

Abstract
The ROS1 tyrosine kinase inhibitor (TKI) crizotinib has shown dramatic effects in patients with non‐small cell lung cancer (NSCLC) harboring ROS1 fusion genes. However, patients inevitably develop resistance to this agent. Therefore, a new treatment strategy is required for lung tumors with ROS1 fusion genes. In the present study, lung cancer cell lines, HCC78 harboring SLC34A2‐ROS1 and ABC‐20 harboring CD74‐ROS1, were used as cell line‐based resistance models. Crizotinib‐resistant HCC78R cells were established from HCC78. We comprehensively screened the resistant cells using a phosphor‐receptor tyrosine kinase array and RNA sequence analysis by next‐generation sequencing. HCC78R cells showed upregulation of HB‐EGF and activation of epidermal growth factor receptor (EGFR) phosphorylation and the EGFR signaling pathway. Recombinant HB‐EGF or EGF rendered HCC78 cells or ABC‐20 cells resistant to crizotinib. RNA sequence analysis by next‐generation sequencing revealed the upregulation of AXL in HCC78R cells. HCC78R cells showed marked sensitivity to EGFR‐TKI or anti‐EGFR antibody treatment in vitro. Combinations of an AXL inhibitor, cabozantinib or gilteritinib, and an EGFR‐TKI were more effective against HCC78R cells than monotherapy with an EGFR‐TKI or AXL inhibitor. The combination of cabozantinib and gefitinib effectively inhibited the growth of HCC78R tumors in an in vivo xenograft model of NOG mice. The results of this study indicated that HB‐EGF/EGFR and AXL play roles in crizotinib resistance in lung cancers harboring ROS1 fusions. The combination of cabozantinib and EGFR‐TKI may represent a useful alternative treatment strategy for patients with advanced NSCLC harboring ROS1 fusion genes.
Keywords: AXL, cabozantinib, HB‐EGF, non‐small lung cancer, ROS1 fusion genes
1. INTRODUCTION
The discovery of oncogenic driver genes and corresponding targeted drugs has changed the clinical treatment of non‐small cell lung cancer (NSCLC) over the past 15 years.1, 2, 3 Fusions in c‐ros oncogene 1 (ROS1) have been identified in approximately 1%‐2% of patients.3, 4 ALK tyrosine kinase inhibitors (TKI, including crizotinib), which have been approved for clinical use, showed inhibitory activity against ROS1 because ROS1 and ALK protein share 49% amino acid sequence identity in their kinase domains.5 The MET/ALK/ROS1 inhibitor crizotinib has been approved for clinical use as an inhibitor of ROS1 in several countries because it confers excellent benefits and shows acceptable tolerance in patients with ROS1 fusion‐positive lung cancer.6, 7 Similar to other oncoprotein inhibitors, however, lung tumors with ROS1 fusion genes inevitably acquire resistance to crizotinib, and further improvements in treatment strategies are, thus, required.8
Many groups have explored the resistance mechanisms in lung tumors with ROS1 fusion genes in attempts to develop new treatment strategies. Similar to the mechanisms of resistance in lung cancers with ALK fusion genes, secondary mutations in the ROS1 kinase domain (eg, G2032R, S1986Y, S1986F, D2033N or L2155S) have been reported.8, 9, 10, 11 However, the resistance mechanisms of ROS1 inhibitors have not been fully clarified in NSCLC harboring ROS1 fusion genes.
To develop a new treatment strategy for lung cancer patients with ROS1 fusion, we investigated the mechanisms of resistance to crizotinib using HCC78 cells harboring the SLC34A2‐ROS1 fusion gene and the newly established lung cancer cell line, ABC‐20 harboring the CD74‐ROS1 fusion gene. Our analyses indicated that HB‐EGF/epidermal growth factor receptor (EGFR) and AXL were attributable to the acquired resistance to crizotinib, and the combination of cabozantinib and gefitinib showed beneficial effects in crizotinib‐resistant cell lines both in vitro and in vivo.
2. MATERIALS AND METHODS
2.1. Cell culture and establishment of a crizotinib‐resistant cell line
HCC78 cells harboring the SLC34A2‐ROS1 fusion gene were kindly provided by Dr William Pao (Vanderbilt University, Nashville, TN, USA). ABC‐20 cells were established in our laboratory from pleural effusion obtained from a Japanese male former smoker who had lung adenocarcinoma harboring the CD74‐ROS1 fusion gene. The experiment regarding ABC‐20 cells was approved by the Institutional Review Board of Okayama University Hospital. Written informed consent was obtained from the patient. PC‐9 cells harboring EGFR 19 del E746_A750 were purchased from the European Collection of Cell Cultures (Salisbury, UK). 293T cells were purchased from the RIKEN Cell Bank (Ibaragi, Japan). Cells were cultured in RPMI 1640 medium (Sigma‐Aldrich, St. Louis, MO, USA) supplemented with 10% heat‐inactivated FBS and 1% penicillin/streptomycin in a tissue culture incubator at 37°C with 5% CO2. To establish a crizotinib‐resistant cell line, HCC78 cells were treated with gradually increasing concentrations of crizotinib, starting at .2 μmol/L (lower than the IC50 of HCC78 cells). After 4 months, the cells grew in the presence of 2 μmol/L crizotinib and were designated as HCC78R cells. HCC78R cells were maintained in culture medium containing 1 μmol/L crizotinib. The resistant cell lines were tested using the PowerPlex 16 STR System (Promega Corporation, Madison, WI, USA).
2.2. Reagents and antibodies
Crizotinib, ceritinib, brigatinib, cabozantinib and afatinib were purchased from Selleck Chemicals (Houston, TX, USA); gefitinib and cetuximab were purchased from Eveleth (Eveleth, MN, USA); erlotinib was purchased from Chemie Tek (Indianapolis, IN, USA); lorlatinib was purchased from Toronto Research Chemicals (Toronto, ON, Canada; and gilteritinib was purchased from Cayman Chemical (Ann Arbor, MI, USA). Recombinant HB‐EGF, EGF, FGF and IGF were purchased from R&D Systems (Minneapolis, MN, USA).
Antibodies against ROS1, phospho‐specific (p) ROS1 (Tyr2274), EGFR, pEGFR (Tyr1068), mitogen‐activated protein kinase (MAPK), pMAPK (Thr202/Tyr204), AKT, pAKT (Ser473), AXL and glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH), and HRP‐conjugated antirabbit antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA).
2.3. MTT assay
Growth inhibition was determined using a modified MTT assay.12 Cells were plated on 96‐well plates at a density of 2000‐4000 cells per well and continuously exposed to each drug for 96 hours. Absorbance values were expressed as percentages relative to those of untreated cells. The drug concentration required to inhibit the growth of tumor cells by 50% (IC50) was used to evaluate the effect of the drug. Each assay was performed in triplicate or more.
2.4. Immunoblotting analysis and phosphor‐receptor tyrosine kinase array
Cells and frozen tissue were lysed in radioimmunoprecipitation assay buffer (1% Triton X‐100, .1% SDS, 50 mmol/L Tris‐HCl, pH 7.4, 150 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 10 mmol/L β‐glycerophosphate, 10 mmol/L NaF, 1 mmol/L sodium orthovanadate) containing protease inhibitor tablets (Roche Applied Sciences, Penzberg, Germany). Proteins were separated by SDS‐PAGE, transferred onto nitrocellulose membranes, and probed with the appropriate antibodies followed by detection with Enhanced Chemiluminescence Plus (GE Healthcare Biosciences, Pittsburgh, PA, USA). A Phospho‐Receptor Tyrosine Kinase Array Kit (ARY002; R&D Systems) was used according to the manufacturer's instructions.
2.5. ELISA
HB‐EGF levels were determined by Human HB‐EGF DuoSet ELISA (R&D Systems) according to the manufacturer's instructions.
2.6. Fluorescence in situ hybridization
FISH was performed on formalin‐fixed, paraffin‐embedded samples using a custom ROS1 break‐apart probe set in the laboratory of SRL (Tokyo, Japan).13 The probe set hybridizes with the neighboring 5′ telomeric (RP11‐48A22, labeled with SpectrumGreen) and 3′ centromeric (RP11‐1036C2, labeled with SpectrumOrange) sequence of ROS1. Cases with >15% split signals in cells were defined as FISH‐positive.
2.7. Quantitative RT‐PCR
RNA samples were prepared using an RNeasy Mini kit (Qiagen, Venlo, The Netherlands) according to the manufacturer's protocol, and cDNA was synthesized using SuperScript II reverse transcriptase (Invitrogen, Carlsbad, CA, USA). Quantitative PCR was performed using TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA, USA) with an ABI 5700 sequence detection system (Applied Biosystems) according to the manufacturer's protocol. Primers and probes used for each gene were as follows: HB‐EGF, Hs00181813_m1; EGF, Hs01099999_m1; transforming growth factor‐alpha (TGF‐α), Hs00608187_m1; epiregulin (EREG), Hs00914313_m1; amphiregulin (AR), Hs00950669_m1; and GAPDH, Hs99999905_m1. The relative expression of the target gene to GAPDH expression was calculated.
2.8. Peptide nucleic acid‐locked nucleic acid PCR clamp assay
Peptide nucleic acid (PNA)‐locked nucleic acid (LNA) PCR clamp assay for EGFR‐activating mutations was performed by LSI Medience (Tokyo, Japan).14 The assay detects the following types of EGFR mutations: EGFR exon18 (G719C, G719S, G719A), EGFR exon19 (E746‐A750del, L747‐A750del T751S, L747‐S752del P753S, L747‐E749del A750P, L747‐S752del E746V, S752‐I759del), EGFR exon21 (L858R, L861Q) and EGFR exon20 (T790M).
2.9. siRNA experiments
siRNA oligos targeted toward AXL, and negative control oligos (Dharmacon, Lafayette, CO, USA) were used at a concentration of 10 nmol/L and were transfected into cells with Lipofectamine RNAimax according to the manufacturer's protocol (Invitrogen).
2.10. Targeted RNA sequencing
RNA was extracted from each cell line using an RNeasy Mini Kit (Qiagen) according to the manufacturer's protocol. Aliquots of 1500 pg of genomic RNA per cell line were used for targeted RNA sequence analysis using a SureSelect RNA Human Kinome Kit (Agilent Technologies, Santa Clara, CA, USA), which targets 612 genes, including 517 protein kinases. Sequencing was performed using the MiSeq Sequencing System with a V2 Reagent Kit (Illumina, San Diego, CA, USA). Sequencing data were analyzed using the CLC Genomics Workbench (CLC Bio, Aarhus, Denmark). RNA samples of each cell line were analyzed 4 times, and the results were averaged.
2.11. Xenograft model
Female NOD.Cg‐PrkdcscidIl2rgtm1Sug/Jic mice (6 weeks of age) were purchased from the Central Institute for Experimental Animals (Kawasaki, Japan). All mice were provided with sterilized food and water and were housed in a barrier facility under a 12‐hour light/dark cycle. Cells (1 × 107) were injected subcutaneously into the back flanks of the mice. Four weeks after injection, the mice were randomly assigned to groups (3‐5 mice per group) and treated with vehicle (p.o.), crizotinib monotherapy (p.o., 100 mg/kg/d), gefitinib monotherapy (p.o., 5 mg/kg/d), cabozantinib monotherapy (p.o., 30 mg/kg/d), or gefitinib/cabozantinib combination therapy (p.o., 5/30 mg/kg/d). These agents were administered once a day, 5 times a week by gavage. Tumor volume (width2 × length/2) was determined every other day. The administration period for each agent was 21 days, and animals were followed up for an additional 28 days. All experiments involving animals were performed with the approval of the Institutional Animal Care and Research Advisory Committee of the Department of Animal Resources, Okayama University Advanced Science Research.
2.12. Statistical analysis
All experiments were performed 3 times, and statistical analyses were performed using STATA software (ver. 13; StataCorp, College Station, TX, USA). Group differences were compared using a 2‐tailed unpaired t test. In the box plots, the center line is the median and whiskers show minimum to maximum values. In all analyses, P < .05 was considered to indicate statistical significance.
3. RESULTS
3.1. Epidermal growth factor receptor activation in crizotinib‐resistant HCC78R cells
To explore the mechanisms of resistance, we used HCC78 cells harboring the SLC34A2‐ROS1 fusion gene as a cell line‐based resistance model. Crizotinib inhibited the proliferation of HCC78 cells at the nanomolar level, and the compound consistently inhibited the phosphorylation of ROS1 protein or the downstream signaling protein in parental HCC78 cells (Figures 1A,D). Resistant cell lines were established from HCC78 cells by continuous exposure to crizotinib for 4 months and were then designated as HCC78R (Figures 1A and S1A). FISH analysis indicated that the resistant cells maintained ROS1 fusion genes to the same extent as the parental HCC78 cells (Figure 1B). However, the RNA expression of ROS1 decreased in HCC78R cells (Figures 1C and S1C). The expression of ROS1 protein also decreased in HCC78R cells compared with that in the parental HCC78 cells (Figure 1D).
Figure 1.
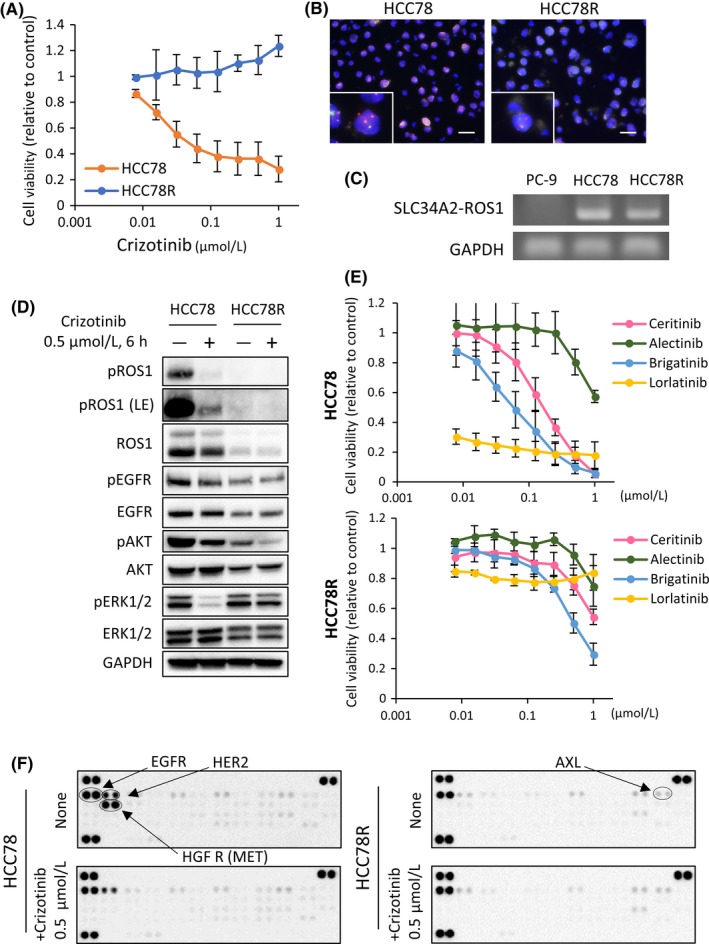
Establishment of the crizotinib‐resistant cell line, HCC78R. A, Cell proliferation assay of HCC78 and HCC78R cells treated with the indicated concentrations of crizotinib. Error bars: SD. All experiments were performed in triplicate. B, FISH analysis of ROS1 fusion gene in HCC78 and HCC78R cells. Red probes hybridized to the 5′ region of ROS1 and green probes to the 3′ region. In the presence of ROS1 rearrangement, the 2 colors are observed separately. HCC78R cells maintained ROS1 fusion genes (98% ROS1 fusion FISH positive) to the same extent as the parental HCC78 cells (94% ROS1 fusion FISH positive) C, Detection of SLC34A2‐ROS1. Complementary DNA (cDNA) derived from PC‐9 (negative control), HCC78 and HCC78R were examined by PCR. D,F. Immunoblots and phospho‐receptor tyrosine kinase (RTK) arrays in HCC78 and HCC78R cells. These cells were cultured in normal medium for 4 d, after which both types of cells were exposed to .5 μmol/L crizotinib for 6 h. E, Cell proliferation assay of HCC78 and HCC78R cells treated with the indicated concentrations of ALK/ROS1 inhibitors, ceritinib, alectinib, brigatinib and lorlatinib. Error bars: SD. All experiments were performed in triplicate
Next, we assessed the effect of next‐generation ALK inhibitors, including ceritinib, brigatinib, alectinib and lorlatinib, on each of the cell lines in vitro. With the exception of alectinib, all the inhibitors inhibited the growth of parental HCC78 cells at the nanomolar level, while these compounds showed limited effects in HCC78R cells (Figures 1E, S4A and Table 1). The ALK/EGFR inhibitor brigatinib exhibited a relatively good inhibitory effect in the resistant cells (Figure 1E, Table 1). None of the HCC78R cell lines possessed acquired resistance mutations, such as ROS1 S1986Y, S1986F, D2033N or G2032R, in the ROS1 kinase domain8, 9, 10, 11 (Figures S1D, S5A and B). Morphological examination yielded no obvious evidence of epithelial‐mesenchymal transition and western blotting experiments confirmed that the expression patterns of E‐cadherin and vimentin were similar between parental HCC78 and HCC78R cells (Figure S1B).
Table 1.
IC50 values on HCC78 and HCC78R cells
| IC50 (μmol/L) values ± SD | ||
|---|---|---|
| Drug | HCC78 | HCC78R |
| Crizotinib | .066 ± .045 | >1.0 |
| Ceritinib | .162 ± .042 | >1.0 |
| Alectinib | >1.0 | >1.0 |
| Brigatinib | .098 ± .069 | .525 ± .112 |
| Lorlatinib | <.01 | >1.0 |
The antiproliferative effects were evaluated using the MTT assay. Data are presented as the mean ± SD from 3 independent experiments.
Subsequently, we comprehensively assessed the phosphorylation of receptor tyrosine kinases (RTK) in HCC78 cells and HCC78R cells using an RTK array. The results indicated that EGFR phosphorylation was relatively well maintained in HCC78R cells under conditions of crizotinib exposure compared with phosphorylation in the parental HCC78 cells (Figures 1F and S1E). The levels of ERBB2 and MET phosphorylation markedly decreased in HCC78R cells compared with that in parental HCC78 cells. In contrast, phosphorylation of AXL increased in HCC78R cells compared with that in parental HCC78 cells (Figures 1F and S1E). Western blotting analysis showed that phosphorylation of EGFR and the downstream signaling protein, ERK1/2, were maintained in HCC78R cells under crizotinib exposure (Figure 1D). Taken together, these observations suggested that EGFR or AXL may play a role in the mechanism of resistance in HCC78R cells.
3.2. Effects of epidermal growth factor receptor inhibitors in HCC78R
We performed an MTT assay using the EGFR‐TKI (gefitinib) in HCC78R cells to examine whether activation of EGFR is responsible for crizotinib resistance. Gefitinib monotherapy had little effect on cell proliferation but co‐treatment with gefitinib and crizotinib showed a superior effect of inhibiting the proliferation of parental HCC78 cells (Figure S4B). The results indicated that the EGFR pathway played an important role in intrinsic sensitivity to crizotinib in HCC78 cells. This was consistent with previous reports.15, 16 In contrast to parental HCC78 cells, crizotinib showed no inhibitory effect (Figure 1A), but gefitinib inhibited the proliferation of HCC78R cells (IC50 ± SD: .136 ± .022 μmol/L) (Figure 2A). Next, we assessed the effects of EGFR‐TKI on the EGFR signaling pathway in each cell line. Phosphorylation of EGFR and its downstream signaling protein, ERK1/2, was not suppressed in HCC78R cells treated with crizotinib, while these signaling pathways were suppressed upon gefitinib exposure (Figure 2B). Adding gefitinib to crizotinib monotherapy showed a great inhibitory effect on the proliferation of HCC78R cells (Figure S4C). In contrast, combination therapy with gefitinib plus crizotinib was not superior to gefitinib monotherapy with regard to the proliferation of HCC78R (Figure 2C). These results suggested that the resistant HCC78R cells were no longer addicted to the oncogenic ROS1 fusion protein but were instead addicted to EGFR. To confirm the effect of EGFR‐TKI, we performed the same experiments using other EGFR‐TKI, erlotinib and afatinib. As expected, similar results were observed with both of these agents in HCC78R cells (Figures S2A,B). Third, we examined the inhibitory effect of the anti‐EGFR antibody cetuximab, in vitro. Interestingly, cetuximab inhibited the proliferation of resistant cell lines to a significantly greater extent than that of the parental cells in vitro (mean IC50 ± SD > 20 μg/mL for HCC78 cells and 11.2 ± 1.33 μg/mL for HCC78R cells (Figure 2D). Western blotting analysis showed that 1 or 5 μg/mL of cetuximab inhibited the phosphorylation of EGFR and its downstream signaling proteins (Figure 2E). Taken together, these observations suggested that the EGFR signaling pathway played an important role in the mechanism of resistance in HCC78R cells.
Figure 2.
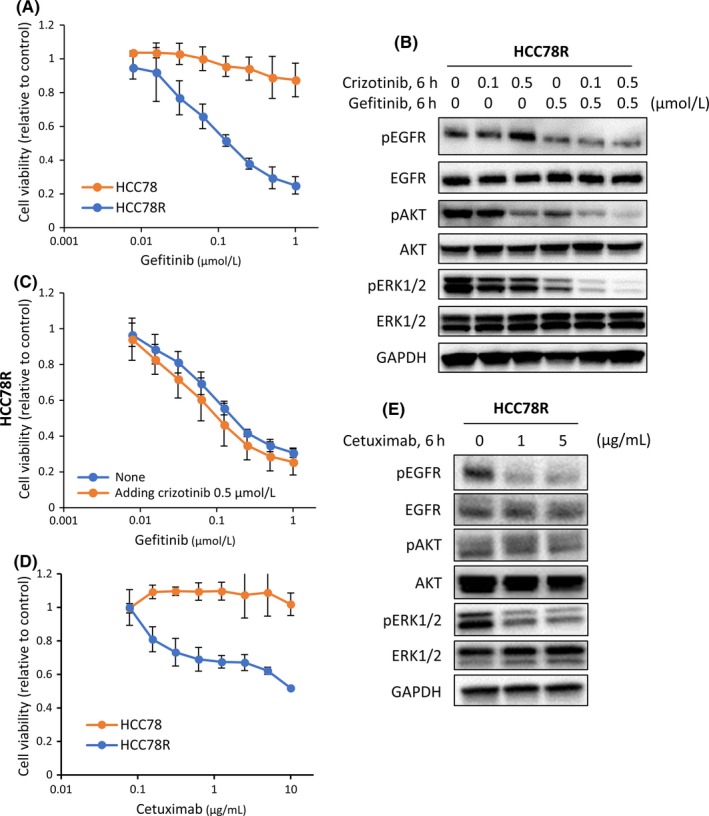
Effects of epidermal growth factor receptor (EGFR) inhibitor treatment in HCC78R cells. A, Cell proliferation assays in HCC78 and HCC78R cells treated with the indicated concentrations of gefitinib. Error bars: SD. All experiments were performed in triplicate. B, Effects of combined treatment with crizotinib and gefitinib on the EGFR signaling pathway in HCC78R cells. Cells were exposed to gefitinib at .5 μmol/L for 6 h, and crizotinib at .1 and .5 μmol/L for 6 h. C, Inhibitory effects of crizotinib and gefitinib on HCC78R cell proliferation upon the addition of .5 μmol/L crizotinib. Error bars: SD. All experiments were performed in triplicate. D, Cell proliferation assays in HCC78 and HCC78R cells treated with the indicated concentrations of cetuximab. Error bars: SD. All experiments were performed in triplicate. E, Effects of cetuximab on the EGFR pathway in HCC78R cells. Cells were exposed to cetuximab at 1 and 5 μg/mL for 6 h
3.3. Heparin‐binding epidermal growth factor‐like growth factor/epidermal growth factor receptor axis signaling confers resistance to crizotinib in lung cancer cells harboring ROS1 fusion genes
Next, we investigated the mechanisms of EGFR activation in HCC78R cells. The level of EGFR protein expression was not significantly increased in HCC78R cells (Figure 1D). Activated EGFR mutations were not detected by PNA‐LNA‐PCR clamp assay14 in HCC78R cells (data not shown). Therefore, we assessed the level of EGFR ligand gene expression in HCC78R cells. We screened for gene expression of EGFR ligands, such as epidermal growth factor receptor (EGF), heparin‐binding EGF‐like growth factor (HB‐EGF), transforming growth factor‐alpha (TGF‐α), epiregulin (EREG) and amphiregulin (AR) by RT‐PCR in HCC78 cells and HCC78R cells. Interestingly, the expression level of HB‐EGF was 2.5‐fold higher in HCC78R cells than in HCC78 cells, whereas the other ligands showed similar expression levels in both HCC78R and HCC78 cells (Figure 3A). Consistent with the results of RT‐PCR analyses, the expression of HB‐EGF protein in the medium from culture dishes of HCC78R cells was approximately 10 times that of the parental HCC78 cells (Figure 3B). As expected, adding recombinant HB‐EGF to the culture medium significantly reduced the inhibitory effect of crizotinib on the proliferation of parental HCC78 cells (Figure 3C). Western blotting analysis indicated that the expression of total EGFR protein decreased, but the phosphorylation of EGFR and its downstream signaling protein, ERK1/2, was maintained under crizotinib exposure by the addition of HB‐EGF to HCC78 cells (Figure 3D). We also examined the effect of the conditioned medium prepared by mixing equal parts fresh medium and the supernatant of HCC78R cells. As expected, the conditioned medium rendered the parental HCC78 cells resistant to crizotinib in vitro (Figure S3E). In addition, we investigated the impact of other growth factors on the sensitivity of HCC78 cells to crizotinib. Consistent with the results for HB‐EGF (Figure 3C), EGF (100 ng/mL) stimulation rendered HCC78 cells resistant to crizotinib in vitro (Figure S3A). Similar to the effects of HB‐EGF, EGF maintained phosphorylation of EGFR and ERK1/2 in HCC78 treated with crizotinib (Figure S3B). In contrast to the results for EGFR ligand, neither insulin‐like growth factor (IGF) nor fibroblast growth factor (FGF) rescued the proliferation of HCC78 cells during crizotinib exposure (Figure S3F). Finally, another lung cancer cell line, ABC‐20, harboring the CD74‐ROS1 fusion gene (Figure S3C) was investigated to reconfirm the roles of HB‐EGF and EGF. Similar to HCC78, HB‐EGF, or EGF, stimulation rendered ABC‐20 cells resistant to crizotinib (Figures 3E and S3D), and the phosphorylation of both EGFR and ERK1/2 was maintained in ABC‐20 cells under crizotinib treatment (Figure 3F).
Figure 3.
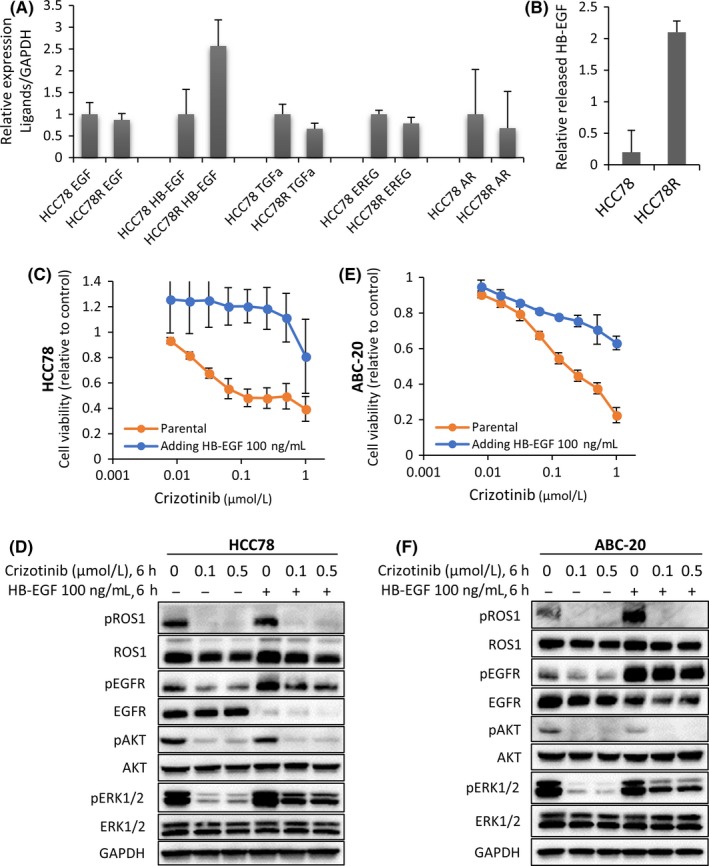
The heparin‐binding epidermal growth factor‐like growth factor (HB‐EGF)/epidermal growth factor receptor (EGFR) axis confers resistance to crizotinib in lung cancer cells with ROS1 fusion genes. A, Quantitative PCR analysis of the EGFR ligand family was performed in HCC78 and HCC78R cells. The relative mRNA expression levels were calculated as ratios to glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) expression. Data are presented as the means ± SD of 3 independent experiments. B, The released HB‐EGF proteins were measured by ELISA of the conditioned medium from HCC78 cells and HCC78R cells. Error bars: SD. C,E, Inhibitory effect of crizotinib on the proliferation of HCC78 and ABC‐20 cells stimulated with recombinant HB‐EGF (100 ng/mL, 6 h). Error bars: SD. All experiments were performed in triplicate. D,F, Inhibitory effects of crizotinib on the EGFR signaling pathway in HCC78 and ABC‐20 cells stimulated with recombinant HB‐EGF. Cells were exposed to recombinant HB‐EGF at 100 ng/mL for 6 h, and to crizotinib at .1 and .5 μmol/L for 6 h
3.4. AXL upregulation and resistance to crizotinib in HCC78R cells
To explore the mechanisms of resistance in more detail, we performed RNA‐targeted sequence analysis using next‐generation sequencing in HCC78 and HCC78R cells. The samples were collected from dishes in which HCC78 cells (n = 4) or HCC78R cells (n = 4) were independently cultured. The mRNA expression level of 612 human kinome genes and kinase‐related genes was comprehensively compared (Figure 4A). The raw data are shown in Table S1. The mRNA expression of 9 genes, PLK1 and 2, PBK, AURKA, AURKB, TTK, CDK1, NEK2 and AXL, showed changes of more than 8‐fold between HCC78 and HCC78R cells (Figure 4A). In contrast, the mRNA levels of KDR, INSR, SBK1, EPHA4 and TNIK decreased 8‐fold between the cell lines (Figure S6). Among these genes, we focused on AXL, because the read frequency of AXL was among the highest and phosphorylation of AXL was increased in HCC78R (Figures 1F and S1E). Consistent with the NGS data, western blotting analysis showed that expression of the AXL protein significantly increased (Figure 4B). The effects of 2 clinically relevant AXL inhibitors,17 cabozantinib18 and gilteritinib,19 were assessed in HCC78R cells. Compared with the effects of the EGFR‐TKI gefitinib, the effects of cabozantinib and gilteritinib were relatively limited in HCC78R cells (Figure 5A). Furthermore, the combination of crizotinib with cabozantinib showed an antagonistic effect. In contrast, the combination of gefitinib with cabozantinib or gilteritinib inhibited cell proliferation to a significantly greater extent than gefitinib monotherapy (Figure 5A). Consistent with these observations, combination therapy more strongly inhibited phosphorylation of ERK1/2 than monotherapy (Figure 5B). In addition, we assessed the effect of combining gefitinib with knockdown of AXL using siRNA. As expected, the combination showed a superior inhibitory effect on cell proliferation than AXL inhibition alone in HCC78R cells (Figure S7). Finally, we examined the effects of combination therapy in vivo. The effects of gefitinib, cabozantinib and combination treatment with gefitinib and cabozantinib were assessed in xenograft mouse tumors of HCC78R cells. The xenograft tumors were treated with vehicle (control), crizotinib (100 mg/kg), gefitinib (5 mg/kg), cabozantinib (30 mg/kg) or a combination of gefitinib (5 mg/kg) and cabozantinib (30 mg/kg). Xenograft tumors treated with vehicle or crizotinib showed similar growth (Figure 5C), while cabozantinib showed a moderate effect on tumor growth in vivo. In contrast, gefitinib led to a significant inhibition of tumors in the mice. Combination therapy with gefitinib and cabozantinib also showed a better inhibitory effect than each of the monotherapies alone in mouse tumors bearing HCC78R cells, although this effect was not statistically significant (Figure 5C). No differences in body weight were observed among any of the mouse groups (Figure 5C).
