Biomol Ther. 2014;22(3):184–192. doi: 10.4062/biomolther.2014.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shang H-S, Chang J-B, Lin J-H, et al. Deguelin Inhibits the Migration and Invasion of U-2 OS Human Osteosarcoma Cells via the Inhibition of Matrix Metalloproteinase-2/-9 in Vitro. Molecules. 2014;19(10):16588–16608. doi: 10.3390/molecules191016588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gille H, Kowalski J, Li B, et al. Analysis of biological effects and signaling properties of Flt-1 (VEGFR-1) and KDR (VEGFR-2). A reassessment using novel receptor-specific vascular endothelial growth factor mutants. J Biol Chem. 2001;276(5):3222. doi: 10.1074/jbc.M002016200. [DOI] [PubMed] [Google Scholar]
- 34.Trisciuoglio D, Iervolino A, Zupi G, del Bufalo D, del BD. Involvement of PI3K and MAPK signaling in bcl-2-induced vascular endothelial growth factor expression in melanoma cells. Mol Biol Cell. 2005;16(9):4153–4162. doi: 10.1091/mbc.E04-12-1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Erdem O, Erdem M, Dursun A, Akyol G, Erdem A. Angiogenesis, p53, and bcl-2 expression as prognostic indicators in endometrial cancer: comparison with traditional clinicopathologic variables. Int J Gynecol Pathol. 2003;22(3):254–260. doi: 10.1097/01.PGP.0000070850.25718.A5. [DOI] [PubMed] [Google Scholar]
- 36.Slomovitz BM, Coleman RL. The PI3K/AKT/mTOR pathway as a therapeutic target in endometrial cancer. Clin Cancer Res. 2012;18(21):5856–5864. doi: 10.1158/1078-0432.CCR-12-0662. [DOI] [PubMed] [Google Scholar]
- 37.Zhang S, Wang M, Li Q, Zhu P. MiR-101 reduces cell proliferation and invasion and enhances apoptosis in endometrial cancer via regulating PI3K/Akt/mTOR. Cancer Biomarkers. 2017;21(1):179–186. doi: 10.3233/CBM-170620. [DOI] [PubMed] [Google Scholar]