

Abstract
This open‐label multicenter phase 1 study evaluated the safety, tolerability, efficacy, pharmacokinetics and pharmacodynamics of weekly carfilzomib and dexamethasone (Cd) in Japanese patients with relapsed or refractory multiple myeloma (RRMM). Carfilzomib was administered by 30‐minute intravenous infusion on Days 1, 8 and 15 in a 28‐day cycle starting at 20 mg/m2 on Day 1/Cycle 1 and 70 mg/m2 thereafter until progressive disease or unacceptable toxicity. Dexamethasone 40 mg was administered on Days 1, 8, 15 and 22 in Cycles 1‐9 and on Days 1, 8 and 15 thereafter. Six patients were enrolled between March 2015 and June 2015. Patients had received a median of 4.5 (range, 4‐8) prior regimens; all patients had previous therapies with bortezomib and immunomodulatory drugs. Of the 6 patients, 1 had a dose‐limiting toxicity (DLT), and tolerability was confirmed. The DLT was grade 3 thrombotic microangiopathy, which was considered serious and occurred on Day 11/Cycle 1. All 6 patients (100%) experienced at least 1 grade ≥3 adverse event (AE). Two patients (33.3%) experienced AE (also considered adverse drug reactions) leading to study discontinuation: thrombotic microangiopathy (Day 11/Cycle 1) and thrombotic thrombocytopenic purpura (Day 6/Cycle 2). The overall response rate was 83.3% (95% confidence interval, 43.6‐97.0). The weekly Cd regimen at a carfilzomib dose of 20/70 mg/m2 was well‐tolerated among Japanese patients with RRMM. Our results could be the basis for the further development of carfilzomib treatment considering safety profiles including microangiopathy‐related events and efficacy.
Keywords: carfilzomib, Japanese, multiple myeloma, phase 1, weekly
1. INTRODUCTION
Given the advances in the treatment of multiple myeloma (MM) in the past decade,1 disease outcomes, including survival rates, have improved for patients with MM.2 However, relapse is still common among these patients and MM remains incurable; thus, new therapeutic approaches are needed.1
Carfilzomib, an epoxyketone proteasome inhibitor, binds selectively and irreversibly to the constitutive proteasome and immunoproteasome. A recent phase 3 (ASPIRE) study evaluated the combination of carfilzomib, lenalidomide and dexamethasone (CLd) and showed that this combination resulted in a 31% decrease in the risk of disease progression or death compared with lenalidomide and dexamethasone, and an increase of 8.7 months in the median progression‐free survival (PFS) of patients with relapsed MM.3 In a phase 3, head‐to‐head comparison study with bortezomib (ENDEAVOR), which included patients from Europe, North America, South America and the Asia–Pacific region, carfilzomib and dexamethasone (Cd) provided a significant and clinically meaningful reduction in the risk of death compared with bortezomib and dexamethasone.4
Both the CLd and Cd twice‐weekly regimens have been approved in the USA, Europe and Japan based on the results of these phase 3 studies. Furthermore, studies with carfilzomib monotherapy,5 Cd6 and CLd7 have been conducted in Japan. Although the efficacy of twice‐weekly carfilzomib, as monotherapy and in combination with other drugs (Ld and dexamethasone), has been demonstrated, the twice‐weekly dosing schedule results in some inconveniences for patients. Therefore, a more convenient dosing schedule can improve treatment compliance, and weekly treatment alternatives are highly sought after.
Recently, a phase 1/2 study (CHAMPION‐1) in the USA evaluated weekly Cd in relapsed or refractory MM (RRMM) patients.8 In that study, the overall response rate (ORR) at 70 mg/m2 was 77%, and the median PFS was 12.6 months, indicating that further assessment of the weekly dosing was warranted. The objectives of this study were to evaluate the safety, tolerability, efficacy, pharmacokinetics (PK) and pharmacodynamics of weekly Cd in Japanese patients with RRMM.
2. MATERIALS AND METHODS
2.1. Study design and ethical considerations
This was an open‐label, multicenter, phase 1 study conducted in 4 centers in Japan. The study started in December 2012 and is currently ongoing. For the weekly Cd cohort, the first patient was enrolled in March 2015. The cutoff date for the present analysis was 30 November 2016. The Institutional Review Board of each participating institution within which the work was undertaken approved the protocol for the present study. All study procedures conformed to the provisions of the Declaration of Helsinki (as revised in Fortaleza, Brazil, October 2013), the Pharmaceutical and Medical Device Act, and the Japanese Good Clinical Practice (GCP) Guidelines (including Ministry of Health and Welfare Ordinance No. 28) and their amended ordinances. All participants provided written informed consent to participate in the study. This clinical trial was registered at the Japan Pharmaceutical Information Center (JAPIC) under the identifier JapicCTI‐122020.
2.2. Participants
The inclusion criteria were as follows: age ≥20 years; Eastern Cooperative Oncology Group Performance Status of 0‐2; symptomatic MM with measurable disease (serum M‐protein level ≥0.5 g/dL or urine M‐protein level ≥200 mg/24 hours); at least 2 previous treatments for MM, including bortezomib and immunomodulatory agents (both refractory and non‐refractory patients could be enrolled); response to at least 1 previous MM treatment (≥25% reduction in M‐protein); and adequate hematologic, hepatic and renal function at screening, evidenced by absolute white blood cell count of ≥2000/mm3, neutrophil count of ≥1000/mm3, platelet count of ≥50 000/mm3, hemoglobin of ≥8.0 g/dL, aspartate aminotransferase and alanine aminotransferase of less than 3 times the upper limits of the normal values, serum bilirubin of less than 2 times the upper limit of the normal value, estimated creatinine clearance value by the Cockcroft–Gault formula of ≥30 mL/min, and serum creatinine of ≤2.0 mg/dL.
The major exclusion criteria were as follows: patients previously treated with carfilzomib, those with grade 3‐4 neuropathy or grade ≥2 neuropathy accompanied by neuropathic pain; a history of interstitial lung disease, New York Heart Association class III or IV congestive heart failure, symptomatic myocardial ischemia or uncontrolled conduction abnormalities; serious amyloidosis or suspicion of serious amyloidosis such as cardiac or intestinal amyloidosis; or poorly controlled hypertension or diabetes.
2.3. Treatment
Carfilzomib was administered by 30‐minute intravenous infusion on Days 1, 8 and 15 in the 28‐day cycle (Figure 1). The starting dose was 20 mg/m2 on Day 1 of Cycle 1, and 70 mg/m2 was given thereafter until progressive disease (PD) or unacceptable toxicity. Dexamethasone was administered orally or intravenously at a dose of 40 mg on Days 1, 8, 15 and 22 in Cycles 1 to 9 and on Days 1, 8 and 15 thereafter. Pre‐treatment and post‐treatment intravenous hydration (250‐500 mL) were provided during Cycle 1.
Figure 1.

Dosage and administration. D, Day
For infection prophylaxis, patients received an antibacterial drug and/or trimethoprimsulfamethoxazole during Cycle 1. Patients with prior history of herpes zoster infection received an anti‐herpes viral drug. Patients receiving oral dexamethasone (40 mg) also received a proton pump inhibitor. Prohibited concomitant agents or therapies were anti‐cancer agents during the study, hematopoietic growth factor, platelet transfusion, bisphosphonates, and denosumab during Cycle 1.
2.4. Study outcomes and assessments
In this study, the primary objectives were to evaluate the tolerability and safety of the weekly Cd regimen with 20/70 mg/m2, based on the incidence of dose‐limiting toxicities (DLT) during Cycle 1 and adverse events (AE) until 30 days after the last dose. All AE were evaluated using National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0. AE and these events were coded using MedDRA Version 19.1J. (International Council for Harmonisation, Geneva, Switzerland)
Efficacy endpoints were ORR, clinical benefit rate (CBR), disease control rate, PFS, overall survival (OS), time to progression and duration of response. Anti‐tumor response was assessed by the investigators according to the International Myeloma Working Group (IMWG) Uniform Response Criteria,9 with minimal response (MR) assessed according to the European Group for Blood and Marrow Transplantation criteria.10 ORR was defined as the proportion of patients who achieved a stringent complete response (sCR), complete response (CR), very good partial response (VGPR), and partial response (PR). CBR was defined as the proportion of patients who achieved sCR, CR, VGPR, PR, and MR. PFS was defined as the time from the day of the first administration of carfilzomib to the day of progression or death from any cause. OS was defined as the time from the day of the first administration of carfilzomib to the day of progression or death from any cause.
Pharmacokinetic analysis involved the calculation of the maximum plasma concentration (Cmax), area under the plasma concentration–time curve from time 0 to the time of last quantifiable concentration (AUClast), and terminal elimination half‐life (T1/2) for each patient. Blood samples for the PK assay were collected on Days 1 and 15 of Cycle 1 at the following time points: before the intravenous infusion; at 5 and 15 minutes after the start of the infusion; immediately before the end of the infusion; and at 5, 15 and 30 minutes and 1, 2 and 4 hours after the end of the infusion.
The pharmacodynamics analysis of carfilzomib was conducted with the evaluation of proteasome inhibition activity in peripheral blood mononuclear cells and whole blood. Blood samples for the pharmacodynamics assay were collected on Days 1 and 8 of Cycle 1 and Day 1 of Cycle 2 before the start of intravenous infusion and at 1 hour after the end of the infusion.
