

Abstract
Sym004 is a 1:1 mixture of two antibodies targeting non‐overlapping epitopes of the epidermal growth factor receptor that antagonizes ligand binding and induces receptor downregulation. In preclinical models, it has superior antitumor activity to cetuximab and panitumumab. Japanese adults aged ≥20 years with an Eastern Cooperative Oncology Group status of 0/1 and life expectancy ≥3 months were eligible. Patients in Part A (dose escalation) had refractory or recurrent late‐stage solid tumors and received Sym004 6 mg/kg/wk (n = 3), 9 mg/kg loading/6 mg/kg/wk (n = 6), 12 mg/kg/wk (n = 6), or 18 mg/kg biweekly (n = 6). Patients in expansion Part B (n = 30) had esophageal squamous cell carcinoma and received Sym004 at the dose recommended from Part A. Fifty‐one patients received Sym004. No dose‐limiting toxicities were observed in Part A. A dose of 12 mg/kg/wk was selected for Part B. All patients in Part B experienced treatment‐related adverse events, most commonly dermatitis acneiform (76.7%). Eighteen grade ≥3 treatment‐related adverse events and five serious adverse events occurred (cardiac arrest, lung infection, interstitial lung disease, toxic skin eruption, blood creatinine increase). Two patients had treatment‐related adverse events resulting in death (cardiac arrest and blood creatinine increase). Five patients in Part B had a best overall response of partial response, 12 stable diseases and 12 disease progression (1 not evaluable). The objective response rate was 16.7% (95% CI: 5.6%‐34.7%). Sym004 therapy was well tolerated with no dose‐limiting toxicities at any dose studied. Evidence of antitumor activity was seen in patients with esophageal squamous cell carcinoma. ClinicalTrials.gov Identifier: NCT01955473.
Keywords: antibody, EGFR, solid tumors, Sym004, therapy
1. INTRODUCTION
Esophageal cancer, most commonly esophageal squamous cell carcinoma (ESCC), is the seventh leading cause of cancer death in Japan. It is an aggressive malignancy with an annual mortality of >11 000. Curative surgery is possible following the early diagnosis of ESCC, but treatment options are otherwise limited. Chemotherapy provides modest improvement in overall survival, but causes considerable toxicity.1 A significant unmet need for more effective therapies for ESCC in Japan and elsewhere therefore remains.
Improved understanding of the molecular biology of cancer has seen the development of targeted therapies in recent years, with some remarkable successes documented. The epidermal growth factor receptor (EGFR) has been identified as an important oncogenic driver in several tumor types including ESCC,2, 3 for which aberrant EGFR activity is associated with aggressive disease and poor clinical outcome.2, 4, 5, 6 Aberrant EGFR activity in tumors can arise through gene amplification, somatic mutation, overexpression of EGFR protein, or increased exposure to ligands.7 EGFR overexpression occurs in 32%‐86% of ESCCs.8, 9, 10
Agents targeting the EGFR signaling pathway have proven effective in the treatment of various solid tumors with aberrant EGFR activity.3 Two anti‐EGFR antibodies, cetuximab and panitumumab, are approved by the US Food and Drug Administration for treatment of colorectal cancer (CRC), and cetuximab is also approved for the treatment of head and neck cancer.11, 12 Each targets a different epitope in the extracellular domain III of EFGR and inhibits signaling by antagonizing ligand binding.13, 14 Cetuximab can also induce antibody‐dependent cellular cytotoxicity (ADCC), an activity which may contribute to its clinical efficacy.15 However, EGFR signaling may persist in tumors exposed to cetuximab or panitumumab through ligand‐independent activation. Furthermore, tumors can acquire resistance to anti‐EGFR antibody therapy through mutation of the EGFR antibody‐binding epitopes. Anti‐EGFR antibodies less susceptible to these limitations may be more effective anticancer therapies.
Sym004 is a 1:1 mixture of 2 recombinant human/mouse chimeric IgG1 antibodies (mAb992 and mAb1024) directed against non‐overlapping epitopes in the extracellular domain of EGFR. This combination was selected following systematic testing of antibody combinations for the ability to inhibit cancer cell growth in vivo and in vitro.16 Unlike cetuximab and panitumumab, Sym004 can induce rapid internalization and degradation of EGFR.17 This leads to inhibition of both ligand‐dependent and ligand‐independent EGFR activity. Sym004 also exhibits potent ADCC and complement‐mediated cytotoxicity in vitro.16 Furthermore, because Sym004 targets two separate EGFR epitopes, complete resistance to Sym004 is unlikely to develop through mutation of EGFR. These properties are thought to contribute to the greater efficacy of Sym004 compared to the comparator cetuximab in preclinical models.18 Sym004 has also demonstrated antitumor activity in models of acquired cetuximab resistance,19 including examples where resistance arises through mutation of EGFR.20 Clinically, Sym004 has shown the evidence of efficacy in patients with squamous cell carcinoma of the head and neck21 and in patients with CRC,22 including one with acquired resistance to cetuximab.20 We therefore hypothesized that Sym004 would be active against ESCC driven by oncogenic EGFR. To test this hypothesis, we conducted a phase I trial to establish the safety, tolerability, pharmacokinetics and preliminary antitumor activity of Sym004 in Japanese patients with solid tumors, with an expansion cohort of patients with ESCC.
2. MATERIALS AND METHODS
2.1. Study design and treatment
This was a phase I, open label, single‐arm study of Sym004 (trial number EMR200637‐001). The trial was conducted in accordance with the Declaration of Helsinki, the International Council for Harmonization guideline for Good Clinical Practice (GCP), the Japanese ministerial ordinance on GCP, and all other applicable regulations.
The trial consisted of two parts, with dose escalation (Part A) based on a traditional “3 + 3” design followed by expansion at the recommended phase II dose (RP2D) established in Part A (Part B). Sym004 was administered intravenously. The Sym004 starting dose in Part A was 6 mg/kg weekly, escalating to a loading dose of 9 mg/kg and a maintenance dose of 6 mg/kg weekly, then 12 mg/kg weekly, and finally 18 mg/kg biweekly. Whether to escalate the dose was decided by a Safety Monitoring Committee (SMC) according to tolerability and the occurrence of dose‐limiting toxicity (DLT) during the 4 weeks following the first dose.
Subjects in Part B received weekly Sym004 doses at the RP2D, which was the maximum tolerated dose (MTD) established in Part A, or a dose that was lower than the MTD and determined to be appropriate by the SMC after safety assessment of Part A. The MTD was defined as the next lower dose level to the dose level that was confirmed as too toxic in Part A (ie, caused a DLT in 2 or more of 6 patients). Dosing was continued until unacceptable toxicity, disease progression, or withdrawal of consent occurred.
The primary objective of the study was to evaluate the safety and tolerability of Sym004 in Japanese patients with advanced solid tumors, including ESCC. Secondary objectives included assessment of antitumor activity and pharmacokinetics.
2.2. Patients
For Part A, Japanese adults aged ≥20 years with refractory or recurrent histologically confirmed late‐stage tumors for which no therapeutic options were available were recruited. Patients were required to have an Eastern Cooperative Oncology Group performance status of 0 or 1, and a life expectancy of ≥3 months.
For Part B, patients were required to have histologically confirmed, refractory or recurrent ESCC for which there was no available standard therapy and with at least one measurable tumor defined by Response Evaluation Criteria in Solid Tumors (RECIST) v1.1.
Key exclusion criteria for both parts included: symptomatic brain metastases; prior total resection or irradiation of the target lesion; prior treatment with cytotoxic or cytostatic anticancer therapy, antibody therapy, tyrosine kinase inhibitors, or any investigational agent within 4 weeks; prior vaccine therapy within 12 weeks; diarrhea grade >1; and skin manifestation grade >1 according to the National Cancer Institute – Common Terminology Criteria for Adverse Events (NCI‐CTCAE) v4.03.
2.3. Assessments
Patients were monitored continually for treatment‐emergent adverse events (TEAEs) throughout the reporting period. These were graded according to NCI‐CTCAE v4.03. A DLT was defined as one of the following toxicities occurring during the first 4 weeks of treatment: grade 4 neutropenia (absolute neutrophil count [ANC] < 0.5 × 109/L for ≥7 days); febrile neutropenia (fever ≥38.5°C with ANC < 1.0 × 109/L); grade 4 thrombocytopenia (platelet count ≤ 25.0 × 109/L); grade 3 thrombocytopenia with bleeding episodes; any nonhematologic toxicity of grade ≥3 except for: grade 3 fatigue that improved to grade ≤2 within 2 weeks; grade 3 nausea or vomiting; grade 3 skin toxicity that recovered to grade ≤2 within 2 weeks; grade 3 diarrhea that recovered within 2 days; and grade 3/4 laboratory liver parameter abnormalities with a duration <3 days. A serious adverse event (SAE) was defined as any untoward medical occurrence that resulted in death, was life‐threatening, required hospitalization, resulted in persistent or significant disability/incapacity, or was otherwise considered medically important. Antitumor activity was assessed according to RECIST v1.1.
Blood samples were taken for pharmacokinetic (PK) assessment and immunogenicity evaluation. In Part B only, paired tumor biopsies were highly recommended but not mandated for patients with accessible tumors. The first biopsy was performed during the screening period or in Week 1 before first administration of Sym004. The second tumor biopsy was performed on treatment at Week 4. Blood samples for PK analysis and skin biopsies for pharmacodynamic (Pd) assessment (EGFR expression) were performed at the same time if possible. EGFR expression and amplification in skin and tumor biopsies were analyzed by immunohistochemistry (IHC; H‐score) and fluorescence in situ hybridization (FISH; positive/negative), respectively.
2.4. Pharmacokinetics
Blood samples for PK analysis were taken in Week 1 and Week 4 (Week 5 for patients receiving Sym004 biweekly) before infusion, after infusion, and at +4, +8, +12, and 24 hours (and +48 hours, Week 1 only). Additional blood samples were taken before infusion in all other weeks up to Week 8 and during follow‐up assessment (4 and 8 weeks after final dose), and after infusion in Week 2 (weekly regimens) and Week 3 (all regimens). Serum concentrations of both Sym004 component antibodies were determined using validated bioanalytical methods.
Non‐compartmental PK parameters were calculated using Phoenix® WinNonlin® V6.3 (Certara USA, Inc., Princeton, NJ, USA) or higher. For each patient, the following parameters were determined as appropriate: the area under the curve (AUC) from start of first infusion to 168 hours (AUC(0‐168 h)); AUC from start of first infusion to the last sampling time at which the concentration was greater than or equal to the lower limit of quantification (AUC (0‐last)); half‐life (t 1/2); terminal first‐order rate constant (λz); apparent total body clearance (CL) and clearance at steady state (CLss); apparent volume of distribution during the terminal phase (V z) and at steady state (V ss); maximum concentration (C max); trough concentration (C trough); and time to C max (t max). For patients receiving the biweekly regimen, AUC from the start of infusion to 336 hours (AUC(0‐336)) and AUC from start of first infusion extrapolated to infinity (AUC(0‐inf)) were determined instead of AUC(0‐168 h) and AUC (0‐last), respectively.
2.5. Statistical plan
The planned sample size of 54 patients (24 in Part A and 30 in Part B) meant that there was a 95% probability of identifying a potentially previously unrecognized TEAE occurring at a true incidence of 5.4%.
The primary endpoints were DLT and TEAEs. Secondary endpoints were PK, Pd, antitumor activity, and immunogenicity. Summary statistics, including mean values, frequencies, percentages, and measures of variation, were calculated for patient cohorts and combined groups as appropriate.
The safety analysis set included all subjects from Parts A and B who received at least 1 dose of Sym004. To be included in the efficacy analysis set, patients were additionally required to have a baseline tumor assessment and at least one tumor assessment according to RECIST v1.1 after the first dose of Sym004.
All statistical analyses were performed using SAS® v9.2 (SAS Institute Inc., Cary, NC, USA). The best overall response rate and the disease control rate were estimated along with 95% confidence intervals using the method of Clopper and Pearson. Progression‐free survival and time to progression were estimated using the Kaplan‐Meier method.
