

Abstract
The sensitivity of breast cancer cells to epirubicin (EPI) is closely related to the efficacy of the drug and the prognosis of patients. A growing body of research has suggested that autophagy is involved in the treatment of a variety of cancers, including breast cancer, and modifies the sensitivity of anticancer drugs. However, the mechanism by which autophagy participates in cancer therapy and modulates drug sensitivity has not been fully elucidated. In this study, we showed that the expression of Autophagy/Beclin 1 regulator 1 (Ambra1), a key protein of autophagy, was negatively correlated with EPI sensitivity in breast cancer cells. In addition, it altered the sensitivity of breast cancer cells to EPI by regulating EPI‐induced autophagy. As a potential mechanism, we demonstrated that autophagy‐related protein 12 (ATG12) was a downstream protein that Ambra1‐regulated EPI‐induced autophagy. Therefore, Ambra1 plays an important role in regulating the sensitivity of breast cancer cells to EPI. And the regulatory effect of Ambra1 on EPI sensitivity is achieved through the regulation of autophagy by targeting ATG12. Overall, we propose a novel mechanism by which autophagy modulates the sensitivity of breast cancer cells to EPI. ATG12 is a novel targeting protein of Ambra1 in regulating EPI‐induced autophagy. In addition, the important role of Ambra1 in modulating the sensitivity of breast cancer cells to EPI is confirmed in vivo. This finding indicates that Ambra1 might be a target for developing breast cancer treatments.
Keywords: Ambra1, ATG12, autophagy, breast cancer, epirubicin
Abbreviations
- Ambra1
Autophagy/Beclin 1 regulator 1
- BAF1
Bafilomycin A1
- LC3
Microtubule‐associated protein 1 light chain 3
- LV‐AMBRA1
Lentiviral vector‐AMBRA1
- LV‐ATG12
Lentiviral vector‐ATG12
- p62
SQSTM1/p62
1. INTRODUCTION
Breast cancer is the most common malignancy and among the leading causes of cancer‐related mortality in women worldwide.1 Epirubicin (EPI), an anthracycline, is one of the most effective drugs for the treatment of breast cancer. Presently, regimens containing EPI are recommended as the first‐line adjuvant therapy for breast cancer.2 The sensitivity of breast cancer cells to EPI significantly influences the effectiveness of this treatment. However, the factors involved in the regulation of chemosensitivity are numerous and the mechanisms have not yet been fully elucidated.3
Macroautophagy (herein referred to as autophagy) is an evolutionarily conserved protein degradation process in eukaryotic cells that helps the cell to overcome adverse conditions by recycling nutrients and energy.4, 5, 6 Recently, an increasing number of studies have linked autophagy to the treatment of a variety of cancers, including breast cancer.7, 8, 9 Nevertheless, the mechanisms by which autophagy participates in cancer treatment and modulates drug sensitivity have not been established. Autophagy/Beclin 1 regulator 1 (Ambra1) is a protein essential for autophagy induction.10, 11, 12 Meanwhile, it is also important for the execution of apoptosis.13, 14, 15 The levels of Ambra1 in cells are critical for determining the rate of apoptosis.16 Therefore, the level of Ambra1 may modify the sensitivity of breast cancer cells to EPI. So far, few studies have reported on the role of Ambra1 in breast cancer; in particular, there is a lack of evidence that Ambra1 is involved in the sensitivity of breast cancer cells to chemotherapy. Autophagy‐related protein 12 (ATG12) is an ubiquitin‐like protein that conjugates to ATG5 to form an ATG12‐ATG5 conjugate. The conjugate promotes the lipidation of ATG8 (LC3) and directs its correct subcellular localization, which is necessary for the elongation of phagophores and the maturation of autophagosomes.17, 18, 19, 20 Thus, ATG12 is an important protein in regulating autophagy. However, it is unclear whether there is an interaction between Ambra1 and ATG12.
In this study, MDA‐MB‐231, SK‐Br‐3 and MCF‐7 breast cancer cells were used as the cell model. It was found that the level of Ambra1 altered the sensitivity of breast cancer cells to EPI by regulating the occurrence of EPI‐induced autophagy. In addition, ATG12 was a downstream protein of Ambra1 in regulating EPI‐induced autophagy. Consequently, Ambra1 is an important factor in modulating the sensitivity of breast cancer cells to EPI.
2. MATERIALS AND METHODS
2.1. Cell culture
MCF‐7, MDA‐MB‐231 and SK‐Br‐3 cells were from the cell bank of the Chinese Academy of Sciences (Shanghai, China). MCF‐7 and SK‐Br‐3 cells were cultured in MEM media (Thermo Fisher, Waltham, MA, USA), and MDA‐MB‐231 cells were cultured in L15 media (Thermo Fisher), supplemented with 10% FBS (Thermo Fisher), 100 units/mL penicillin and 100 μg/mL streptomycin at 37°C in a humidified incubator with 5% CO2. Before the study, the cells were passaged for 6 generations. The identity of the cell lines was determined by short tandem repeat profiling.
2.2. Agents and antibodies
Epirubicin was from Pfizer Pharmaceuticals (Dalian, China). Bafilomycin A1 (BAF1) was from Sigma‐Aldrich (Shanghai) Trading (Shanghai, China). The annexin V‐PE/7‐AAD Apoptosis Assay Kit was from Nanjing KeyGen Biotech (Nanjing, China). The caspase‐9 activity assay kit, the lactate dehydrogenase (LDH) cytotoxicity assay kit, and the Cell Counting Kit‐8 (CCK‐8) were from the Beyotime Institute of Biotechnology (Shanghai, China). Anti‐MAPLC3β (H50) and anti‐Ambra1 antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti‐GAPDH antibody was from MultiSciences (Lianke) Biotech (Shanghai, China). Anti‐p62, ATG12, and ATG5 were from Abcam (HongKong, China). Anti‐Beclin 1 was from CST (Danvers, MA, USA).
2.3. Cell viability and death assay
For the cell viability and death assay, cells were seeded at 8 × 103 cells per well in 96‐well flat‐bottomed plates and were allowed to attach overnight at 37°C. Afterward, medium containing the assay agents was added to each well, and cells were further cultured at 37°C for the indicated times. The number of viable cells was estimated by CCK‐8 assay. The absorbance was measured at 450 nm with a microplate reader. The number of dead cells was estimated by LDH cytotoxicity assay kit according to the manufacturer's instructions.
2.4. Apoptosis assay
For the apoptosis assay, cells were seeded at 5 × 105 cells in 12.5 cm2 tissue culture flasks and were treated as described for the CCK‐8 assay for the indicated times. Afterward, cells were trypsinized at the indicated time and dyed with annexin V‐PE and 7‐AAD according to the manufacturer's instructions. Then, the apoptosis was detected with a flow cytometer.
2.5. Caspase‐9 activity assay
Cells were collected after treatment with the assay agents for the indicated time, and 30 μL of lysis buffer was added to the collected cells. The cells were resuspended in the lysis buffer and incubated on ice with light agitation for 30 minutes. Lysates were centrifuged at 1500 g for 5 minutes; 10 μL of supernatant was used to assay the protein concentration with Bradford reagent, and another 10 μL was used to assay caspase‐9 activity. The activity of caspase‐9 was assayed with Ac‐LEHD‐pNA as a substrate; the samples were incubated at 37°C for 2 hours, and the OD values were detected at 405 nm with a microplate reader.
2.6. Western blotting
For western blotting analyses, cells were seeded in 25‐cm2 tissue culture flasks and were allowed to reach approximately 80% confluency in fresh medium before treatment with the agents. After treatment, detached and attached cells were collected by centrifugation, and whole‐cell lysates were obtained using a lysis buffer (1 × PBS pH 7.6, 1% NP‐40, 0.1% sodium dodecyl sulfate and 0.5% sodium deoxycholate supplemented with inhibitor cocktails). Approximately 30‐50 μg of total protein from each group was electrophoretically separated on 12% or 15% SDS‐PAGE gels and electrotransferred to polyvinylidene fluoride membranes (PVDF membranes, Pierce, Rockford, USA). The PVDF membranes were blocked with 5% nonfat dry milk in Tris‐buffered saline‐Tween 20 (TBST, pH 7.6) for 1 hour at room temperature, incubated with the primary antibodies diluted in 5% nonfat dry milk in TBST with light agitation overnight at 4°, washed with TBST 3 times, and incubated with the secondary antibodies diluted in 5% nonfat dry milk in TBST with light agitation for 1 hour at room temperature; the proteins were then detected with electrochemiluminescence (Bio‐Rad, Hercules, CA, USA).
2.7. Lentiviral vector and shRNA construction and transfection
A lentiviral vector‐AMBRA1 transfected with full‐length human AMBRA1 cDNA (LV‐AMBRA1), a lentiviral vector‐ATG12 transfected with full‐length human ATG12 cDNA (LV‐ATG12) and an empty vector (EV) were constructed by Genechem (Shanghai, China). Two specific‐target AMBRA1 shRNA (2450 and 3388), a specific‐target BECLIN 1 shRNA, a specific‐target ATG12 shRNA and control scrambled plasmids were synthesized by GenePharma (Shanghai, China). The sequence of 2450 was GCT GGA ATC TTC CCT CAT TTC, the 3388 was GGA GAC ATG TCA GTA TCA ACT, sh‐BECLIN 1 was CAG TTT GGC ACA ATC AAT A,21 and sh‐ATG12 was GCA AAT CCT CTA TGC CTT CTT. ShRNA plasmids were transfected into cells by Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA), the transfection was performed according to the instructions of the manufacturer.
2.8. Autophagy assay
Microtubule‐associated protein 1 light chain 3 (LC3) puncta was monitored by RFP‐GFP‐LC3 tandem fluorescent probe (Genechem). Autophagosomes have both RFP and GFP signals, whereas autolysosomes emit only an RFP signal because GFP is quenched in the acidic lysosomal environment.22 The protein levels of LC3 (LC3‐I/LC3‐II) and SQSTM1/p62 (p62) were detected by western blotting.
2.9. Real‐time quantitative PCR
RNA was extracted by using Trizol Reagent (Generay Biotech [Shanghai], Shanghai, China) as indicated by the supplier. CDNA synthesis was generated using a reverse transcription kit (Vazyme Biotech, Shanghai, China) according to the manufacturer's recommendations. CDNA from cell samples were amplified by quantitative RT‐PCR (qRT‐PCR) with specific primers for AMBRA1 (upper: 5′‐TGGGGAGGTTAGGATTTGGGA‐3′, lower: 5′‐GAGCCGTAGGGTGGAAAGC‐3′), ATG12 (upper: 5′‐GAGACACTCCCATAATGAA‐3′, lower: 5′‐GTAGGACCAGTTTACCATC‐3′), ATG5 (upper: 5′‐ATGTGCTTCGAGATGTGTGG‐3′, lower: 5′‐TGGTTCTGCTTCCCTTTCAG‐3′) and ACTIN (upper: 5′‐TGACGTGGACATCCGCAAAG‐3′, lower: 5′‐CCAAGAAGGAAGGCTGGAAA‐3′) with the ChamQ SYBR Color qPCR Master Mix (Vazyme Biotech). The primers were synthesized by Shanghai Sunny Biotechnology (Shanghai, China). Data were normalized to ACTIN expression.
2.10. Mice xenograft models
To generate murine subcutaneous tumors, 1 × 107 MDA‐MB‐231 cells transfected with scramble or 2450 were injected subcutaneously to the right of the forelimb armpits in BALB/c nude mice (Shanghai SLAC Laboratory Animal, Shanghai, China). Upon the subcutaneous tumor size reaching a diameter of approximately 5 mm, the mice received i.p. injections of EPI (5 mg/kg). Tumor volumes were calculated using the following formula: length × width2 × ∏/6. All animal experiments conformed to the provisions of the Declaration of Helsinki (as revised in Fortaleza, Brazil, October 2013) and were approved by the Ethics Committee of the Second Affiliated Hospital of Guangxi Medical University.
2.11. Statistical analyses
Statistical comparisons of the mean values were performed using ANOVA. P < 0.05 was considered statistically significant.
3. RESULTS
3.1. Knockdown of Ambra1 increases the sensitivity of breast cancer cells to epirubicin
First, the expression of Ambra1 was examined by western blotting in MCF‐7, MDA‐MB‐231 and SK‐Br‐3 breast cancer cells (Figure 1A). To investigate the potential role of Ambra1 in EPI‐induced cell death in breast cancer cells, 2 target‐specific AMBRA1 shRNA (2450 and 3388) were constructed to knock down Ambra1 and an irrelevant shRNA (scramble) as a control. qRT‐PCR and western blotting revealed that after transfection with 2450 or 3388 for 48 hours, Ambra1 decreased significantly in both mRNA and protein (Figure 1B,C, *P < 0.05). After knocking down of Ambra1, the cells were treated with 2.2 μmol/L EPI for another 24 hours. The knockdown of Ambra1 resulted in a dramatic decrease in the viability and an increase in mortality (Figure 1D, *P < 0.05). Along with the increase of cell death, caspase‐9 activity and apoptosis also increased obviously (Figure 1E, *P < 0.05). Therefore, knockdown of Ambra1 increases EPI‐induced cell death in breast cancer cells by activating caspase‐9 and spurring apoptosis.
Figure 1.
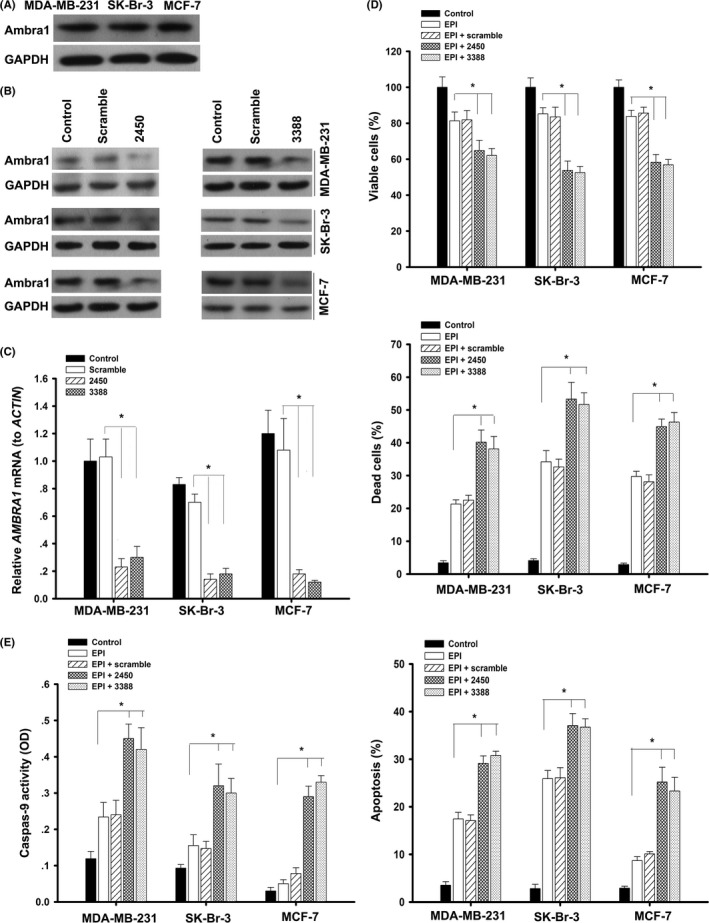
Knockdown of Ambra1 increased epirubicin (EPI)‐induced apoptosis. A, The basal expression of Ambra1 in MCF‐7, MDA‐MB‐231 and SK‐Br‐3 cells was detected by western blotting. B, Cells were incubated with scrambled shRNA or target‐specific AMBRA1 shRNA (2450 or 3388) for 48 h, the protein of Ambra1 was examined by western blotting, and the mRNA was examined by quantitative RT‐PCR (C). The results (mean ± SE) are from 3 independent experiments (*P < 0.05). D, Cells were transfected with scrambled shRNA or 2450 or 3388 for 48 h, following treatment with 2.2 μmol/L EPI for another 24 h; then, cell viability and mortality were analyzed. At the same time, caspase‐9 activity and apoptosis were measured (E). The results (mean ± SE) are from 3 independent experiments (*P < 0.05)
3.2. Overexpression of Ambra1 increases the resistance of breast cancer cells to epirubicin
To further characterize the role of Ambra1 in EPI‐induced cell death, a lentiviral vector‐AMBRA1 transfected with full‐length human AMBRA1 cDNA and an empty vector (EVam, as a control) were constructed to overexpress Ambra1 in MCF‐7, MDA‐MB‐231 and SK‐Br‐3 cells. After transfection with LV‐AMBRA1 for 48 hours, the expression of Ambra1 was significantly increased in both mRNA and protein (Figure 2A,B, *P < 0.05). In addition, the overexpression of Ambra1 led to a significant increase in cell viability as well as decreases in cell death, caspase‐9 activity and apoptosis after EPI treatment (Figure 2C,D, *P < 0.05). Thus, overexpression of Ambra1 increases EPI‐resistance by inhibiting apoptosis in breast cancer cells. LC3 is currently the most widely used autophagosome marker.22 Overexpression of Ambra1 resulted in more LC3 puncta formations in MCF‐7, MDA‐MB‐231 and SK‐Br‐3 cells at basal level and after EPI treatment, and the increased LC3 puncta was more pronounced in the presence of BAF1, a potent V‐ATPase inhibitor that blocks the fusion of autophagosomes and lysosomes22 (Figure 2E, *P < 0.05).
Figure 2.
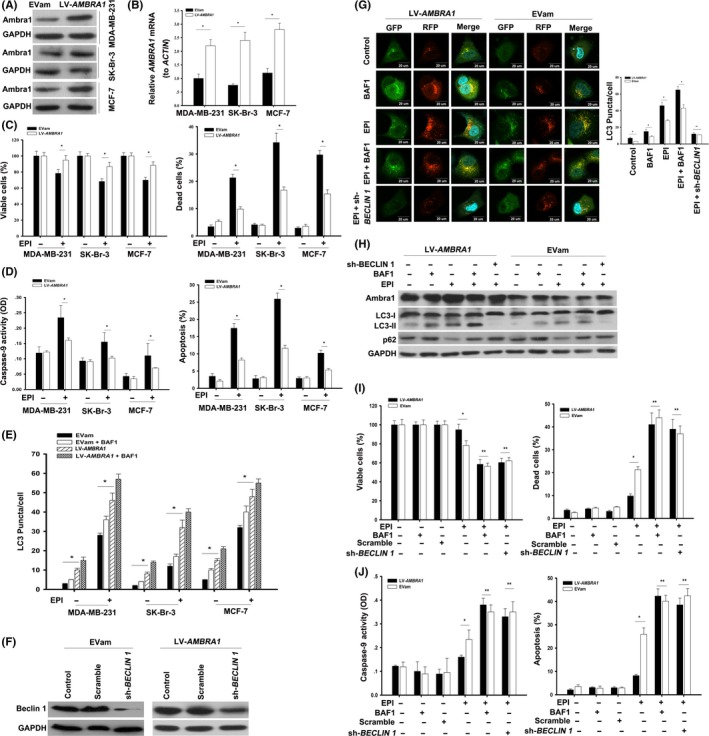
Overexpression of Ambra1 increased the resistance of breast cancer cells to epirubicin (EPI). A, MCF‐7, MDA‐MB‐231 and SK‐Br‐3 cells were transfected with LV‐AMBRA1 or empty vector (EVam) for 48 h, and the protein of Ambra1 was tested by western blotting. Meanwhile, the mRNA of AMBRA1 was analyzed by quantitative RT‐PCR (B). The results (mean ± SE) are from 3 independent experiments (*P < 0.05). C, Cells incubated with LV‐AMBRA1 or EVam for 48 h, following treatment with 2.2 μmol/L EPI for 24 h; then, cell viability and mortality were analyzed. Moreover, caspase‐9 activity and apoptosis were assayed (D). The results (mean ± SE) are from 3 independent experiments (*P < 0.05). E, Cells expressing RFP‐GFP‐LC3 were incubated with LV‐AMBRA1 or EVam for 48 h. After that, these cells were treated with 2.2 μmol/L EPI for 24 h in the presence or absence of Bafilomycin A1 (BAF1, 20 nmol/L). Autophagy was assessed with the LC3 puncta. The results (mean ± SE) are from 3 independent experiments (*P < 0.05). F, MDA‐MB‐231 cells were transfected with LV‐AMBRA1 or EVam for 48 h and then incubated with scrambled shRNA or sh‐BECLIN 1 for additional 48 h. Then, the protein of Beclin 1 was detected by western blotting. G, MDA‐MB‐231 cells expressing RFP‐GFP‐LC3 were incubated with LV‐AMBRA1 or EVam for 48 h. Then, the cells were transfected with scrambled shRNA or sh‐BECLIN 1 for 48 h, followed by treatment with 2.2 μmol/L EPI for another 24 h in the presence or absence of Bafilomycin A1 (BAF1, 20 nmol/L). After treatment, the fluorescence of RFP and GFP was observed with a fluorescence microscope (600×) and LC3 puncta were counted. The nucleus was stained with Hoechst. The results (mean ± SE) are from 3 independent experiments (*P < 0.05, **P > 0.05). In parallel, the proteins of Ambra1, LC3‐I/II and p62 were detected by western blotting (H). Then, (I) and (J), cell viability, cell death, caspase‐9 activity and apoptosis were analyzed. The results (mean ± SE) are from 3 independent experiments (*P < 0.05, **P > 0.05)
To explore whether autophagy mediated the effects of Ambra1‐mediated resistance to the apoptotic response after treatment with EPI, we knocked down Beclin 1 by sh‐BECLIN 1 in MDA‐MB‐231 cells and corresponding cells that highly expressed Ambra1 (Figure 2F). Beclin 1 is a critical autophagic regulator in mammalian cells.23 The knockdown of Beclin 1 not only inhibited the formation of LC3 puncta induced by EPI in MDA‐MB‐231 cells but also inhibited the formation of LC3 puncta in cells with high expression of Ambra1 (Figure 2G, *P < 0.05, **P > 0.05). In addition, silencing of Beclin 1 inhibited the conversion of LC3‐I into LC3‐II and the degradation of p62 by EPI treatment (Figure 2H). During autophagy, LC3‐I is converted to LC3‐II, which is used as an indicator of autophagy.24, 25 P62 forms protein aggregates that are degraded by autophagy and is another indicator of autophagy.26, 27 BAF1 was used to inhibit the lysosome‐dependent degradation of LC3 and p62 and block autophagy in late stages. At the same time, the inhibition of autophagy by Beclin 1‐knockdown and BAF1 also increased the sensitivity of cells overexpressing Ambra1 to EPI (Figure 2I,J, *P < 0.05, **P > 0.05). Therefore, autophagy is required for Ambra1‐mediated EPI‐resistance.