2.5. Statistical analysis
The planned sample size was 6 patients. For this cohort, the sample size was selected considering the number of patients required to successfully evaluate tolerability and PK.
Study populations for the analysis were defined as follows: The safety set (SAF) comprised the patients who received at least 1 dose of the study drug. The full analysis set comprised the patients in the SAF who were GCP compliant, met the main inclusion criteria, and in whom efficacy could be analyzed at least once post‐dose. The population for the PK analysis comprised the patients who received at least 1 dose of the drug, were GCP compliant, and had a plasma concentration measurement result. PK parameters were calculated using Phoenix WinNonlin version 6.2.1 (Certara L.P., Princeton, NJ, USA). The population for the pharmacodynamics analysis comprised the patients who received at least 1 dose of the study drug, were GCP compliant, and had a pharmacodynamics assay result. A specific level of significance was not established, as formal statistical testing was not performed. For the efficacy parameters, 95% confidence intervals (CI) were calculated using the Wilson method. There was no imputation for missing data. All statistical analyses were performed using SAS version 9.3 and 9.4 (SAS Institute, Cary, NC, USA) and were conducted by Ono Pharmaceutical.
High‐risk cytogenetics were defined as positive for del(17p) in ≥20% of screened plasma cells, t(4;14), t(14;16) or hypodiploidy. The presence of del(17p), t(4;14) and t(14;16) was evaluated by fluorescence in situ hybridization, and hypodiploidy was evaluated using the G‐banded karyotype analyzed at a central laboratory. The relative dose intensity of carfilzomib was calculated as the actual total dose (mg/m2)/planned dose (mg/m2) during the study period.
3. RESULTS
3.1. Patients and treatment
A total of 6 patients with RRMM were enrolled between March 2015 and June 2015. All 6 patients were evaluable for safety and efficacy analyses. Baseline demographic and clinical characteristics of patients are presented in Table 1. Four out of 6 patients (4/6, 66.7%) were male, and the median age was 68.0 (range, 63‐80) years. The median number of previous regimens was 4.5 (range, 4‐8). All patients had previous therapies with bortezomib and immunomodulatory drugs, including lenalidomide and/or thalidomide. One out of 5 evaluable patients had t(4;14) and was categorized as having high‐risk cytogenetics.
Table 1.
Baseline demographic and clinical characteristics of patients
| Characteristic | Category | Cd 20/70 mg/m2 (N = 6) |
|---|---|---|
| Gender | Male | 4 (66.7) |
| Female | 2 (33.3) | |
| Age (years) | Median (range) | 68.0 (63‐80) |
| ECOG performance status | 0 | 5 (83.3) |
| 1 | 1 (16.7) | |
| ISS Stage | 1 | 1 (16.7) |
| 2 | 2 (33.3) | |
| 3 | 3 (50.0) | |
| Presence of neuropathy | Yes | 6 (100.0) |
| Severity of neuropathya | Grade 1 | 4 (66.7) |
| Grade 2 | 2 (33.3) | |
| Number of prior treatments (number of times) | Median | 4.5 (4‐8) |
| 4 | 3 (50.0) | |
| 5 | 1 (16.7) | |
| ≥6 | 2 (33.3) | |
| High‐risk cytogeneticsb | Yes | 1 (16.7) |
| No | 4 (66.7) | |
| Unevaluable | 1 (16.7) | |
| Plasma cell ratio in bone marrow (%) | Median | 10.80 |
| Hemoglobin (g/dL) | Median | 10.30 |
| <10.5 | 3 (50.0) | |
| ≥10.5 | 3 (50.0) | |
| Neutrophil count (/mm3) | Median | 1689.5 |
| <1500 | 2 (33.3) | |
| ≥1500 | 4 (66.7) | |
| Lymphocyte count (/mm3) | Median | 1164.3 |
| <800 | 1 (16.7) | |
| ≥800 | 5 (83.3) | |
| Platelet count (104/mm3) | Median | 13.80 |
| <15 | 3 (50.0) | |
| ≥15 | 3 (50.0) |
Data in the table are presented as n (%), unless otherwise indicated.
Cd, carfilzomib and dexamethasone; ECOG, Eastern Cooperative Oncology Group; ISS, International Staging System.
In the case of more than 1 neuropathy, the most severe was reported.
High‐risk cytogenetics were defined as positive for del(17p) in ≥20% of screened plasma cells, t(4;14), t(14;16), or hypodiploidy.
Patient exposure to the study drug is summarized in Table 2. The median number of cycles was 15 (range, 1‐19). The median carfilzomib treatment duration was 421.5 (range, 8‐582) days. Three out of 6 patients remained under treatment with the study drug at the cutoff date (30 November 2016).
Table 2.
Exposure to study treatment
| Cd 20/70 mg/m2 (N = 6) | ||
|---|---|---|
| Median | Minimum‐maximum | |
| Number of cycles | 15.0 | 1‐19 |
| Duration of exposure (days) | 421.5 | 8‐582 |
| Number of doses (number of times) | 38.0 | 2‐51 |
| Total dosage (mg) | 4602.0 | 121‐6019 |
| Relative dose intensitya (%) | 83.12 | 56.3‐94.4 |
Cd, carfilzomib and dexamethasone.
Relative dose intensity was calculated as follows: Relative dose intensity = Actual total dose (mg/m2)/planned dose (mg/m2) during the study period.
3.2. Tolerability/safety
One out of 6 patients experienced a DLT, and the tolerability of the weekly Cd regimen with 20/70 mg/m2 was confirmed. The DLT was grade 3 thrombotic microangiopathy, which occurred on Day 11 of Cycle 1 and was considered a serious event. All 6 patients (100%) experienced at least 1 grade ≥3 AE. Five out of 6 patients (83.3%) experienced serious AE, which were also considered serious adverse drug reactions (ADR). All AE of any grade with incidence ≥20% and grade ≥3 AE are shown in Table 3.
Table 3.
Adverse events of any grade with an incidence ≥20% in the safety analysis set or grade ≥3 adverse events
| Event | All grades | Grade ≥3 |
|---|---|---|
| N | 6 | |
| Thrombocytopenia | 4 (66.7) | 3 (50.0) |
| Pyrexia | 4 (66.7) | 0 (0.0) |
| Pneumonia | 3 (50.0) | 2 (33.3) |
| Weight increased | 3 (50.0) | 0 (0.0) |
| Hypophosphatemia | 3 (50.0) | 2 (33.3) |
| Upper respiratory tract inflammation | 3 (50.0) | 0 (0.0) |
| Lymphopenia | 2 (33.3) | 1 (16.7) |
| Constipation | 2 (33.3) | 0 (0.0) |
| Malaise | 2 (33.3) | 0 (0.0) |
| Edema peripheral | 2 (33.3) | 0 (0.0) |
| Bronchitis | 2 (33.3) | 0 (0.0) |
| Nasopharyngitis | 2 (33.3) | 0 (0.0) |
| Headache | 2 (33.3) | 0 (0.0) |
| Insomnia | 2 (33.3) | 0 (0.0) |
| Dry skin | 2 (33.3) | 0 (0.0) |
| Leukopenia | 1 (16.7) | 1 (16.7) |
| Thrombotic microangiopathy | 1 (16.7) | 1 (16.7) |
| Thrombotic thrombocytopenic purpura | 1 (16.7) | 1 (16.7) |
| Large intestine polyp | 1 (16.7) | 1 (16.7) |
| Hypertriglyceridemia | 1 (16.7) | 1 (16.7) |
Data in the table are presented as n (%).
Adverse events reported by physicians were coded using MedDRA Version 19.1J.
Cd, carfilzomib and dexamethasone.
The most common AE (occurring in more than 3 patients) included thrombocytopenia (66.7%), pyrexia (66.7%), pneumonia (50.0%), upper respiratory tract inflammation (50.0%), weight increase (50.0%) and hypophosphatemia (50.0%). The most common grade ≥3 AE (occurring in more than 2 patients) were thrombocytopenia (50.0%), pneumonia (33.3%) and hypophosphatemia (33.3%). The most common serious AE occurring in more than 2 patients was pneumonia (33.3%). One patient (16.7%) experienced grade 1 peripheral neuropathy. Two patients (33.3%) experienced AE (also considered ADR) leading to study discontinuation. These AE were thrombotic microangiopathy (Day 11 of Cycle 1) and thrombotic thrombocytopenic purpura (Day 6 of Cycle 2). Both patients were treated with plasmapheresis, and they recovered from their conditions.
No cases of cardiac failure or interstitial lung disease were observed in this study. No patients died until 30 days after the last dose. One patient died after the study period ended; the cause of death was gastric carcinoma.
3.3. Efficacy
All 6 patients were evaluable for response. The ORR and CBR are presented in Table 4. The ORR (partial response or better) was 83.3% (95% CI, 43.6‐97.0). One patient (16.7%) experienced a VGPR and 4 patients experienced PR. The CBR was also 83.3%.
Table 4.
Overall response rate and clinical benefit rate
| Cd 20/70 mg/m2 (N = 6) | ||
|---|---|---|
| N (%) | 95% confidence intervala | |
| ORR (sCR + CR + VGPR + PR) | 5 (83.3) | (43.6‐97.0) |
| CBR (sCR + CR + VGPR + PR + MR) | 5 (83.3) | ‐ |
| sCR | 0 (0.0) | ‐ |
| CR | 0 (0.0) | ‐ |
| VGPR | 1 (16.7) | ‐ |
| PR | 4 (66.7) | ‐ |
| MR | 0 (0.0) | ‐ |
| SD | 0 (0.0) | ‐ |
| PD | 0 (0.0) | ‐ |
| NE | 1 (16.7) | ‐ |
sCR, CR, VGPR and PR were evaluated by international uniform response criteria by IMWG, while MR was evaluated according to criteria of EBMT.
CBR, clinical benefit rate; Cd, carfilzomib and dexamethasone; CR, complete response; MR, minimal response; NE, not evaluable; ORR, overall response rate; PD, progressive disease; PR, partial response; sCR, stringent complete response; SD, stable disease; VGPR, very good partial response.
Normal approximation by the Wilson method.
Figure 2 shows a spider plot of changes in M‐protein during the treatment. One patient was excluded from this analysis because of missing M‐protein data. M‐protein decreased rapidly after beginning the treatment and continued decreasing during the treatment.
Figure 2.

Change in M‐protein during the study treatment
At the data cutoff (30 November 2016), 3 out of 6 patients continued dosing without PD, and 5 out of 6 patients remained alive. Table 5 summarizes the OS, PFS, time to progression and duration of response of all 6 patients included in the full analysis set. The OS ranged from 463 to 604 days. The PFS ranged from 1 day to 603 days. Similarly, the time to progression ranged from 1 to 603 days. The duration of response (evaluated in 5 patients) ranged from 35 to 560 days. These data were censored at the data cutoff.
Table 5.
Summary of survival data for each of the 6 patients
| Cd 20/70 mg/m2 (N = 6) | ||||
|---|---|---|---|---|
| Patient's number | Overall survival (days) | Progression‐free survival (days) | Time to progression (days) | Duration of response (days) |
| Patient 1 | 603a | 603a | 603a | 547a |
| Patient 2 | 604a | 63a | 63a | 35a |
| Patient 3 | 526a | 526a | 526a | 498a |
| Patient 4 | 463 | 296 | 296 | 239 |
| Patient 5 | 540a | 1a | 1a | ‐ |
| Patient 6 | 588a | 588a | 588a | 560a |
Cd, carfilzomib and dexamethasone.
Indicates censored.
3.4. PK
The PK parameters of carfilzomib are shown in Table 6. After a 30‐minute infusion of 20 mg/m2 on Day 1, the carfilzomib plasma concentration decreased rapidly, with a mean T1/2 (0.684 ± 0.251 hours) ≤1 hour (Figure 3). Similar results were obtained even after intravenous administration of 70 mg/m2, with a T1/2 of 0.900 ± 0.0740 hours (Figure 3). The Cmax of carfilzomib after administration of 70 mg/m2 was 2500 ± 777 ng/mL, and AUClast was 1250 ± 285 ng*h/mL. The Cmax and AUClast increased in a dose‐dependent manner.
Table 6.
Pharmacokinetic parameters of carfilzomib
| Day 1 of Cycle 1 | Day 15 of Cycle 1 | |
|---|---|---|
| Carfilzomib 20 mg/m2 N = 6 | Carfilzomib 70 mg/m2 N = 5 | |
| Cmax a (ng/mL) | 924 ± 452 | 2500 ± 777 |
| AUClast a (ng*h/mL) | 355 ± 120 | 1250 ± 285 |
| Tmax b (h) | 0.575 (0.250‐0.617) | 0.467 (0.250‐0.617) |
| T1/2 a (h) | 0.684 ± 0.251 | 0.900 ± 0.0740 |
AUClast, area under the plasma concentration–time curve from time 0 to the time of last quantifiable concentration; Cmax, maximum plasma concentration; T1/2, terminal elimination half‐life, systemic clearance; Tmax, time at which the Cmax is observed.
Mean ± standard deviation.
Median (minimum‐maximum).
Figure 3.
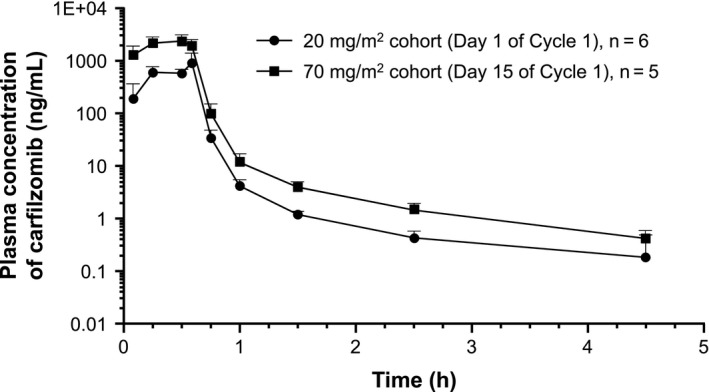
Plasma concentration of carfilzomib after administration of 20 mg/m2 (Day 1 of Cycle 1) and 70 mg/m2 (Day 15 of Cycle 1)
3.5. Pharmacodynamics
The proteasome activities in whole blood and peripheral blood mononuclear cells are shown in Figure 4A,B. The proteasome activity was inhibited 1 hour after administration on Days 1 and 8 of Cycle 1 and on Day 1 of Cycle 2 with ≥90% inhibition.
Figure 4.
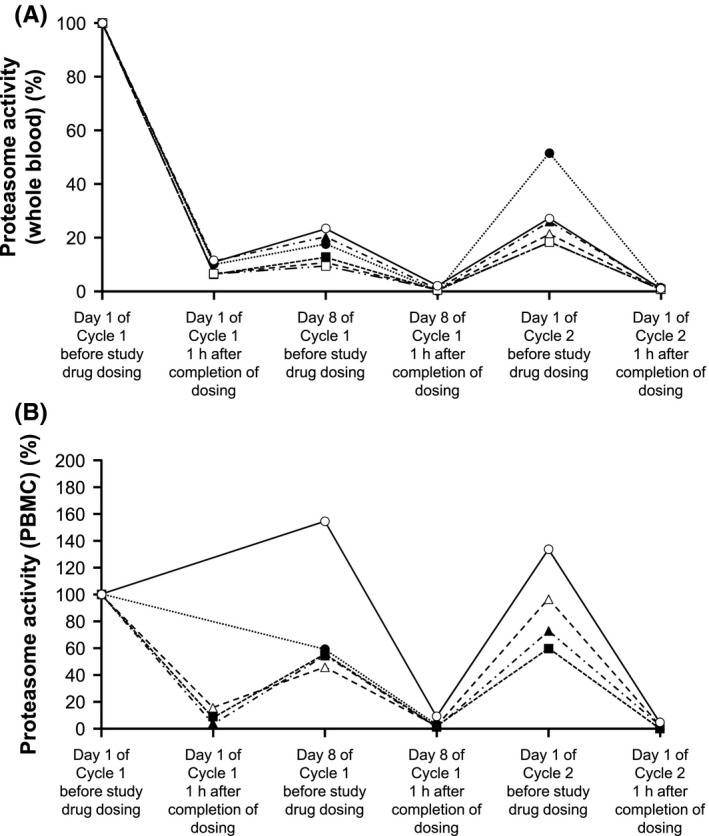
Proteasome activity in whole blood (A) and peripheral blood mononuclear cells (B). PBMC, peripheral blood mononuclear cells
4. DISCUSSION
This was the first study in which a weekly Cd regimen with carfilzomib at a dose of 20/70 mg/m2 was administrated to Japanese RRMM patients. The weekly Cd regimen was generally well‐tolerated and showed an improved efficacy in heavily pretreated Japanese RRMM patients.
The US phase 1/2 study (CHAMPION‐1) was the first study to evaluate weekly dosing of Cd.8 In the CHAMPION‐1 study, the maximum tolerated dose of weekly Cd was determined to be 20/70 mg/m2 , and this dose was shown to be effective with an ORR of 77%. When comparing the present efficacy findings with those of CHAMPION‐1, the ORR results were similar in both studies at 83.3% and 77%, respectively. Regarding the safety profile, the incidences of pyrexia, pneumonia, weight increase, upper respiratory tract infection and hypophosphatemia were higher in the present study. This difference may be influenced by the number of patients in both studies and the status of previous therapies.
Although cross‐trial comparisons are difficult, when we compare the findings of the present study with those of previous Japanese studies, the ORR of 83.3% with Cd in the present study was similar to that in a previous study by Suzuki et al7 (88.5%) in which carfilzomib was administered at a dose of 20/27 mg/m2 in combination with lenalidomide and dexamethasone. In contrast, the ORR was comparable to the data reported in previous studies of twice weekly carfilzomib monotherapy at a dose of 20/27 mg/m2 (22.5%)5 and 20/45 or 20/56 mg/m2 (57.1%).6 The present efficacy findings indicate that the weekly Cd regimen with carfilzomib at a dose of 70 mg/m2 has promising antitumor activity in heavily pretreated MM patients who had previously received bortezomib, lenalidomide or thalidomide. On the other hand, the incidences of infection‐related events (pneumonia, upper respiratory tract infection and nasopharyngitis), pyrexia and weight increase seem higher compared with those in previous Japanese phase 1 and 1/2 studies of carfilzomib.5, 6, 7 This indicates that patients should be carefully monitored for these AE when carfilzomib is administered at a dose of 20/70 mg/m2.
Bortezomib is the first proteasome inhibitor to be tested and is widely used in clinical practice, and ixazomib is a novel oral proteasome inhibitor. The efficacy of carfilzomib was shown in the present study, which was conducted in a population that included patients resistant to bortezomib. In addition, the superiority of carfilzomib over bortezomib in terms of PFS was previously demonstrated in a head‐to‐head comparison in ENDEAVOR, in which OS was also confirmed to be superior based on follow‐up data.4 In a study of ixazomib, PFS was reported to be 20.6 months in patients who received ixazomib with lenalidomide and dexamethasone.11 In the ASPIRE study, PFS was reported to be 26.3 months in patients who received CLd.3 These data suggest that the efficacy of carfilzomib is comparable to that of ixazomib. As ixazomib is an oral agent, it may be considered more convenient than drugs that require intravenous infusion. However, the weekly Cd regimen used in the present study is more convenient than twice‐weekly regimens and may improve carfilzomib compliance in this respect.
New and clinically significant safety findings were raised in the present study as 2 patients experienced microangiopathy‐related events (thrombotic microangiopathy and thrombotic thrombocytopenic purpura). In both patients, a sudden and severe thrombocytopenia with fragmented erythrocytes triggered this diagnosis. To the best of our knowledge, this is the first study to report such microangiopathy‐related events when a high dose carfilzomib (70 mg/m2) with Cd was administered to RRMM patients. Although the incidence was low, microangiopathy‐related events were considered ADR potentially related to carfilzomib treatment. Thrombotic microangiopathy occurred on Day 11 of Cycle 1 and thrombotic thrombocytopenic purpura occurred on Day 6 of Cycle 2, so the timings of both were during the early phase of treatment. Both patients were immediately withdrawn from carfilzomib treatment, followed by treatment with plasmapheresis, and they recovered from their condition. Therefore, we consider that these events may be reversible with an appropriate diagnosis and early therapeutic intervention, including plasmapheresis. Physicians should be especially aware of the possible occurrence of such microangiopathy‐related events and manage symptoms of hemolysis, such as a decrease in haptoglobin levels or schizocyte count. No case of cardiac failure or interstitial lung disease was observed in this study.
The PK parameters of carfilzomib, Cmax and AUClast increased in a dose‐dependent manner. These findings are consistent with those of 3 previous studies assessing PK parameters of carfilzomib at different doses in Japanese subjects.5, 6, 7 Furthermore, the pharmacodynamics analysis revealed that proteasome activities were well inhibited (≥90%) by the carfilzomib administration, which is also consistent with previous Japanese studies.5, 6
This study had several limitations: notably, the small sample size, the lack of a comparator and the fact that it enrolled only Japanese patients.
In conclusion, the weekly Cd regimen at a dose of 20/70 mg/m2 was well‐tolerated among Japanese patients with RRMM. However, careful attention needs to be paid to the occurrence of serious AE, such as infectious events and thrombotic microangiopathy. Further studies should be conducted to confirm the efficacy of high‐dose weekly carfilzomib in combination with dexamethasone and to determine the incidence of AE related to this regimen. Although this was a phase 1 study with a small number of patients, we believe that our results could be the basis for the further development of carfilzomib treatment in view of the safety profile, including microangiopathy‐related events, and efficacy.
CONFLICT OF INTEREST
Dai Maruyama has received honoraria from Takeda, Janssen, Eisai, Biomedis International, Celgene, Kyowa Hakko Kirin, Fujifilm, Ono Pharmaceutical, Mundipharma International, Chugai Pharmaceutical, MSD and Zenyaku Kogyo, and research funds from Takeda, Janssen, Eisai, Biomedis International, Celgene, Kyowa Hakko Kirin, Fujifilm, Ono Pharmaceutical, Mundipharma International, Chugai Pharmaceutical, MSD, Zenyaku Kogyo, GlaxoSmithKline, Abbvie, Astellas Pharma, Amgen Astellas BioPharma, Otsuka, Novartis, Nippon Boehringer Ingelheim, Pfizer, Solasia Pharma and AstraZeneca. Kensei Tobinai has received consulting fees from Celgene, and research funds from Celgene, Janssen, Ono Pharmaceutical and Takeda. Takaaki Chou has received honoraria from Ono Pharmaceutical, Takeda, BMS, Celgene and Janssen, and research funds from Ono Pharmaceutical, Takeda and Celgene. Masafumi Taniwaki has received research funds from Ono Pharmaceutical. Yoshihisa Shumiya is an employee of Ono Pharmaceutical. Shinsuke Iida has received honoraria from Ono Pharmaceutical, Takeda, Janssen, Celgene, BMS and Novartis, and research funds from Ono Pharmaceutical, Celgene, Kyowa Hakko Kirin, Takeda, Janssen, MSD, Sanofi, Daiichi‐Sankyo, Novartis, BMS and Chugai Pharmaceutical. The study was designed under the responsibility of Ono Pharmaceutical. The study was funded by Ono Pharmaceutical. Carfilzomib was provided by Ono Pharmaceutical. Ono Pharmaceutical contributed to the study design, the collection, analysis and interpretation of data, the writing of the manuscript and the decision to submit the manuscript for publication.
ACKNOWLEDGMENTS
We would like to thank all the patients who participated in this study and their families, as well as all investigators, physicians, nurses and clinical research coordinators who helped with this study. We would also like to thank Dr Hirokazu Murakami (Gunma University Graduate School of Health Science, Maebashi), who was the medical consultant, as well as Dr Yutaka Ariyoshi (Aichi Cancer Center Aichi Hospital, Okazaki), Dr Chihiro Shimazaki (Japan Community Health care Organization Kyoto‐Kuramaguchi Medical Center, Kyoto), Dr Masahiro Kizaki (Saitama Medical Center, Saitama Medical University, Saitama), Dr Takao Katoh (International University of Health and Welfare, Mita Hospital, Tokyo), Dr Masahiro Endo (Shizuoka Cancer Center, Shizuoka) and Dr Terufumi Kato (Kanagawa Cardiovascular and Respiratory Center, Yokohama) for their strict review of the clinical data as members of the Efficacy and Safety Monitoring Committee. We also acknowledge the statistical support of Naokazu Gion and Toshiaki Ozaki (Ono Pharmaceutical, Osaka) and critical review of the manuscript by Amgen (Thousand Oaks). We thank Keyra Martinez Dunn, MD, of Edanz Medical Writing for providing medical writing services.
Maruyama D, Tobinai K, Chou T, Taniwaki M, Shumiya Y, Iida S. Weekly carfilzomib and dexamethasone in Japanese patients with relapsed or refractory multiple myeloma: A phase 1 and PK/PD trial. Cancer Sci. 2018;109:3245‐3252. 10.1111/cas.13753
Clinical trial registration: JapicCTI‐122020.
REFERENCES
- 1. Genadieva Stavric S, Bonello F, Bringhen S, Boccadoro M, Larocca A. How is patient care for multiple myeloma advancing? Expert Rev Hematol. 2017;10:551‐561. [DOI] [PubMed] [Google Scholar]
- 2. Kumar SK, Rajkumar SV, Dispenzieri A, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111:2516‐2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stewart AK, Rajkumar SV, Dimopoulos MA, et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med. 2015;372:142‐152. [DOI] [PubMed] [Google Scholar]
- 4. Dimopoulos MA, Goldschmidt H, Niesvizky R, et al. Carfilzomib or bortezomib in relapsed or refractory multiple myeloma (ENDEAVOR): an interim overall survival analysis of an open‐label, randomised, phase 3 trial. Lancet Oncol. 2017;18:1327‐1337. [DOI] [PubMed] [Google Scholar]
- 5. Watanabe T, Tobinai K, Matsumoto M, et al. A phase 1/2 study of carfilzomib in Japanese patients with relapsed and/or refractory multiple myeloma. Br J Haematol. 2016;172:745‐756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Iida S, Tobinai K, Taniwaki M, Shumiya Y, Nakamura T, Chou T. Phase I dose escalation study of high dose carfilzomib monotherapy for Japanese patients with relapsed or refractory multiple myeloma. Int J Hematol. 2016;104:596‐604. [DOI] [PubMed] [Google Scholar]
- 7. Suzuki K, Ri M, Chou T, et al. Carfilzomib, lenalidomide and dexamethasone in patients with heavily pretreated multiple myeloma: a phase 1 study in Japan. Cancer Sci. 2017;108:461‐468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Berenson JR, Cartmell A, Bessudo A, et al. CHAMPION‐1: a phase 1/2 study of once‐weekly carfilzomib and dexamethasone for relapsed or refractory multiple myeloma. Blood. 2016;127:3360‐3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Durie BG, Harousseau JL, Miguel JS, et al. International uniform response criteria for multiple myeloma. Leukemia. 2006;20:1467‐1473. [DOI] [PubMed] [Google Scholar]
- 10. Bladé J, Samson D, Reece D, et al. Criteria for evaluating disease response and progression in patients with multiple myeloma treated by high‐dose therapy and haemopoietic stem cell transplantation. Br J Haematol. 1998;102:1115‐1123. [DOI] [PubMed] [Google Scholar]
- 11. Moreau P, Masszi T, Grzasko N, et al. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;374:1621‐1634. [DOI] [PubMed] [Google Scholar]