Figure 4.
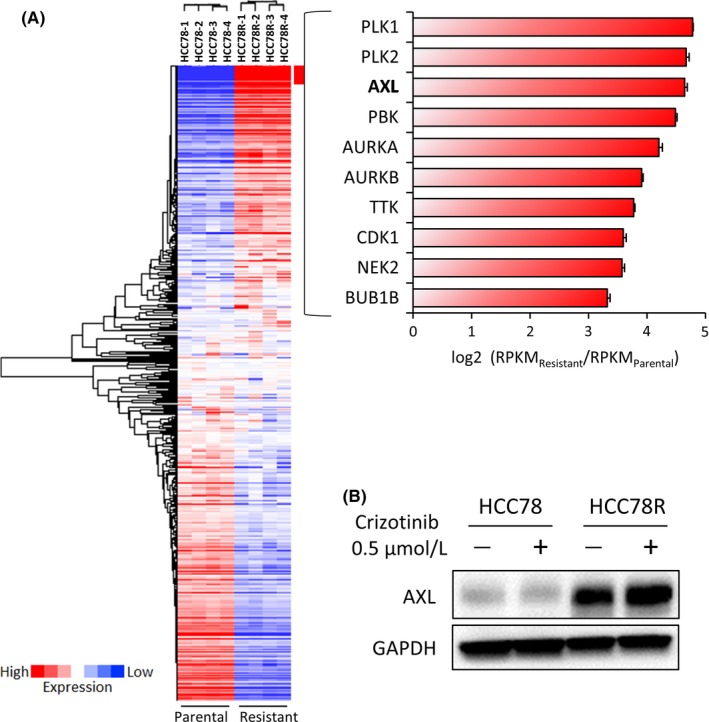
AXL upregulation in HCC78R cells. A, Heat map of targeted RNA sequencing analysis of 612 kinase and kinase‐related genes from parental and resistant cells. RNA samples for each cell line were analyzed in 4 independent experiments. The gene expression profile of HCC78R cells was compared with that of HCC78 cells. The top 10 upregulated genes are listed. B, Immunoblots of HCC78 and HCC78R cells. These cells were cultured in normal medium for 4 d, after which both cell types were exposed to crizotinib at .5 μmol/L for 6 h
Figure 5.
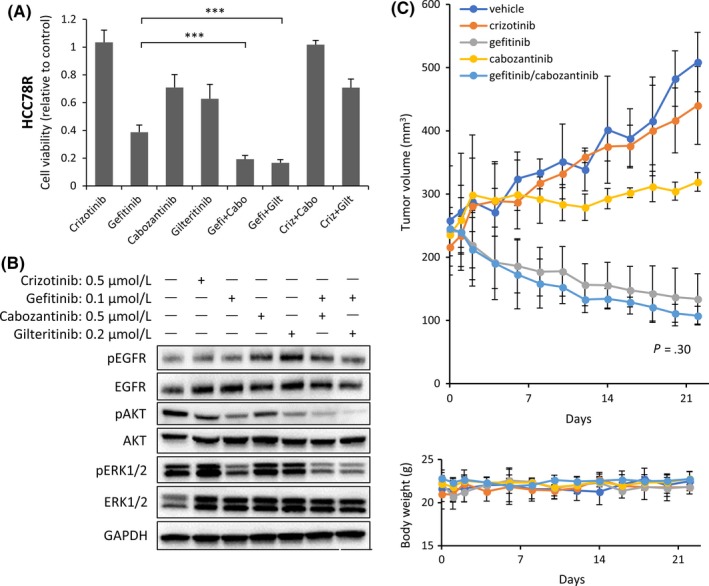
Beneficial effects of combination therapy with AXL inhibitor and epidermal growth factor receptor (EGFR) inhibitor in HCC78R cells. A, Inhibitory effects of combining an AXL inhibitor, cabozantinib or gilteritinib, with gefitinib on the proliferation of HCC78R cells. Cells were exposed to crizotinib or cabozantinib at .5 μmol/L, to gilteritinib at .2 μmol/L, and to gefitinib at .1 μmol/L, all for 96 h. Data are presented as the means ± SD of 3 independent experiments. ***P < .001. Cabo, cabozantinib; Criz, crizotinib; Gefi, gefitinib; Gilt, gilteritinib. B, Effects of combined treatment with AXL inhibitors and gefitinib on EGFR pathway signaling in HCC78R cells. Cells were exposed to all drugs for 6 h. C, Effects of combined treatment with cabozantinib and gefitinib on tumor growth and body weight in HCC78R cell xenograft models. Mice were treated with 100 mg/kg/d crizotinib or 5 mg/kg/d gefitinib, 30 mg/kg/d cabozantinib, or 5 mg/kg/d gefitinib with 30 mg/kg/d cabozantinib. Statistical analysis of the data from the vehicle and treated groups was performed on day 14. Tumor volume (top) and body weight (bottom) curves
4. DISCUSSION
The development of resistance to targeted therapy is critical for patients with lung cancers harboring driver oncogenes. We demonstrated that HB‐EGF/EGFR and AXL play roles in the mechanism of resistance of lung cancers harboring ROS1 fusions treated with crizotinib. Using a cell line‐based model, we also found that dual inhibition of EGFR and AXL has the potential to overcome crizotinib resistance in vitro and in vivo. These findings could be clinically relevant, as EGFR inhibitors and AXL inhibitors17 have been approved for clinical use, and the efficacy and safety of combination therapy with cabozantinib and erlotinib have been reported in clinical trials.20, 21
Up to 50% of lung tumors harboring ROS1 fusion genes treated with crizotinib develop a secondary mutation, ROS1 G2032R, in the kinase domain.8 Several next‐generation ROS1 inhibitors have been developed for patients with lung cancers harboring ROS1 fusion genes.22 Preclinical studies showed that lorlatinib, foretinib and cabozantinib can inhibit drug resistance.23, 24, 25 The MET/VEGFR2/RET/ROS1/AXL inhibitor, cabozantinib, has already been approved for use in patients with advanced renal cell carcinoma, and clinical trials have been conducted for lung cancer with RET gene fusion26 or ROS1 fusion genes (NCT01639508). Therefore, cabozantinib could be a clinically relevant drug to overcome crizotinib resistance in lung cancers containing ROS1 fusion genes.
In addition to acquired mutations in the ROS1 kinase domain, activation of bypass track signaling, such as the EGFR, HER2, KIT, β‐catenin or BRAF pathways, was reported as a mechanism of resistance against crizotinib.9, 27, 28, 29, 30 Several preclinical studies, including our study, showed that stimulation of EGFR by EGF or HB‐EGF resulted in resistance in multiple lung cancer cell lines harboring ROS1 fusions; ie, HCC78 with SLC34A2‐ROS1, CUTO‐2, and ABC‐20 with CD74‐ROS1. In addition, a recent preclinical study showed that inhibiting oncogenic fusion proteins, such as ALK, ROS1 and RET, induced a shift in the adaptor protein, GRB2, from the fusion oncoprotein to EGFR, resulting in activation of bypass track EGFR signaling.16 We also showed that IGF and FGF did not affect crizotinib sensitivity in HCC78 cells (Figure S3F). Therefore, the EGFR pathway may be especially important for persister cells to survive drug exposure in lung cancer cells harboring ROS1 fusion genes.
In this study, we found that the AXL RNA expression level significantly increased in crizotinib‐resistant HCC78R cells. AXL is thought to play a role in acquired resistance to oncoprotein inhibitors,31, 32, 33 but its role regarding crizotinib resistance has not yet been reported. We examined the effects of 2 clinically relevant AXL inhibitors in HCC78R cells. Monotherapy with AXL inhibitors showed only moderate effects, but combining AXL inhibitors with EGFR‐TKI resulted in a superior inhibitory effect compared with monotherapies. This suggested that AXL plays some role, in concert with EGFR, in resistance to crizotinib.
“Oncogene swap” has been reported as a resistance mechanism in lung cancer with EGFR mutations.34 In this situation, the activation of other oncogenes acts not as a “bypass”, but rather as a “main” oncoprotein. In our study, HCC78R cells maintained ROS1 fusion genes (Figure 1C) but their RNA or protein expression decreased in HCC78R cells due to unknown mechanisms compared with that in parental HCC78R cells (Figures 1D, S1C and Table S1). Furthermore, the dependency on ROS1 protein seems to be almost lost (Figures 1A, 2C and S4C). Taken together, “oncogene swap” may have occurred in HCC78R cells.
Alternative treatment strategies are required to improve the management of lung tumors harboring ROS1 fusion genes. Considering the rarity of these lesions, it is difficult to compare each of the ROS1 inhibitors, and, therefore, a preclinical study may be useful to develop a rationale for new treatment strategies. Although it is necessary to consider the limitations of this study, in which we used only a cell line‐based model and performed no clinical sample analysis, our preclinical observations could provide a rationale for the development of a new treatment strategy for lung cancer with ROS1 fusion genes. Given the importance of the EGFR pathway and the inhibitory profile of cabozantinib, which can inhibit not only AXL but also wild‐type ROS1 and ROS1 G2032R, combination treatment with cabozantinib and EGFR‐TKI may be a reasonable option for lung cancers with ROS1 fusion genes. The clinical assessment of this combination therapy is worth considering for patients with lung cancers harboring ROS1 fusion genes.
CONFLICTS OF INTEREST
All authors declare no conflict of interests regarding this study.
Supporting information
ACKNOWLEDGMENTS
We are grateful to Hiromi Nakashima and Kyoko Maeda for the technical support. We also thank Dr Takehiro Matsubara (Division of Biobank, Center for Comprehensive Genomic Medicine, Okayama University Hospital) for analyzing next‐generation sequencing data, and our laboratory colleagues for the useful discussions. This work received a poster award, ESMO 2014 (Madrid, Spain).
Kato Y, Ninomiya K, Ohashi K, et al. Combined effect of cabozantinib and gefitinib in crizotinib‐resistant lung tumors harboring ROS1 fusions. Cancer Sci. 2018;109:3149‐3158. 10.1111/cas.13752
Kato and Ninomiya contributed equally to this project.
REFERENCES
- 1. Kris MG, Johnson BE, Berry LD, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA. 2014;311(19):1998‐2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ohashi K, Maruvka YE, Michor F, Pao W. Epidermal growth factor receptor tyrosine kinase inhibitor‐resistant disease. J Clin Oncol. 2013;31(8):1070‐1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Takeuchi K, Soda M, Togashi Y, et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med. 2012;18(3):378‐381. [DOI] [PubMed] [Google Scholar]
- 4. Bergethon K, Shaw AT, Ou SH, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. 2012;30(8):863‐870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ou SH, Tan J, Yen Y, Soo RA. ROS1 as a ‘druggable’ receptor tyrosine kinase: lessons learned from inhibiting the ALK pathway. Expert Rev Anticancer Ther. 2012;12(4):447‐456. [DOI] [PubMed] [Google Scholar]
- 6. Shaw AT, Ou SH, Bang YJ, et al. Crizotinib in ROS1‐rearranged non‐small‐cell lung cancer. N Engl J Med. 2014;371(21):1963‐1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wu YL, Yang JC, Kim DW, et al. Phase II study of Crizotinib in East Asian patients with ROS1‐positive advanced non‐small‐cell lung cancer. J Clin Oncol. 2018;36(14):1405‐1411. [DOI] [PubMed] [Google Scholar]
- 8. Awad MM, Katayama R, McTigue M, et al. Acquired resistance to Crizotinib from a mutation in CD74‐ROS1. N Engl J Med. 2013;368(25):2395‐2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Song A, Kim TM, Kim DW, et al. Molecular changes associated with acquired resistance to Crizotinib in ROS1‐rearranged non‐small cell lung cancer. Clin Cancer Res. 2015;21(10):2379‐2387. [DOI] [PubMed] [Google Scholar]
- 10. Drilon A, Somwar R, Wagner JP, et al. A novel crizotinib‐resistant solvent‐front mutation responsive to cabozantinib therapy in a patient with ROS1‐rearranged lung cancer. Clin Cancer Res. 2016;22(10):2351‐2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Facchinetti F, Loriot Y, Kuo MS, et al. Crizotinib‐resistant ROS1 mutations reveal a predictive kinase inhibitor sensitivity model for ROS1‐ and ALK‐rearranged lung cancers. Clin Cancer Res. 2016;22(24):5983‐5991. [DOI] [PubMed] [Google Scholar]
- 12. Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1–2):55‐63. [DOI] [PubMed] [Google Scholar]
- 13. Yoshida A, Tsuta K, Wakai S, et al. Immunohistochemical detection of ROS1 is useful for identifying ROS1 rearrangements in lung cancers. Mod Pathol. 2014;27(5):711‐720. [DOI] [PubMed] [Google Scholar]
- 14. Nagai Y, Miyazawa H, Huqun Tanaka T, et al. Genetic heterogeneity of the epidermal growth factor receptor in non‐small cell lung cancer cell lines revealed by a rapid and sensitive detection system, the peptide nucleic acid‐locked nucleic acid PCR clamp. Cancer Res. 2005;65(16):7276‐7282. [DOI] [PubMed] [Google Scholar]
- 15. Davies KD, Le AT, Theodoro MF, et al. Identifying and targeting ROS1 gene fusions in non‐small cell lung cancer. Clin Cancer Res. 2012;18(17):4570‐4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vaishnavi A, Schubert L, Rix U, et al. EGFR mediates responses to small molecule drugs targeting oncogenic fusion kinases. Cancer Res. 2017;77(13):3551‐3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Myers SH, Brunton VG, Unciti‐Broceta A. AXL inhibitors in cancer: a medicinal chemistry perspective. J Med Chem. 2016;59(8):3593‐3608. [DOI] [PubMed] [Google Scholar]
- 18. Yakes FM, Chen J, Tan J, et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther. 2011;10(12):2298‐2308. [DOI] [PubMed] [Google Scholar]
- 19. Lee LY, Hernandez D, Rajkhowa T, et al. Preclinical studies of gilteritinib, a next‐generation FLT3 inhibitor. Blood. 2017;129(2):257‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Neal JW, Dahlberg SE, Wakelee HA, et al. Erlotinib, cabozantinib, or erlotinib plus cabozantinib as second‐line or third‐line treatment of patients with EGFR wild‐type advanced non‐small‐cell lung cancer (ECOG‐ACRIN 1512): a randomised, controlled, open‐label, multicentre, phase 2 trial. Lancet Oncol. 2016;17(12):1661‐1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wakelee HA, Gettinger S, Engelman J, et al. A phase Ib/II study of cabozantinib (XL184) with or without erlotinib in patients with non‐small cell lung cancer. Cancer Chemother Pharmacol. 2017;79(5):923‐932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dagogo‐Jack I, Shaw AT. Expanding the roster of ROS1 inhibitors. J Clin Oncol. 2017;35(23):2595‐2597. [DOI] [PubMed] [Google Scholar]
- 23. Zou HY, Li Q, Engstrom LD, et al. PF‐06463922 is a potent and selective next‐generation ROS1/ALK inhibitor capable of blocking crizotinib‐resistant ROS1 mutations. Proc Natl Acad Sci USA. 2015;112(11):3493‐3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Davare MA, Saborowski A, Eide CA, et al. Foretinib is a potent inhibitor of oncogenic ROS1 fusion proteins. Proc Natl Acad Sci USA. 2013;110(48):19519‐19524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Katayama R, Kobayashi Y, Friboulet L, et al. Cabozantinib overcomes crizotinib resistance in ROS1 fusion‐positive cancer. Clin Cancer Res. 2015;21(1):166‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Drilon A, Rekhtman N, Arcila M, et al. Cabozantinib in patients with advanced RET‐rearranged non‐small‐cell lung cancer: an open‐label, single‐centre, phase 2, single‐arm trial. Lancet Oncol. 2016;17(12):1653‐1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Davies KD, Mahale S, Astling DP, et al. Resistance to ROS1 inhibition mediated by EGFR pathway activation in non‐small cell lung cancer. PLoS ONE. 2013;8(12):e82236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dziadziuszko R, Le AT, Wrona A, et al. An activating KIT mutation induces crizotinib resistance in ROS1‐positive lung cancer. J Thorac Oncol. 2016;11(8):1273‐1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Watanabe J, Furuya N, Fujiwara Y. Appearance of a BRAF mutation conferring resistance to crizotinib in non‐small cell lung cancer harboring oncogenic ROS1 fusion. J Thorac Oncol. 2018;13(4):e66‐e69. [DOI] [PubMed] [Google Scholar]
- 30. McCoach CE, Le AT, Gowan K, et al. Resistance mechanisms to targeted therapies in ROS1+ and ALK+ non‐small cell lung cancer. Clin Cancer Res. 2018;24(14):3334‐3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Scaltriti M, Elkabets M, Baselga J. Molecular pathways: AXL, a membrane receptor mediator of resistance to therapy. Clin Cancer Res. 2016;22(6):1313‐1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang Z, Lee JC, Lin L, et al. Activation of the AXL kinase causes resistance to EGFR‐targeted therapy in lung cancer. Nat Genet. 2012;44(8):852‐860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nelson‐Taylor SK, Le AT, Yoo M, et al. Resistance to RET‐inhibition in RET‐rearranged NSCLC is mediated by reactivation of RAS/MAPK signaling. Mol Cancer Ther. 2017;16(8):1623‐1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mizuuchi H, Suda K, Murakami I, et al. Oncogene swap as a novel mechanism of acquired resistance to epidermal growth factor receptor‐tyrosine kinase inhibitor in lung cancer. Cancer Sci. 2016;107(4):461‐468. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials