

Abstract
Granulocyte-macrophage colony-stimulating factor (GM-CSFor Csf-2) is a pro-inflammatory mediator implicated in the pathogenesis of various autoimmune diseases. In this issue of Immunity, Spath et al. show that the dysregulated production of GM-CSF rather than IL-17 induces spontaneous immunopathology in a mouse model of CNS inflammation.
Originally discovered as a hematopoietic growth factor, granulocyte-macrophage colony-stimulating factor (GM-CSFor Csf-2) is now considered a mediator for T cells to communicate with myeloid populations during tissue inflammation (Becher et al., 2016). In particular, GM-CSF has been shown to play a crucial role in the pathogenesis of the experimental autoimmune encephalomyelitis (EAE), an animal model for multiple sclerosis. GM-CSF is a major cytokine produced by the encephalitogenic IL-17-producing T helper cells (Th17 cells) and required for Th17 cell-mediated EAE pathogenesis (Codarri et al., 2011) (El-Behi et al., 2011). GM-CSF expression is driven by the Th17 cell signature transcription factor RORγt (Codarri et al., 2011) and is known to be induced by interleukin-23 (IL-23) and IL-1 (El-Behi et al., 2011), both of which promote Th17 cell expansion and render them encephalitogenic. The importance of GM-CSF in mediating Th17 cell-induced EAE is highlighted by a fate mapping study showing that the Th17 cells have enhanced GM-CSF production when they become interferon-γ (IFN-γ)-producing “ex-Th17” cells in the CNS (Hirota et al., 2011). Th17 and ex-Th17 cells produce a group of cytokines (including IL-17A, IL-17F, IL-21, GM-CSF, TNF-α and IFN-γ) that all have been implicated in CNS inflammation. However, the specific function of these cytokines in disease process remains unclear. In this issue of Immunity, Spath et al. reports that dysregulated GM-CSF, rather than IL-17 production from T cells initiates an inflammatory cascade that leads to immunopathology and functional impairment in the brain and spinal cord.
To control the overexpression of GM-CSF in T cells, the authors targeted a Csf2 transgene into the ROSA26 locus, which directs the expression of GM-CSF upon Cre-mediated recombination. Together with the Cd4-creER, the transgene enabled the inducible overexpression of GM-CSF specifically in CD4+ T helper cells in mice (Csf2CD4). Following the induction of GM-CSF expression, Csf2CD4 mice showed significant increased frequencies of neutrophils and monocytes in the periphery. Importantly, CD11c+MHC+CD11b+CCR2+ myeloid cells, termed inflammatory monocyte-derived cells (MdCs), also underwent dramatic expansion. Of note, a prior study from the same group showed that GM-CSF signaling in CCR2+ monocytes is required for the induction of EAE (Croxford et al., 2015). Interestingly, Csf2CD4 micedeveloped spontaneous neurological deficits around 12 weeks after the induction of GM-CSF overexpression, exhibiting profound lack of balance and occasional paralysis. Histology analysis showed accumulation of leukocytes, primarily Mac3+ myeloid cells, in the meninges, cerebellum and the brain stem in the diseased Csf2CD4 mice. The infiltration of myeloid cells coincided with substantial loss of myelin and nerve fibers, indicative of inflammation-induced tissue damage. These results demonstrate that the overexpression of GM-CSF in T cells triggers the initiation of CNS inflammation and immunopathology.
The initiation of CNS inflammation is believed to begin with primed T cells that survey the CNS. A “two-wave hypothesis” has been proposed for the T cell-mediated effector stage of EAE (Ransohoff, 2009). After priming in the peripheral lymph nodes, the activated T cells traffic through the choroid plexus into the subarachnoid space (first wave). Subsequently, the T cells are re-stimulated by meningeal antigen presenting cells (APCs) to undergo clonal expansion. Productive T cell-APC interactions lead to the expression of inflammatory cytokines that activate blood-brain barrier to trigger the second wave of leukocyte infiltration. However, in the Csf2CD4 mice, CNS inflammation was initiated without priming. In fact, GM-CSF overexpression in T cells recognizing ovalbumin, a foreign antigen absent in the CNS, also induced spontaneous CNS inflammation. The onset of neurological symptoms was preceded by GM-CSF-induced the expansion of myeloid populations in the periphery followed by the influx of inflammatory MdCs in the CNS. Notably, the infiltration of T cells in the CNS was only detected after the disease had already manifested. Therefore, the antigen-specific T activation in the periphery and CNS required for the initiation of CNS inflammation (“two-wave model”) was completely bypassed in the Csf2CD4 mice. The aggressive myeloid populations in the Csf2CD4 mice seemed to be able to directly breach the blood-brain barrier. The authors noted that the inflammatory MdCs from the Csf2CD4 mice were producing IL-1β and reactive oxygen species (ROS), both of which have been implicated in influencing the blood-brain barrier.
Regarding how GM-CSF-induced myeloid cells initiate the inflammatory cascade, the study implicates the activation of microglial during the early stage of the disease. Prior to the onset of the symptoms, the expression of CCL2 chemokine, TNF-α and IL-1β in the microglia cells were slightly but noticeably elevated. CCL2 is a high affinity ligand for CCR2, which is expressed by the monocyte-derived cells in the periphery that are expanded by GM-CSF. Thus, it is possible that the GM-CSF-induced CCR2+ inflammatory MdCs may infiltrate into CNS and then activate the microglial cells; the activated microglial cells then increase their expression of CCL2 to further recruit the inflammatory MdCs, forming a positive feedback loop. The authors indeed observed ameboid-shaped microglia in the inflamed CNS of Csf2CD4 mice, a morphology indicative of activation of microglia. Moreover, it was noted a down-regulation of CCR2 and up-regulation of IL-1β in the infiltrating MCs, implicating the differentiation of monocytes towards inflammatory macrophages. Following the accumulation of inflammatory MdCs, the endothelial cells of the blood-brain barrier might be further activated to allow the entry of GM-CSF-overexpressing CD4+ T cells into the CNS, exacerbating the CNS inflammation and tissue damage. Thus, the Csf2CD4 mice could be an excellent model to study the cross-talk between monocyte-derived cells and microglial cells in promoting CNS inflammation and immunopathology.
In the current study, the authors also attempted to overexpress IL-17 in the T cells using the same strategy. The Il17CD4 mice failed to develop spontaneous CNS inflammation. Notably, previous studies have clearly demonstrated that IL-17 signaling is required for effector stage of EAE (Hu et al., 2010) (Kang et al., 2010; Kang et al., 2013). Cell-specific deletion of Il17r or Act1 (an adaptor of IL-17R) demonstrates that IL-17 signaling primarily acts on tissue cells (including CNS resident cells, such as oligodendrocytes and astrocytes) to produce neutrophil mobilizing cytokines and chemokines (Kang et al., 2013) (Kang et al., 2010). As a consequence, IL-17-mediated inflammatory response can only take place when the IL-17-producing T cells enter the tissue (such as CNS). Thus, the forced expression of IL-17 in polyclonal T cells may not effectively deliver IL-17 to its target cells in the CNS. Interestingly, a fate mapping study demonstrates a temporal regulation of T cell function and cytokine production during the course of EAE. It has been shown that after MOG35–55 immunization, classical IFN-γ–producing Th1 cells are present at an ~1:1 ratio with respect to Th17 cell–lineage IL-17A–producing cells in the draining lymph nodes. However, after arriving in the CNS, cells of the classical Th1 cell lineage rapidly disappear after immunization. Meanwhile, large numbers of Th17 cells are converted to IFN-γ+ cells, also known as ex-Th17 cells, which secret GM-CSF in the spinal cord. Therefore, IL-17, GM-CSF and IFN-γ produced by T cells may play a specific temporal role during the course of CNS inflammation, which might not be precisely recapitulated by the strategy of overexpression of these cytokines in the CD4+ cells. As such, information provided by the Csf2CD4 and Il17 CD4 mice should be viewed as a snapshot of the disease pathogenesis.
In addition to CNS inflammation, the authors also observed opacity of the eyes with immune cell infiltrates in the retina and noticeable destruction of the retinal layer in Csf2CD4 mice. Intriguingly, in spite of an equally extensive infiltration of myeloid cells in kidney, lung and the liver in the Csf2CD4 mice, the functionality and tissue integrity in those sites were not substantially affected compared to immune-privileged retina and the CNS. The authors found that the myeloid populations in the CNS were distinct from those in the other sites. Transcriptomic profiling suggested that the myeloid cells in the CNS exhibited features of phagocytes expressing phagocytosis markers such as Mertk and CD64 and as well as genes involved in ROS generation. Indeed, the authors observed that the CNS infiltrating myeloid cells can take up myelin debris and were capable of producing ROS, which may contribute to the lesion formation in the CNS of Csf2CD4 mice. A plausible explanation for the more pro-inflammatory phenotype of myeloid cells in the CNS, as the authors pointed out, might be the absence of regulatory T cells (Treg) in the immune-privileged sites. Treg cells are known to be a critical population for maintaining the homeostasis of the tissue microenvironment by quenching excessive inflammation. Future studies are required to elucidate the intimate interaction between infiltrating myeloid cells and the local environment, which may shed light on organ-specific impact of GM-CSF-mediated myelopoiesis.
Overall, the study by Spath and colleagues has provided insight into the role of exaggerated production of GM-CSF from T cells in triggering CNS inflammation. T cells armed with GM-CSF can aggressively expand and deploy myeloid cells from the periphery to the CNS for tissue destruction. Inhibiting GM-CSF may provide an opportunity to interrupt the dysregulated T cells’ chain of command and halt the tissue-damaging inflammatory cascade.
Figure.1. GM-CSF drives the expansion of CNS-invading phagocytes.
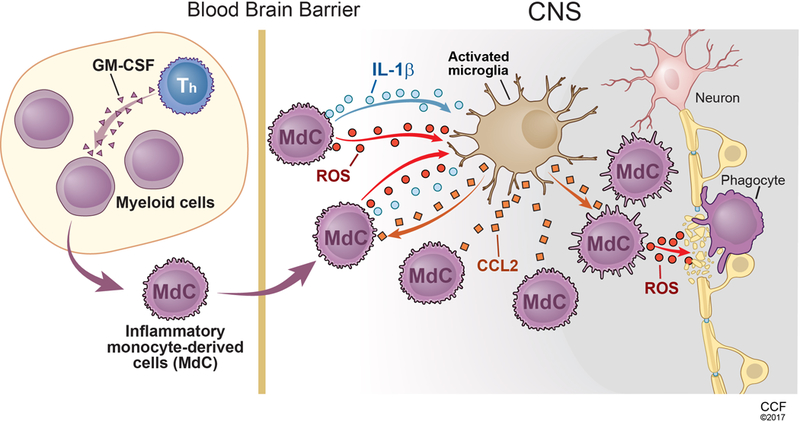
Dysregulated production of GM-CSF by T helper cells induces the expansion of inflammatory monocyte-derived cells (MdCs) in the periphery. The GM-CSF-expanded inflammatory MdCs are capable of producing IL-1β and ROS, which breaches the blood-brain barrier and activate the microglia. Activated microglia secret CCL2 and further recruit the CCR2 expressing inflammatory MCs into the CNS. The infiltrating inflammatory MCs then differentiate into ROS-producing phagocytes, resulting in demyelination and loss of nerve fiber.
References:
- 1.Becher B, Tugues S, and Greter M (2016). GM-CSF: From Growth Factor to Central Mediator of Tissue Inflammation. Immunity 45, 963–973. [DOI] [PubMed] [Google Scholar]
- 2.Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, Suter T, and 3. Becher B (2011). RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nature immunology 12, 560–567. [DOI] [PubMed] [Google Scholar]
- 3.Croxford AL, Lanzinger M, Hartmann FJ, Schreiner B, Mair F, Pelczar P, Clausen BE, Jung S, Greter M, and Becher B (2015). The Cytokine GM-CSF Drives the Inflammatory Signature of CCR2+ Monocytes and Licenses Autoimmunity. Immunity 43, 502–514. [DOI] [PubMed] [Google Scholar]
- 4.El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, Zhang GX, Dittel BN, and Rostami A (2011). The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nature immunology 12, 568–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, Ahlfors H, Wilhelm C, Tolaini M, Menzel U, et al. (2011). Fate mapping of IL-17-producing T cells in inflammatory responses. Nature immunology 12, 255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu Y, Ota N, Peng I, Refino CJ, Danilenko DM, Caplazi P, and Ouyang W (2010). IL-17RC is required for IL-17A- and IL-17F-dependent signaling and the pathogenesis of experimental autoimmune encephalomyelitis. Journal of immunology 184, 4307–4316. [DOI] [PubMed] [Google Scholar]
- 7.Kang Z, Altuntas CZ, Gulen MF, Liu C, Giltiay N, Qin H, Liu L, Qian W, Ransohoff RM, Bergmann C, et al. (2010). Astrocyte-restricted ablation of interleukin-17-induced Act1-mediated signaling ameliorates autoimmune encephalomyelitis. Immunity 32, 414–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kang Z, Wang C, Zepp J, Wu L, Sun K, Zhao J, Chandrasekharan U, DiCorleto PE, Trapp BD, Ransohoff RM, and Li X (2013). Act1 mediates IL-17-induced EAE pathogenesis selectively in NG2+ glial cells. Nature neuroscience 16, 1401–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ransohoff RM (2009). Immunology: In the beginning. Nature 462, 41–42. [DOI] [PubMed] [Google Scholar]
- 10.Spath S, Komuczki J, Hermann M, Pelczar P, Mair F, Schreiner B, Bcher B (2017). GM-CSF-driven expansion of phagocytes induces spontaneous immunopathology in the central nervous system. Immunity [DOI] [PubMed]