3.3. Ambra1 regulates epirubicin‐induced autophagy in breast cancer cells
As a key factor in autophagy, we explored whether Ambra1 was also involved in EPI‐induced autophagy in breast cancer cells. To do this, we used 2450 or 3388 to knock down Ambra1 in MDA‐MB‐231 cells (Figure 3A). After 48 hours of transfection with either 2450 or 3388, the cells were treated with EPI for additional 24 hours. The knockdown of Ambra1 inhibited the conversion of LC3‐I into LC3‐II and p62 degradation caused by EPI (Figure 3B). In parallel, the increase of LC3 puncta caused by EPI treatment was also suppressed by 2450 or 3388 (Figure 3C, *P < 0.05). BAF1 was used to inhibit the degradation of LC3 and p62 and to analyze autophagic flux. Hence, EPI‐induced autophagy is Ambra1‐dependent in breast cancer cells; that is, Ambra1 regulates EPI‐induced autophagy.
Figure 3.

Ambra1 regulated epirubicin (EPI)‐induced autophagy in breast cancer cells. A, MDA‐MB‐231 cells were incubated with scrambled shRNA or target‐specific AMBRA1 shRNA (2450 or 3388) for 48 h. The protein of Ambra1 was detected by western blotting. B, MDA‐MB‐231 cells were incubated with scrambled shRNA or 2450 or 3388 for 48 h, followed by treatment with EPI for another 24 h in the presence or absence of Bafilomycin A1 (BAF1, 20 nmol/L); then, the protein of LC3‐I/II and p62 was detected by western blotting. C, MDA‐MB‐231 cells expressing RFP‐GFP‐LC3 were incubated with scrambled shRNA or 2450 or 3388 for 48 h. Then, the cells were treated with EPI for another 24 h in the presence or absence of Bafilomycin A1 (BAF1, 20 nmol/L). Autophagy was assessed with the LC3 puncta. The results (mean ± SE) are from 3 independent experiments (*P < 0.05)
3.4. ATG12 is a downstream protein of Ambra1 in epirubicin‐induced autophagy
To explore the underlying mechanism of Ambra1 in the regulation of EPI‐induced autophagy in breast cancer cells, we analyzed the expression of ATG12 in response to Ambra1 knockdown. ATG12 is an ubiquitin‐like protein involved in the formation of autophagic vesicles, which is crucial for autophagy.17, 18, 19, 20 Ambra1 was knocked down by 2450 in MDA‐MB‐231 cells (Figure 4A). Interestingly, 2450 also caused a decrease in ATG12 protein and mRNA (Figure 4A, *P < 0.05, upper histogram). Correspondingly, the overexpression of Ambra1 by LV‐AMBRA1 increased the expression of ATG12 protein and mRNA (Figure S1, *P < 0.05). To further confirm the relationship between ATG12 and Ambra1, we constructed a lentiviral vector‐ATG12 (LV‐ATG12) transfected with full‐length human ATG12 cDNA to overexpress ATG12, and an empty vector (EVat) served as a control. LV‐ATG12 caused ATG12 overexpression in MDA‐MB‐231 cells, but it did not affect the expression of Ambra1 (Figure 4B, *P < 0.05, **P > 0.05). Therefore, Ambra1 positively regulates the expression of ATG12. Because ATG12 is closely related to ATG5, we also detected the expression of ATG5 and ATG12‐ATG5. Similar to ATG12, the knockdown of Ambra1 also resulted in decreased expression of ATG12‐ATG5 (Figure 4A). However, there were no significant changes in ATG5 protein and mRNA (Figure 4A, **P > 0.05, lower histogram). It was worth noting that the downregulation of Beclin1 did not affect the expressions of ATG12 and ATG5 (Figure 4A, **P > 0.05, upper and lower histograms). Thus, ATG12 is a target protein of Ambra1 in breast cancer cells.
Figure 4.

ATG12 was a downstream protein of Ambra1 in epirubicin (EPI)‐induced autophagy. A, MDA‐MB‐231 cells were incubated with scrambled shRNA or sh‐BECLIN 1 or 2450 for 48 h. The protein of Ambra1, ATG12, ATG12‐ATG5 and ATG5 was detected by western blotting. At the same time, the mRNA of ATG12 and ATG5 were examined by quantitative RT‐PCR (qRT‐PCR). The results (mean ± SE) are from 3 independent experiments (*P < 0.05, **P > 0.05). B, MDA‐MB‐231 cells were transfected with LV‐ATG12 or empty vector (EVat) for 48 h. The proteins and mRNA of ATG12 and Ambra1 were detected by western blotting and qRT‐PCR, respectively. The results (mean ± SE) are from 3 independent experiments (*P < 0.05, **P > 0.05). C, MDA‐MB‐231 cells expressing RFP‐GFP‐LC3 were incubated with LV‐ATG12 or empty vector (EVat) for 48 h. After that, the cells were treated with EPI for another 24 h in the presence or absence of Bafilomycin A1 (BAF1, 20 nmol/L); then, autophagy was assessed with the LC3 puncta. The results (mean ± SE) are from 3 independent experiments (*P < 0.05). D, MDA‐MB‐231 cells were transfected with scramble shRNA or 2450 for 48 h, and then incubated with LV‐ATG12 or empty vector (EVat) for another 48 h. Subsequently, the cells were treated with EPI for 24 h. After treatment, the proteins of ATG12, LC3‐I/II and p62 were detected by western blotting (D). In parallel, autophagy was assessed with LC3 puncta in MDA‐MB‐231 cells that expressed RFP‐GFP‐LC3 (E). The results (mean ± SE) are from 3 independent experiments (*P < 0.05, **P > 0.05). Moreover, (F) and (G), cell viability, cell death, caspase‐9 activity and apoptosis were measured, respectively. The results (mean ± SE) are from 3 independent experiments (*P < 0.05, **P > 0.05)
In addition, overexpression of ATG12 also resulted in more LC3 puncta formations (Figure 4C, *P < 0.05), and more LC3‐I conversion into LC3‐II and p62 degradation regardless of EPI treatment (Figure 4D). Therefore, ATG12 is also a proautophagic protein in breast cancer cells. Meanwhile, overexpression of ATG12 dramatically increased the cell viability of EPI treatment and decreased cell death, caspase‐9 activity and apoptosis (Figure 4F,G, *P < 0.05). This suggests that ATG12 plays an important role in regulating EPI sensitivity.
Next, to affirm the role of ATG12 in Ambra1‐mediated autophagy, we knocked down ATG12 by sh‐ATG12 in MDA‐MB‐231 cells and corresponding cells with high expression of Ambra1 (Figure S2A). Even if Ambra1 was overexpressed, the knockdown of ATG12 reduced the number of LC3 puncta (Figure S2B, *P < 0.05, **P > 0.05). Thus, ATG12 is a key protein of Ambra1‐mediated autophagy. Subsequently, we simultaneously knocked down Ambra1 and re‐expressed ATG12 in MDA‐MB‐231 cells. Unfortunately, the re‐expression of ATG12 did not restore EPI‐induced autophagy inhibited by Ambra1 knockdown (Figure 4D,E, **P > 0.05); moreover, the re‐expression of ATG12 did not disturb the increased sensitivity of cells to EPI induced by Ambra1 knockdown (Figure 4F,G, **P > 0.05). The autophagy was detected by LC3‐I conversion into LC3‐II, p62 degradation and LC3 puncta. Therefore, ATG12 is a downstream protein of the Ambra1‐mediated autophagy pathway and is an important protein for Ambra1 to modulate EPI sensitivity.
3.5. Knockdown of Ambra1 enhances epirubicin sensitivity in vivo
To confirm whether downregulation of Ambra1 also increased the sensitivity of breast cancer cells to EPI in vivo, we inoculated BALB/c nude mice with MDA‐MB‐231 cells that had previously been transfected with 2450 and scramble shRNA. The expression of Ambra1 was inhibited by 2450 (Figure 5A,B, *P < 0.05, **P > 0.05). On days 24, 29, 36 and 43 after inoculation of the cells, 5 mg/kg EPI was intraperitoneally administered to the mice. After EPI treatment, the growth of 2450‐transfected tumors was significantly inhibited compared to scramble shRNA transfected tumors (Figure 5C, *P < 0.05). Therefore, Ambra1 is important in modulating EPI sensitivity in breast cancer cells in vivo.
Figure 5.
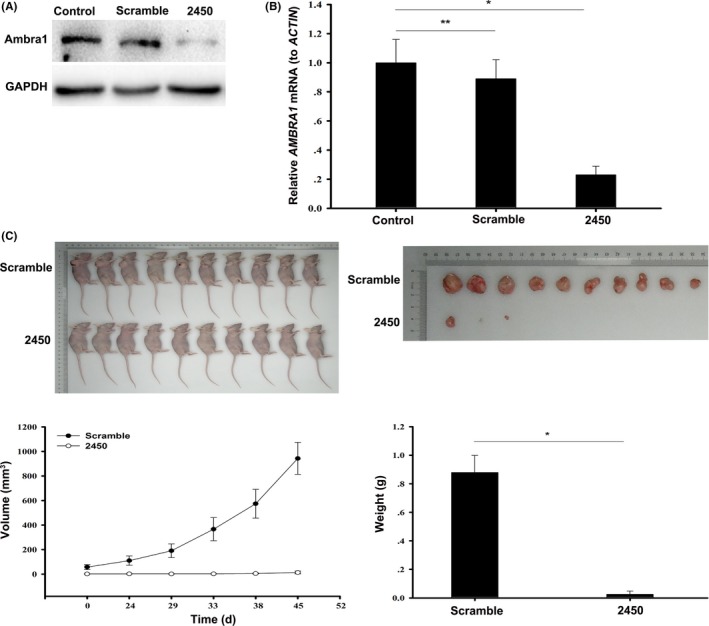
Downregulation of Ambra1 increased sensitivity of breast cancer to epirubicin (EPI) in vivo. A, MDA‐MB‐231 cells were transfected with scrambled shRNA or 2450 for 48 h, and then the protein of Ambra1 was detected by western blotting. At the same time, the mRNA of AMBRA1 was tested by quantitative RT‐PCR (B). The results (mean ± SE) are from 3 independent experiments (*P < 0.05, **P > 0.05). C, After knocking down of Ambra1, MDA‐MB‐231 cells we inoculated BALB/c nude mice. Then, the mice were treated with 5 mg/kg EPI for indicated time, and the volume and weight of the tumor were measured (*P < 0.05)
4. DISCUSSION
Based on the results in vitro and in vivo, we propose that Ambra1 plays an important role in modulating the sensitivity of breast cancer cells to EPI. Furthermore, ATG12 is a downstream protein of Ambra1 in the regulation of EPI‐induced autophagy.
Ambra1 is a key protein in the crosstalk between autophagy and apoptosis. Its expression may determine the fate of cells.13, 14, 15, 16 Studies on several cell lines have shown that high expression of Ambra1 is beneficial for cell survival.10, 14, 16, 28 In this study, we found that downregulation of Ambra1 resulted in an increase in EPI‐induced apoptosis in breast cancer cells; in contrast, overexpression of Ambra1 led to the resistance of breast cancer cells to EPI‐induced apoptosis. Therefore, the expression of Ambra1 determines the apoptosis rate of breast cancer cells treated with EPI. In fact, overexpression of Ambra1 resulted in more LC3 puncta formations, while blocking autophagy by sh‐BECLIN 1 or BAF1 increased the sensitivity of cells overexpressing Ambra1 to EPI. Therefore, autophagy is involved in Ambra1‐mediated EPI‐resistance in breast cancer cells. Recently, autophagy is widely concerned because of its close relationship with tumorigenesis and cancer treatment.7, 8, 9 Our previous study has found that EPI can induce autophagy in MCF‐7 breast cancer cells, which protects the cells from apoptosis.29 Guo et al30 subsequently obtained similar results in MDA‐MB‐231 and SK‐BR‐3 cells. However, Garbar et al31 report that multiple chemotherapy drugs, including EPI, induced an increase of autophagy in MCF‐7 cells, but not in MDA‐MB‐231 cells. Indeed, we confirmed that EPI could induce an increase of autophagy in MDA‐MB‐231 cells by detecting the conversion of LC3‐I into LC3‐II, p62 degradation and LC3 puncta formations. The same was true in MCF‐7 and SK‐Br‐3 cells. This is consistent with our previous findings and those of Guo et al. Although a great deal of studies have been published on the relationship between autophagy and cancer therapy, the mechanism by which autophagy participates in the treatment of cancer has not yet been fully established.7, 8, 9 Here, we found that knockdown of Ambra1 blocked EPI‐induced autophagy and increased the sensitivity of breast cancer cells to EPI. Therefore, Ambra1 is a key protein that autophagy regulates the sensitivity of breast cancer cells to EPI.
Interestingly, we found that Ambra1 positively regulated the expression of ATG12, but not ATG5. ATG12 is a key protein in the elongation and maturation of autophagic vesicles.17, 18, 19, 20 In fact, overexpression of ATG12 increased EPI‐induced autophagy and EPI resistance. This suggests that ATG12 is also a proautophagic protein in breast cancer cells and is involved in the regulation of EPI sensitivity. In contrast, knockdown of ATG12 inhibited EPI‐induced autophagy of the cells overexpressing Ambra1. Thus, ATG12 plays an important role in Ambra1‐mediated autophagy. However, in the presence of ATG12 overexpression, knockdown of Ambra1 inhibited EPI‐induced autophagy and increased cell sensitivity to EPI. This indicates that ATG12 is a downstream protein of Ambra1 during EPI‐induced autophagy, and it is an important protein through which Ambra1 regulates the sensitivity of breast cancer cells to EPI.
In summary, Ambra1 is an important protein that determines whether EPI‐treated breast cancer cells undergo apoptosis or autophagy. Its level modulates the sensitivity of breast cancer cells to EPI. In addition, ATG12 is a downstream protein of Ambra1 during EPI‐induced autophagy and plays an important role in regulating EPI sensitivity.
We conclude that Ambra1 regulates EPI‐induced autophagy in breast cancer cells by targeting ATG12, thereby modulating EPI sensitivity. This finding also indicates that Ambra1 might be a potential target for breast cancer treatment.
CONFLICT OF INTEREST
The authors have no conflict of interest.
Supporting information
Sun W‐L, Wang L, Luo J, Zhu H‐W, Cai Z‐W. Ambra1 modulates the sensitivity of breast cancer cells to epirubicin by regulating autophagy via ATG12. Cancer Sci. 2018;109:3129–3138. 10.1111/cas.13743
Contributor Information
Wei‐Liang Sun, Email: swl20022001@hotmail.com.
Zheng‐Wen Cai, Email: czw001967@sohu.com.
REFERENCES
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics. CA Cancer J Clin. 2017;67:7‐30. [DOI] [PubMed] [Google Scholar]
- 2. NCCN clinical practice guidelines in oncology™, breast cancer V.1.2017. http://www.nccn.org. Accessed March 10, 2017.
- 3. Pan ST, Li ZL, He ZX, Qiu JX, Zhou SF. Molecular mechanisms for tumour resistance to chemotherapy. Clin Exp Pharmacol Physiol. 2016;43:723‐737. [DOI] [PubMed] [Google Scholar]
- 4. Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931‐937. [DOI] [PubMed] [Google Scholar]
- 5. Noda NN, Inagaki F. Mechanisms of autophagy. Annu Rev Biophys. 2015;44:101‐122. [DOI] [PubMed] [Google Scholar]
- 6. Yin ZY, Pascual C, Klionsky DJ. Autophagy: machinery and regulation. Microb Cell. 2016;12:588‐596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5:726‐734. [DOI] [PubMed] [Google Scholar]
- 8. Lorina S, Hamaïb A, Mehrpourb M, Codognob P. Autophagy regulation and its role in cancer. Semin Cancer Biol. 2013;23:361‐379. [DOI] [PubMed] [Google Scholar]
- 9. Sui X, Chen R, Wang Z, et al. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 2013;4:e838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fimia GM, Stoykova A, Romagnoli A, et al. Ambra1 regulates autophagy and development of the nervous system. Nature. 2007;447:1121‐1125. [DOI] [PubMed] [Google Scholar]
- 11. Cianfanelli V, Nazio F, Cecconi F. Connecting autophagy: AMBRA1 and its network of regulation. Mol Cell Oncol. 2015;2:e970059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cianfanelli V, Zio DD, Bartolomeo SD, Nazio F, Strappazzon F, Cecconi F. Ambra1 at a glance. J Cell Sci. 2015;128:2003‐2008. [DOI] [PubMed] [Google Scholar]
- 13. Fimia GM, Corazzari M, Antonioli M, Piacentini M. Ambra1 at the crossroad between autophagy and cell death. Oncogene. 2013;32:3311‐3318. [DOI] [PubMed] [Google Scholar]
- 14. Gu W, Wan DW, Qian QY, et al. Ambra1 is an essential regulator of autophagy and apoptosis in SW620 cells: pro‐survival role of Ambra1. PLoS ONE. 2014;9:e90151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sun WL. Ambra1 in autophagy and apoptosis: implications for cell survival and chemotherapy resistance (Review). Oncol Lett. 2016;12:367‐374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pagliarini V, Wirawan E, Romagnoli A, et al. Proteolysis of Ambra1 during apoptosis has a role in the inhibition of the autophagic pro‐survival response. Cell Death Differ. 2012;19:1495‐1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tanida I, Nishitani T, Nemoto T, Ueno T, Kominami E. Mammalian Apg12p, but not the Apg12p.Apg5p conjugate, facilitates LC3 processing. Biochem Biophys Res Commun. 2002;296:1164‐1170. [DOI] [PubMed] [Google Scholar]
- 18. Geng J, Klionsky DJ. The Atg8 and Atg12 ubiquitinlike conjugation systems in macroautophagy. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep. 2008;9:859‐864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tang B, Li N, Gu J, et al. Compromised autophagy by MIR30B benefits the intracellular survival of Helicobacter pylori . Autophagy. 2012;8:1045‐1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Otomo C, Metlagel Z, Takaesu G, Otomo T. Structure of the human ATG12~ATG5 conjugate required for LC3 lipidation in autophagy. Nat Struct Mol Biol. 2013;20:59‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yu L, Alva A, Su H, et al. Regulation of an ATG7‐beclin 1 program of autophagic cell death by caspase‐8. Science. 2004;304:1500‐1502. [DOI] [PubMed] [Google Scholar]
- 22. Yoshii SR, Mizushima N. Monitoring and measuring autophagy. Int J Mol Sci. 2017;18:18091865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liang XH, Jackson S, Seaman M, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672‐676. [DOI] [PubMed] [Google Scholar]
- 24. Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720‐5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1‐222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bjorkoy G, Lamark T, Brech A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin‐induced cell death. J Cell Biol. 2005;171:603‐614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131‐24145. [DOI] [PubMed] [Google Scholar]
- 28. Yazdankhah M, Farioli‐Vecchioli S, Tonchev AB, Stoykova A, Cecconi F. The autophagy regulators Ambra1 and Beclin 1 are required for adult neurogenesis in the brain subventricular zone. Cell Death Dis. 2014;5:e1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sun WL, Chen J, Wang YP, Zheng H. Autophagy protects breast cancer cells from epirubicin‐induced apoptosis and facilitates epirubicin‐resistance development. Autophagy. 2011;7:1035‐1044. [DOI] [PubMed] [Google Scholar]
- 30. Guo W, Wang Y, Wang Z, Wang YP, Zheng H. Inhibiting autophagy increases epirubicin's cytotoxicity in breast cancer cells. Cancer Sci. 2016;107:1610‐1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Garbar C, Mascaux C, Giustiniani J, Merrouche Y, Bensussan A. Chemotherapy treatment induces an increase of autophagy in the luminal breast cancer cell MCF7, but not in the triple‐negative MDA‐MB231. Sci Rep. 2017;7:7201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials