

Abstract
Vasculogenic mimicry (VM) refers to the unique capability of aggressive tumor cells to mimic the pattern of embryonic vasculogenic networks. Cancer stem cells (CSC) represent a subpopulation of tumor cells endowed with the capacity for self‐renewal and multilineage differentiation. Previous studies have indicated that CSC may participate in the formation of VM. With the advance of high‐resolution microarrays and massively parallel sequencing technology, long noncoding RNAs (lncRNAs) are suggested to play a critical role in tumorigenesis and, in particular, the development of human hepatocellular carcinoma (HCC). Currently, no definitive relationship between lncRNA and VM formation has been described. In the current study, we demonstrated that expression of the lncRNA, n339260, is associated with CSC phenotype in HCC, and n339260 level correlated with VM, metastasis, and shorter survival time in an animal model. Overexpression of n339260 in HepG2 cells was associated with a significant increase in CSC. Additionally, the appearance of VM and vascular endothelial (VE)‐cadherin, a molecular marker of VM, was also induced by n339260 overexpression. Using a short hairpin RNA approach, n339260 was silenced in tumor cells, and knockdown of n339260 was associated with reduced VM and CSC. The results of this study indicate that n339260 promotes VM, possibly by the development of CSC. The related molecular pathways may be used as novel therapeutic targets for the inhibition of HCC angiogenesis and metastasis.
Keywords: cancer stem cell, hepatocellular carcinoma, lncRNA n339260, metastasis, vasculogenic mimicry
Abbreviations
- CSC
cancer stem cell
- EET
epithelium‐endothelium transition
- EMT
epithelial‐mesenchymal transition
- FFPE
formalin‐fixed, paraffin‐embedded
- HCC
hepatocellular carcinoma
- HCS
HepG2‐c‐Myc‐SOX2
- HTA
Human Transcriptome Array
- LncRNAs
long noncoding RNAs
- TGF‐β
transforming growth factor beta
- VE
vascular endothelial
- VM
vasculogenic mimicry
1. INTRODUCTION
Hepatocellular carcinoma has a relatively high incidence rate and a poor prognosis, and there is increased interest in the discovery of therapeutic targets to help fight this illness. The process by which a vessel is formed from tumor cells has been termed VM, and this term refers to the unique ability of aggressive tumor cells to express an endothelial phenotype and to form vessel‐like networks in 3‐D cultures.1, 2, 3, 4, 5, 6 Research has shown that VM is closely associated with high tumor grade, increased degree of invasiveness, metastases, and shorter overall survival in patients.7, 8, 9 However, the cellular and molecular events underlying VM are not well understood.
Cancer stem cells have characteristics associated with normal stem cells, specifically the ability to give rise to all cell types found in a particular cancer tissue. Recently, Yao et al10 proposed the concept of CSC plasticity, whereby CSC may differentiate/transdifferentiate and form branching lumens and tubes to provide nutrition for tumor mass, resembling VM. A previous study from our laboratory showed that CSC potentially participated in the process of VM.3
Differentiated cells can be reprogrammed to acquire pluripotency by transcription regulators such as Oct3/4, SOX2, Klf4, and c‐Myc. These regulators may play a role in HCC and contribute to the acquisition of stem cell‐like properties in HCC. Recently, liver CSC were observed in c‐Myc‐driven tumors, suggesting a role for c‐Myc in liver CSC development and maintenance.11 In particular, our previous study showed that SOX2 functioned in the maintenance of CSC phenotype and VM formation induced by slug overexpression in HCC.12, 13
Mammalian genome encodes numerous lncRNAs of more than 200 nucleotides in length that lack an open reading frame.14 LncRNAs constitute a very heterogeneous group of RNA molecules that participate in a broad spectrum of molecular and cellular functions.15, 16, 17 LncRNAs are deregulated in several human cancers,18 and the mechanisms through which lncRNAs contribute to the regulatory networks that underpin cancer development are diverse.19, 20
LncRNAs have been implicated in the development of human HCC21 and dysregulation of many HCC‐related lncRNAs has been identified.22 In the present study, we explore the role of the lncRNA, n339260, in CSC development and VM formation of HCC to provide better understanding of the molecular mechanism in HCC‐related lncRNAs.
2. MATERIALS AND METHODS
2.1. Cell culture and lentiviral transduction
Human liver cancer cell lines HepG2 cells (ATCC, Rockville, MD, USA) were cultured in DMEM supplemented with 10% FBS (Invitrogen, Carlsbad, CA, USA). Gene transfer was carried out using the pReceiver‐M56 Expression Clone vector for cMyc and pReceiver‐M03 Expression Clone vector for SOX2 overexpression (GeneCopoeia, Rockville, MD, USA) and psi‐LVRH1GP vector (GeneCopoeia) was used for n339260 silencing in HepG2‐c‐Myc/SOX2 overexpressing cells. The shRNA target sequence for n339260 was 5′‐gaaatgattctggcctaccct‐3′. The pReceiver‐Lv105R vector was used for n339260 overexpression. Lentivirus was produced by transient transfection of 293T cells with plasmids.
2.2. Wound‐healing assay
Cells were implanted into 6‐well plates for 70%‐90% confluence, and then a sterilized tip was used to draw a line with the same width on the bottom of the dishes. The medium in the plates was replaced with 1% FBS medium, and the cells were cultured for 48 hours. Images were captured at 0, 24 and 48 hours after the wounding. Experiments were carried out in three replicates.
2.3. Cell migration and invasion assays
Cell migration and invasion assays were carried out using Transwell cell culture inserts (Invitrogen). Transwell chambers were coated with Matrigel for invasion assays. The cells were allowed to migrate for 24‐72 hours. The passed cells were stained with crystal violet solution and counted using 40× objective. Each experiment was carried out in triplicate.
2.4. Three‐dimensional cell culture
The assay was carried out according to previous literature.1, 2, 3 Matrigel (BD Biosciences, Franklin Lakes, NJ, USA) was thawed at 4°C and added to a 96‐well culture plate. Then, the plate was moved to a 37°C incubator with 5% CO2 for 12 hours. Tumor cells in regular medium were seeded on top of the gel for 24 hours on Matrigel culture. While in Matrigel culture, tumor cells were mixture‐seeded with Matrigel for 72‐120 hours. Addition of regular medium was carried out during the incubation. Cells were photographed using a phase contrast microscope (Nikon USA, Garden City, NY, USA).
2.5. Immunofluorescence
Cells on the coverslips were fixed in absolute methanol at −20°C for 20 minutes, blocked in 5% normal goat serum, and incubated with primary antibody against the protein VE‐cadherin (1:100; Abcam, Cambridge, UK), followed by secondary antibody conjugated with Alexa 568 (Molecular Probes, Eugene, OR, USA). After immunolabeling, cells were washed, stained with DAPI (Sigma, St Louis, MO, USA), mounted, and then viewed and captured with fluorescent microscopy (Nikon).
2.6. RNA fluorescence in situ hybridization
Stellaris RNA FISH Probes (Biosearch Technologies Inc., Petaluma, CA, USA) for n339260 and VE‐cadherin mRNA were used in the following way. Frozen tissue sections were fixed in 95% ethanol for 6 minutes. FFPE tissue was deparaffinized by immersion in 100% xylene for 10 minutes followed by 100% ethanol for 10 minutes and wash buffer for 2‐5 minutes. Hybridization buffer containing the appropriate FISH probe was added to the slide at a volume of 100 μL. A clean 18‐mm square coverglass was placed over the hybridization solution to allow for even distribution of the hybridization solution. The slide was then placed in a humidified chamber, covered, and sealed where it was allowed to incubate in the dark at 37°C for at least 4‐16 hours. Tissue sections were stained with DAPI (Sigma), mounted, and then examined using confocal scanning laser microscopy.
2.7. Western blot analysis
Sodium dodecyl sulfate‐PAGE was carried out on whole cell lysates, followed by transfer to polyvinylidene difluoride membranes (Millipore, Burlington, MA, USA). Blots were blocked and incubated with monoclonal antibody (c‐Myc 1:500, SOX2 1:200, VE‐cadherin 1:400, CD90 1:200, CD133 1:50 and Nanog 1:200), followed by incubation with a secondary antibody (1:2000; Santa Cruz Biotechnology, Dallas, TX, USA). Blots were developed using an ECL detection kit (Amersham Pharmacia Biotech, Piscataway, NJ, USA). For protein‐loading analyses, a monoclonal β‐actin antibody (1:2000; Santa Cruz) was used.
2.8. Spheroid formation in 3‐D culture
Cells were seeded at 1 × 105 into 6‐well, ultra‐low adherent plates covered with poly 2‐hydroxyethyl methacrylate (Sigma). Each well also contained 2 mL serum‐free suspension medium or DMEM/F12 (1:1; Gibco, Gaithersburg, NJ, USA) supplemented with 2% B27 (Gibco), 0.5% epidermal growth factor (EGF; Pepro Tech, Rocky Hill, NJ, USA), and 0.5% basic fibroblast growth factor (bFGF; Pepro Tech). Cell growth was observed daily under an inverted microscope.
2.9. Animal model and xenografting
Care of all laboratory animals followed guidelines established by Tianjin Medical University, China. All of the experimental protocols were conducted in accordance with our Institutional Animal Care and Use Committee (IACUC) guidelines and were approved by the Tianjin Medical University IACUC committee. Four‐week‐old non‐ovariectomized female BALB/c nude mice were injected s.c. with 5 × 106 cells suspended in 100 μL PBS. Tumor volume was monitored weekly using digital calipers. Tumor volume (TV) was calculated with the following formula: TV = 1/2 × a × b 2 (where a is the length and b is the width of tumor). After 4 weeks, mice were killed and xenograft tumors were processed for histology and immunohistochemistry analyses.
2.10. Tissue specimens
Through the Tumor Tissue Bank of Tianjin Cancer Hospital, tissue specimens were obtained from 239 patients who underwent hepatectomy for HCC between 2001 and 2014. All methods were carried out in accordance with the guidelines and regulations of Tianjin Medical University, China. All experimental protocols were approved by the Ethical Committee of Tianjin Medical University, China.
2.11. Immunohistochemistry
Information on the staining methods may be found in the literature.1, 2, 3 Tissue sections (5 μm) were deparaffinized and hydrated using standard procedures. Heat‐induced antigen retrieval with citrate buffer pH 6 or Tris‐EDTA pH 9 was done. Primary antibodies against the following proteins: c‐myc (LifeSpan BioSciences, Seattle, WA, USA), Sox2 (GeneTex, Irvine, CA, USA), Nanog (Novus Biologicals, Littleton, CO, USA), VE‐cadherin (Abcam), CD133 (Biorbyt, Cambridge, UK), endomucin (Abcam) and CD31 (Beijing Zhongshan Golden Bridge Biotechnology Co., Ltd, Beijing, China) were applied to the sections. The staining systems used in this study were PicTure PV6000 and Elivision Plus (Zhongshan Chemical Co., Beijing, China).
2.12. Microarray analysis and quantitative real‐time PCR
Samples were sent to Oebiotech (Shanghai, China) for microarray analysis and quantitative real‐time PCR (qRT‐PCR).
2.13. Statistical analysis
Data analysis was carried out with SPSS16.0 software (IBM). All P‐values were two‐sided, and statistical significance was measured at the .05 level.
3. RESULTS
3.1. Coexpression of c‐Myc and SOX‐2 was associated with increased cell invasion, migration, and formation of VM in vitro
Wound healing, invasion, and migration were analyzed after ectopic expression of SOX‐2 and c‐Myc, as confirmed by western blot (Figure 1A). In wound‐healing assays (Figure 1B), a quantitative analysis suggested a significant difference in the speed of wound healing between the c‐Myc, SOX2 and the control empty vector groups. Importantly, c‐Myc and SOX2 cotransfected cells displayed the fastest speed of wound healing. In the migration and invasion assays, the increased migration and invasion ability was most remarkable in the c‐Myc‐ and SOX2‐cotransfected cells (Figure 1C).
Figure 1.
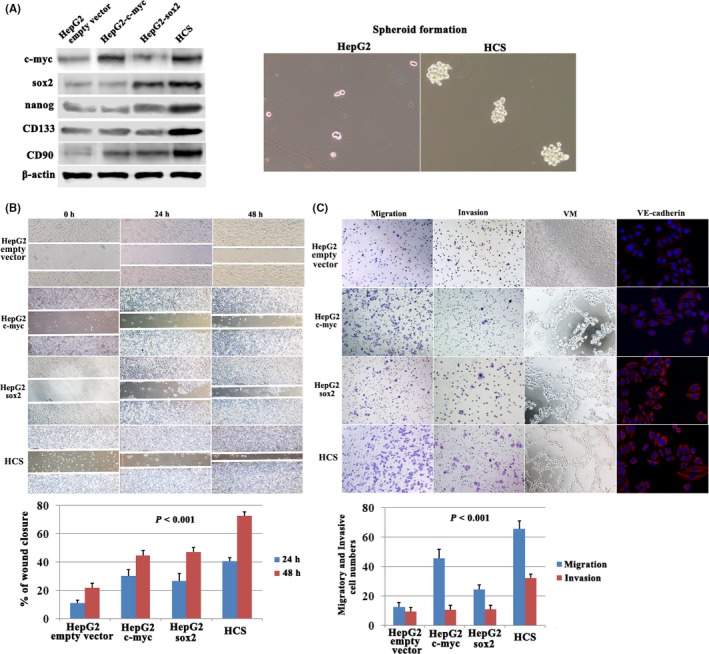
Effect of c‐Myc and SOX2 coexpression on cell invasion, migration, and vasculogenic mimicry (VM) formation in hepatocellular carcinoma (HCC) cells. A, Western blotting showed c‐Myc, SOX2, Nanog, CD133 and CD90 expression. HepG2‐c‐Myc‐SOX2 (HCS) cells successfully developed spheroid formation compared with HepG2 cells. B, HCS cells showed the highest rate of wound healing. C, Cell migration and invasion assays, VM formation by 3‐D culture and VE‐cadherin expression by immunofluorescence in c‐Myc or SOX2‐transfected HepG2 cells
An in vitro model of 3‐D culture was used for investigating VM formation. Control groups showed a lack of VM; however, pipe‐like structure formation and cellular plasticity were observed in HepG2‐c‐Myc and HepG2‐SOX2 cells. In the HepG2‐c‐Myc‐SOX2 (HCS) group, typical pipe‐like structures indicating VM formation were also observed (Figure 1C). Previously, our laboratory showed that VE‐cadherin was a marker of VM formation. Consistently, VE‐cadherin showed increased expression in HepG2‐c‐Myc, HepG2‐SOX2, and HCS cells (Figure 1C).
In addition, western blot showed that expression of reprogramming factor Nanog and CSC markers CD133 and CD9013, 23 was increased following c‐Myc and SOX2 coexpression (Figure 1A). Meanwhile, spheroid formation in 3‐D culture was carried out for HCS and HepG2 cells to grow under nonadherent conditions and to generate spheroids from single‐cell suspensions. Notably, HCS cells successfully developed spheroid formation and showed a significant increase in the size of spheroids compared with HepG2 (Figure 1A). These results suggested that the cotransfection of c‐Myc and SOX2 in HepG2 cells might initiate reprogramming and induce HCS cells harboring CSC characteristics.
3.2. c‐Myc and SOX2 expression promote vascular mimicry in vivo
We developed a xenograft model using the following 4 cell lines: HepG2, HepG2‐c‐Myc, HepG2‐SOX2, and HCS, and these cell lines were each inoculated s.c. into 10 nude mice. Xenografts in HepG2‐c‐Myc and HepG2‐SOX2 showed a higher rate of tumor growth compared to control HepG2 cells (Figure 2A). The highest rate of tumor growth was observed in HCS (Figure 2A). Immunohistochemistry for endomucin and periodic acid‐Schiff (PAS) was carried out using a double‐staining technique to identify VM channels. VM was defined as a vessel‐like structure containing red blood cells formed by endomucin‐negative tumor cells (Figure 2B; black arrow, VM; green arrow, endomucin‐positive authentic blood vessel; star, red blood cells). Xenograft tumor tissue that had at least 1 VM structure was deemed as having the capacity to form VM. VM formation was not observed in xenografts of the HepG2 group (0/10) although there might be small numbers of CSC in HepG2 cells that were too few to contribute to VM formation. In contrast, there was VM formation in 3 xenografts of the HepG2‐c‐Myc group (3/10, 30%), in 4 xenografts of the HepG2‐SOX2 group (4/10, 40%), and in 9 xenografts of the HCS group (9/10, 90%) (Figure 2C).
Figure 2.
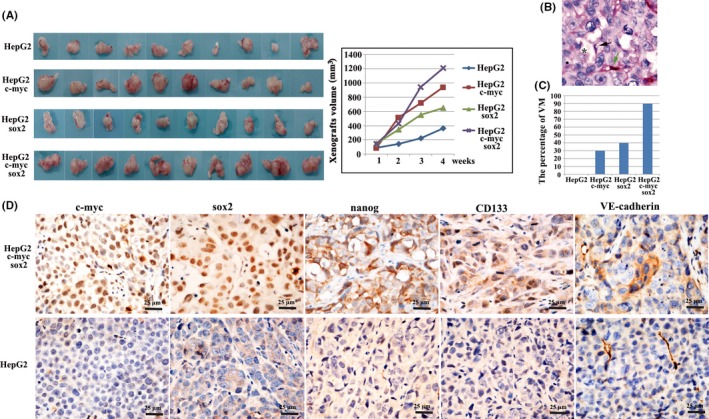
Vasculogenic mimicry (VM) formation after c‐Myc and SOX2 upregulation in vivo. A, Rate of tumor growth in xenografts. B, Immunohistochemistry for endomucin/periodic acid‐Schiff (PAS) double staining was carried out for detecting VM formation in HepG2‐c‐Myc‐SOX2 (HCS) xenografts (black arrow, VM; green arrow, endomucin‐positive authentic blood vessel; star, red blood cells). C, Percentage of VM in 4 groups. D, Immunohistochemistry showing positive expression of c‐Myc, SOX2, Nanog, CD133 and VE‐cadherin in HCS xenografts and negative expression in HepG2 control xenografts
Immunohistochemistry confirmed elevated expression of c‐Myc and SOX2 in the HCS xenografts (Figure 2D) and, importantly, protein levels of Nanog and CD133 were increased following c‐Myc and SOX2 upregulation in vivo (Figure 2D). Consistent with VM in HCS xenografts, VE‐cadherin also showed positive expression. These results indicate that c‐Myc and SOX2 upregulation is associated with VM formation and CSC phenotype development in vivo.
3.3. Molecular profiling of HCS cells and control HepG2 cells
Affymetrix GeneChip HTA 2.0 (Affymetrix Inc., Santa Clara, CA, USA) was used to compare global genome expression in HCS compared to HepG2 cells. This analysis identified 2326 genes that were differentially expressed following c‐Myc and SOX2 upregulation, which included 1813 upregulated genes and 513 downregulated genes (Figure S1; Table S1).
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and gene ontology (GO) biological process showed that the most represented category was the TGF‐β signaling pathway, cell cycle, gene expression, DNA repair, mitotic cell cycle, transcription from RNA polymerase II promoter, chromatin modification (Figure 3A) etc. Expectantly, c‐Myc/SOX2 upregulation targets were enriched with cancer‐related functions such as the TGF‐β signaling pathway (Table S2) which played an essential role for the maintenance of CSC, and the mitotic cell cycle (Table S3) involved in cell proliferation regulation.
Figure 3.
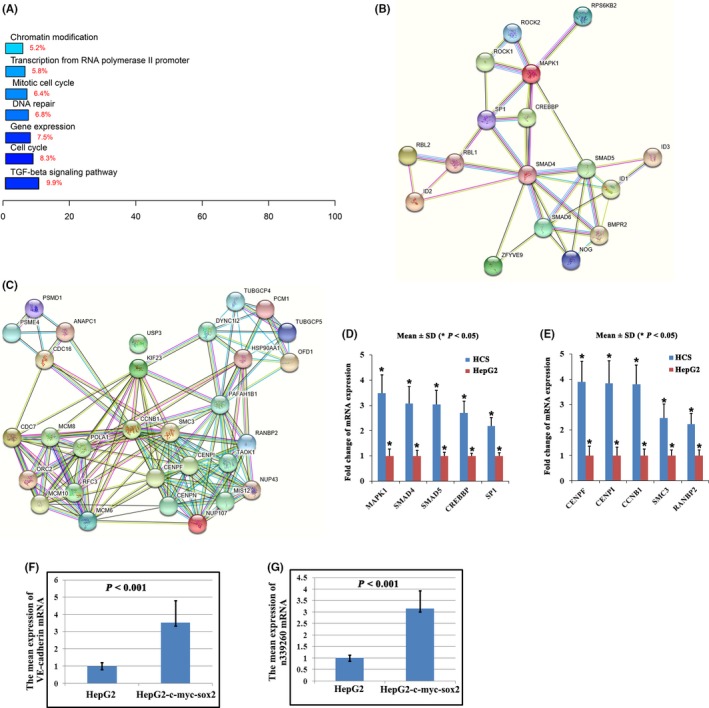
Molecular profiling of HepG2‐c‐Myc‐SOX2 (HCS) cells and control HepG2 cells. A, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and gene ontology (GO) analysis of c‐Myc/SOX2‐regulated targets. B‐C, Functional association network of genes in transforming growth factor beta (TGF‐β) signaling pathway (B) and mitotic cell cycle (C) by Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) analysis. D‐E, Expression changes validation of the 5 selected genes in the TGF‐β signaling pathway (D) and mitotic cell cycle (E) by qRT‐PCR. F, qRT‐PCR indicates that vascular endothelial (VE)‐cadherin mRNA shows a higher expression in HCS than in control cells. G, Noncoding RNA n339260 was confirmed to show a higher expression in HCS cells than in control cells by qRT‐PCR
By Search Tool for the Retrieval of Interacting Genes/Proteins (STRING)‐Known and Predicted Protein‐Protein Interactions analysis, the c‐Myc/SOX2‐regulated targets involved in the TGF‐β signaling pathway (Figure 3B; Table S2) and mitotic cell cycle (Figure 3C; Table S3) were functionally connected into well‐linked interaction networks. Such functional connection was not surprising, suggesting that c‐Myc/SOX2 upregulation had the potential to promote cancer cell proliferation. Importantly, the enrichment of TGF‐β signaling molecules suggested that c‐Myc/SOX2 upregulation could promote CSC development. We also arbitrarily selected 5 genes in the TGF‐β signaling pathway and mitotic cell cycle for qRT‐PCR validation. Relative changes of selected genes obtained from qRT‐PCR were highly correlated to those observed by microarray data (Figure 3D,E; Tables S2 and S3). Meanwhile, qRT‐PCR showed that VE‐cadherin mRNA was elevated in HCS cell lines compared with HepG2‐empty vector (Figure 3F).
Besides mRNA, HTA 2.0 analysis in the present study identified 748 differentially expressed noncoding RNA genes (Table S1). LncRNAs had been shown to play a critical role in twist‐induced EMT and LncRNA‐Hh strengthened CSC generation associated with twist‐induced EMT.24 Other lncRNAs such as LncTCF7, lncRNA H19, LncRNA‐hPVT1, and HOTAIR had been noted to be responsible for acquisition of stem cell‐like properties in HCC cells and contributed to promote liver CSC growth.25 Our previous study showed that CSC were associated with VM formation and VE‐cadherin expression as a result of their plasticity and transdifferentiation ability. Therefore, we focused on noncoding RNA to show their role in CSC phenotype development and VM formation. Based on GeneChip data, noncoding RNA n346018, n377754, n384393, LOC100268168, XLOC_009777, n339260, n333444, and n383953 were selected for qRT‐PCR verification. Noncoding RNA n339260 was confirmed to show a higher expression in the HCS cell line compared with HepG2‐empty vector (Figure 3G).
3.4. Expression of n339260 correlates with expression of CSC markers in human HCC tumor samples
Cancer stem cell marker CD133 was used to detect the presence of CSC in HCC tissues. Two distinct CD133 staining patterns were observed. The first pattern was characterized by apical/endoluminal cell surface staining of CD133 (Figure 4A, black arrow). The second pattern showed abundant cytoplasmic and membrane positivity of CD133 staining (Figure 4A, black arrowhead). Percentage of CD133+ cells in the total tumor cells ranged from 5% to 30%. Immunohistochemistry of 239 HCC tissues showed moderate‐to‐strong CD133 expression in 73 (30.5%) cases.
Figure 4.
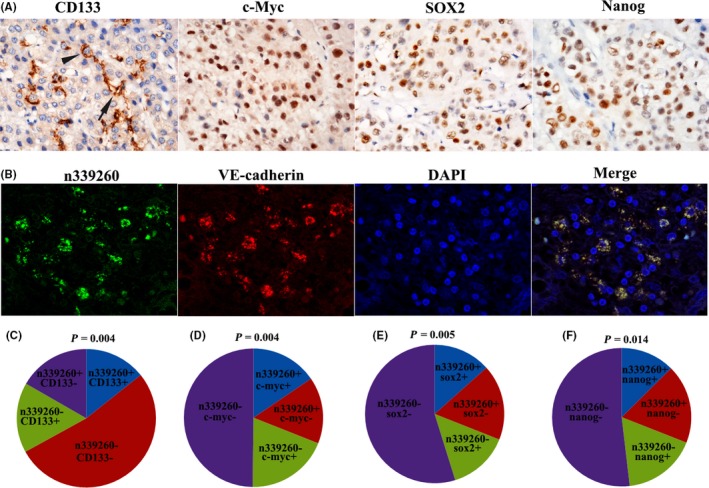
Expression of n339260 correlates with cancer stem cell (CSC) phenotype in human hepatocellular carcinoma (HCC) tissue. A, Immunohistochemical staining of CD133, c‐Myc, SOX2 and Nanog in HCC. B, By RNA FISH, n339260 expression was identified in the cytoplasm. There was colocalization of the spatial overlap between n339260 expression and vascular endothelial (VE)‐cadherin mRNA. C‐F, There was correlation between n339260 and CD133 expression (C), n339260 and c‐Myc (D), n339260 and SOX2 (E), n339260 and Nanog (F)
Cytoplasmic expression of n339260 was identified in frozen HCC tissues by RNA FISH (Figure 4B). Cases with ≥10% positive cell staining were considered as positive. In this cohort, 74 (31.0%) of 239 cases showed positive expression of n339260. By statistical analysis, there was a positive correlation between n339260 and CD133 expression (r = 0.183, P = .004) (Figure 4C). Patients with positive n339260 expression also showed positive CD133 expression (45.9%, 34/74), whereas low n339260 expression showed only 23.6% (39/165) of cases with CD133‐positive expression.
Expression of reprogramming factors (SOX2, Nanog, c‐Myc) was also assessed by immunohistochemistry. Positive cells contained positive staining in the nucleus and cytoplasm. c‐Myc (Figure 4A), SOX2 (Figure 4A), and Nanog (Figure 4A) were detected in 34.7% (83/239), 27.6% (66/239), and 29.7% (71/239) of HCC patients, respectively. Importantly, there was a positive correlation between n339260 and c‐Myc expression (r = 0.186, P = .004) (Figure 4D), n339260 and SOX2 expression (r = 0.181, P = .005) (Figure 4E), and n339260 and Nanog expression (r = 0.159, P = .014) (Figure 4F) (Table 1).
Table 1.
Correlation of n339260 expression and metastasis, VM, VE‐cadherin, CD133, Nanog, c‐myc, sox2 expressions
| n339260+ | n339260− | χ2 | P | |
|---|---|---|---|---|
| VM+ | 51 | 4 | 127.499 | <.001 |
| VM− | 23 | 161 | ||
| VE‐cadherin+ | 48 | 14 | 84.528 | <.001 |
| VE‐cadherin− | 26 | 151 | ||
| CD133+ | 34 | 39 | 11.986 | <.001 |
| CD133− | 40 | 126 | ||
| Nanog+ | 30 | 41 | 6.024 | .014 |
| Nanog− | 44 | 124 | ||
| c‐myc+ | 37 | 46 | 11.029 | .001 |
| c‐myc− | 37 | 119 | ||
| Sox2+ | 32 | 34 | 13.097 | <.001 |
| Sox2− | 42 | 131 | ||
| Metastasis | ||||
| Yes | 40 | 41 | 13.097 | <.001 |
| No | 34 | 124 | ||
VE, vascular endothelial; VM, vascular mimicry.
3.5. n339260 colocalizes and correlates with the VM marker, VE‐cadherin, by RNA FISH
Based on previous studies, CD31/PAS double‐staining is useful to identify VM in HCC tissue.1, 2, 3 A CD31‐negative, PAS‐positive vascular‐like pattern of HCC containing red blood cells was deemed VM (Figure 5A; black arrow, VM; star, red blood cells). HCC cells formed extracellular matrix‐rich channels (PAS‐positive), with the absence of necrosis and inflammatory cells infiltrating around the channels. VM channels were not lined by endothelial cells, as evidenced by the lack of CD31 (brown) staining (Figure 5A; red arrow, typical blood vessel). Interestingly, we observed that n339260 positivity had a close relationship with VM, suggesting that n339260 played an important role in tumor vasculature. Many patients with n339260 expression showed VM (68.9%, 51/74), whereas low n339260 expression showed VM in only 5.4% (4/74) of cases (χ2 = 127.499, P < .001) (Figure 5C; Table 1). Statistically significant correlations were also found between VM and n339260 expression (r = 0.730, P < .001).
Figure 5.
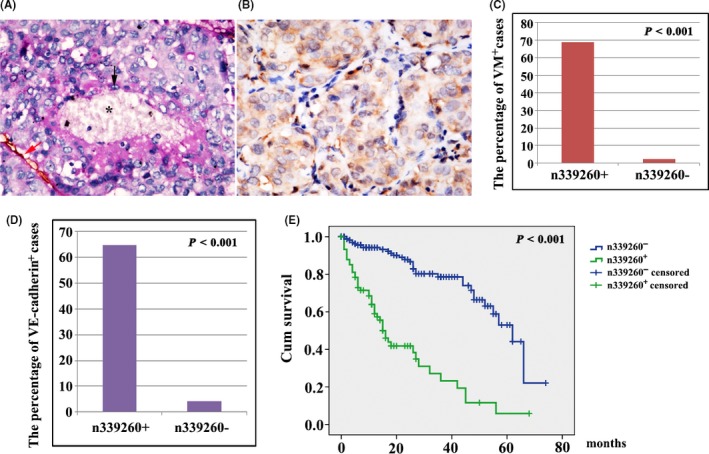
Expression of n339260 in correlation with vascular mimicry (VM) formation, metastasis, and shorter survival time in hepatocellular carcinoma (HCC) patients. A, CD31/periodic acid‐Schiff (PAS) double staining shows VM in HCC tissue (black arrow, VM; red arrow, CD31‐positive blood vessel; star, red blood cells). B, Vascular endothelial (VE)‐cadherin‐positive expression by immunohistochemistry. C‐D, Statistically significant correlations were found between VM and n339260 expression (C), VE‐cadherin and n339260 expression (D). E, Survival analysis indicates that patients with n339260 positivity in HCC were significantly associated with poor overall survival. Censored means that the individual was out of touch or alive during the follow‐up period in either n339260‐positive or ‐negative cases
Vascular endothelial‐cadherin is a transmembrane glycoprotein that is expressed in adherens junctions between vascular endothelial cells. Our previous study showed that HCC tumor cells which had the potential to form VM could express VE‐cadherin. In the present study, VE‐cadherin expression of tumor cells with ≥10% cell positive staining were considered VE‐cadherin‐positive patient specimens, regardless of vascular endothelial cell staining. In this study, VE‐cadherin expression was found to be positive in 62 (25.9%) of 239 HCC tissue samples (Figure 5B). VE‐cadherin positively correlated with VM formation (r = 0.882, P < .001). Importantly VE‐cadherin expression was positively associated with n339260 expression (r = 0.666, P < .001) (Figure 5D) (Table 1).
By fluorescence microscopy, there was marked colocalization of the spatial overlap between n339260 expression and VE‐cadherin mRNA (Figure 4B).
3.6. Expression of n339260 is associated with metastasis and shorter overall survival in HCC patients
Within the patient cohort, 81 cases were associated with metastasis, whereas 158 were nonmetastatic (Table 1). There was positive expression of n339260 in 49.4% (40/81) of samples from metastatic groups, higher than that of the nonmetastasis group (21.5%) (34/158). The difference was statistically significant (χ2 = 19.449, P < .001).
Survival analysis indicated that patients with n339260 positivity in HCC were significantly associated with poor overall survival (Figure 5E). Mean (95% CI) overall survival time was 29.061 (23.527‐34.596) and 46.433 (41.725‐51.140) months, respectively, for patients with and without n339260‐positive expression in HCC (P < .001).
3.7. Expression of n339260 promotes VM formation in vitro
HepG2 cells transfected with n339260 plasmid expressed higher levels of n339260 than HepG2‐empty‐vector cells as analyzed by qRT‐PCR (Figure 6A). Remarkably, HepG2 cells overexpressing n339260 showed a higher vasculogenic capacity and VM marker VE‐cadherin expression than control cells (Figure 6B,C). HepG2‐empty‐vector cells without VM ability formed typical pipe‐like structures on and in the 3‐D Matrigel medium following n339260 expression (Figure 6C). These results provide further support for a role of n339260 in promoting VM formation.
Figure 6.
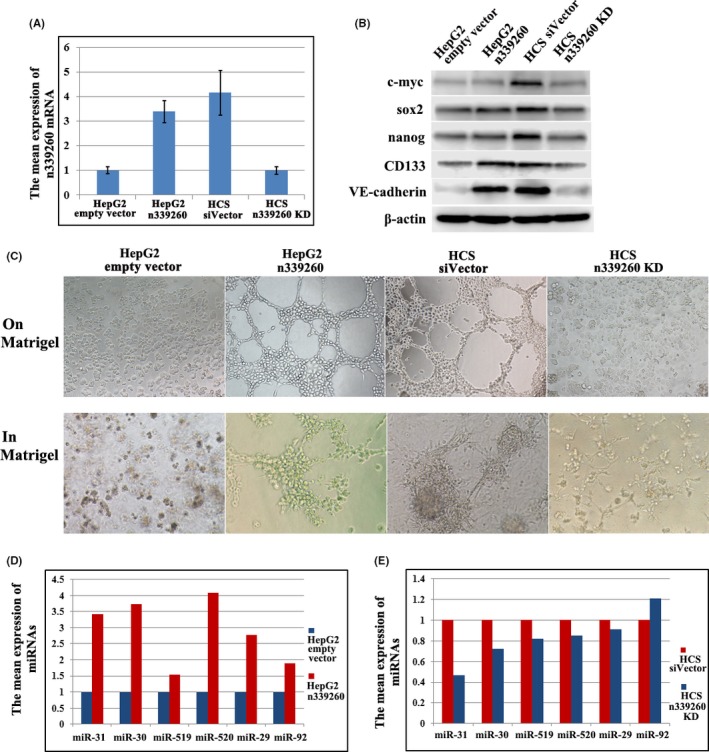
Expression of n339260 promoted vascular mimicry (VM) formation, cancer‐related microRNAs (miRNAs) expression in vitro. A, HepG2 n339260 expressed higher levels of n339260 than control cells as analyzed by qRT‐PCR; knockdown of n339260 expression in HepG2‐c‐Myc‐SOX2 (HCS) cells. B‐C, Expression of c‐myc, sox2, Nanog, CD133 and vascular endothelial (VE)‐cadherin (B) in HepG2 n339260, and tubular channel formation on Matrigel and in Matrigel in these cells (C). Effect of n339260 knockdown on c‐myc, sox2, Nanog, CD133 and VE‐cadherin expression (B) and VM formation (C). D‐E, Cancer‐related miRNAs expression following n339260 ectopic expression in HepG2 (D) and n339260 knockdown in HCS (E) by qPCR
To further evaluate whether n339260 plays a role in VM formation and CSC phenotype development, we knocked down n339260 expression in HCS cells using a siRNA approach. Stable knockdown of n339260 was confirmed by qRT‐PCR (Figure 6A). Vascular channel formation was inhibited when n339260 expression was downregulated (Figure 6B,C). In addition, although c‐myc, sox2, and Nanog expression did not show a significant increase in HepG2‐overexpressing n339260 cells, their expression was impaired following n339260 knockdown in HCS cells. CD133 and VE‐cadherin expression increased with the overexpression of n339260 in HepG2 cells and decreased with n339260 knockdown in HCS cells (Figure 6B). These data provide strong evidence that an intimate relationship exists between n339260 expression, CSC phenotype development, and VM in HCC cells.
3.8. MicroRNA profile of n339260 in HCC cells
To predict the microRNAs (miRNAs) regulated by n339260 in HCC cells, we used Affymetrix miRNA Chip 4.0 (Affymetrix Inc.) to assess miRNAs expression in HepG2‐empty vector, HepG2 n339260 (Table S4), HCS siVector and HCS sin339260 cells (Table S5). Our analysis identified 50 miRNAs that were differentially expressed following n339260 upregulation in HepG2 cells, which included 12 upregulated miRNAs and 38 downregulated miRNAs. Twenty‐eight miRNAs were found to be differentially expressed following n339260 downregulation in HCS cells, which included 18 upregulated miRNAs and 10 downregulated miRNAs.
AnnoLnc is a web server to provide on‐the‐fly analysis for novel human lncRNAs, and we used AnnoLnc as the tool to identify the miRNAs regulated by n339260 based on the miRNA profiles in HepG2‐empty vector, HepG2 n339260, HCS siVector and HCS sin339260 cells. miR‐31‐3p, miR‐30e‐5p, miR‐519c‐5p, miR‐520c‐5p, miR‐29b‐1‐5p, and miR‐92a‐1‐5p that were abnormally expressed in our study were identified as downstream miRNAs regulated by n339260 (Figure 6D,E).
4. DISCUSSION
In the present study, c‐Myc and SOX2 coexpression induced elevated Nanog expression in HepG2 cells, suggesting that CSC in HCC may arise through a reprogramming‐like mechanism promoted by c‐Myc and SOX2. Further in vivo and in vitro experiments showed that c‐myc and sox2 coexpression promoted CSC subpopulation development and VM formation. These results suggested that EET occurred in accordance with CSC phenotype development and that CSC might have potential in promoting VM formation.
Further microarray analysis showed that the genes in the TGF‐β signaling pathway and mitotic cell cycle regulation were activated by c‐Myc and SOX2 expression. Interestingly, HepG2 cells contained a higher lncRNA n339260 expression when transfected with c‐Myc and SOX2, suggesting that n339260 overexpression could be induced with the development of CSC phenotype and VM formation.
Formation of VM networks by tumor cells is a feature associated with a pluripotent gene expression pattern in aggressive tumor cells.6 CSC showing pluripotent stemness might have a unique ability to express an endothelial phenotype and form vessel‐like networks.26, 27 In the present study, there was a correlation between n339260 expression and CSC marker CD133, or between n339260 expression and pluripotency‐maintaining factors c‐Myc, SOX2, Nanog, or between n339260 expression and VM marker VE‐cadherin. Additionally, we found that VM was present in patients with overexpression of n339260. Interestingly, there was colocalization for n339260 and VE‐cadherin. Although the specific role of n339260 in the regulation of VE‐cadherin is not completely clear, our results show that n339260 is critical to induce stem‐like properties and VM formation. Importantly, statistical analysis showed that expression of n339260 was also correlated with metastasis and shorter overall survival in HCC patients. Therefore, our study suggests that n339260 overexpression may lead to a poor prognosis through promoting VM that could be induced by an increased population of cells displaying CSC characteristics.
Further in vitro experiments reinforced the results in human HCC tissue. There was a significant difference in the formation of vascular network in Matrigel and on Matrigel between HepG2 n339260 and control HepG2. With the upregulation of n339260 in HepG2 cells, those cells with n339260 overexpression formed typical pipe‐like structures and showed increased VE‐cadherin expression, whereas a lack of tube formation was observed in the parental cells. Remarkably, our study showed that n339260 silencing induced decreased VE‐cadherin expression and VM formation inhibition in HepG2‐c‐myc‐sox2 cells. Therefore, our study provides further support for the role of n339260 in promoting VM formation.
Recently, the influence of lncRNAs on miRNA function has been rapidly emerging. These miRNA‐lncRNA interactions modulate gene expression patterns that drive major cellular processes.28, 29, 30 In this study, miR‐31‐3p, miR‐30e‐5p, miR‐519c‐5p, miR‐520c‐5p, miR‐29b‐1‐5p, and miR‐92a‐1‐5p were identified as the downstream miRNAs regulated by n339260. Considering that miR‐31‐3p, miR‐30e‐5p, miR‐520c‐5p, miR‐519c‐5p, and miR‐29b‐1‐5p expression increased/decreased following lncRNA n339260 upregulation/downregulation, we speculated that n339260 were processed to yield these miRNAs, or n339260 could serve as a key regulatory factor to modulate other RNAs that were processed to yield these miRNAs. Interestingly, miR‐92a‐1‐5p expression increased following n339260 knockdown in HCS cells, suggesting that n339260 might function as a miRNA sponge and repress miR‐92a‐1‐5p expression. miR‐31‐3p seems to be a new metastatic cancer biomarker, and its expression level allows for the identification of patients with wild‐type KRAS who are more likely to respond to anti‐epidermal growth factor receptor (EGFR) therapy than non‐wild‐type KRAS patients for colon cancer.31 miR‐30‐5p functions as a tumor suppressor and novel therapeutic tool by targeting the oncogenic Wnt/β‐catenin/BCL9 pathway.32 Serum miR‐519c‐5p can predict disease risk in prostate cancer patients.33 miR‐520c promotes breast cancer cell invasion and metastasis by suppression of CD44.34 miR‐29b‐1‐5p could be regulated by hypoxia in cancer cells.35 Downregulation of miR‐92a is associated with aggressive breast cancer features.36 Specifically, among these identified miRNAs, miR‐29b‐1‐5p could upregulate Akt‐dependent nuclear factor kappa B (NF‐κB) signaling leading to activation of MMP2 and MMP9,37 which had been found to play key roles in VM formation of HCC in our previous study.1, 2 Collectively, our study suggested that the miRNAs regulated by n339260 were cancer‐related miRNAs that might be involved in HCC aggressiveness.
In summary, the present study showed that n339260 promoted VM in HCC by the induction of CSC‐like phenotype. VM represent an important survival mechanism contributing to the failure of currently available angiogenesis inhibitors. The results of our study suggest that the molecular target of n339260 might act on a specific CSC subpopulation, offering a tempting new therapeutic perspective for HCC.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest for this article.
Supporting information
ACKNOWLEDGMENTS
This study was partially supported by the following grants: The National Natural Science Foundation of China (No. 81572872); Key project of the National Natural Science Foundation of China (No. 81230050).
Zhao X, Sun B, Liu T, et al. Long noncoding RNA n339260 promotes vasculogenic mimicry and cancer stem cell development in hepatocellular carcinoma. Cancer Sci. 2018;109:3197–3208. 10.1111/cas.13740
Xiulan Zhao, Baocun Sun and Tieju Liu contributed equally to this study.
REFERENCES
- 1. Sun T, Zhao N, Zhao XL, et al. Expression and functional significance of Twist1 in hepatocellular carcinoma: its role in vasculogenic mimicry. Hepatology. 2010;51:545‐556. [DOI] [PubMed] [Google Scholar]
- 2. Sun T, Sun BC, Zhao XL, et al. Promotion of tumor cell metastasis and vasculogenic mimicry by way of transcription coactivation by Bcl‐2 and Twist1: a study of hepatocellular carcinoma. Hepatology. 2011;54:1690‐1706. [DOI] [PubMed] [Google Scholar]
- 3. Liu TJ, Sun BC, Zhao XL, et al. CD133+ cells with cancer stem cell characteristics associates with vasculogenic mimicry in triple‐negative breast cancer. Oncogene. 2013;32:544‐553. [DOI] [PubMed] [Google Scholar]
- 4. Liu T, Sun B, Zhao X, et al. HER2/neu expression correlates with vasculogenic mimicry in invasive breast carcinoma. J Cell Mol Med. 2013;17:116‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Maniotis AJ, Folberg R, Hess A, et al. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am J Pathol. 1999;155:739‐752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Seftor RE, Hess AR, Seftor EA, et al. Tumor cell vasculogenic mimicry: from controversy to therapeutic promise. Am J Pathol. 2012;181:1115‐1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sun B, Zhang S, Zhang D, et al. Vasculogenic mimicry is associated with high tumor grade, invasion and metastasis, and short survival in patients with hepatocellular carcinoma. Oncol Rep. 2006;16:693‐698. [PubMed] [Google Scholar]
- 8. Liu T, Sun B, Zhao X, et al. OCT4 expression and vasculogenic mimicry formation positively correlate with poor prognosis in human breast cancer. Int J Mol Sci. 2014;15:19634‐19649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kirschmann DA, Seftor EA, Hardy KM, Seftor RE, Hendrix MJ. Molecular pathways: vasculogenic mimicry in tumor cells: diagnostic and therapeutic implications. Clin Can Res. 2012;18:2726‐2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yao XH, Ping YF, Bian XW. Contribution of cancer stem cells to tumor vasculogenic mimicry. Protein Cell. 2011;2:266‐272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yamashita T, Wang XW. Cancer stem cells in the development of liver cancer. J Clin Invest. 2013;123:1911‐1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhao X, Sun B, Sun D, et al. Slug promotes hepatocellular cancer cell progression by increasing sox2 and nanog expression. Oncol Rep. 2015;33:149‐156. [DOI] [PubMed] [Google Scholar]
- 13. Sun D, Sun B, Liu T, et al. Slug promoted vasculogenic mimicry in hepatocellular carcinoma. J Cell Mol Med. 2013;17:1038‐1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mattick JS, Makunin IV. Non‐coding RNA. Hum Mol Genet. 2006;15(Spec No. 1):R17‐R29. [DOI] [PubMed] [Google Scholar]
- 15. Kretz M, Siprashvili Z, Chu C, et al. Control of somatic tissue differentiation by the long non‐coding RNA TINCR. Nature. 2013;493:231‐235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yoon JH, Abdelmohsen K, Gorospe M. Posttranscriptional gene regulation by long noncoding RNA. J Mol Biol. 2013;425:3723‐3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhu J, Fu H, Wu Y, Zheng X. Function of lncRNAs and approaches to lncRNA‐protein interactions. Sci China Life Sci. 2013;56:876‐885. [DOI] [PubMed] [Google Scholar]
- 18. Isin M, Dalay N. LncRNAs and neoplasia. Clin Chim Acta. 2015;444:280‐288. [DOI] [PubMed] [Google Scholar]
- 19. Hajjari M, Salavaty A. HOTAIR: an oncogenic long non‐coding RNA in different cancers. Cancer Biol Med. 2015;12:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hu L, Wu Y, Tan D, et al. Up‐regulation of long noncoding RNA MALAT1 contributes to proliferation and metastasis in esophageal squamous cell carcinoma. J Exp Clin Can Res. 2015;34:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li C, Chen J, Zhang K, Feng B, Wang R, Chen L. Progress and Prospects of Long Noncoding RNAs (lncRNAs) in Hepatocellular Carcinoma. Cell Physiol Biochem. 2015;36:423‐434. [DOI] [PubMed] [Google Scholar]
- 22. He Y, Meng XM, Huang C, et al. Long noncoding RNAs: Novel insights into hepatocellular carcinoma. Cancer Lett. 2014;344:20‐27. [DOI] [PubMed] [Google Scholar]
- 23. Yamashita T, Honda M, Nakamoto Y, et al. Discrete nature of EpCAM+ and CD90+ cancer stem cells in human hepatocellular carcinoma. Hepatology. 2013;57:1484‐1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhou M, Hou Y, Yang G, et al. LncRNA‐Hh strengthen cancer stem cells generation in twist‐positive breast cancer via activation of hedgehog signaling pathway. Stem Cells. 2016;34:55‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Parasramka MA, Patel T. Long non‐coding RNA regulation of liver cancer stem cell self‐renewal offers new therapeutic targeting opportunities. Stem Cell Invest. 2016;3:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jhaveri N, Chen TC, Hofman FM. Tumor vasculature and glioma stem cells: Contributions to glioma progression. Cancer Lett. 2016;380:545‐551. [DOI] [PubMed] [Google Scholar]
- 27. Fan YL, Zheng M, Tang YL, Liang XH. A new perspective of vasculogenic mimicry: EMT and cancer stem cells (Review). Oncol Letters. 2013;6:1174‐1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353‐358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Karreth FA, Tay Y, Perna D, et al. In vivo identification of tumor‐ suppressive PTEN ceRNAs in an oncogenic BRAF‐induced mouse model of melanoma. Cell. 2011;147:382‐395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yoon JH, Abdelmohsen K, Gorospe M. Functional interactions among microRNAs and long noncoding RNAs. Semin Cell Dev Biol. 2014;34:9‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Manceau G, Imbeaud S, Thiebaut R, et al. Hsa‐miR‐31‐3p expression is linked to progression‐free survival in patients with KRAS wild‐type metastatic colorectal cancer treated with anti‐EGFR therapy. Clin Cancer Res. 2014;20:3338‐3347. [DOI] [PubMed] [Google Scholar]
- 32. Zhao JJ, Lin J, Zhu D, et al. miR‐30‐5p functions as a tumor suppressor and novel therapeutic tool by targeting the oncogenic Wnt/beta‐catenin/BCL9 pathway. Can Res. 2014;74:1801‐1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang SY, Shiboski S, Belair CD, et al. miR‐19, miR‐345, miR‐519c‐5p serum levels predict adverse pathology in prostate cancer patients eligible for active surveillance. PLoS One. 2014;9:e98597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu B, Wu X, Liu B, et al. MiR‐26a enhances metastasis potential of lung cancer cells via AKT pathway by targeting PTEN. Biochem Biophys Acta. 2012;1822:1692‐1704. [DOI] [PubMed] [Google Scholar]
- 35. Camps C, Saini HK, Mole DR, et al. Integrated analysis of microRNA and mRNA expression and association with HIF binding reveals the complexity of microRNA expression regulation under hypoxia. Mol Cancer. 2014;13:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nilsson S, Moller C, Jirstrom K, et al. Downregulation of miR‐92a is associated with aggressive breast cancer features and increased tumour macrophage infiltration. PLoS One. 2012;7:e36051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Datta C, Subuddhi A, Kumar M, et al. Genome‐wide mRNA‐miRNA profiling uncovers a role of the microRNA miR‐29b‐1‐5p/PHLPP1 signalling pathway in Helicobacter pylori‐driven matrix metalloproteinase production in gastric epithelial cells. Cell Microbiol. 2018;e12859. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials