

Abstract
Fibroblast growth factor receptors (FGFR) play a significant role in both embryonic development and in adults. Upon binding with ligands, FGFR signaling is activated and triggers various downstream signal cascades that are implicated in diverse biological processes. Aberrant regulations of FGFR signaling are detected in numerous cancers. Although FGFR4 was discovered later than other FGFR, information on the involvement of FGFR4 in cancers has significantly increased in recent years. In this review, the recent findings in FGFR4 structure, signaling transduction, physiological function, aberrant regulations, and effects in cancers as well as its potential applications as an anticancer therapeutic target are summarized.
Keywords: aberrant signaling pathway, cancer, dysregulation, fibroblast growth factor receptor 4, therapeutic target
Abbreviations
- ACC2
acetyl CoA carboxylase‐2
- CCl4
carbon tetrachloride
- CYP7A1
cholesterol 7α‐hydroxylase
- EGFR
epidermal growth factor receptor
- EMT
epithelial‐mesenchymal transformation
- Erk
extracellular signal‐regulated kinases
- FGFR
fibroblast growth factor receptor
- FRS2
FGFR substrate 2
- HCC
hepatocellular carcinoma
- HNSCC
head and neck squamous cell carcinoma
- HSPG
heparan sulfate proteoglycans
- LPA
lysophosphatidic acid
- MAPK
mitogen‐activated protein kinase
- OSCC
oral squamous cell carcinoma
- PI3K
phosphoinositide 3‐kinase
- PKC
protein kinase C
- PLCγ
phospholipase γ
- SCD1
stearoyl CoA desaturase1
- SNP
single‐nucleotide polymorphism
- STAT3
signal transducer and activator of transcription 3
- VEGF
vascular endothelial growth factor
1. INTRODUCTION
Cancer is a major threat to public health nowadays. Breast cancer, colorectal cancer, hepatocarcinoma and head and neck squamous cell carcinoma (HNSCC) rank among the most frequent cancers worldwide. The mechanisms underlying cancer development, however, are far from being fully understood.1
Dysregulation of fibroblast growth factor receptor (FGFR)‐dependent signaling has been observed in various tumors and is considered as an oncogenic signaling pathway. Several mechanisms are responsible for aberrant regulation of FGFR signaling, including altered expression, mutation, chromosomal rearrangement, and aberrant FGFR splicing. There is compelling evidence that dysregulated FGFR4 is also involved in the pathogenesis of many cancer types (Table 1).2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18
Table 1.
Alterations of FGFR4 in diverse cancers
| Alteration | Cancer type | References |
|---|---|---|
| Overexpression | Breast cancer, liver cancer, colon cancer, prostate cancer, rhabdomyosarcoma | 2, 3, 4 |
| SNP | ||
| Arg388 | Breast cancer, colorectal cancer, prostate cancer, OSCC, HNSCC | 5, 6, 7, 8, 9, 10, 11, 12 |
| Mutation | ||
| Y367C | Breast cancer | 13 |
| K535 | Breast cancer | 14 |
| E550 | Rhabdomyosarcoma | 14 |
| Aberrant regulation of FGFR4 ligand | ||
| FGF19‐FGFR4 | Hepatocellular carcinoma, colorectal cancer | 15, 16, 17, 18 |
FGF, fibroblast growth factor; FGFR, fibroblast growth factor receptor; HNSCC, head and neck squamous cell carcinoma; OSCC, oral squamous cell carcinoma; SNP, single‐nucleotide polymorphism.
2. STRUCTURE OF FGFR4
All FGFR contain an extracellular domain, a transmembrane domain, and a cytoplasmic domain (Figure 1A).19 The extracellular domain contains the leader peptide and 3 immunoglobulin domains (D1‐3). After FGFR are translocated on the cell membrane, the leader peptide is cleaved off. There is a serine‐rich sequence between D1 and D2, termed acid box, which, together with the D1 domain, functions in automatic inhibition of FGFR.20 D2 and D3 are sufficient for ligand binding and determining the ligand specificity. A highly conserved sequence is found in the D2 domain, which is considered as the binding site for heparan sulfate proteoglycans (HSPG). Through the variable splicing, FGFR1‐3 codes for IIIb and IIIc isoforms, respectively, with the distinct D3 domain but the identical N‐terminal and different C‐terminus. The cytoplasmic domain is a tyrosine kinase domain.20
Figure 1.

Fibroblast growth factor receptor (FGFR) structure, FGFR signaling, and specificity of the FGF–FGFR interaction. A, FGFR consist of an extracellular domain that contains 3 immunoglobulin domains (D1‐D3), a transmembrane domain, and an intracellular tyrosine kinase domain. B, The formation of a 2:2:2 FGFR‐FGF‐heparan sulfate proteoglycan (HSPG) complex results in receptor dimerization and transphosphorylation at several tyrosine residues in the intracellular portion of FGFR. Activated FGFR causes the activation of the RAS–RAF–MAPK and PI3K‐AKT signaling pathway. Some other signaling pathways can also be activated by FGFR, including signal transducer and activator of transcription (STAT)‐dependent signaling. Several negative regulators are involved in FGFR signaling, such as SPRY and MKP3. C, Specificity of FGF ligands for FGFR‐IIIb and FGFR‐IIIc isoforms. DAG, diacylglycerol; PIP2, phosphatidylinositol‐4,5‐biphosphate; PIP3, phosphatidylinositol‐3,4,5‐triphosphate; PLCγ, phospholipase γ; SPRY, sprouty
FGFR4 shares the conserved structure with other FGFR, but the FGFR4 gene codes only for IgIIIc isoform.20
3. SIGNAL TRANSDUCTION
The formation of a 2:2:2 complex of FGFR‐FGF‐HSPG leads to receptor dimerization and transphosphorylation. Activated FGFR phosphorylates 2 major intracellular targets, phospholipase γ (PLCγ) and FGFR substrate 2 (FRS2; Figure 1B).21
PLCγ binds to the autophosphorylated‐tyrosines in the carboxyl terminus of the activated receptor, resulting in PLCγ being phosphorylated and activated by FGFR4.21 Activated PLCγ produces 2 secondary signals, named diacylglycerol and phosphatidylinositol‐3,4,5‐triphosphate (PIP3), by hydrolyzing phosphatidylinositol‐4,5‐biphosphate (PIP2), which then causes the release of intracellular calcium and protein kinase C (PKC) activation. Protein kinase C triggers the activation of the mitogen‐activated protein kinase (MAPK) pathway, partly by phosphorylating RAF.22
In contrast, FRS2 is recruited to the juxtamembrane region of FGFR4 and then phosphorylated. FRS2 activation results in recruitment of growth factor receptor bound 2 (GRB2) and son of sevenless (SOS), which can further activate Ras/MAPK signaling pathways and the phosphoinositide 3‐kinase (PI3K)‐AKT pathway.23
4. LIGAND‐RECEPTOR BINDING
Fibroblast growth factor receptor 1–4 are specific for various ligands (Figure 1C). The binding specificity of FGF with FGFR is determined by temporal and spatial expression patterns of FGF and FGFR, alternative splicing of FGFR as well as cell surface or secreted molecules.24
Alternative splicing of FGFR is a significant determinant of binding specificity, which is actually regulated in a tissue‐specific manner. Alternative splicing leads to 2 isoforms of FGFR: IIIb and IIIc, which have distinct ligand binding specificity. FGFR IIIb isoform is mainly expressed in epithelial tissues, whereas the IIIc isoform is mainly expressed in mesenchymal tissues. The isoforms expressed in epithelial tissues usually prefer to interact with the ligands expressed in mesenchymal tissues. In contrast, the isoforms expressed in mesenchymal tissues usually interact with the ligands expressed in epithelial tissues (Figure 2).24, 25 In addition, binding specificity is also modulated by cell surface or secreted molecules that can facilitate ligand‐receptor interactions, such as heparan sulfate and heparin‐like molecules, which are required for the canonical FGF to bind with their receptors. In the presence of heparan sulfate, FGF bind with FGFR to form a 2:2:2 FGF‐FGFR‐heparan sulfate complex, which is necessary for subsequent activation of FGFR and stimulation of the downstream signaling pathway.21
Figure 2.

Tissue distribution of fibroblast growth factor receptor (FGFR) isoforms. The FGFR IIIb isoform is mainly expressed in epithelial tissues, whereas the FGFR IIIc isoform is mainly expressed in mesenchymal tissues
Fibroblast growth factor receptor 4 can bind to distinct FGF, including FGF1, FGF2, FGF4, FGF6, FGF8, FGF9, FGF17–19, FGF21, FGF23. Except for FGF19, FGF21 and FGF23, the rest of them are all canonical FGF that rely on heparan sulfate as essential tissue‐selective cofactors for binding to FGFR4. Given that FGFR4 has no alternative splicing pattern, alternative splicing is not the mechanism of its ligand‐binding specificity.22
Although FGF19 is not entirely specific for FGFR4, FGFR4 is considered as the predominant receptor of FGF19.20 FGF19 is a kind of hormone‐like FGF that has a low affinity for heparin‐like molecules. Instead, Klotho beta (KLB) serves as an essential tissue‐selective cofactor that is required for FGF19 binding to FGFR4 and is necessary for the activation of the downstream signaling pathway. Further, FGF19 is primarily expressed in the ileum and circulates to the liver to exert its liver‐specific functions. Liver is also the only tissue where KLB and FGFR4 are highly expressed in human and mouse.16, 26 This suggests that tissue‐specific expression also regulates the specific binding of FGF19 to FGFR4.
5. PHYSIOLOGICAL FUNCTION OF FGFR4
In general, FGFR4 is highly expressed in embryonic tissues and is involved in embryonic development, angiogenesis, and tissue differentiation.27 In adult tissues, the expression of FGFR4 is limited in the actively growing tissues. The physiological effects of FGFR4 in adults include regulating bile acid production, metabolism, muscle differentiation, and tissue repair.
5.1. Regulation of bile acid production
Bile acid is synthesized in the liver and is stored in the gallbladder. Bile acid is secreted into the intestine, contributing to the absorption of nutrients in the intestine. FGF19 is the target gene of farnesoid X receptor. Postprandial intestinal bile acid is capable of activating farnesoid X receptor in the intestinal epithelium, resulting in expression and secretion of FGF19.28 In turn, FGF19 activates the hepatic FGFR4, and reduces bile acid synthesis by inhibiting cholesterol 7α‐hydroxylase (CYP7A1) expression, the rate‐limiting enzyme in bile acid synthesis. The regulatory role of FGFR4 in bile acid production was also observed in FGFR4 knockout mice. CYP7A1 mRNA and protein in the liver of FGFR4 knockout mice are significantly higher than that in wild‐type mice. In addition, liver 3‐hydroxy‐3‐methyl glutaryl coenzyme A reductase (HMG‐CoA reductase), the rate‐limiting enzyme in cholesterol biosynthesis, is also upregulated. Accordingly, the bile acid pool and bile acid excretion in FGFR4 knockout mice are enhanced compared with that of wild‐type mice.29 These observations suggest an important role of FGFR4 in regulating bile acid homeostasis.
5.2. Regulation of metabolism
The role of FGF19 in metabolism was identified by studying the phenotype of FGF19 transgenic mice or FGF19‐treated mice. FGF19 transgenic mice show a phenotype of losing weight, decreased blood glucose, triglyceride and insulin, and enhanced metabolic rate, glucose tolerance, and insulin sensitivity compared with wild‐type mice.30 Decreased expression of acetyl CoA carboxylase‐2 (ACC2), serving as a repressor in β‐oxidation, is observed in FGF19 transgenic mice. FGF19 also downregulates the lipogenic enzyme stearoyl CoA desaturase1 (SCD1). Mice with depleted SCD1 or ACC2 are resistant to adiposity and have a lower bodyweight.31, 32 These studies indicate that the FGF19‐FGFR4 axis promotes glucose metabolism.
5.3. Muscle differentiation and tissue repair
In chick embryos, myoblasts from skeletal muscle masses have a high level of FGFR4 expression. Inhibition of FGFR4 impairs differentiation of muscle progenitor and results in a loss of limb muscles, which is seen as altered expression of muscle cell markers, such as myogenic factor 5 (Myf5), myogenic differentiation factor (MyoD), and the embryonic myosin heavy chain (MHC).33 In addition, a soluble dominant‐negative FGFR4‐containing adenovirus was used to infect mouse myoblasts, the differentiation of these cells was markedly retarded in 4‐day culture, accompanied by interruption of extracellular signal‐regulated kinases (Erk) 1/2 phosphorylation and aberrant expression of MHC, suggesting that muscle differentiation is suppressed when the FGFR4 signaling pathway is blocked.34 In normal adults, FGFR4 is expressed in myofibroblast during tissue repair after injury. The MyoD‐Tead2‐Fgfr4 pathway has been reported to be involved in muscle regeneration. Fgfr4−/− mice have an impaired muscle regeneration, manifesting as slow maturation of regenerating fibers, and development of calcifications and intramuscular adipose.35
The FGFR4‐deficient mice also have a lower resistance to acute and chronic liver injury induced by carbon tetrachloride (CCl4). Acute CCl4 exposure causes accelerated liver injury, delayed hepatolobular repair, and liver mass increase in the FGFR4‐deficient mice, compared with their normal counterparts. After chronic CCl4 exposure, the FGFR4‐deficient mice exhibit severe fibrosis.36
6. MECHANISMS OF ONCOGENIC FGFR4 SIGNALING
The FGFR4 signaling pathway has gradually come to be seen as a significant oncogenic pathway in various cancers and as being involved in carcinogenesis (Figure 3). Dysregulation of FGFR4 in cancers is summarized in the following section, including altered expression, single‐nucleotide polymorphism (SNP), mutations, and aberrant regulation of FGFR4 ligands (Figure 4A).
Figure 3.

Deregulated fibroblast growth factor receptor (FGFR) signaling in carcinogenesis. Aberrant FGFR4 signaling is implicated in both tumor cells and the stroma. 1, FGFR4 promotes tumor cell proliferation; 2, FGFR4 accelerates metastasis and invasion of tumor cells; 3, FGFR4 contributes to radio‐resistance and chemo‐resistance in cancer therapy; and 4, FGFR4 promotes stroma‐induced epithelial‐to‐mesenchymal transformation (EMT) in cancer
Figure 4.
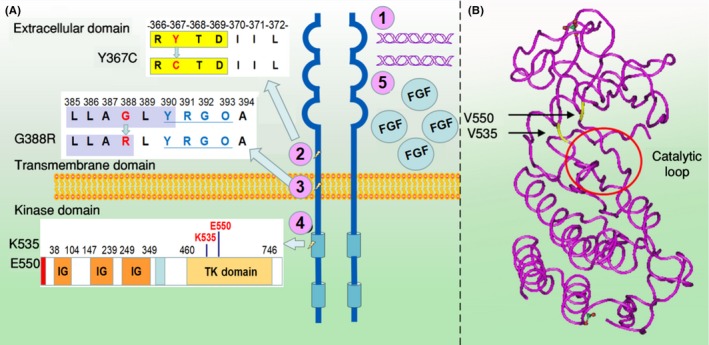
Fibroblast growth factor receptor 4 (FGFR4) dysregulation in cancers and crystal structures of the FGFR4 codon 535 and 550 mutations. A, Mechanisms of activation of oncogenic FGFR4 signaling. 1, gene amplification of the receptor, which can cause overexpression of FGFR4. 2, FGFR4 mutation in the extracellular juxtamembrane domain (Y367C) promotes FGFR4 dimerization. 3, G388R SNP in the transmembrane domain exposes a membrane‐proximal signal transducer and activator of transcription 3 (STAT3) binding site Y(390)‐(P)XXQ(393) and increases downstream STAT3 signaling cascade. 4, FGFR4 mutation in kinase domain (N535K and V550E) leads to increased FGFR4 kinase activity. 5, Overexpression of FGF ligands. B, Human FGFR4 structure (PDB ID: 4TYJ) shows the relative locations of N535, V550 (yellow) and FGFR4 catalytic loop (red cycle)
6.1. Aberrant expression of FGFR4
Overexpression of FGFR4 has been detected in multiple types of human cancer, such as breast cancer, liver cancer, colon cancer, prostate cancer, rhabdomyosarcoma, and is associated with decreased survival time.2, 3, 4 In addition, high FGFR4 expression is associated with resistance to chemotherapy and radiotherapy in cancer. Overexpression of the FGFR4 may result from gene amplification. FGFR4 amplification approximately 2–4‐fold has been detected in 10% of breast cancer samples, especially in tumors with high lymph node metastases and in estrogen receptor‐ and progesterone receptor‐positive tumors.4
6.2. Single‐nucleotide polymorphism
The most well‐known SNP of FGFR4 is codon 388 in exon 9, where the first base mutates from G to A. This mutation causes the coded amino residue to be changed from glycine (Gly388) into arginine (Arg388) in the transmembrane domain.5
The incidence of Arg388 is associated with several prognostic parameters in cancer. A statistically significant association of FGFR4 Arg388 with the prognosis of multiple cancers was noted in a meta‐analysis and pooled analysis.6 Some other studies have come to similar results. For example, Arg388 allele carriers were found to have a significantly reduced disease‐free survival time compared with Gly388 homozygous allele carriers.2 Moreover, the FGFR4 Arg388 allele carriers with colon cancer had early lymph node metastasis and advanced tumor‐node‐metastasis (TNM) stage, an indicator of poor cancer prognosis.2 Another study focusing on HNSCC showed that only the HNSCC patients with the FGFR4 Arg388 allele died in the follow‐up period, and showed a trend to a reduced overall survival.7 Accordingly, in a study specifically focusing on the association between the FGFR4 Arg388 allele with oral squamous cell carcinoma (OSCC) development, it was shown that, compared with OSCC patients carrying the Gly/Gly genotype, the patients carrying the FGFR4 Arg/Arg or Arg/Gly genotype had advanced nodal stage (pathologic N2 + N3). Further, FGFR4 Arg388 allele and mutations in TP53 were associated with shorter survival time. These 2 parameters influenced the survival of OSCC patients synergistically.8
The effects of FGFR4 388 variants on cancer progression were investigated in the MDA‐MB‐231 human breast cancer cell models.9 It was found that expression of FGFR4 Gly388 allele suppressed cancer cell invasiveness and motility. This was probably mediated by regulating the expression of genes involved in invasiveness and motility. Further, FGFR4 Gly388 allele inhibited the process of PI3K‐dependent lysophosphatidic acid‐induced AkT activation and cell migration by downregulating lysophosphatidic acid receptor endothelial cell differentiation gene‐2 (Edg‐2). Moreover, FGFR4 Gly388 allele attenuated the invasion ability of the breast cancer cell line by inhibiting small Rho GTPase.9
Codon 385 of the mouse FGFR4 gene corresponds to codon 388 in humans. Mouse embryonic fibroblasts with Arg385 allele knock‐in have been found to show accelerated cell transformation.10 The cells after transformation exhibited enhanced motility and invasion. Further, in a transforming growth factor‐α (TGF‐α)‐induced mammary cancer mouse model harboring Arg385 allele, the quantity and size of the breast lump were significantly increased, and the development of the lump as well as cancer metastases to lung was also obviously accelerated.10
Although the underlying mechanism is not fully clear yet, some studies indicate that Arg388 SNP prolongs FGFR4 half‐life and increases its stability.11 In other studies, the Arg388 variant recruiting signal transducer and activator of transcription 3 (STAT3) to the inner cell membrane has been suggested to be one of the molecular mechanisms.12 The FGFR4 Arg388‐variant changes transmembrane spanning segment, and also exposes the membrane‐proximal cytoplasmic STAT3 binding site Y(390)‐(P)XXQ(393). By recruiting STAT3 proteins to the inner cell membrane, membrane‐proximal STAT3 binding motifs, FGFR4 phosphorylates STAT3.12
6.3. Activating mutations
The Y367C FGFR4 mutation is the tyrosine‐to‐cysteine mutation at position 367 of FGFR4 in the extracellular juxtamembrane domain. On analysis of FGFR4 expression in 318 different cancer cell lines, MDA‐MB453, which harbors the Y367C mutation, had the highest FGFR4 expression and most activated downstream pathways of all cell lines tested. Moreover, in MDA‐MB361 cells, the Y367C FGFR4 mutation could constitutively activate downstream signaling, such as Erk, leading to an activation of the MAPK cascade. Further, ectopic expression of the FGFR4 Y367C mutant in HEK293 cells, which has endogenous WT FGFR4, was sufficient to induce Erk phosphorylation and promote cell proliferation.13
The effects of the Y367C mutation on FGFR4 signaling are based on its promotion impact in FGFR4 dimerization. Adjacent cysteine residues facilitate the formation of disulphide bond connecting protein chains, so this mutation increases the incidence of FGFR4 dimerization on the cell surface, thereby leading to activation of receptor tyrosine kinase and ligand‐independent activation of signaling pathways.13
Several mutations in the FGFR4 kinase domain have been reported in approximately 7%‐8% of rhabdomyosarcoma, such as N535K and V550E mutations.14 N535K and V550E mutations cluster in proximity to the FGFR4 tyrosine kinase domain (Figure 4B). The N535K mutation disrupts FGFR4 R‐group hydrogen bonds between N535 and residues H530 and I533, which inhibit receptor autophosphorylation. V550E mutation may alter the ATP binding cleft. In addition, the V550E mutation is considered an FGFR4 gatekeeper mutation, which increases the size of the gatekeeper residue and stabilizes the active state of FGFR4, thus leading to enhanced kinase activity. N535K and V550E mutations cause receptor autophosphorylation, then activate the STAT3 signal pathway, resulting in increased tumor cell proliferation and metastasis when expressed in murine rhabdomyosarcoma cell lines.14
6.4. Aberrant regulation of FGFR4 ligands
Fibroblast growth factor 19 has been reported to promote the development of hepatocellular carcinoma (HCC).15 Recombinant FGF19 can increase proliferation, and also inhibit apoptosis in HCC cell lines, while siRNA‐mediated knockdown of FGF19 has the opposite effect. FGFR4 is the predominant FGF receptor expressed in liver that mediates the liver‐specific function of FGF19. To study the role of FGFR4 in FGF19‐induced hepatocyte proliferation, FGF19dCTD, a C‐terminally truncated variant of FGF19 protein, was designed to activate FGFR4 but not FGFR 1c, 2c, and 3c. It was found that this specific activator of FGFR4 could induce hepatocyte proliferation.16 This suggests that hepatic FGFR4 activation alone results in increased hepatocyte proliferation, which may be a prerequisite for neoplastic transformation.16
To study the direct effect of FGFR4 on hepatocarcinogenesis, researchers bred the FGF19 transgenic (FGF19‐TG) mice with FGFR4 knockout (FGFR4‐KO) mice or FGFR4 wild‐type (FGFR4‐WT) mice. Liver tumorigenesis was not found in the progeny of FGF19 transgenic mice bred with FGFR4 knockout mice. Moreover, hepatocellular neoplasia and hepatocellular proliferation were found only in FGF19‐TG mice with an FGFR4‐WT background. These studies suggest that FGFR4 is necessary for FGF19‐mediated hepatocarcinogenesis.17
Epithelial‐mesenchymal transformation (EMT) plays an important role in embryonic development, chronic inflammation, tissue remodeling, various fibroid diseases and cancer metastasis. Through EMT, epithelial cells lose epithelial phenotype‐like cell polarity and connectivity to the basement membrane, but show a stronger ability of migration and invasion, similar to mesenchymal cells.37 CCL2 derived from tumor‐associated fibroblasts induced FGFR4 expression in colorectal cancer cells.18 In contrast, tumor‐associated fibroblasts could largely produce FGF19, which in turn activated FGFR4 in tumor cells. Activated FGFR4 directly phosphorylated membranous b‐catenin at Y142, leading to b‐catenin translocation into the nucleus. Nuclear b‐catenin activated Snail expression and reduced E‐cadherin expression, which promoted EMT in colorectal cancer cell and facilitated tumor metastasis.18
7. FGFR4 AS A POTENTIAL THERAPEUTIC TARGET
Given the important roles of FGFR4 in the development of cancer, anticancer agents such as RNA interference (RNAi), small‐molecule FGFR inhibitors, and monoclonal antibodies, have been designed to target FGFR4 (Table 2).
Table 2.
Small‐molecule tyrosine kinase inhibitors of FGFR in clinical development
| Compound | Company | Target | Clinical development |
|---|---|---|---|
| Small‐molecule tyrosine kinase inhibitors: non‐selective FGFR TKI | |||
| TK1258 | Novartis | FGFR, PDGFR, VEGFR, FLT3, KIT | Phase II |
| AZ2171 | AstraZeneca | FGFR, VEGFR, KIT | Phase I |
| BIBF1120 | Boehringer Ingelheim | FGFR, PDGFR, VEGFR, FLT3, LCK, SRC | Phase III |
| BMS‐582,664 | Bristol‐Myers Squibb | FGFR, VEGFR | Phase II |
| E7080 | Eisai | FGFR, PDGFR, VEGFR | Phase I |
| TSU‐68 | Taiho Pharmaceutical | FGFR, PDGFR, VEGFR | Phase I/II |
| Small‐molecule tyrosine kinase inhibitors: selective FGFR TKI | |||
| AZD4547 | AstraZeneca | FGFR1‐3 | Phase II |
| BGJ398 | Novartis | FGFR1‐3 | Phase I |
| LY2874455 | Eli Lilly | FGFR1‐4 | Phase I |
FGFR, fibroblast growth factor receptor; FLT3, fms‐like tyrosine kinase 3; KIT, v‐kit Hardy‐Zuckerman 4 feline sarcoma viral oncogene homolog; LCK, lymphocyte‐specific protein tyrosine kinase; PDGFR, platelet‐derived growth factor receptors; SRC, sarcoma gene; TKI, tyrosine‐kinase inhibitor; VEGFR, vascular endothelial growth factor receptor.
7.1. RNA interference
RNA interference is a phenomenon of degradation of homologous mRNA induced by double‐stranded RNA (dsRNA).38 So far, RNAi targeting at different oncogenes, including survivin, epidermal growth factor receptor (EGFR), vascular endothelial growth factor (VEGF), and FGFR, has produced marked inhibitory effects in tumors cells. In SW480 and SW48 colorectal cancer cell lines, shRNA‐mediated FGFR4‐silencing inhibited the activation of FGFR4‐signaling pathways and led to a significant reduction in cell proliferation, adhesion, migration, and invasion. Notably, the cells showed altered expression of some EMT marker proteins, including an increased expression of E‐cadherin and decreased expression of Snail, Twist, and TGF‐β, and exhibited a reversion toward an epithelial phenotype. In addition, FGFR4‐silencing reduced tumor growth in vivo.39 Similar results were also observed in prostate cancer cells.40 These data suggest that shRNA‐mediated FGFR4 knockdown exerts its anti‐tumor function partly by blocking the process of EMT.
7.2. Small‐molecule FGFR inhibitors
As the first generation of FGFR small‐molecule inhibitors, mixed kinase inhibitors have a remarkably broad substrate specificity. These inhibitors showed even better effects on anti‐VEGF receptor (VEGFR) and/or antiplatelet‐derived growth factor receptor (PDGFR) compared with FGFR, and more side‐effects and higher toxicity.41
The second‐generation compounds are potent FGFR inhibitors, such as AZD4547, BGJ398, and LY2874455, which have a greater selectivity compared with mixed kinase inhibitors. The first 2 are inhibitors of FGFR1‐3 with moderate‐weak activity against FGFR4, and the third is a pan‐FGFR inhibitor. These inhibitors exhibit an antitumor effect in cancer involving dysregulation of FGFR1 ,2, or 3 in clinical trials.42 BLU9931 is a potent and highly selective small‐molecule inhibitor of FGFR4 that spares other FGFR family members and all other kinases. BLU9931 treatment inhibits the proliferation of HCC cell lines harboring an activated FGFR4 signaling pathway. It also shows significant antitumor activity in mice bearing an HCC tumor xenograft.43 Therefore, BLU9931 may be an effective therapy targeting FGFR4 in HCC patients with the activated FGFR4 signaling pathway.
7.3. Monoclonal antibody
The first anti‐FGFR4 monoclonal antibody tested in an animal tumor model is called LD1(chLD1). LD1 binds mouse, cynomolgus monkey, and human FGFR4 with high affinity but does not bind to mouse or human FGFR1, FGFR2, or FGFR3. LD1 blocks FGRF4, so that it prevents FGF19 binding to FGFR4 and inhibits FGFR4 signaling, leading to decreased cell proliferation and colony formation in liver cancer cell lines and tumor growth in the HUH7 HCC xenograft model.17
Another monoclonal antibody, 1A6, targeting FGF19, is now under preclinical development. 1A6 can bind to FGF19 and inhibits FGF19 binding with FGFR4. 1A6 has been reported to prevent tumor formation and inhibit the growth of colon cancer xenografts in FGF19 transgenic mice.44
8. CONCLUSION
Recent efforts have enabled us to identify various oncogenic alterations of FGFR4 signaling involved in cancer. A large body of studies link aberrant FGFR4 signaling, due to either aberrant expression, SNP, activating mutations or alterations in FGFR4 ligands, with the development of cancer. Thus, targeting of FGFR4 is a potential therapeutic strategy. Substantial progress is being made in FGFR4‐targeting agents, which show promising antitumor activity, and some of them are currently in preclinical development or in the early phase of clinical trials. More work is still required in the future to overcome the challenges in lowering the toxicity of FGFR4‐targeting agents, and in selection of patients suitable for FGFR4‐targeting therapy.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (81520108009 [Q.‐M.C.], 81621062 [Q.‐M.C.] and 81672674 [R.L.]), 111 Project of MOE B14038 (Q.‐M.C.), and the Young Elite Scientist Sponsorship Program by CAST 2016QNRC001 (R.L.).
Tang S, Hao Y, Yuan Y, Liu R, Chen Q. Role of fibroblast growth factor receptor 4 in cancer. Cancer Sci. 2018;109:3024–3031. 10.1111/cas.13759
References
- 1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet‐Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87‐108. [DOI] [PubMed] [Google Scholar]
- 2. Bange J, Prechtl D, Cheburkin Y, et al. Cancer progression and tumor cell motility are associated with the FGFR4 Arg(388) allele. Cancer Res. 2002;62(3):840‐7. [PubMed] [Google Scholar]
- 3. Gowardhan B, Douglas DA, Mathers ME, et al. Evaluation of the fibroblast growth factor system as a potential target for therapy in human prostate cancer. Br J Cancer. 2005;92(2):320‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jaakkola S, Salmikangas P, Nylund S, et al. Amplification of fgfr4 gene in human breast and gynecological cancers. Int J Cancer. 1993;54(3):378‐82. [DOI] [PubMed] [Google Scholar]
- 5. Marme F, Hielscher T, Hug S, et al. Fibroblast growth factor receptor 4 gene (FGFR4) 388Arg allele predicts prolonged survival and platinum sensitivity in advanced ovarian cancer. Int J Cancer. 2012;131(4):E586‐91. [DOI] [PubMed] [Google Scholar]
- 6. Frullanti E, Berking C, Harbeck N, et al. Meta and pooled analyses of FGFR4 Gly388Arg polymorphism as a cancer prognostic factor. Eur J Cancer Prev. 2011;20(4):340‐7. [DOI] [PubMed] [Google Scholar]
- 7. Streit S, Bange J, Fichtner A, Ihrler S, Issing W, Ullrich A. Involvement of the FGFR4 Arg388 allele in head and neck squamous cell carcinoma. Int J Cancer. 2004;111(2):213‐7. [DOI] [PubMed] [Google Scholar]
- 8. Tanuma J, Izumo T, Hirano M, et al. FGFR4 polymorphism, TP53 mutation, and their combinations are prognostic factors for oral squamous cell carcinoma. Oncol Rep. 2010;23(3):739‐44. [PubMed] [Google Scholar]
- 9. Stadler CR, Knyazev P, Bange J, Ullrich A. FGFR4 GLY388 isotype suppresses motility of MDA‐MB‐231 breast cancer cells by EDG‐2 gene repression. Cell Signal. 2006;18(6):783‐94. [DOI] [PubMed] [Google Scholar]
- 10. Seitzer N, Mayr T, Streit S, Ullrich A. A single nucleotide change in the mouse genome accelerates breast cancer progression. Cancer Res. 2010;70(2):802‐12. [DOI] [PubMed] [Google Scholar]
- 11. Wang J, Yu W, Cai Y, Ren C, Ittmann MM. Altered fibroblast growth factor receptor 4 stability promotes prostate cancer progression. Neoplasia. 2008;10(8):847‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ulaganathan VK, Sperl B, Rapp UR, Ullrich A. Germline variant FGFR4 p. G388R exposes a membrane‐proximal STAT3 binding site. Nature. 2015;528(7583):570‐4. [DOI] [PubMed] [Google Scholar]
- 13. Roidl A, Foo P, Wong W, et al. The FGFR4 Y367C mutant is a dominant oncogene in MDA‐MB453 breast cancer cells. Oncogene. 2010;29(10):1543‐52. [DOI] [PubMed] [Google Scholar]
- 14. Taylor JGt, Cheuk AT, Tsang PS, et al. Identification of FGFR4‐activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J Clin Invest. 2009;119(11):3395‐407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Miura S, Mitsuhashi N, Shimizu H, et al. Fibroblast growth factor 19 expression correlates with tumor progression and poorer prognosis of hepatocellular carcinoma. BMC Cancer. 2012;12:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wu X, Ge H, Lemon B, et al. FGF19‐induced hepatocyte proliferation is mediated through FGFR4 activation. J Biol Chem. 2010;285(8):5165‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. French DM, Lin BC, Wang M, et al. Targeting FGFR4 inhibits hepatocellular carcinoma in preclinical mouse models. PLoS ONE. 2012;7(5):e36713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu R, Li J, Xie K, et al. FGFR4 promotes stroma‐induced epithelial‐to‐mesenchymal transition in colorectal cancer. Cancer Res. 2013;73(19):5926‐35. [DOI] [PubMed] [Google Scholar]
- 19. Haugsten EM, Wiedlocha A, Olsnes S, Wesche J. Roles of fibroblast growth factor receptors in carcinogenesis. Mol Cancer Res. 2010;8(11):1439‐52. [DOI] [PubMed] [Google Scholar]
- 20. Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov. 2009;8(3):235‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Klint P, Claesson‐Welsh L. Signal transduction by fibroblast growth factor receptors. Front Biosci. 1999;4:D165‐77. [DOI] [PubMed] [Google Scholar]
- 22. Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10(2):116‐29. [DOI] [PubMed] [Google Scholar]
- 23. Gotoh N. Regulation of growth factor signaling by FRS2 family docking/scaffold adaptor proteins. Cancer Sci. 2008;99(7):1319‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang X, Ibrahimi OA, Olsen SK, Umemori H, Mohammadi M, Ornitz DM. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J Biol Chem. 2006;281(23):15694‐700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ornitz DM, Xu J, Colvin JS, et al. Receptor specificity of the fibroblast growth factor family. J Biol Chem. 1996;271(25):15292‐7. [DOI] [PubMed] [Google Scholar]
- 26. Lin BC, Wang M, Blackmore C, Desnoyers LR. Liver‐specific activities of FGF19 require Klotho beta. J Biol Chem. 2007;282(37):27277‐84. [DOI] [PubMed] [Google Scholar]
- 27. Thisse B, Thisse C. Functions and regulations of fibroblast growth factor signaling during embryonic development. Dev Biol. 2005;287(2):390‐402. [DOI] [PubMed] [Google Scholar]
- 28. Inagaki T, Choi M, Moschetta A, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2(4):217‐25. [DOI] [PubMed] [Google Scholar]
- 29. Yu C, Wang F, Kan M, et al. Elevated cholesterol metabolism and bile acid synthesis in mice lacking membrane tyrosine kinase receptor FGFR4. J Biol Chem. 2000;275(20):15482‐9. [DOI] [PubMed] [Google Scholar]
- 30. Tomlinson E, Fu L, John L, et al. Transgenic mice expressing human fibroblast growth factor‐19 display increased metabolic rate and decreased adiposity. Endocrinology. 2002;143(5):1741‐7. [DOI] [PubMed] [Google Scholar]
- 31. Abu‐Elheiga L, Wu H, Gu Z, Bressler R, Wakil SJ. Acetyl‐CoA carboxylase 2‐/‐ mutant mice are protected against fatty liver under high‐fat, high‐carbohydrate dietary and de novo lipogenic conditions. J Biol Chem. 2012;287(15):12578‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Miyazaki M, Flowers MT, Sampath H, et al. Hepatic stearoyl‐CoA desaturase‐1 deficiency protects mice from carbohydrate‐induced adiposity and hepatic steatosis. Cell Metab. 2007;6(6):484‐96. [DOI] [PubMed] [Google Scholar]
- 33. Marics I, Padilla F, Guillemot JF, Scaal M, Marcelle C. FGFR4 signaling is a necessary step in limb muscle differentiation. Development. 2002;129(19):4559‐69. [DOI] [PubMed] [Google Scholar]
- 34. Yu S, Zheng L, Trinh DK, Asa SL, Ezzat S. Distinct transcriptional control and action of fibroblast growth factor receptor 4 in differentiating skeletal muscle cells. Lab Invest. 2004;84(12):1571‐80. [DOI] [PubMed] [Google Scholar]
- 35. Zhao P, Caretti G, Mitchell S, et al. Fgfr4 is required for effective muscle regeneration in vivo. Delineation of a MyoD‐Tead2‐Fgfr4 transcriptional pathway. J Biol Chem. 2006;281(1):429‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yu C, Wang F, Jin C, Wu X, Chan WK, McKeehan WL. Increased carbon tetrachloride‐induced liver injury and fibrosis in FGFR4‐deficient mice. Am J Pathol. 2002;161(6):2003‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Skovierova H, Okajcekova T, Strnadel J, Vidomanova E, Halasova E. Molecular regulation of epithelial‐to‐mesenchymal transition in tumorigenesis (Review). Int J Mol Med. 2018;41(3):1187‐200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Castanotto D, Rossi JJ. The promises and pitfalls of RNA‐interference‐based therapeutics. Nature. 2009;457(7228):426‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pelaez‐Garcia A, Barderas R, Torres S, et al. FGFR4 role in epithelial‐mesenchymal transition and its therapeutic value in colorectal cancer. PLoS ONE. 2013;8(5):e63695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sahadevan K, Darby S, Leung HY, Mathers ME, Robson CN, Gnanapragasam VJ. Selective over‐expression of fibroblast growth factor receptors 1 and 4 in clinical prostate cancer. J Pathol. 2007;213(1):82‐90. [DOI] [PubMed] [Google Scholar]
- 41. Lim SM, Kim HR, Shim HS, Soo RA, Cho BC. Role of FGF receptors as an emerging therapeutic target in lung squamous cell carcinoma. Future Oncol. 2013;9(3):377‐86. [DOI] [PubMed] [Google Scholar]
- 42. Gavine PR, Mooney L, Kilgour E, et al. AZD4547: an orally bioavailable, potent, and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family. Cancer Res. 2012;72(8):2045‐56. [DOI] [PubMed] [Google Scholar]
- 43. Hagel M, Miduturu C, Sheets M, et al. First Selective Small Molecule Inhibitor of FGFR4 for the Treatment of Hepatocellular Carcinomas with an Activated FGFR4 Signaling Pathway. Cancer Discov. 2015;5(4):424‐37. [DOI] [PubMed] [Google Scholar]
- 44. Desnoyers LR, Pai R, Ferrando RE, et al. Targeting FGF19 inhibits tumor growth in colon cancer xenograft and FGF19 transgenic hepatocellular carcinoma models. Oncogene. 2008;27(1):85‐97. [DOI] [PubMed] [Google Scholar]