3. RESULTS
3.1. Patient population
A total of 51 patients were enrolled. The first patient's first visit occurred on 30 October 2013; the last patient's last visit was 30 October 2015. Baseline patient characteristics are shown in Table 1.
Table 1.
Baseline characteristics (safety population)
| Part A | Part B | ||||
|---|---|---|---|---|---|
| Sym004 6 mg/kg | Sym004 9/6 mg/kg | Sym004 12 mg/kg | Sym004 18 mg/kg | Sym004 12 mg/kg | |
| Patients, n | 3 | 6 | 6 | 6 | 30 |
| Mean age (years) ± SD | 59.7 ± 3.51 | 62.5 ± 5.09 | 66.5 ± 5.96 | 59.0 ± 9.32 | 61.2 ± 7.2 |
| Male/female | 1/2 | 5/1 | 3/3 | 6/0 | 24/6 |
| ECOG performance status, n | |||||
| 0 | 3 | 5 | 4 | 4 | 16 |
| 1 | 0 | 1 | 2 | 2 | 14 |
| >1 | 0 | 0 | 0 | 0 | 0 |
| Tumor type, n (%) | |||||
| Adenocarcinoma | 2 (66.7) | 4 (66.6) | 4 (66.6) | 5 (83.3) | 0 |
| Squamous cell carcinoma | 1 (33.3) | 2 (33.3) | 2 (33.3) | 0 | 30 (100.0) |
| Other | 0 | 0 | 0 | 1 (16.7) | 0 |
| Prior therapy, n (%) | 3 (100.0) | 6 (100.0) | 6 (100.0) | 6 (100.0) | 30 (100.0) |
| Surgery | 2 (66.7) | 4 (66.7) | 4 (66.7) | 4 (66.7) | 18 (60.0) |
| Radiotherapy | 0 | 1 (16.7) | 1 (16.7) | 2 (33.3) | 21 (70.0) |
| Chemotherapy | 3 (100.0) | 6 (100.0) | 6 (100) | 6 (100.0) | 30 (100.0) |
| Monoclonal antibodya | 2 (66.7) | 3 (50%) | 0 | 2 (33.3) | 0 |
| Protein kinase inhibitorb | 1 (33.3) | 2 (33.3) | 1 (16.7) | 3 (50.0) | 0 |
ECOG, Eastern Cooperative Oncology Group; SD, standard deviation.
Monoclonal antibodies included bevicuzumab, cetuximab, panitumumab, ramucirumab, and trastuzumab.
Protein kinase inhibitors included erlotinib, gefitinib, regorafenib, and sorafenib.
3.2. Safety
The incidence of TEAEs is shown in Table 2. No DLTs occurred during Part A. All patients experienced at least 1 treatment‐related TEAE, the most frequently reported being dermatitis acneiform (n = 43, 84.3%), hypomagnesemia (n = 39, 76.5%), and dry skin (n = 25, 49%). A total of 24 (47.1%) patients experienced at least 1 grade ≥3 treatment‐related TEAE, and 5 (9.8%) experienced at least 1 grade ≥4 treatment‐related TEAE. Those occurring in ≥2 patients are listed in Table 2.
Table 2.
Summary of TEAEs (safety population)
| Number of patients, n (%) | Part A | Part B | |||
|---|---|---|---|---|---|
| Sym004 6 mg/kg (n = 3) | Sym004 9/6 mg/kg (n = 6) | Sym004 12 mg/kg (n = 6) | Sym004 18 mg/kg (n = 6) | Sym004 12 mg/kg (n = 30) | |
| With TEAEs | |||||
| ≥1 event (any grade) | 3 (100) | 6 (100) | 6 (100) | 6 (100) | 30 (100) |
| ≥1 grade ≥3 event | 3 (100.0) | 1 (16.7) | 3 (50.0) | 1 (16.7) | 21 (70.0) |
| ≥1 serious event | 0 | 0 | 2 (33.3) | 0 | 9 (30.0) |
| ≥1 event leading to treatment withdrawal | 0 | 0 | 0 | 0 | 4 (13.3) |
| ≥1 event leading to trial termination | 0 | 0 | 0 | 0 | 4 (13.3) |
| ≥1 event leading to death | 0 | 0 | 0 | 0 | 3 (10.0) |
| ≥1 event leading to dose reduction | 1 (33.3) | 0 | 0 | 0 | 3 (10.0) |
| ≥1 event leading to dose interruption | 3 (100.0) | 3 (50.0) | 4 (66.7) | 0 | 15 (50.0) |
| With treatment‐related TEAEs | |||||
| ≥1 event (any grade) | 3 (100.0) | 6 (100.0) | 6 (100.0) | 6 (100.0) | 30 (100.0) |
| ≥1 grade ≥3 event | 3 (100.0) | 1 (16.7) | 2 (33.3) | 0 | 18 (60.0) |
| ≥1 serious event | 0 | 0 | 0 | 0 | 5 (16.7) |
| ≥1 event leading to death | 0 | 0 | 0 | 0 | 2 (6.7) |
| Grade ≥3 treatment‐related TEAEs occurring in ≥2 patients | |||||
| Fatigue (grade 3) | 1 (33.3) | 0 | 0 | 0 | 1 (3.3) |
| Hypocalcemia (grade 3) | 0 | 0 | 0 | 0 | 2 (6.7) |
| Hypomagnesemia (grade 3) | 0 | 0 | 0 | 0 | 2 (6.7) |
| Hypomagnesemia (grade 4) | 0 | 0 | 0 | 0 | 3 (10.0) |
| Blood magnesium decreased (grade 3) | 0 | 0 | 0 | 0 | 2 (6.7) |
| Dermatitis acneiform (grade 3) | 1 (33.3) | 0 | 2 (33.3) | 0 | 9 (30.0) |
| Dry skin (grade 3) | 1 (33.3) | 0 | 0 | 0 | 1 (3.3) |
| Skin fissures (grade 3) | 1 (33.3) | 1 (16.7) | 0 | 0 | 0 |
TEAE, treatment‐emergent adverse events.
Eleven patients (21.6%) experienced a total of 14 SAEs (all in patients receiving 12 mg/kg Sym004: 2 in Part A, 9 in Part B). Five treatment‐related SAEs occurred, all in Part B. These were: cardiac arrest (1); lung infection (1); interstitial lung disease (1); skin eruptions (1); and blood creatinine increase (1). Two of these (cardiac arrest and blood creatinine increase) resulted in death. The patient who died due to cardiac arrest received 4 doses of Sym004 12 mg/kg weekly and experienced other Sym004‐related TEAEs including rash and mucositis, both grade 1. The patient who died due to blood creatinine increase also received Sym004 12 mg/kg weekly; the investigator assessed the event as suspected to be related to Sym004, but disease and medical history were also listed as causative factors. This patient also experienced a number of other Sym004‐related TEAEs, including pain, palmar‐plantar erythrodysesthesia syndrome, and hypomagnesemia (all grade 2) and grade 3 rash. No evidence of differences in pharmacokinetic parameters between these patients and the overall group of patients receiving Sym004 12 mg/kg/wk were observed (data not shown).
Most patients (n = 45, 88.2%) completed study treatment and discontinued the trial due to progressive disease (n = 44, 86.3%) or death (n = 1, 2.0%) at the time of data cut‐off. The other reasons of discontinuation were TEAEs (n = 3, 5.9%), withdrawal of consent (n = 2, 3.9%) or other reasons (n = 1, 2.0%).
Based on these data, the SMC set the Sym004 dose for Part B at 12 mg/kg weekly, below the highest dose tested of 18 mg/kg biweekly, to reduce the risk of severe TEAEs.
3.3. Antitumor activity
Sym004 showed signs of efficacy, with an overall objective response rate (complete plus partial response) in Parts A and B of 13.7% and an objective response rate in patients in Part B of 16.7% (Table 3). The duration of response for patients in Part B is shown in Figure 1. In Part B, median progression‐free survival (defined as the time from first dose until disease progression or death) was 9.2 weeks (95% CI: 6.0, 12.3), and the median time to progression (defined as the time from first dose until disease progression) was 10.3 weeks (95% CI: 6.0, 16.0).
Table 3.
Best overall response to Sym004 and disease response/control rates (efficacy analysis set)
| Number of patients | Part A | Part B | |||
|---|---|---|---|---|---|
| Sym004 6 mg/kg (n = 3) | Sym004 9/6 mg/kg (n = 6) | Sym004 12 mg/kg (n = 6) | Sym004 18 mg/kg (n = 6) | Sym004 12 mg/kg (n = 30) | |
| Best overall response, n (%) | |||||
| Complete response (CR) | 0 | 0 | 0 | 0 | 0 |
| Partial response (PR) | 0 | 0 | 2 (33.3) | 0 | 5 (16.7) |
| Stable disease (SD) | 2 (66.7) | 4 (66.7) | 1 (16.7) | 1 (16.7) | 12 (40.0) |
| Progressive disease (PD) | 1 (33.3) | 2 (33.3) | 3 (50.0) | 5 (83.3) | 12 (40.0) |
| Not evaluable | 0 | 0 | 0 | 0 | 1 (3.3) |
| Objective response rate, CR + PR, n (%) [95% CI, %] | 0 [0.0, 70.8] | 0 [0.0, 45.9] | 2 (33.3) [4.3, 77.7] | 0 [0.0, 45.9] | 5 (16.7) [5.6, 34.7] |
| Disease control rate, CR + PR + SD, n (%) [95% CI, %] | 2 (66.7) [9.4, 99.2] | 4 (66.7) [22.3, 95.7] | 3 (50.0) [11.8, 88.2] | 1 (16.7) [0.4, 64.1] | 17 (56.7) (37.4, 74.5] |
| Median duration of disease control, wks (range) | 6.1 (6.1, 6.1) | 5.4 (4.0, 6.1) | 24.6 (4.1, 27.1) | 6.1 (6.1, 6.1) | 5.9 (0.1, 22.1) |
Figure 1.
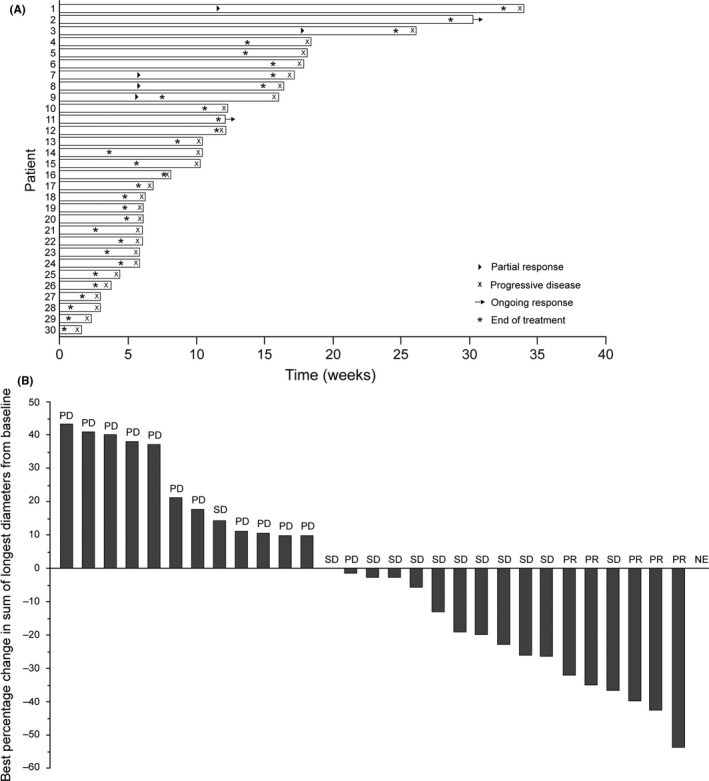
Response to Sym004 therapy of patients in Part B of the trial. (A) Swimmer plot and (B) waterfall plot. NE, not evaluated; PD, progressive disease; PR, partial response; SD, stable disease
3.4. Pharmacokinetics
Following the first dose and multiple doses of Sym004, dose‐normalized exposure measures for the constituent antibodies mAb992 and mAb1024 were consistent among patients in Part A and Part B, indicating reasonable dose proportionality. Following multiple dosing, the geometric mean t1/2 for mAb992 ranged from 85.2 to 136 hours, and for mAb1024 from 117.8 to 163.8 hours (Tables 4,5). The median t max for mAb992 ranged from 5.1 to 10.1 hours, and for mAb 1024 from 2.1 to 7.1 hours (Figure 2). CLss and V ss for both constituent antibodies were dose‐independent and were consistent between Parts A and B.
Table 4.
Summary of PK parameters for mAb992 after a single dose of Sym004 at wk 1 and wk 4 (weekly regimens) or wk 5 (biweekly regimens)
| Geometric mean (GCV%) | |||||||
|---|---|---|---|---|---|---|---|
| AUC0‐inf (μg.h/mL) | AUC0‐inf/dose (μg.h/mL/mg) | C max (μg/mL) | t max (h)a | t 1/2 (h) | CL (L/h) | V z (L) | |
| Wk 1 | |||||||
| 6 mg/kg weekly (n = 3) | 4471 (35.0) | 27.30 (26.7) | 50.13 (29.2) | 2.07 (2.0, 14) | 66.48 (10.0) | 0.037 (26.7) | 3.51 (36.1) |
| 9/6 mg/kg weekly (n = 6) | 7693 (38.3) | 30.33 (37.7) | 85.09 (25.3) | 5.12 (3.1, 7.2) | 79.04 (18.8) | 0.033 (37.7) | 3.76 (21.9) |
| 12 mg/kg weekly, Part A (n = 6) | 11 970 (34.2) | 34.47 (22.5) | 120.37 (33.6) | 5.73 (3.0, 11) | 82.87 (21.1) | 0.029 (22.5) | 3.47 (17.8) |
| 18 mg/kg biweekly (n = 6) | 19 896 (28.9) | 33.77 (30.3) | 157.71 (18.6) | 7.21 (3.2, 15) | 111.7 (18.9) | 0.030 (30.3) | 4.77 (23.3) |
| 12 mg/kg weekly, Part B (n = 7) | 9677 (22.3) | 31.05 (24.6) | 88.59 (15.0) | 6.30 (3.2, 11) | 87.26 (21.1) | 0.032 (24.6) | 4.05 (10.8) |
| Wk 4 or 5 | |||||||
| 6 mg/kg weekly (n = 3) | ND | ND | 76.09 (16.4) | 10.1 (6.0, 14) | ND | ND | ND |
| 9/6 mg/kg weekly (n = 5) | 9662.7 (51.4) | 59.65 (37.3) | 86.76b (34.0) | 5.94b (2.0, 10) | 85.23 (24.4) | 0.023 (27.3) | 2.79 (15.4) |
| 12 mg/kg weekly, Part A (n = 6) | 27 409 (79.0) | 77.86 (65.2) | 181.6 (31.6) | 9.13 (7.1, 11) | 136.0 (52.5) | 0.023 (29.3) | 4.48 (26.2) |
| 18 mg/kg biweekly (n = 5) | 30 127 (36.6) | 51.67 (36.2) | 187.4b (24.3) | 7.03b (3.1, 11) | 130.4 (26.4) | 0.024 (29.9) | 4.40 (24.4) |
| 12 mg/kg weekly, Part B (n = 5) | 22 240 (14.4) | 72.22 (17.1) | 143.8b (23.4) | 5.13b (2.9, 11) | 132.1 (13.9) | 0.024 (15.0) | 4.44 (17.9) |
AUC0‐inf, area under the curve from start of first infusion extrapolated to infinity; C max, maximum concentration; t max, time to C max; t 1/2, half‐life; CL, clearance; V z, apparent volume of distribution during the terminal phase; GCV%, geometric coefficient of variation; ND, not determined.
Median and range are reported.
n = 6.
Table 5.
Summary of PK parameters for mAb1024 after a single dose of Sym004 at week 1 and week 4 (weekly regimens) or week 5 (biweekly regimens)
| Geometric mean (GCV%) | |||||||
|---|---|---|---|---|---|---|---|
| AUC0‐inf (μg.h/mL) | AUC0‐inf/dose (μg.h/mL/mg) | C max (μg/mL) | t max (h)a | t 1/2 (h) | CL (L/h) | V z (L) | |
| Wk 1 | |||||||
| 6 mg/kg weekly (n = 3) | 5217.9 (26.6) | 31.86 (21.9) | 54.66 (15.6) | 2.05 (2.0, 2.1) | 74.08 (8.8) | 0.031 (21.9) | 3.35 (16.7) |
| 9/6 mg/kg weekly (n = 6) | 9182.6 (38.8) | 36.20 (37.4) | 89.44 (26.2) | 7.20 (3.2, 11) | 91.30 (21.7) | 0.028 (37.4) | 3.64 (17.6) |
| 12 mg/kg weekly, Part A (n = 6) | 15 075 (34.6) | 43.41 (23.1) | 116.2 (31.2) | 6.66 (3.1,11) | 100.1 (24.3) | 0.023 (23.1) | 3.32 (3.4) |
| 18 mg/kg biweekly (n = 6) | 27 554 (30.7) | 46.77 (30.9) | 187.0 (23.9) | 7.15 (3.2, 7.6) | 134.5 (27.8) | 0.021 (30.9) | 4.15 (29.1) |
| 12 mg/kg weekly, Part B (n = 6) | 14 781 (17.4) | 47.48 (17.5) | 146.9b (56.3) | 7.13b (3.2, 11) | 100.6 (34.8) | 0.021 (17.6) | 3.06 (47.0) |
| Wk 4 or 5 | |||||||
| 6 mg/kg weekly (n = 3) | ND | ND | 89.26 (10.2) | 2.07 (2.0, 2.1) | ND | ND | ND |
| 9/6 mg/kg weekly (n = 4) | 15 631 (51.4) | 94.91 (44.7) | 91.70c (33.4) | 6.13c (2.0, 18) | 117.8 (27.5) | 0.017 (30.6) | 2.85 (20.6) |
| 12 mg/kg weekly, Part A (n = 6) | 41 581 (71.2) | 118.1 (57.5) | 221.3 (21.1) | 5.18 (3.0, 7.3) | 155.9 (37.4) | 0.016 (32.9) | 3.71 (21.7) |
| 18 mg/kg biweekly (n = 5) | 43 308 (37.8) | 74.28 (36.3) | 224.0c (24.7) | 5.28c (3.1, 11) | 163.6 (36.3) | 0.018 (25.6) | 4.20 (27.7) |
| 12 mg/kg weekly, Part B (n = 4) | 41 351 (42.0) | 136.9 (46.8) | 231.9c (32.8) | 7.10c (3.1, 11) | 163.8 (27.5) | 0.014 (28.2) | 3.40 (24.9) |
AUC0‐inf, area under the curve from start of first infusion extrapolated to infinity; C max, maximum concentration; t max, time to C max; t 1/2, half‐life; CL, clearance; V z, apparent volume of distribution during the terminal phase; GCV%, geometric coefficient of variation; ND, not determined.
Median and range are reported.
n = 7.
n = 6.
Figure 2.
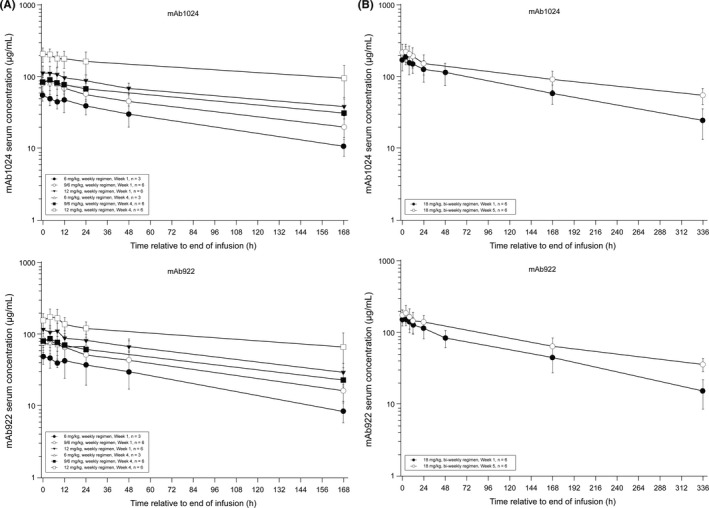
Mean (±SD) serum concentration‐time profiles for mAb1024 and mAb992 after (A) the 1st (wk 1) and 4th (wk 4) weekly infusions and (B) the 1st (wk 1) and 3rd (wk 5) weekly infusions (semilogarithmic scale)
3.5. Pharmacodynamics
No detectable Pd effects of Sym004 were observed from the analysis of EGFR expression/amplification in paired biopsies of either skin or tumor.
3.6. Biomarkers
Epidermal growth factor receptor was assessed as an exploratory biomarker by IHC and FISH. In part B, 13 paired tumor samples were available. The H‐score of 7 predose samples was in the range 200‐300, of 1 sample was 100 to <200, and was missing for 5. At week 4, the H‐score of 2 samples was 200‐300, of 4 was 100 to <200, and was missing for 7. No tumor samples were EGFR FISH‐positive, 7 were negative, and data for 6 were missing.
3.7. Immunogenicity
No anti‐Sym004 antibodies were found in any of the 170 patient serum samples that were screened.
4. DISCUSSION
In this phase I study conducted in Japanese patients with solid tumors, doses of Sym004 up to 18 mg/kg biweekly were explored. No DLTs were observed at any dose, so an MTD was not established. Although the trial protocol permitted a dose of 18 mg/kg biweekly to be used in the extension study (Part B), a dose of 12 mg/kg weekly was chosen to limit the severity and incidence of treatment‐related TEAEs, which occurred in all patients (dermatitis acneiform, hypomagnesemia, and dry skin) and were consistent with anti‐EGFR antibody class effects.23, 24, 25 These events were generally mild (grade ≤2), although 18 grade ≥3 TEAEs and 5 SAEs occurred in Part B, with 2 (cardiac arrest and blood creatinine increase) leading to death. No association between the use of other EGFR inhibitors and cardiotoxicity has been reported, suggesting that this may be an incidental finding. However, further investigation and monitoring of such events in patients receiving Sym004 is warranted.
In patients with previously treated ESCC (Part B) there was evidence of antitumor activity, with a partial response recorded for 5 patients (16.7%). It is possible that a higher response rate may have been achieved if patients had been selected for the trial based on the expression of molecular biomarkers. EGFR amplification and EGFR overexpression are known to increase the sensitivity of tumors to EGFR antibody therapy;26, 27 with increased EGFR gene copy number (GCN) seen in 51.5%27 and overexpression in 32%‐86% of ESCCs,8, 9, 10 selection of patients according to EGFR GCN status or EGFR expression status, determined by in situ hybridization and IHC, respectively,28 has the potential to improve response rates.29 The exclusion of patients with oncogenic alterations potentially conferring resistance to EGFR therapy, eg, KRAS mutations, could also improve response rates.27 Whether Sym004 is more effective in tumors with EGFR amplification or EGFR overexpression requires further study.
The pharmacokinetics of Sym004 determined in this study were comparable with those obtained in non‐Japanese patients with CRC22 and its tolerability at all dose levels was confirmed. Both the 12 mg/kg weekly and 18 mg/kg biweekly doses were candidate RP2Ds for both Japanese and global patients. The 12 mg/kg dose provides the highest drug exposure and, while unsurprisingly all SAEs were observed in patients treated at this dose, it was selected as the RP2D to be adopted in phase II trials of Sym004.
Monoclonal anti‐EGFR antibodies have demonstrated activity in preclinical models of ESCC,30, 31, 32 and when given in combination with chemotherapy and radiotherapy have shown signs of efficacy in patients with esophageal cancer.33, 34 Nevertheless, phase II/III trials of gefitinib alone and cetuximab in combination with chemoradiotherapy for the treatment of patients with esophageal cancer failed to demonstrate efficacy.35, 36 In the context of the results of clinical trials of single‐agent therapy in patients with refractory esophageal cancer, which have shown response rates of 15%‐17% (nivolumab),37, 38 the antitumor activity of Sym004 therapy reported in this study is promising but needs to be confirmed. We hypothesize that Sym004 may be superior to other anti‐EGFR therapies tested in ESCC because Sym004 is a mixture of 2 anti‐EGFR monoclonal antibodies targeting different epitopes, providing the specificity of a monoclonal antibody therapy combined with a degree of diversity. This provides antitumor activity in preclinical models that is superior to that of monoclonal antibodies. Unlike engineered antibody‐like molecules (eg, bispecific antibodies) diversity is not associated with an increased uncertainty regarding immunogenicity. Further trials are needed to establish whether Sym004 can provide significantly greater benefit to patients with ESCC and other types of tumor compared to currently available therapies.
In conclusion, Sym004 represents a novel antibody‐mediated approach to EGFR inhibition. In this trial, we have shown that Sym004 has an acceptable tolerability and safety profile in Japanese patients with solid tumors, including ESCC, and that this is similar to that established previously in Western patients.22 The findings demonstrate that further study of Sym004 therapy for the treatment of patients with ESCC may be warranted.
CONFLICT OF INTEREST
This study was designed under the responsibility of Merck Serono Co., Ltd., Tokyo, Japan, a subsidiary of Merck KGaA, Darmstadt, Germany in conjunction with the steering committee. It was funded by Merck KGaA, Darmstadt, Germany. Toshihiko Doi has received research funding from Merck Serono. Thomas Goddemeier and Stefan Kuffel are employees of Merck KGaA, Darmstadt, Germany. Morihiro Watanabe is an employee of Merck Serono Co., Ltd., Meguro‐ku, Tokyo, Japan. The other authors declare no conflict of interest.
ACKNOWLEDGEMENTS
The authors would like to thank patients and their families, investigators, co‐investigators, and the study teams at each of the participating centers and at Merck KGaA, Darmstadt, Germany and Merck Serono Co. Ltd. Tokyo, Japan. This study was funded by Merck KGaA, Darmstadt, Germany. Medical writing assistance was provided by Russell Huby, Bioscript Science, Macclesfield, UK, and funded by Merck KGaA, Darmstadt, Germany.
Kojima T, Yamazaki K, Kato K, et al. Phase I dose‐escalation trial of Sym004, an anti‐EGFR antibody mixture, in Japanese patients with advanced solid tumors. Cancer Sci. 2018;109:3253–3262. 10.1111/cas.13767
REFERENCES
- 1. Kothari N, Almhanna K. Current status of novel agents in advanced gastroesophageal adenocarcinoma. J Gastrointest Oncol. 2015;6:60‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mendelsohn J, Baselga J. Status of epidermal growth factor receptor antagonists in the biology and treatment of cancer. J Clin Oncol. 2003;21:2787‐2799. [DOI] [PubMed] [Google Scholar]
- 3. Sawada G, Niida A, Hirata H, et al. An integrative analysis to identify driver genes in esophageal squamous cell carcinoma. PLoS ONE. 2015;10:e0139808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jiang D, Li X, Wang H, et al. The prognostic value of EGFR overexpression and amplification in esophageal squamous cell carcinoma. BMC Cancer. 2015;15:377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kaneko K, Kumekawa Y, Makino R, et al. EGFR gene alterations as a prognostic biomarker in advanced esophageal squamous cell carcinoma. Front Biosci (Landmark Ed). 2010;15:65‐72. [DOI] [PubMed] [Google Scholar]
- 6. Lin G, Sun XJ, Han QB, et al. Epidermal growth factor receptor protein overexpression and gene amplification are associated with aggressive biological behaviors of esophageal squamous cell carcinoma. Oncol Lett. 2015;10:901‐906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hanawa M, Suzuki S, Dobashi Y, et al. EGFR protein overexpression and gene amplification in squamous cell carcinomas of the esophagus. Int J Cancer. 2006;118:1173‐1180. [DOI] [PubMed] [Google Scholar]
- 8. Jia J, Cui Y, Lu M, et al. The relation of EGFR expression by immunohistochemical staining and clinical response of combination treatment of nimotuzumab and chemotherapy in esophageal squamous cell carcinoma. Clin Transl Oncol. 2016;18:592‐598. [DOI] [PubMed] [Google Scholar]
- 9. Li JC, Zhao YH, Wang XY, et al. Clinical significance of the expression of EGFR signaling pathway‐related proteins in esophageal squamous cell carcinoma. Tumour Biol. 2014;35:651‐657. [DOI] [PubMed] [Google Scholar]
- 10. Yang YL, Xu KL, Zhou Y, Gao X, Chen LR. Correlation of epidermal growth factor receptor overexpression with increased epidermal growth factor receptor gene copy number in esophageal squamous cell carcinomas. Chin Med J (Engl). 2012;125:450‐454. [PubMed] [Google Scholar]
- 11. FDA . FDA Approval for cetuximab. http://www.cancer.gov/about-cancer/treatment/drugs/fda-cetuximab. Accessed July 1, 2018.
- 12. FDA . FDA approval for panitumumab. http://www.cancer.gov/about-cancer/treatment/drugs/fda-panitumumab. Accessed July 1, 2018.
- 13. Li S, Schmitz KR, Jeffrey PD, et al. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7:301‐311. [DOI] [PubMed] [Google Scholar]
- 14. Cohenuram M, Saif MW. Panitumumab the first fully human monoclonal antibody: from the bench to the clinic. Anticancer Drugs. 2007;18:7‐15. [DOI] [PubMed] [Google Scholar]
- 15. Kawaguchi Y, Kono K, Mimura K, et al. Cetuximab induce antibody‐dependent cellular cytotoxicity against EGFR‐expressing esophageal squamous cell carcinoma. Int J Cancer. 2007;120:781‐787. [DOI] [PubMed] [Google Scholar]
- 16. Koefoed K, Steinaa L, Soderberg JN, et al. Rational identification of an optimal antibody mixture for targeting the epidermal growth factor receptor. MAbs. 2011;3:584‐595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dienstmann R, Salazar R, Tabernero J. Overcoming resistance to anti‐EGFR therapy in colorectal cancer. Am Soc Clin Oncol Educ Book. 2015:e149‐e156. https://meetinglibrary.asco.org/record/105303/edbook. Accessed July 1, 2018. [DOI] [PubMed] [Google Scholar]
- 18. Pedersen MW, Jacobsen HJ, Koefoed K, et al. Sym004: a novel synergistic anti‐epidermal growth factor receptor antibody mixture with superior anticancer efficacy. Cancer Res. 2010;70:588‐597. [DOI] [PubMed] [Google Scholar]
- 19. Iida M, Brand TM, Starr MM, et al. Sym004, a novel EGFR antibody mixture, can overcome acquired resistance to cetuximab. Neoplasia. 2013;15:1196‐1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sanchez‐Martin FJ, Bellosillo B, Gelabert M, et al. The first‐in‐class anti‐EGFR antibody mixture Sym004 overcomes cetuximab‐resistance mediated by EGFR extracellular domain mutations in colorectal cancer. Clin Cancer Res. 2016;22(13):3260‐3267. [DOI] [PubMed] [Google Scholar]
- 21. Machiels JP, Specenier P, Krauss J, et al. A proof of concept trial of the anti‐EGFR antibody mixture Sym004 in patients with squamous cell carcinoma of the head and neck. Cancer Chemother Pharmacol. 2015;76:13‐20. [DOI] [PubMed] [Google Scholar]
- 22. Dienstmann R, Patnaik A, Garcia‐Carbonero R, et al. Safety and activity of the first‐in‐class Sym004 anti‐EGFR antibody mixture in patients with refractory colorectal cancer. Cancer Discov. 2015;5:598‐609. [DOI] [PubMed] [Google Scholar]
- 23. Kozuki T. Skin problems and EGFR‐tyrosine kinase inhibitor. Jpn J Clin Oncol. 2016;46:291‐298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Petrelli F, Borgonovo K, Cabiddu M, Ghilardi M, Barni S. Risk of anti‐EGFR monoclonal antibody‐related hypomagnesemia: systematic review and pooled analysis of randomized studies. Expert Opin Drug Saf. 2012;11(Suppl 1):S9‐S19. [DOI] [PubMed] [Google Scholar]
- 25. FDA . Erbitux® Prescribing Information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/125084s0228lbl.pdf. Accessed July 1, 2018. [Google Scholar]
- 26. Fukuoka S, Kojima T, Koga Y, et al. Preclinical efficacy of Sym004, novel anti‐EGFR antibody mixture, in esophageal squamous cell carcinoma cell lines. Oncotarget. 2017;8:11020‐11029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Guo K, Wang WP, Jiang T, et al. Assessment of epidermal growth factor receptor mutation/copy number and K‐ras mutation in esophageal cancer. J Thorac Dis. 2016;8:1753‐1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sunpaweravong P, Suwiwat S, Sunpaweravong S, Puttawibul P, Mitarnun W. Correlation of epidermal growth factor receptor mutation, immunohistochemistry, and fluorescence in situ hybridization in esophageal squamous cell carcinoma. J Med Assoc Thai. 2009;92:1136‐1142. [PubMed] [Google Scholar]
- 29. Dienstmann R, Salazar R, Tabernero J. The evolution of our molecular understanding of colorectal cancer: what we are doing now, what the future holds, and how tumor profiling is just the beginning. Am Soc Clin Oncol Educ Book. 2014:91‐99. https://meetinglibrary.asco.org/record/89089/edbook. Accessed July 1, 2018. [DOI] [PubMed] [Google Scholar]
- 30. Wu X, Zhang J, Zhen R, et al. Trastuzumab anti‐tumor efficacy in patient‐derived esophageal squamous cell carcinoma xenograft (PDECX) mouse models. J Transl Med. 2012;10:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mimura K, Kono K, Maruyama T, et al. Lapatinib inhibits receptor phosphorylation and cell growth and enhances antibody‐dependent cellular cytotoxicity of EGFR‐ and HER2‐overexpressing esophageal cancer cell lines. Int J Cancer. 2011;129:2408‐2416. [DOI] [PubMed] [Google Scholar]
- 32. Sutter AP, Hopfner M, Huether A, Maaser K, Scherubl H. Targeting the epidermal growth factor receptor by Erlotinib (Tarceva) for the treatment of esophageal cancer. Int J Cancer. 2006;118:1814‐1822. [DOI] [PubMed] [Google Scholar]
- 33. Wang C, Fu X, Cai X, et al. High‐dose nimotuzumab improves the survival rate of esophageal cancer patients who underwent radiotherapy. Onco Targets Ther. 2016;9:117‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen Y, Wu X, Bu S, et al. Promising outcomes of definitive chemoradiation and cetuximab for patients with esophageal squamous cell carcinoma. Cancer Sci. 2012;103:1979‐1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dutton SJ, Ferry DR, Blazeby JM, et al. Gefitinib for oesophageal cancer progressing after chemotherapy (COG): a phase 3, multicentre, double‐blind, placebo‐controlled randomised trial. Lancet Oncol. 2014;15:894‐904. [DOI] [PubMed] [Google Scholar]
- 36. Crosby T, Hurt CN, Falk S, et al. Chemoradiotherapy with or without cetuximab in patients with oesophageal cancer (SCOPE1): a multicentre, phase 2/3 randomised trial. Lancet Oncol. 2013;14:627‐637. [DOI] [PubMed] [Google Scholar]
- 37. Kudo T, Hamamoto Y, Kato K, et al. Nivolumab treatment for oesophageal squamous‐cell carcinoma: an open‐label, multicentre, phase 2 trial. Lancet Oncol. 2017;18:631‐639. [DOI] [PubMed] [Google Scholar]
- 38. Burkart C, Bokemeyer C, Klump B, et al. A phase II trial of weekly irinotecan in cisplatin‐refractory esophageal cancer. Anticancer Res. 2007;27:2845‐2848. [PubMed] [Google Scholar]