

Abstract
Compensatory activation of the signal transduction pathways is one of the major obstacles for the targeted therapy of non‐small cell lung cancer (NSCLC). Herein, we present the therapeutic strategy of combined targeted therapy against the MEK and phosphoinositide‐3 kinase (PI3K) pathways for acquired resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) in NSCLC. We investigated the efficacy of combined trametinib plus taselisib therapy using experimentally established EGFR‐TKI‐resistant NSCLC cell lines. The results showed that the feedback loop between MEK/ERK and PI3K/AKT pathways had developed in several resistant cell lines, which caused the resistance to single‐agent treatment with either inhibitor alone. Meanwhile, the combined therapy successfully regulated the compensatory activation of the key intracellular signals and synergistically inhibited the cell growth of those cells in vitro and in vivo. The resistance mechanisms for which the dual kinase inhibitor therapy proved effective included (MET) mesenchymal‐epithelial transition factor amplification, induction of epithelial‐to‐mesenchymal transition (EMT) and EGFR T790M mutation. In further analysis, the combination therapy induced the phosphorylation of p38 MAPK signaling, leading to the activation of apoptosis cascade. Additionally, long‐term treatment with the combination therapy induced the conversion from EMT to mesenchymal‐to‐epithelial transition in the resistant cell line harboring EMT features, restoring the sensitivity to EGFR‐TKI. In conclusion, our results indicate that the combined therapy using MEK and PI3K inhibitors is a potent therapeutic strategy for NSCLC with the acquired resistance to EGFR‐TKIs.
Keywords: acquired resistance, compensatory activation, MEK inhibitor, non‐small cell lung cancer, PI3K inhibitor
Abbreviations
- EGFR
epidermal growth factor receptor
- EMT
epithelial‐to‐mesenchymal transition
- NSCLC
non‐small cell lung cancer
- PI3K
phosphoinositide‐3 kinase
- TKI
tyrosine kinase inhibitor
1. INTRODUCTION
Mutations of the epidermal growth factor receptor (EGFR) were first reported in 2004, and EGFR‐tyrosine kinase inhibitors (TKIs) have since been found to exert significant antitumor activity against non‐small cell lung cancer (NSCLC) harboring EGFR mutations, representing a breakthrough in the treatment of NSCLC patients.1, 2 However, NSCLC patients initially showing response to EGFR‐TKI treatment often eventually acquire resistance to TKIs, resulting in relapse and cancer‐related death.
A number of diverse mechanisms have been shown to underlie the development of acquired resistance to EGFR‐TKIs in NSCLC, which makes it difficult to overcome the drug resistance to EGFR‐TKIs. Starting with the report of the appearance of a secondary EGFR T790M mutation in 2005, numerous resistance mechanisms have been reported by our group and others, such as MET amplification, activation of the mesenchymal‐epithelial transition factor/hepatocyte growth factor axis, induction of epithelial‐to‐mesenchymal transition (EMT), acquisition of stem cell properties, and transformation from NSCLC into small cell lung cancer.3, 4, 5, 6, 7, 8 Recently, osimertinib, a third‐generation EGFR‐TKI, was developed to overcome the resistance associated with the EGFR T790M mutation, and is expected to play an important role in the treatment of advanced NSCLC.9 However, the emergence of resistance to osimertinib by various mechanisms, including the appearance of the EGFR C797S mutation, has already become a serious problem.10, 11, 12 These phenomena demand the development of novel therapeutic strategies for advanced NSCLC with acquired resistance to EGFR‐TKIs.
In attempting to overcome acquired resistance to EGFR‐TKIs caused by “receptor tyrosine kinase (RTK)‐targeted” therapy, the downstream pathways could be viewed as reasonable next targets. The emergence of the EGFR T790M mutation is known to lead to reactivation of the MEK/ERK or PI3K/AKT pathway.13, 14, 15 Several studies have also demonstrated that MET amplification promotes resistance to TKIs by reactivating both the PI3K/AKT and MEK/ERK pathways.4, 16 Thus, most of the resistance mechanisms were associated with unexpected aberrant re‐awakening of the key intracellular signals that were basically inhibited by the TKIs. However, although these pathways are attractive therapeutic targets, it is well known that the inhibition of one pathway can lead to compensatory activation of the other pathway, which leads to diminished efficacy of single‐agent therapies,17 and overcoming the feedback loop is one of the major issues for molecular targeted therapy in many types of cancer. Among such intrinsic mutual compensation systems of intracellular signal transduction networks in cancer, the tight relationship between MEK/ERK and PI3K/AKT pathways has been of particular interest.18, 19, 20, 21 Indeed, there are reports describing the efficacy of combined inhibition of MEK and PI3K signaling in several types of cancers.22, 23, 24, 25 Furthermore, several clinical trials evaluating the feasibility of MEK plus PI3K dual blockade therapy for advanced solid tumors are currently ongoing.26 A recent search on ClinicalTrials.gov (https://clinicaltrials.gov/, accessed on June 30, 2018) yielded 10 clinical trials for investigating the efficacy of the combined use of MEK and PI3k inhibitors. Among them, 2 trials for patients with solid tumors were terminated due to the lack of tolerability, suggesting the necessity for further consideration of it in some issues, such as knowing the treatment indication, optimal types of MEK and PI3K inhibitors and their doses to be used at not only clinical settings but also basic in vitro contexts. To the best of our knowledge, the efficacy of the combined therapy with MEK and PI3K inhibitors for NSCLC after TKI failure has not been fully elucidated. In this study, we examined the effect of MEK plus PI3K dual inhibition on the cell growth of NSCLC with acquired resistance to EGFR‐TKIs using experimentally established EGFR‐TKI‐resistant cell lines,7, 8 and explored the therapeutic potential of MEK/PI3K dual blockade therapy.
2. MATERIALS AND METHODS
2.1. Cell lines and reagents
We used three human NSCLC cell lines (HCC827, HCC4006, and PC‐9), five gefitinib‐resistant (GR) cell lines (HCC827‐GR‐step [GRS], HCC827‐GR‐high [GRH], HCC4006‐GRS, HCC4006‐GRH, and PC‐9‐GRS), and four afatinib‐resistant (AR) cell lines (HCC827‐ARS, HCC827‐ARH, HCC4006‐ARS, and HCC4006‐ARH). The characteristics of these resistant cell lines are described in our previous reports and summarized in Table 1.7, 8 We used trametinib, which is a potent and selective MEK1/2 inhibitor that was approved by the Food and Drug Administration for the treatment of BRAF V600E‐mutant NSCLC in combination with the BRAF inhibitor dabrafenib.27 For PI3K inhibition, we selected taselisib, which has been shown in early‐phase clinical trials to be of clinical benefit for solid tumors harboring PIK3CA alterations.28 The details about these cell lines, the reagents, and the antibodies for western blot analysis are included in the Data S1.
Table 1.
IC50 values (mmol/L) against trametinib, taselisib, and their combination in EGFR‐mutant cell lines
| Cell lines | Mechanisms of the acquired resistance to TKIs | Trametinib (μmol/L) | Taselisib (μmol/L) | Combination (μmol/L) | |||
|---|---|---|---|---|---|---|---|
| MET amplification | EMT | Stem cell markers | EGFR T790M | ||||
| HCC827 | N/A | N/A | N/A | N/A | >10 | 0.83 | 0.012 |
| HCC827‐GRS | Yes | >10 | 1.06 | 0.0002 | |||
| HCC827‐GRH | Yes | Yes | >10 | >10 | >10 | ||
| HCC827‐ARS | Yes | Yes | >10 | 4.44 | 0.073 | ||
| HCC827‐ARH | Yes | 0.037 | 0.57 | 0.0046 | |||
| HCC4006 | N/A | N/A | N/A | N/A | 0.022 | 1.73 | 0.0052 |
| HCC4006‐GRS | Yes | 0.53 | 4.96 | 0.0045 | |||
| HCC4006‐GRH | Yes | 0.020 | 3.35 | 0.0062 | |||
| HCC4006‐ARS | Yes | Yes | 1.11 | 1.05 | 0.0065 | ||
| HCC4006‐ARH | Yes | 0.048 | 0.125 | 0.0037 | |||
| PC‐9 | N/A | N/A | N/A | N/A | 7 | >10 | 0.0026 |
| PC‐9‐GRS | Yes | >10 | 6.4 | 0.0004 | |||
EGFR, epidermal growth factor receptor; EMT, epithelial‐to‐mesenchymal transition; IC50, 50% inhibitory concentration; MET, mesenchymal‐epithelial transition factor;TKIs, tyrosine kinase inhibitors.
2.2. Determination of cell proliferation
Cell proliferative ability was determined by a modified 3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2H‐tetrazolium assay with CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega, Fitchburg, WI, USA). The detailed protocol was described in our previous report.29 The antiproliferative effects are shown as the 50% inhibitory concentration (IC50). Drug sensitivity was evaluated based on plasma concentration. In summary, the maximum human plasma concentrations of each inhibitor when used in combination with other drugs were reported to be 32.5 ng/mL (approximately 0.053 μmol/L, trametinib) and 35.2 ng/mL (approximately 0.076 μmol/L, taselisib).30, 31 According to these results, we classified drug sensitivity into 3 groups based on the IC50. IC50 values under 0.05 μmol/L, between 0.05 and 0.1 μmol/L, and over 0.1 μmol/L were defined as highly sensitive, moderately sensitive, and insensitive to each drug, respectively.
2.3. Combination effect
The combination effect of two drugs was evaluated using the combination index (CI). The CI values were calculated using the Calcusyn software (Biosoft, Cambridge, UK), as previously described.29 The combination effect is classified as follows: CI < 1: synergistic effect; CI = 1: additive effect; CI > 1: antagonistic effect. Values of CI of <0.4, 0.4‐0.8, and >0.8 indicate strong, moderate, and slight synergistic effect, respectively.
2.4. Detection of apoptotic cells by Hoechst staining and cell cycle analysis
Cells were seeded onto 6‐cm dishes and treated with trametinib (100 nmol/L) alone, taselisib (100 nmol/L) alone, or a combination of the two for 48 hours. The nuclear morphology and the cell cycle distribution were assessed as described in Data S1.
2.5. Western blot analysis
The detailed protocol for western blot analysis has been previously described.7 The relative band intensity was assessed by densitometric analysis using ImageJ (National Institute of Health, Bethesda, MD, USA).
2.6. Human Phospho‐MAP Kinase Array
HCC827‐GRS and HCC4006‐GRS cells were treated with trametinib (100 nmol/L) plus taselisib (100 nmol/L). After 48‐hour treatment, the total protein was purified, and the cell lysates were applied to the Human Phospho‐MAPK Array Kit (R&D Systems, Minneapolis, MN, USA). The relative phosphorylation levels of 26 different MAPKs and serine/threonine kinases were determined in accordance with the manufacturer's instructions.
2.7. Animal experimental procedure
The protocol was approved by the Animal Care and Use Committee of Okayama University. Six‐week‐old female BALB/c nu/nu mice were purchased from CLEA Japan (Tokyo, Japan). Cells (1 × 106) were suspended in 50 μL of RPMI‐1640 medium mixed with 50 μL of Matrigel Basement Membrane Matrix (Corning, Corning, NY, USA) and subcutaneously injected into the backs of the mice. When the tumors grew to approximately 50‐100 mm3 in volume, the mice were randomly divided into four groups, as follows: taselisib (5 mg/kg/day) group, trametinib (0.5 mg/kg/day) group, combined therapy (taselisib; 5 mg/kg/day, trametinib; 0.5 mg/kg/day), and control group (n = 5 for each group). The tumor volume was calculated using the empirical formula V = 1⁄2 × ([shortest diameter]2 × [the longest diameter]). The trametinib and taselisib dosing solutions were prepared in 0.5% (w/v) methyl cellulose. The vehicles and drugs were administered orally by gavage, 5 days per week for 3 weeks. At the end of the experiment, the mice were sacrificed, and tumors were harvested, measured, and photographed.
2.8. mRNA and microRNA expression analyses by quantitative reverse‐transcription‐PCR
The details about cDNA and microRNA (miR) synthesis are described in Data S1 and our previous report 7. The TaqMan assays were purchased from Thermo Fisher Scientific (San Jose, CA, USA), and the catalog numbers are shown in Table S1.
2.9. Microarray and Gene Set Enrichment Analysis (GSEA)
Purified total RNA samples extracted from the resistant cell lines were hybridized on the Human Whole Genome DNA Microarray system (SurePrint G3 Human 8x60K ver. 3.0, Agilent Technologies, Santa Clara, CA, USA) to analyze the alterations in the gene expression profiles. The specific enrichments of gene sets were further analyzed using the GSEA software (GSEA ver. 2.0).32, 33
2.10. Statistical analyses
All in vitro experiments except for a phospho‐MAP Kinase Array were performed at least three times. Data are expressed as the mean ± standard deviation. Statistical analyses were performed using the JMP® 9.0.0 software for Windows (SAS Institute, Inc., Cary, NC, USA). All statistical tests were two‐sided, and probability values <.05 indicated statistically significant differences.
3. RESULTS
3.1. Effect of MEK plus PI3K dual inhibition on the cell growth activity in NSCLC cell lines
We first examined the growth‐inhibitory effect of trametinib, taselisib, and the combination of trametinib plus taselisib on the cell growth activity of three EGFR‐mutated parental NSCLC cell lines and nine EGFT‐TKI‐resistant cell lines. The characteristics of the cell lines and the IC50 values are summarized in Table 1. The IC50 values of the combination of trametinib plus taselisib were <0.05 μmol/L for all the cell lines examined, except HCC827‐GRH and HCC827‐ARS, indicating that the MEK plus PI3K dual blockade therapy is highly sensitive against both the resistant cell lines and the parental cell lines. As for HCC827‐ARS, the IC50 value of the drug combination was 0.073 μmol/L, higher than the values for the other cell lines, but lower than the IC50 values of either drug used alone, indicating the synergistic effect of the drug combination. Taken together, the resistance mechanisms for which this combination therapy was effective included MET amplification (HCC827‐GRS and HCC827‐ARH), induction of EMT (HCC827‐ARS, HCC4006‐GRS, GRH, ARS, and ARH), and the EGFR T790M mutation (PC‐9‐GRS), suggesting that the effect of the dual inhibitor therapy may not be influenced by the resistance mechanism per se. Among the resistant cell lines, we focused on the five cell lines (HCC827‐GRS and ARS, HCC4006‐GRS and ARS, and PC‐9‐GRS) against which the dual kinase inhibitor therapy was particularly effective, as compared to the effect of single‐agent therapies. The survival curves for these cell lines are shown in Figure 1A, and the effects of the drug combination were evaluated by the CI values. As shown in Figure 1B, a strong synergistic effect of combined trametinib plus taselisib therapy was observed even at low concentrations.
Figure 1.
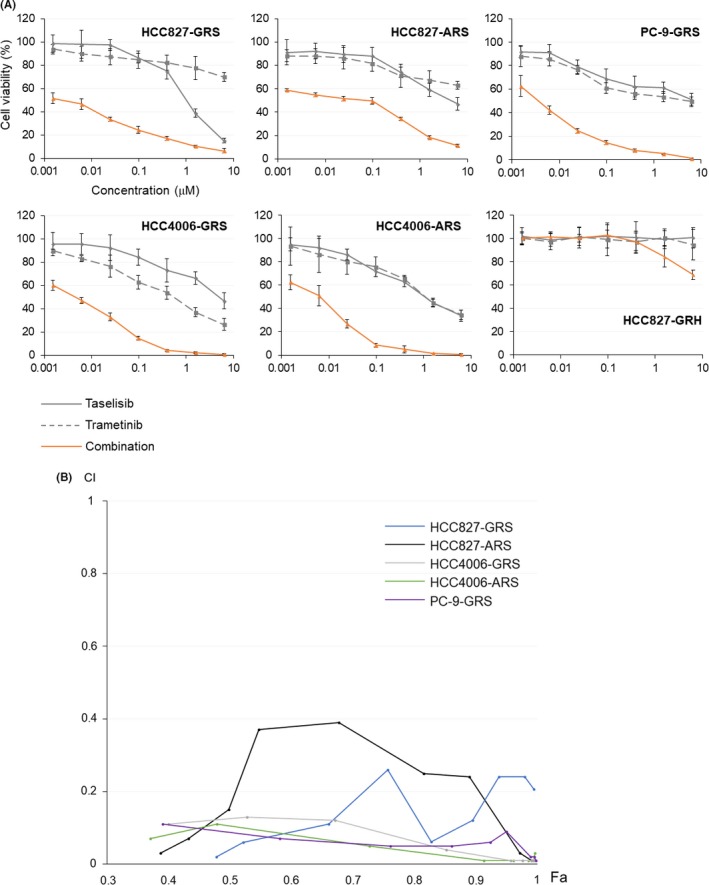
Growth‐inhibitory effect of combined trametinib plus taselisib therapy on non‐small cell lung cancer (NSCLC) cell lines with acquired resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs). (A) Survival curves of each resistant cell line treated with trametinib, taselisib, or a combination of the two drugs. When the drug combination was used, equal concentrations of the two drugs were used. (B) Synergistic effect of the combined kinase inhibitor therapy. CI, combination index; Fa, fraction affected
3.2. Combined trametinib plus taselisib therapy suppressed the activation of downstream signaling in EGFR‐TKI‐resistant NSCLC cell lines
To gain insight into the intracellular signaling events involved in the growth suppression caused by concurrent use of trametinib plus taselisib, we examined the alterations in the expression levels of key protein molecules associated with cancer progression. As shown in Figure 2, inhibition of MAPK and AKT phosphorylation was confirmed in all five cell lines treated with the drug combination. On the other hand, the single‐agent therapy with trametinib induced the upregulation of AKT. Similarly, taselisib alone induced the upregulation of MAPK. These results suggest that compensatory activation of other pathways is one of the reasons why single‐agent therapies fail to sufficiently suppress cell proliferation in these resistant cell lines.
Figure 2.
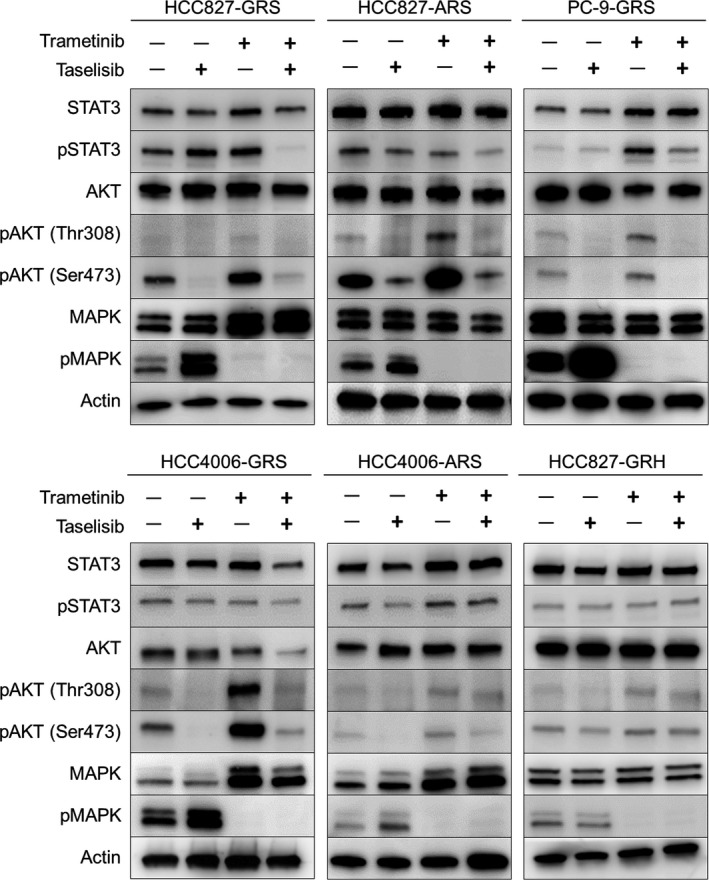
Effect of combined trametinib plus taselisib therapy on the intracellular transduction pathways. Cells were treated with trametinib (100 nmol/L), taselisib (100 nmol/L), or a combination of the two drugs. After 48 h, the cell extracts were analyzed by western blot analysis
3.3. Combined therapy with trametinib plus taselisib inhibited the tumor growth in a mouse xenograft model of EGFR‐TKI‐resistant NSCLC
We investigated the antitumor effect of trametinib, taselisib, and a combination of the two on the growth of HCC827‐GRS and PC9‐GRS cells in vivo. As shown in Figure 3A,B, the tumor growth in the dual inhibitor therapy group was significantly suppressed during the observation period, as compared to that in the groups treated with trametinib or taselisib alone. There was no apparent toxicity, eg, weight loss or behavioral changes, in any of the groups. As for the signal transduction pathway, phosphorylation of MAPK and AKT was assessed to confirm the efficacy of the drug regimen by western blot analysis at the time point of sacrifice (Figure 3C). In the xenografts of the HCC827‐GRS and PC‐9‐GRS cells, combined therapy with trametinib plus taselisib suppressed the phosphorylation of both MAPK and AKT. In addition, treatment with trametinib induced marked phosphorylation of AKT, and taselisib induced slight phosphorylation of MAPK, consistent with the in vitro data, reinforcing the contention that this was one of the reasons for the insufficient growth‐inhibitory effect of either inhibitor agent used alone.
Figure 3.
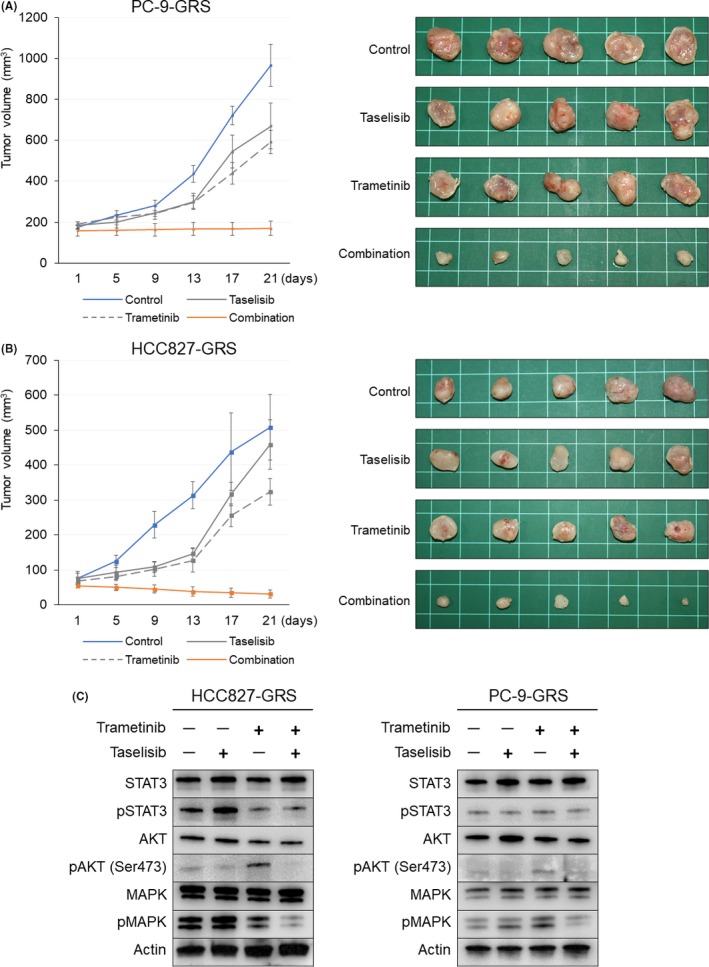
Therapeutic effect of combined trametinib plus taselisib therapy on the tumor growth in vivo. (A,B) The mean volumes of the subcutaneous xenograft tumors were calculated for 5 tumors in each group. The combined inhibitor therapy significantly inhibited the tumor growth in the mouse xenograft models of PC9‐GRS (A) and HCC827‐GRS (B). Time‐dependent changes of the tumor volumes are shown on the left side and the appearances of the tumors at the time of sacrifice are shown on the right. (C) At the time point of sacrifice, protein was extracted from the HCC827‐GRS tumor specimens and subjected to western blot analysis
3.4. Combined therapy with trametinib plus taselisib induced apoptosis or cell cycle arrest in EGFR‐TKI‐resistant NSCLC cell lines
Next, to elucidate the mechanism underlying the growth inhibition, we examined the effect of the combined inhibitor therapy on cell apoptosis by western blot analysis. After 48 hours of combined trametinib plus taselisib treatment, marked induction of the expression of c‐caspase3 and c‐PARP (poly[ADP‐ribose] polymerase) was observed in the HCC827‐GRS, HCC4006‐GRS, and PC9‐GRS cells, suggestive of activation of an early apoptosis cascade (Figure 4A). Next, when the cells were treated with the combination of the two drugs at concentrations ranging from 1 to 100 nmol/L, increased c‐PARP expression was detected, suggesting the dose‐dependent effect of the combination therapy (Figure S1A). In addition, in order to investigate the effect of trametinib, taselisib, and a combination of the two drugs on DNA fragmentation and morphology, the morphological changes of the nuclei were visualized by Hoechst 33342 staining. As shown in Figure 4B, cells treated with the drug combination exhibited the typical apoptotic nuclear morphology (chromatin condensation, nuclear fragmentation) of the later stages of apoptosis. These phenomena were not observed in the cells treated with either kinase inhibitor alone, which was concordant with the results of the cell proliferation assay. To further verify the mechanism underlying the activation of the apoptosis cascade, we performed a phospho‐MAP kinase array analysis using HCC827‐GRS and HCC4006‐GRS cells (Figure S1B). Based on these results, the alterations of the relative values of the key molecules related to the MAPK cascade were calculated by densitometric analysis. As expected, the phosphorylation of ERK1/2 and AKT was suppressed (Figure S1C). In addition, the combined inhibitor therapy induced the phosphorylation of p38α, p38β and p38δ, and their downstream molecule, heat shock protein 27, whereas it had no effect on the phosphorylation level of pan‐JNK (Figure 4C). In HCC827‐GRS and HCC4006‐GRS, western blot analysis also revealed that the combined inhibitor therapy induced the PARP cleavage displayed by c‐PARP appearance concomitant with the phosphorylation of p38 but not of JNK, to which a pan‐p38 inhibitor SB203580 reduced the expression of c‐PARP. On the other hand, this phenomenon was not observed in HCC827‐ARS cells that were resistant to the same combined therapy in apoptotic events (Figure 4D). These results may indicate that the combined kinase inhibitor therapy induces cellular apoptosis through the activation of the p‐38 MAPK signal transduction pathway. On the other hand, overexpression of c‐caspase3 and c‐PARP was not observed in the HCC827‐ARS or HCC4006‐ARS cells even after 48‐hour treatment (Figure 4A). We then conducted cell cycle analysis. The combined inhibitor therapy induced significant G1 cycle arrest in the HCC827‐ARS and HCC4006‐ARS cells, as compared to the observations in the cells treated with either inhibitor agent alone (Figure 4E). Furthermore, drug exposure for more than 5 days induced the expression of c‐PARP in the HCC4006‐ARS cells (Figure S1D). To clarify the groups of genes involved in the apoptotic process induced by the combined kinase inhibitor therapy, we carried out gene expression microarray analysis. Five cell lines (HCC827‐GRS and ARS, HCC4006‐GRS and ARS, and PC‐9‐GRS) and HCC827‐GRH were divided into two groups based on the degree of apoptosis induced: the apoptosis‐resistant group (HCC827‐GRH and ARS, and HCC4006‐ARS) and the apoptosis‐sensitive group (HCC827‐GRS, HCC4006‐GRS, and PC‐9‐GRS), and a GSEA was performed. The data provided us interesting information that the enriched genes in the apoptosis‐resistant group of cells at a significant level were relevant to tumor growth factor (TGF)β1 (HALLMARK_TGF_BETA_SIGNALING) and those in the apoptosis‐sensitive group of cells were relevant to the PI3K/AKT/mechanistic target of rapamycin (mTOR) pathways (HALLMARK_PI3K_AKT‐MTOR_SIGNALING) (Figure 4F). Knowing this, we performed western blot analysis to examine the phosphorylation status of key molecules on the PI3K/AKT/mTOR pathway. As a result, in the apoptosis‐sensitive group, all the phosphorylation levels of AKT, mTOR, and S6 showed tendency to be higher than those in the apoptosis‐resistant group, which was consistent with the result of GSEA (Figure S2). The activation of PI3K/AKT/mTOR cascade is a renowned pathway to give cancer cells an anti‐apoptotic force; therefore, it is reasonable to consider that the tight regulation of the pathway by combined therapy results in higher efficacy in the apoptosis‐sensitive group in comparison to the apoptosis‐resistant group.
Figure 4.
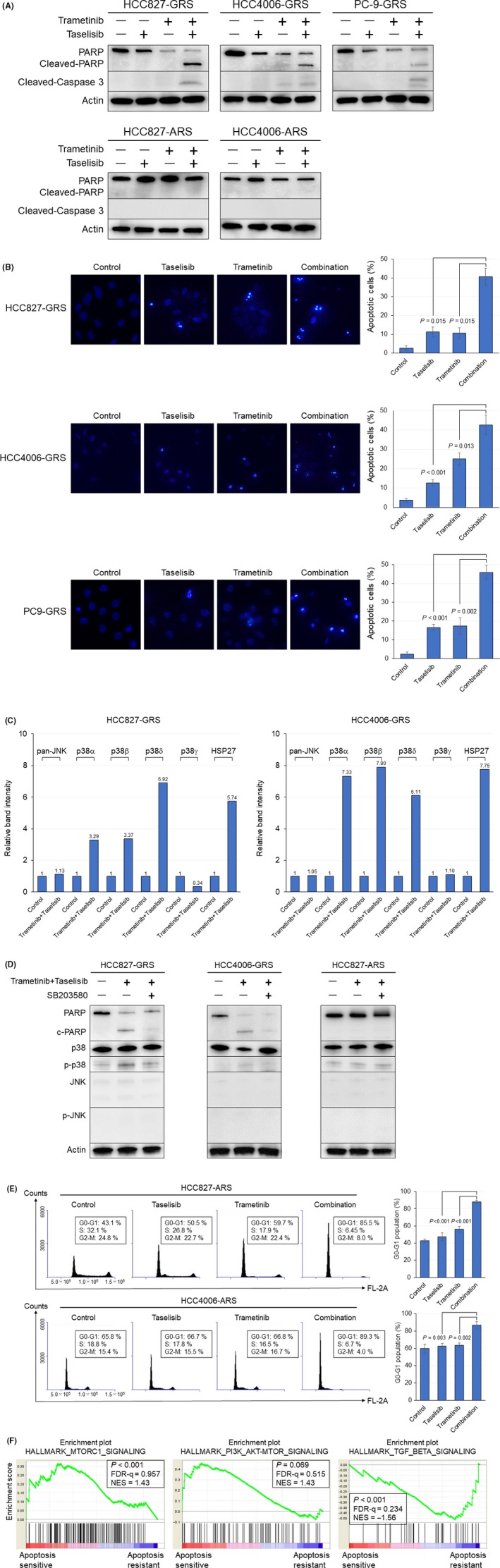
Mechanism underlying the growth inhibition induced by combined trametinib plus taselisib therapy. (A) Cells were treated with trametinib (100 nmol/L), taselisib (100 nmol/L), or a combination of the two drugs for 48 h. The effect of the combined inhibitor therapy on cellular apoptosis was analyzed by western blot analysis. (B) Nuclear staining of HCC827‐GRS, HCC4006‐GRS and PC9‐GRS cells with Hoechst. The percentage of apoptotic cells was calculated by counting the number of cells with condensed chromatin. (C) The results of the phospho‐MAPK protein array analysis using HCC827‐GRS and HCC4006‐GRS cells treated or not treated with the drug combination of trametinib plus taselisib. The phosphorylation statuses of JNK, four p38 isoforms, and het shock protein (HSP)27 were evaluated by densitometric analysis. (D) The combined inhibitor therapy induced the phosphorylation of p38, and p38 inhibitor, SB203580, suppressed the expression of c‐PARP (poly[ADP‐ribose] polymerase) in HCC827‐GRS and HCC4006‐GRS cells. Cells were treated with the combination of trametinib plus taselisib (100 nmol/L each) ± SB203580 (1 μmol/L) for 48 h. (E) The combined inhibitor therapy induced significant G1 cycle arrest in the HCC827‐ARS and HCC4006‐ARS cells. (F) Representative enriched pathways in each group are shown. FDR, false discovery rate; NES, normalized enrichment score
3.5. Effect of combined therapy with trametinib plus taselisib on the EGFR‐TKI‐resistant NSCLC cell line harboring EMT features
It is well known that TGF‐β signaling promotes the acquisition of the EMT phenotype. The results of the GSEA analysis raised the question of what effect the combined kinase inhibitor therapy might exert on the resistant cell lines harboring evidence of EMT. As described above, cell death was not observed in the HCC827‐ARS cells that exhibited the evidence of EMT, although cell cycle arrest was induced. Then, we examined the effect of sustained exposure to trametinib plus taselisib in the HCC827‐ARS cells. After 7‐day exposure, marked morphological changes were observed, and on day 7 after discontinuation of the drug exposure, the cell morphologies were restored to those observed before the drug exposure (Figure 5A). We subsequently examined the alterations of the EMT‐related markers in this setting. In western blot analysis, the HCC827‐ARS cells treated with the combination of trametinib plus taselisib for 7 days showed evidence of MET, with upregulation of E‐cadherin and downregulation of vimentin and zinc finger E‐box binding homeobox 1 (ZEB)1 (Figure 5B). Moreover, after discontinuation of exposure to the drugs, the features of EMT were restored in the HCC827‐ARS cells, accompanied by morphological changes of the cells. In addition, 7‐day treatment with trametinib plus taselisib restored the expression of miR‐200c, which has been implicated in EMT through the reciprocally linked feedback loop with ZEB1 and ZEB2,34 and on day 7 after discontinuation of exposure to the drugs, the expression of miR‐200c returned to its original level (Figure 5C). Meanwhile, the combined inhibitor therapy did not affect the expression of aldehyde dehydrogenase 1 family member A1 (ALDH1A1), which is one of the cancer stem cell markers (Figure 5D). We further investigated the change in the sensitivity of HCC827‐ARS to afatinib, when the drug was used concurrently with trametinib and taselisib. The results revealed that the IC50 values of afatinib changed from 3.82 to 0.72 μmol/L, suggesting the possibility that the combination therapy restored the sensitivity to EGFR‐TKIs in the cell lines with acquired TKI resistance harboring EMT features (Figure 5E).
Figure 5.
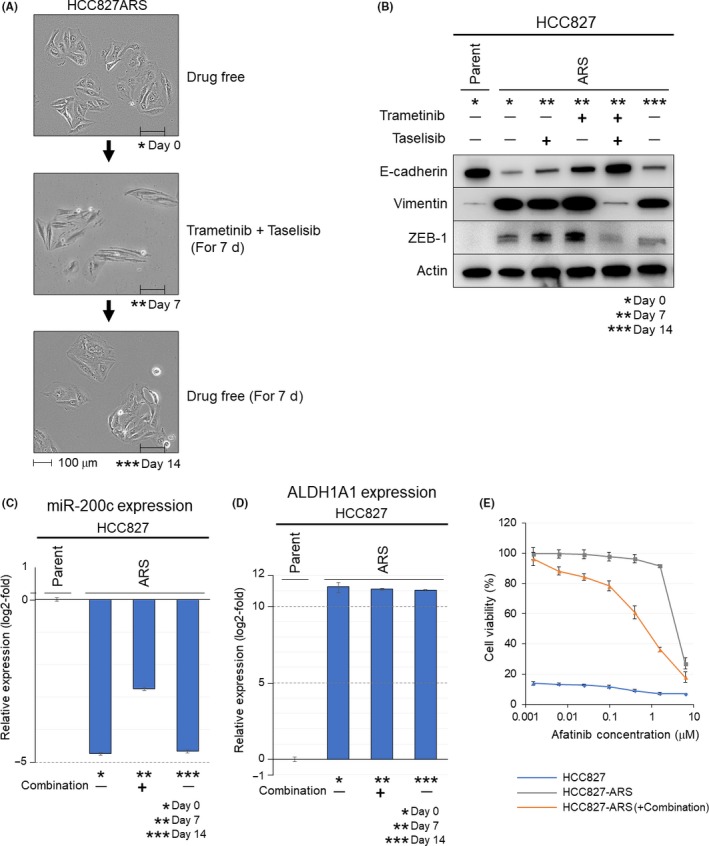
Effects of combined trametinib plus taselisib therapy in HCC827‐ARS cells harboring epithelial‐to‐mesenchymal transition (EMT) features. (A) HCC827‐ARS cells were treated with trametinib plus taselisib (100 nmol/L) for 7 d. Thereafter, the cells were cultured in a drug‐free condition for 7 d. The morphologies of the cells at the indicated times (* Day 0, ** Day 7, *** Day 14) are shown. Scale bars, 100 μm. (B) Alterations of EMT‐related markers. Combined trametinib plus taselisib treatment, but not treatment with either inhibitor used alone, induced conversion from EMT to mesenchymal‐epithelial transition (MET) in the HCC827‐ARS cells. (C) Relative miR‐200c expression level measured by quantitative reverse‐transcription PCR (qRT‐PCR) assay in the HCC827‐ARS cells. Treatment with the drug combination restored the expression of miR‐200c. (D) Relative aldehyde dehydrogenase 1 family member A1 (ALDH1A1) expression level measured by qRT‐PCR assay in the HCC827‐ARS cells. Exposure of the cells to the drug combination did not affect the expression levels of ALDH1A1. (E) The sensitivity of cells (HCC827, HCC827‐ARS [no treatment], HCC827‐ARS (after the 7‐d treatment]) to afatinib were examined. For HCC827‐ARS cells after the 7‐d combined treatment, afatinib sensitivity was investigated in the presence of trametinib (100 nmol/L) and taselisib (100 nmol/L) to maintain MET features. The 50% inhibitory concentration (IC50) values of afatinib were <1 nmol/L for the HCC827 cells, 3.82 μmol/L for the HCC827‐ARS cells (no treatment), and 0.72 μmol/L for the HCC827‐ARS cells (treated with trametinib and taselisib)
4. DISCUSSION
In this study, we demonstrated that the concurrent use of trametinib and taselisib synergistically suppressed cell proliferation through the activation of p38‐mediated apoptosis in NSCLC cell lines with acquired EGFR‐TKI resistance. The combined therapy was effective against both gefitinib‐ and afatinib‐resistant cell lines, and the resistance mechanisms against which the combined regimen showed efficacy included EGFR T790M mutation, MET amplification, and acquisition of EMT features. In addition, in a mouse xenograft model, the dual inhibitor therapy proved feasible, from the viewpoint of tumor growth inhibition and side effects. Recently, some clinical trials examining the efficacy of MEK plus PI3K inhibitors have failed because of toxicity. However, the efficacy of the combined therapy with other types of MEK and PI3K inhibitors, trametinib and taselisib, have not been clinically examined. Our results suggest that the dual inhibition of MEK plus PI3K pathways using our selected trametinib and taselisib may be helpful with a potent therapeutic strategy for NSCLC with acquired resistance to EGFR‐TKI.
One of the major obstacles in single‐agent therapies, targeting only one intracellular pathway, is compensatory activation of another intracellular pathway in a negative feedback loop.17 In our experiment, in 5 out of 9 resistant cell lines, the feedback loop between MEK/ERK and PI3K/AKT pathways had already developed before treatment, causing resistance to single‐agent therapy with inhibitors of either pathway. This result indicates that the compensatory activation between the central oncogenic pathways is one of the important issues that need to be overcome also in NSCLC after TKI failure.
We have pointed out the possibility that the p38 MAPK pathway plays an important role in the activation of apoptosis induced by the MEK plus PI3K dual blockade therapy. The p38 MAPK is a family of stress‐activated protein kinases that are usually activated by a variety of extracellular stresses. In human tissues, four isoforms (p38α, p38β, p38γ, and p38δ) have been identified, are expressed ubiquitously at different levels in tissues, and have different functions.35 Conventionally, the p38 MAPK cascade has been regarded to function as a tumor‐suppressive pathway by negatively regulating cell survival and proliferation.36, 37 In contrast, recent studies have reported that the activation of p38 MAPK pathway also promotes the acquisition of drug resistance, tumor progression, and metastasis in several types of cancers.38, 39 According to our results, SB203580, a pan‐p38 inhibitor, simultaneously inhibited activation of the apoptosis cascade and phosphorylation of p38, suggesting that the p38 MAPK pathway interferes with tumor growth in NSCLC cells with acquired EGFR‐TKI resistance. Further investigation on the underlying mechanism about the activation of p38 MAPK caused by the concurrent MEK/ERK and PI3K/AKT pathways inhibition is necessary to reveal the complex apoptotic mechanisms in NSCLC with acquired resistance to TKIs.
As we reported, EMT features are strongly associated with the acquisition of EGFR‐TKI resistance, and development of a new therapeutic strategy for overcoming EMT is desired.7, 8 Among resistant cells harboring EMT features examined in this study, in HCC827‐ARS cells, long‐term exposure to this treatment caused reversible conversion from EMT to MET, which was accompanied by restoration of miR‐200c expression and sensitivity to afatinib. It is well known that the activation of the MEK/ERK pathway represses the expression of e‐cadherin and plays a crucial role in the induction of EMT.40, 41 Several studies have also reported that the PI3K/AKT pathway is involved in the induction of EMT via upregulating the expression of the transcription factors Twist, Snail, and Slug.42, 43 Thus, activation of the MEK/ERK and PI3K/AKT pathways plays a crucial role in the induction of the EMT phenotype in cancer cells, and our results suggest the possibility that simultaneous inhibition of these two pathways could be effective to overcome the drug resistance caused by the induction of EMT. We previously reported that the expression level of miR‐200c is negatively correlated with EMT features in NSCLC cells with acquired TKI resistance through the reciprocally linked feedback loop with ZEB1 and ZEB2.7, 44 Therefore, the conversion from EMT to MET was considered due to the involvement of the negative feedback loop of ZEB1 and miR‐200c. On the other hand, the dual inhibitor therapy proved to be insufficient/of limited efficacy for three resistant cell lines harboring EMT features; namely, no inhibition of cell growth was observed in the HCC827‐GRH cells, and in the HCC827‐ARS and HCC4006‐ARS cells, while growth inhibition was observed, the resistance to cell apoptosis was implied. Although the detailed mechanism underlying this resistance to the dual MEK plus PI3K inhibition remains unclear, the stem cell properties of the cells may provide some clue. As we have reported previously, a putative stem cell marker, ALDH1A1, was expressed in all three resistant cell lines,7, 8 although there were obvious differences in the level of expression (Figure S3), and the resistant cell lines with higher expression levels of ALDH1A1 also showed a higher resistance level to the combined inhibitor therapy. These results may imply that not only the appearance of EMT, but also that of stem cell properties may underlie the resistance of the cells to the combined inhibitor therapy. Several studies indicated the involvement of multiple pathways for the acquisition and maintenance of cancer stem cell (CSC) properties. Cordenonsi and colleagues reported that Hippo pathway, which is a conserved regulator of organ size, plays an essential role for acquisition of CSC properties.45 Takebe and colleagues also mentioned that CSC is characterized by its aberrant activation of various signal transduction pathways, such as the Notch, Hedgehog, and Wnt pathways.46 Thus, CSC dynamics is governed by the more complex interaction of several pathways, compared to differentiated cancer cells, implying that the inhibition of only two pathway, MEK/ERK and PI3K/AKT pathways, may be insufficient to impair the viability of CSC. The inhibitory role of the concurrent MEK plus PI3K pathways on the cancer EMT and stemness are not fully elucidated in this study, therefore, further investigation about the issue is warranted.
In conclusion, we demonstrated that MEK plus P13K dual blockade therapy with the combination of trametinib plus taselisib significantly inhibits the growth of cells with acquired resistance to EGFR‐TKIs, both in vitro and in vivo. This therapeutic strategy could overcome the tough mutual compensation systems in cancer and be developed as one of the treatment options for patients with advanced NSCLC after EGFR‐TKI failure. Future investigation of this therapy may lead to a marked advance in the treatment of NSCLC.
CONFLICT OF INTEREST
Shinichi Toyooka received research funding from Boehringer‐Ingelheim. The other authors have no conflict of interest to declare.
Supporting information
ACKNOWLEDGMENTS
This work was supported by a Grant‐in‐Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS KAKENHI grant number: 16H05431 to S.T.).
Sato H, Yamamoto H, Sakaguchi M, et al. Combined inhibition of MEK and PI3K pathways overcomes acquired resistance to EGFR‐TKIs in non‐small cell lung cancer. Cancer Sci. 2018;109:3183–3196. 10.1111/cas.13763
REFERENCES
- 1. Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non‐small‐cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129‐2139. [DOI] [PubMed] [Google Scholar]
- 2. Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497‐1500. [DOI] [PubMed] [Google Scholar]
- 3. Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non‐small‐cell lung cancer to gefitinib. N Engl J Med. 2005;352:786‐792. [DOI] [PubMed] [Google Scholar]
- 4. Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039‐1043. [DOI] [PubMed] [Google Scholar]
- 5. Yano S, Wang W, Li Q, et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor‐activating mutations. Cancer Res. 2008;68:9479‐9487. [DOI] [PubMed] [Google Scholar]
- 6. Sequist LV, Waltman BA, Dias‐Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3:75ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shien K, Toyooka S, Yamamoto H, et al. Acquired resistance to EGFR inhibitors is associated with a manifestation of stem cell‐like properties in cancer cells. Cancer Res. 2013;73:3051‐3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hashida S, Yamamoto H, Shien K, et al. Acquisition of cancer stem cell‐like properties in non‐small cell lung cancer with acquired resistance to afatinib. Cancer Sci. 2015;106:1377‐1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Soria JC, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR‐mutated advanced non‐small‐cell lung cancer. N Engl J Med. 2017;378(2):113‐125. [DOI] [PubMed] [Google Scholar]
- 10. Thress KS, Paweletz CP, Felip E, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non‐small cell lung cancer harboring EGFR T790M. Nat Med. 2015;21:560‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang S, Song Y, Yan F, Liu D. Mechanisms of resistance to third‐generation EGFR tyrosine kinase inhibitors. Front Med. 2016;10:383‐388. [DOI] [PubMed] [Google Scholar]
- 12. Ham JS, Kim S, Kim HK, et al. Two cases of small cell lung cancer transformation from EGFR mutant adenocarcinoma during AZD9291 treatment. J Thorac Oncol. 2016;11:e1‐e4. [DOI] [PubMed] [Google Scholar]
- 13. Kobayashi S, Ji H, Yuza Y, et al. An alternative inhibitor overcomes resistance caused by a mutation of the epidermal growth factor receptor. Cancer Res. 2005;65:7096‐7101. [DOI] [PubMed] [Google Scholar]
- 14. Gendreau SB, Ventura R, Keast P, et al. Inhibition of the T790M gatekeeper mutant of the epidermal growth factor receptor by EXEL‐7647. Clin Cancer Res. 2007;13:3713‐3723. [DOI] [PubMed] [Google Scholar]
- 15. Ercan D, Xu C, Yanagita M, et al. Reactivation of ERK signaling causes resistance to EGFR kinase inhibitors. Cancer Discov. 2012;2:934‐947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Turke AB, Zejnullahu K, Wu YL, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 2010;17:77‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Niederst MJ, Engelman JA. Bypass mechanisms of resistance to receptor tyrosine kinase inhibition in lung cancer. Sci Signal. 2013;6:re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ebi H, Corcoran RB, Singh A, et al. Receptor tyrosine kinases exert dominant control over PI3K signaling in human KRAS mutant colorectal cancers. J Clin Invest. 2011;121:4311‐4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Muranen T, Selfors LM, Worster DT, et al. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix‐attached cancer cells. Cancer Cell. 2012;21:227‐239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Turke AB, Song Y, Costa C, et al. MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors. Cancer Res. 2012;72:3228‐3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zmajkovicova K, Jesenberger V, Catalanotti F, Baumgartner C, Reyes G, Baccarini M. MEK1 is required for PTEN membrane recruitment, AKT regulation, and the maintenance of peripheral tolerance. Mol Cell. 2013;50:43‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Engelman JA, Chen L, Tan X, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351‐1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Valentino JD, Li J, Zaytseva YY, et al. Cotargeting the PI3K and RAS pathways for the treatment of neuroendocrine tumors. Clin Cancer Res. 2014;20:1212‐1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Garcia‐Garcia C, Rivas MA, Ibrahim YH, et al. MEK plus PI3K/mTORC1/2 therapeutic efficacy is impacted by TP53 mutation in preclinical models of colorectal cancer. Clin Cancer Res. 2015;21:5499‐5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. ElMokh O, Ruffieux‐Daidie D, Roelli MA, et al. Combined MEK and Pi3’‐kinase inhibition reveals synergy in targeting thyroid cancer in vitro and in vivo. Oncotarget. 2017;8:24604‐24620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Britten CD. PI3K and MEK inhibitor combinations: examining the evidence in selected tumor types. Cancer Chemother Pharmacol. 2013;71:1395‐1409. [DOI] [PubMed] [Google Scholar]
- 27. Planchard D, Smit EF, Groen HJM, et al. Dabrafenib plus trametinib in patients with previously untreated BRAF(V600E)‐mutant metastatic non‐small‐cell lung cancer: an open‐label, phase 2 trial. Lancet Oncol. 2017;18:1307‐1316. [DOI] [PubMed] [Google Scholar]
- 28. Juric D, Krop I, Ramanathan RK, et al. Phase I dose‐escalation study of taselisib, an oral PI3K inhibitor, in patients with advanced solid tumors. Cancer Discov. 2017;7:704‐715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ikeda H, Taira N, Nogami T, et al. Combination treatment with fulvestrant and various cytotoxic agents (doxorubicin, paclitaxel, docetaxel, vinorelbine, and 5‐fluorouracil) has a synergistic effect in estrogen receptor‐positive breast cancer. Cancer Sci. 2011;102:2038‐2042. [DOI] [PubMed] [Google Scholar]
- 30. Ding X, Faber K, Shi Y, et al. Validation and determination of taselisib, a beta‐sparing phosphoinositide 3‐kinase (PI3K) inhibitor, in human plasma by LC‐MS/MS. J Pharm Biomed Anal. 2016;126:117‐123. [DOI] [PubMed] [Google Scholar]
- 31. Yamazaki N, Tsutsumida A, Takahashi A, et al. Phase 1/2 study assessing the safety and efficacy of dabrafenib and trametinib combination therapy in Japanese patients with BRAF V600 mutation‐positive advanced cutaneous melanoma. J Dermatol. 2018;45(4):397‐407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mootha VK, Lindgren CM, Eriksson KF, et al. PGC‐1alpha‐responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267‐273. [DOI] [PubMed] [Google Scholar]
- 33. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545‐15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brabletz S, Brabletz T. The ZEB/miR‐200 feedback loop–a motor of cellular plasticity in development and cancer? EMBO Rep. 2010;11:670‐677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. Biochem J. 2010;429:403‐417. [DOI] [PubMed] [Google Scholar]
- 36. Bulavin DV, Fornace AJ Jr. p38 MAP kinase's emerging role as a tumor suppressor. Adv Cancer Res. 2004;92:95‐118. [DOI] [PubMed] [Google Scholar]
- 37. Han J, Sun P. The pathways to tumor suppression via route p38. Trends Biochem Sci. 2007;32:364‐371. [DOI] [PubMed] [Google Scholar]
- 38. del Barco Barrantes I, Nebreda AR. Roles of p38 MAPKs in invasion and metastasis. Biochem Soc Trans. 2012;40:79‐84. [DOI] [PubMed] [Google Scholar]
- 39. Igea A, Nebreda AR. The stress kinase p38alpha as a target for cancer therapy. Cancer Res. 2015;75:3997‐4002. [DOI] [PubMed] [Google Scholar]
- 40. Lemieux E, Bergeron S, Durand V, Asselin C, Saucier C, Rivard N. Constitutively active MEK1 is sufficient to induce epithelial‐to‐mesenchymal transition in intestinal epithelial cells and to promote tumor invasion and metastasis. Int J Cancer. 2009;125:1575‐1586. [DOI] [PubMed] [Google Scholar]
- 41. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial‐mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. Phosphatidylinositol 3‐kinase function is required for transforming growth factor beta‐mediated epithelial to mesenchymal transition and cell migration. J Biol Chem. 2000;275:36803‐36810. [DOI] [PubMed] [Google Scholar]
- 43. Xu W, Yang Z, Lu N. A new role for the PI3K/Akt signaling pathway in the epithelial‐mesenchymal transition. Cell Adh Migr. 2015;9:317‐324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sato H, Shien K, Tomida S, et al. Targeting the miR‐200c/LIN28B axis in acquired EGFR‐TKI resistance non‐small cell lung cancer cells harboring EMT features. Sci Rep. 2017;7:40847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cordenonsi M, Zanconato F, Azzolin L, et al. The Hippo transducer TAZ confers cancer stem cell‐related traits on breast cancer cells. Cell. 2011;147:759‐772. [DOI] [PubMed] [Google Scholar]
- 46. Takebe N, Miele L, Harris PJ, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. 2015;12:445‐464. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials