

Abstract
Background
Given that diabetes-associated complications are closely associated with neuroinflammation, it is imperative to study potential changes in neuroinflammatory modulators in the central nervous system of diabetic primates.
Methods
The mRNA levels of pro-inflammatory and anti-inflammatory cytokines, toll-like receptors (TLR), growth factors, and cannabinoid receptors were compared in the spinal dorsal horn (SDH) and thalamus of naturally occurring type 2 diabetic monkeys and an age-matched control group using reverse transcription and quantitative real-time polymerase chain reaction.
Results
In the SDH of diabetic monkeys, we observed increases in the mRNA levels of pro-inflammatory cytokines (i.e., interleukin-1β and tumor necrosis factor-α (TNFα)), TLR1, and TLR2, and decreases in the mRNA levels of interleukin-10, an anti-inflammatory cytokine. No changes were observed in the mRNA levels of growth factors and cannabinoid receptors. In line with the mRNA data, TNFα-immunoreactivity was also significantly increased in diabetic monkeys. Moreover, the mRNA expression levels of interleukin-1β, TNFα, TLR1, and TLR2 in the SDH were positively correlated with plasma glucose levels in all monkeys.
Conclusions
Several ligands and receptors involved in neuroinflammation are simultaneously dysregulated in the spinal cord of diabetic monkeys. This primate disease model will facilitate the design of novel treatment approaches to ameliorate neuroinflammation-driven adverse effects in diabetic patients.
Keywords: cytokine, spinal dorsal horn, nonhuman primate, interleukin, type 2 diabetes
Introduction
Type 2 diabetes mellitus (T2DM), a long-term metabolic disorder, severely affects the eyes, kidneys, and peripheral and autonomic nervous systems, accounting for vision loss, end-stage kidney disease, dismemberment, and the increased risk of cardiovascular disease1,2. Neuropathy is one of the intractable symptoms of T2DM, and more than 50% of T2DM patients experience sensory dysfunction3–5. Given that T2DM-associated complications are closely associated with neuroinflammation6–10, characterization of neuroinflammatory events (e.g., altered expression of inflammatory ligands and receptors for sensory processing in the central nervous system (CNS)) in diabetic primates could substantially facilitate the development of novel treatment options.
Glial activation and upregulation of inflammatory mediators such as chemokines CC-chemokine ligand 2 (CCL2) and CXC-chemokine ligand 10 (CXCL10) have been reported in rodents with T2DM11,12. Compared with rodent models, there are numerous changes in the spinal dorsal horn (SDH) of monkeys with T2DM. In addition to activated microglia and astrocytes, three chemokine ligand-receptor systems, CCL2-CCR2, CCL3-CCR1/5, and CCL4-CCR5, and multiple chemokine receptors (i.e., CCR4, CCR6, CCR8, CCR10, CXCR3, CXCR5, and CXCR6) are simultaneously upregulated13. In addition, mRNA expression levels of classical opioid receptor subtypes, μ, κ, and δ, are downregulated. These findings demonstrate that there is potential crosstalk between chemokines and opioids in the spinal cord of diabetic monkeys13. However, potential changes in other ligand-receptor systems associated with neuroinflammation in primates with T2DM are virtually unknown.
Neuroinflammatory processes are classified as either pro-inflammatory or anti-inflammatory events, and are dynamically regulated by several mediators14–16. Maintaining steady balance between pro- and anti-inflammatory mediators is required for homeostasis in the CNS17,18. Among these modulators, cytokines, toll-like receptors (TLRs), growth factors, and cannabinoid receptors differentially affect inflammatory processes in rodents15,19–25. Pro-inflammatory cytokines, such as interleukin-1β (IL-1β) and tumor necrosis factor-α (TNFα), are well characterized in the SDH. Pharmacological and genetic manipulations of these mediators in rodents relieve or modulate intractable inflammatory diseases26–28. Although rodent studies have documented the functional significance of these neuroinflammatory modulators, the translatability of their functional changes from rodents to primates with chronic diseases remains unclear.
In monkeys with T2DM, considerable changes in cutaneous innervation were shown to be consistent with sensory deficits experienced by patients with diabetic neuropathy29,30. Thus, diabetic monkeys can be a valuable model for studying T2DM pathophysiology31,32. In this study, we examined potential changes in neuroinflammatory modulators in the CNS of diabetic monkeys. Because the SDH and thalamus play a fundamental role in sensory processing33,34, we specifically analyzed pro-inflammatory and anti-inflammatory cytokines, TLRs, growth factors, and cannabinoid receptors in the SDH and thalamus of monkeys with T2DM. These findings will facilitate the design of novel treatments which may ameliorate adverse events associated with T2DM.
Methods
Animals
All experimental procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the US National Institutes of Health (Bethesda, MD, USA) and approved by the Institutional Animal Care and Use Committee of Wake Forest University School of Medicine (Winston-Salem, NC, USA). Seven (one male and six females) cynomolgus macaques (Macaca fascicularis) diagnosed with 3-6 years of spontaneously occurring type 2 diabetes and six (one male and five females) non-diabetic (control) cynomolgus macaques were used. Animals were housed individually in a room with a temperature of 21–25 °C, 40–60% relative humidity, and a 12-h light/12-h dark cycle at Wake Forest University Primate Center. They were fed with regular chow (LabDiet 5038, St. Louis, MO, USA), fresh fruits, and water ad libitum. General characteristics of all subjects are shown in Supplementary Table S1. Subjects were diagnosed with diabetes by veterinarians. These subjects showed increased glucose, cholesterol, triglycerides, and hemoglobin A1c (HbA1C) levels, which are similar to those of other diabetic monkeys32,35,36.
Reverse transcription and quantitative real-time polymerase chain reaction
Detailed procedures for tissue collection, and reverse transcription and quantitative real-time polymerase chain reaction (RT-qPCR) were previously described13. Briefly, the skull and spinal column were cut open to expose the brain and spinal cord. The ventral posterior thalamus and L4 spinal dorsal horn tissues were dissected according to the histology atlas of the Rhesus monkey brain37. The TRIzol® Plus RNA Purification Kit (Thermo Fisher Scientific, Waltham, MA) was used to isolate total RNA from tissues. One microgram of purified total RNA was converted to cDNA by reverse transcription using Random primers (Promega, Madison, WI), dNTP mix (Thermo Fisher Scientific), and M-MLV reverse transcriptase (Promega). qPCR was performed by using the synthesized cDNA, gene-specific primers (Table 1, Sigma-Aldrich, St. Louis, MO), and iTaq™ Universal SYBR® Green Supermix with iCycler and iQ Real-Time PCR systems (Bio-Rad, Hercules, CA). Reactions were performed under the following conditions: 10 min at 95 °C, followed by 50 cycles of two steps, 20 sec at 95 °C, and 40 sec at 60 or 62 °C, depending on the Tm value of primers. According to the previously characterized comparative CT method38, the threshold cycle (CT) value was defined as the PCR cycle at which the relative fluorescent unit (RFU) crossed a threshold line (100 RFU). The mRNA expression level of each gene was quantified based on the average CT value from three replicates and was normalized to β-actin (ACTB) using the formula 2−(CTtarget gene – CTACTB) (2−ΔCT). All data from individual monkeys are presented as mean ± standard error of mean (SEM). Data were analyzed using unpaired t-tests. Correlation analysis was performed using the Pearson’s correlation coefficient. The criterion for significance was set at P<0.05.
Table 1.
Primer sequences for RT-qPCR
| Gene | Forward (5′ to 3′) | Reverse (5′ to 3′) |
|---|---|---|
| ACTB | TCTTCCAACCTTCCTTCCTG | TGTGTTGGCGTACAGGTCTT |
| IL-1β | GCACCTTGATTCCCTTCATC | TGCAGTGCAGTGATCGTACA |
| IL-6 | TCAGCCCTGAGAAAGGAGAC | TTTCAGCCATCTTTGGAAGG |
| IL-18 | ATCGGCCCCTATTTGAAGAT | CCATACCTCTAGGCTGGCTAT |
| TNFα | TGTCTGCTGCACTTTGGAGT | CTTGGGGTTCGAGAAGATGA |
| IFNγ | GTCCAACGCAAAGCAGTACA | TCGACCTCGAAACATCTGAC |
| IL-1RN | GGAAGATGTGCCTGTCCTGT | AGCGCTTGTCCTGTTTTCTG |
| IL-4 | AACTGCCATATCGCCTTACG | GCAGCAAGGATGTCCGTTAT |
| IL-10 | GCGCTGTCATCGATTTCTTC | TGGCTTTGTAGACGCCTTTC |
| IL-13 | ATGGTGTGGAGCATCAACCT | TTCAGCATCCTCTGGGTCTT |
| IL-18BP | ACTGAATGGAACGCTGACCT | GGAGGTGCTCAATGAAGGAA |
| TLR1 | TTCTGGGGTTGAGCACTACA | TTGTCCCCATAATGCTCTCC |
| TLR2 | GCCTCTCCAAGGAAGAATCC | GAGCACTGAGGGAATGGAGT |
| TLR3 | TTGCTTGGCTTCCACAACTA | TCAGGTACCGCACATTGAAA |
| TLR4 | AATCCCCTGAGGCATTTAGG | CCCCATCTTCAATTGTCTGG |
| TLR5 | GACAAGGAGGCCTTCAGAAA | GGAACAGTCCCTGAAAAGCA |
| TLR6 | CGCAGACAGCATTGTACACC | GAATGTGCTTGGTGCATGAG |
| TLR7 | TAAGTGGAAATTGCCCTCGT | TTCTGTCAGCGCATCAAAAG |
| TLR8 | CTGAGCAACACCCAGATCAA | ATTGAAGCACCTCGGACAAT |
| TLR9 | CACAAGAGGGTGTCCTTTGC | ATCGAGTGAGCGGAAGAAGA |
| TLR10 | TTCTGTCTTCAGGTGCTTGC | AGCTCTCGTAAGGCCATCAG |
| CSF1 | TCAGAGACAACACCCCCAAT | GGCCTTGTCATGCTCTTCAT |
| BDNF | CAAACATCCGAGGACAAGGT | GATGTCAGGCCTCTTGAACC |
| NGF | GGGAGCAGCTTTCTATCCTG | GTGTGGTTCTGCCTGTATGC |
| VEGF | TCTTCAAGCCATCCTGTGTG | CTGCATGGTGATGTTGGACT |
| GDNF | CCGGGGTTGTGTCTTAACTG | AGAGCCGCTGCAGTACCTAA |
| TGFβ1 | CAGCTCCACGGAGAAGAACT | CAGAAGTTGGCATGGTAGCC |
| CB1 | ATCCTAGATGGCCTTGCAGA | TGCCATGTCACCTTTGATGT |
| CB2 | TCCACTGATTCCCAACGACT | TGATGGGCTTTCCAGAGAAC |
Immunohistochemistry
Spinal cord tissues were fixed in 4% paraformaldehyde and cryoprotected in phosphate buffered saline (PBS) containing 30% sucrose. They were subsequently embedded in an optimal cutting temperature compound (Sakura Finetek USA, Torrance, CA) and kept at −80 °C until use. Frozen tissues were cut at a thickness of 30 μm with a cryostat (Leica CM3050S; Leica Biosystems GmbH, Wetzlar, Germany). The tissue sections were treated with 0.3% H2O2 for 15 min, washed with PBS and blocked with PBS containing 0.3% Triton X-100 and 3% normal donkey serum at room temperature for 30 min. The sections were incubated with a primary antibody against TNFα (rabbit polyclonal, 1:500; ab6671, Abcam, Cambridge, MA) at 4 °C overnight. The next day, sections were rinsed in PBS containing 0.1% Triton X-100 (PBST), and incubated with biotinylated secondary antibody (1:400; Jackson ImmunoResearch Laboratories, West Grove, PA) at room temperature for 1 h. All antibodies were diluted in PBST containing 1% normal donkey serum. Sections were rinsed in PBST, and incubated with Vectastain ABC kit (PK-6100; Vector Laboratories, Burlingame, CA) for 30 min. After rinsing in PBST, sections were stained using a DAB (3,3′-diaminobenzidine) substrate kit (SK-4100; Vector Laboratories), mounted on glass slides to air dry, dehydrated through a graded series of ethanol and xylene, and cover-slipped with mounting medium DPX (Sigma-Aldrich, St. Louis, MO, USA).
Image analysis
Images of spinal cord sections were acquired at 10x magnification using a Nikon Eclipse Ni fluorescent and brightfield microscope (Nikon, Tokyo, Japan). The Image J software (National Institutes of Health, Bethesda, MD) was used to quantify TNFα immunostaining in the superficial laminae (I-II) of four randomly selected lumbar spinal cord sections from each animal. The data were presented as the fractional area, which reflects the percentage of the sampled area that fell within a defined density threshold. The individual who analyzed the images was blind to the experimental conditions.
Results
Upregulation of pro-inflammatory cytokines in the SDH of diabetic monkeys
The mRNA expression levels of IL-1β, IL-6, IL-18, TNFα, and interferon-γ (IFNγ), were evaluated using RT-qPCR in order to determine if these pro-inflammatory cytokines were altered in the SDH and thalamus of diabetic monkeys. In the SDH of non-diabetic monkeys, the basal mRNA expression level of IL-18 was the highest, followed by IL-1β and TNFα (Figure 1A). We found that the mRNA expression levels of IL-1β (1.3-fold, 95% confidence interval=−0.563 to 0.057, t=1.80, degrees of freedom (df)=11, r2=0.23, P=0.049) and TNFα (1.3-fold, 95% confidence interval=−0.691 to 0.039, t=1.97, df=11, r2= 0.26, P=0.038) were upregulated in the SDH of diabetic monkeys compared to those of non-diabetic monkeys (Figure 1B). In the thalamus of non-diabetic monkeys, basal mRNA expression patterns of these cytokines were similar to those of cytokines in the SDH (Figure 1C). However, none of these cytokine genes showed significant changes in their mRNA expression levels between diabetic and non-diabetic monkeys (Figure 1D). Interestingly, the mRNA expression levels of IL-1β and TNFα in the SDH were positively correlated with plasma glucose levels of all subjects (Supplementary Figure S1). To examine whether there was a change in protein level in the SDH of diabetic monkeys, immunohistochemistry for TNFα was performed and analyzed. In line with qPCR data, TNFα-immunoreactivity was significantly increased in diabetic monkeys than in non-diabetic monkeys (1.8-fold, 95% confidence interval=−2.074 to 33.99, t=1.95, df=11, r2=0.26, P=0.039) (Figure 2).
Figure 1. Upregulation of pro-inflammatory cytokines in the spinal dorsal horn of diabetic monkeys.
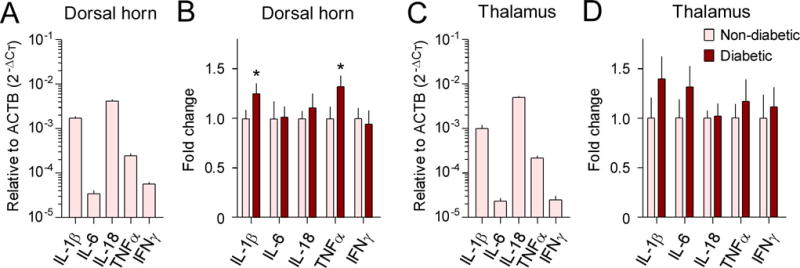
The mRNA expression levels of IL-1β, IL-6, IL-18, TNFα, and IFNγ in the spinal dorsal horn (A, B) and thalamus (C, D) were evaluated with RT-qPCR using the comparative CT method (2−ΔCT). Basal mRNA expression levels of these cytokines relative to β-actin (ACTB) in the non-diabetic group are shown in A and C. Fold changes of mRNA expression levels in the type 2 diabetic group compared to that in the non-diabetic control group are shown in B and D. Each value is presented as the mean ± SEM (n=6–7). *P < 0.05 vs. non-diabetic.
Figure 2. Upregulation of TNFα immunoreactivity in the spinal dorsal horn of diabetic monkeys.
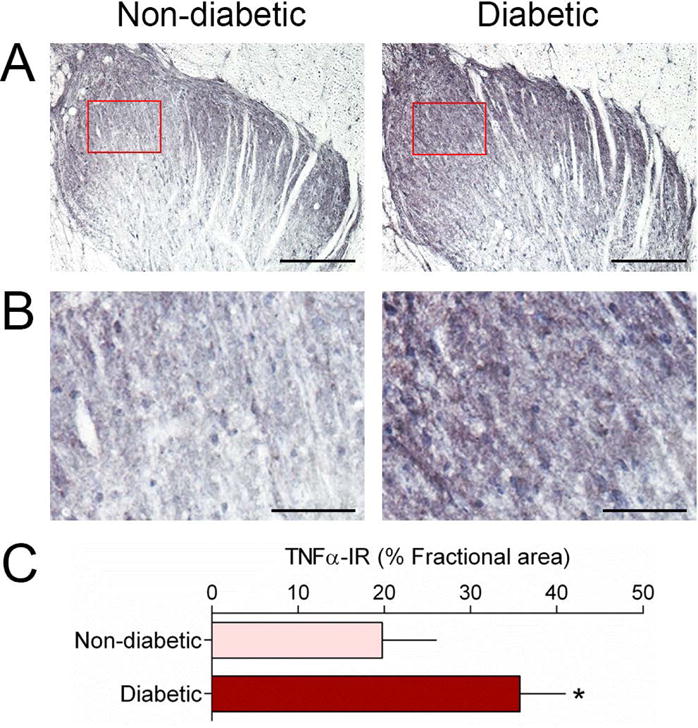
Representative images of TNFα immunohistochemistry in non-diabetic and diabetic monkeys (A). Higher magnification of the boxed area in panel A showing a denser immunostaining in the diabetic group (B). Quantification of TNFα immunostaining was expressed as the fractional area which reflects the percentage of the sampled area that fell within a defined density threshold (C). Scale bars, 200 μm (A) and 50 μm (B). Each value is presented as the mean ± SEM. (n = 6–7). *P < 0.05 vs. non-diabetic.
Altered expression of anti-inflammatory cytokines in the SDH and thalamus of diabetic monkeys
The mRNA expression levels of the IL-1 receptor antagonist (IL-1RN), IL-4, IL-10, IL-13, and IL-18 binding protein (IL-18BP) were quantified and compared using RT-qPCR in order to investigate whether these anti-inflammatory cytokines were changed in the SDH and thalamus of diabetic monkeys. The basal mRNA expression level of IL-18BP was the highest, followed by IL-1RN and IL-10 in the SDH of non-diabetic monkeys (Figure 3A). We found that the mRNA expression level of IL-18BP (1.2-fold, 95% confidence interval=−0.505 to 0.013, t=2.09, df=11, r2=0.28, P=0.030) was upregulated, while the expression of IL-10 (0.7-fold, 95% confidence interval=−0.039 to 0.553, t=1.91, df=11, r2=0.25, P=0.041) was downregulated in the SDH of diabetic monkeys than in non-diabetic monkeys (Figure 3B). In the thalamus of non-diabetic monkeys, the basal mRNA expression level of IL-18BP was much higher than that of the other four molecules (Figure 3C). As observed for the SDH, IL-10 expression (0.6-fold, 95% confidence interval=−0.019 to 0.773, t=2.10, df=11, r2=0.29, P=0.030) was also downregulated in the thalamus of diabetic monkeys. However, the level of expression of IL-18BP did not increase significantly in the thalamus of diabetic monkeys (Figure 3D). The mRNA expression levels of IL-10 and IL-18BP in the SDH, or IL-10 in the thalamus did not correlate with plasma glucose levels in any of the subjects (Supplementary Figures S2 & S3).
Figure 3. Altered expression of anti-inflammatory cytokines in the spinal dorsal horn and thalamus of diabetic monkeys.
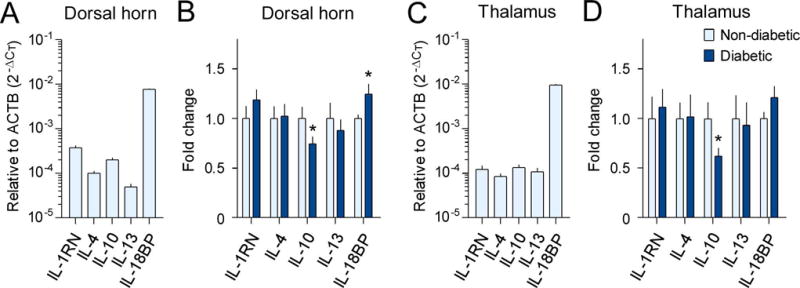
The mRNA expression levels of IL-1RN, IL-4, IL-10, IL-13, and IL-18BP in the spinal dorsal horn (A, B) and thalamus (C, D) were evaluated with RT-qPCR using the comparative CT method (2−ΔCT). The basal mRNA expression levels of these cytokines relative to β-actin (ACTB) in the non-diabetic group are shown in A and C. Fold changes in mRNA expression levels in the type 2 diabetic group compared to those in the non-diabetic control group are shown in B and D. Each value is presented as the mean ± SEM (n=6–7). *P < 0.05 vs. non-diabetic.
Upregulation of toll-like receptors in the SDH of diabetic monkeys
In order to examine whether TLRs were altered in the SDH and thalamus of diabetic monkeys, we used the RT-qPCR to determine the mRNA expression levels of TLR subtypes (i.e., from TLR1 to TLR10). The basal mRNA expression level of TLR4 was the highest, followed by TLR1 and TLR2 in the SDH of non-diabetic monkeys (Figure 4A). We found that the mRNA expression levels of TLR1 (1.2-fold, 95% confidence interval=−0.432 to 0.008, t=2.12, df=11, r2=0.29, P=0.029) and TLR2 (1.4-fold, 95% confidence interval=−0.733 to 0.019, t=2.09, df=11, r2=0.28, P=0.030) were upregulated in the SDH of diabetic monkeys, compared with non-diabetic monkeys (Figure 4B). In the thalamus of non-diabetic monkeys, basal mRNA expression patterns of these ten TLR subtypes were similar to those in the SDH (Figure 4C). Nonetheless, none of these ten TLR genes displayed significant changes in their mRNA expressions between diabetic and non-diabetic monkeys (Figure 4D). The mRNA expression levels of TLR1 and TLR2 in the SDH from all subjects were positively correlated with plasma glucose levels (Supplementary Figure S4).
Figure 4. Upregulation of toll-like receptors in the spinal dorsal horn of diabetic monkeys.
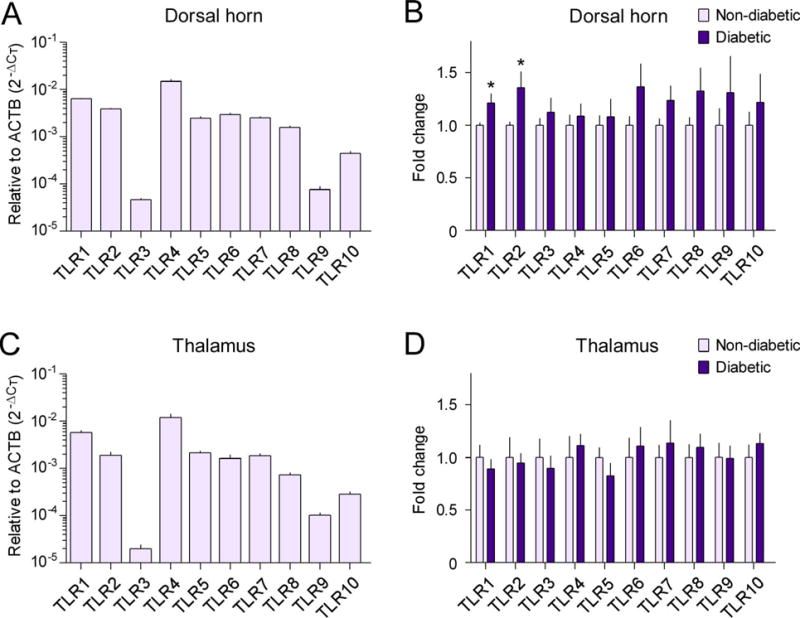
The mRNA expression levels of TLR subtypes, TLR1-TLR10, in the spinal dorsal horn (A, B) and thalamus (C, D) were evaluated with RT-qPCR using the comparative CT method (2−ΔCT). The basal mRNA expression levels of these receptors relative to β-actin (ACTB) in the non-diabetic group are shown in A and C. Fold changes of mRNA expression levels in the type 2 diabetic group compared to those in the non-diabetic control group are shown in B and D. Each value is presented as the mean ± SEM (n=6–7). *P < 0.05 vs. non-diabetic.
Expression of growth factors in the SDH and thalamus of diabetic monkeys
Next, we further investigated whether the expression of growth factors was altered in the SDH and thalamus of diabetic monkeys by determining mRNA expression levels of colony stimulating factor 1 (CSF1), brain-derived neurotrophic factor (BDNF), nerve growth factor (NGF), vascular endothelial growth factor (VEGF), glial cell-derived growth factor (GDNF), and transforming growth factor β1 (TGFβ1). In the SDH of non-diabetic monkeys, basal mRNA expression levels of CSF1, VEGF, and TGFβ1 were more abundant than other growth factors, such as BDNF (Figure 5A). Surprisingly, the mRNA expression levels of these genes did not change significantly between the SDH of diabetic and non-diabetic monkeys (Figure 5B). The basal mRNA expression levels of CSF1, VEGF, and TGFβ1 were relatively higher than those of GDNF, BDNF, and NGF, in the thalamus of non-diabetic monkeys (Figure 5C). However, none of these six genes had mRNA expression levels that were significantly altered in the thalamus of diabetic monkeys, compared with those in non-diabetic monkeys (Figure 5D).
Figure 5. Expression of growth factors in the spinal dorsal horn and thalamus of diabetic monkeys.
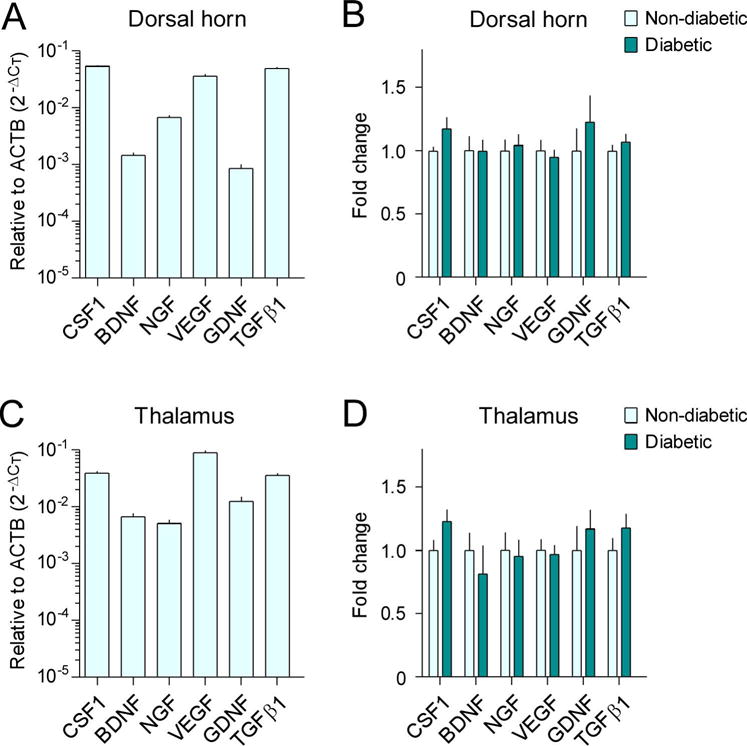
The mRNA expression levels of CSF1, BDNF, NGF, VEGF, GDNF, and TGFβ1 in the spinal dorsal horn (A, B) and thalamus (C, D) were evaluated with RT-qPCR using the comparative CT method (2−ΔCT). The basal mRNA expression levels of these growth factors relative to β-actin (ACTB) in the non-diabetic group are shown in A and C. Fold changes of mRNA expression levels in the type 2 diabetic group compared to those in the non-diabetic control group are shown in B and D. Each value is presented as the mean ± SEM (n=6–7).
Expression of cannabinoid receptors in the SDH and thalamus of diabetic monkeys
Finally, we evaluated the mRNA expression levels of CB1 and CB2 receptors in order to determine whether the expression of cannabinoid receptors were altered in the SDH and thalamus of diabetic monkeys. In both the SDH and thalamus of non-diabetic monkeys, the basal mRNA expression level of the CB1 receptor was much higher than that of the CB2 receptor (Figure 6A, C). Neither the mRNA expression level of the CB1 receptor nor that of the CB2 receptor was significantly changed in the SDH and thalamus of diabetic monkeys, compared to that in non-diabetic monkeys (Figure 6B, D).
Figure 6. Expression of cannabinoid receptors in the spinal dorsal horn and thalamus of diabetic monkeys.
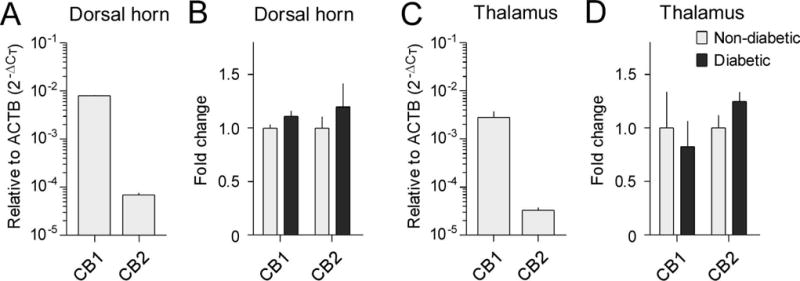
The mRNA expression levels of CB1 and CB2 in the spinal dorsal horn (A, B) and thalamus (C, D) were evaluated with RT-qPCR using the comparative CT method (2−ΔCT). The basal mRNA expression levels of these cannabinoid receptors relative to β-actin (ACTB) in the non-diabetic group are shown in A and C. Fold changes of mRNA expression levels in the type 2 diabetic group as compared with the non-diabetic control group are shown in B and D. Each value is presented as the mean ± SEM (n=6–7).
Discussion
Here we report for the first time that upregulation of IL-1β and TNFα gene expression occurs in the spinal cord of diabetic monkeys. IL-1β and TNFα, and their cognate receptors, are located on microglia, astrocytes, and neurons. They contribute to neuroinflammation in the CNS16,23,39,40. Particularly, inhibition of these pro-inflammatory cytokines using pharmacological or genetic approaches alleviated the symptoms arising from spinal neuroinflammation in rodents15,41–43. Given that microglia and astrocytes were activated in the SDH of monkeys with T2DM13, and that neuron-glia interactions are bidirectional26,27, the upregulation of IL-1β and TNFα is closely related to glial activation and spinal neuroinflammation. Future studies to investigate whether the inhibition of both glial activation and pro-inflammatory cytokines can effectively alleviate neuroinflammation-associated symptoms in patients with T2DM are warranted.
Anti-inflammatory cytokines play a critical role in the suppression and resolution of inflammation44,45, which is a key component in the homeostatic control of neuroinflammation. We found that the mRNA expression level of IL-10 was downregulated in the SDH and thalamus of diabetic monkeys. According to previous findings, the IL-10 expression level in the SDH could control neuroinflammation in different rodent models46,47. Intrathecal delivery of IL-10 prevented the expression of pro-inflammatory cytokines such as IL-1β and TNFα, and relieved symptoms due to spinal neuroinflammation48–50. On the other hand, the functional role of IL-10 in the thalamus has not been characterized in animals. To our knowledge, this is the first report showing a correlation between the central IL-10 expression level and T2DM in primates. It will be essential to understand the function of IL-10 in modulating spinal neuroinflammation in diabetic monkeys. Unlike the expression of IL-10, IL-18BP expression was upregulated in the SDH of diabetic monkeys. Given that IL-18BP counteracts the function of IL-18, which involves glial activation and neuroinflammation51, the upregulation of IL-18BP in the SDH of diabetic monkeys may be one of the homeostatic mechanisms for suppressing the function of IL-1852. Overall, the balance between pro- and anti-inflammatory cytokines is critical for modulating neuroinflammation in the CNS of diabetic monkeys.
Among all the TLR subtypes examined herein, we found that mRNA expression levels of TLR1 and TLR2 were upregulated in the SDH of diabetic monkeys. Growing evidence demonstrates that TLR2 was upregulated in the SDH and participated in glial activation and cytokine expression in rodents53,54. Importantly, TLR2 and TLR4 were upregulated and accounted for spinal neuroinflammation in patients with amyotrophic lateral sclerosis55. TLR2 forms heterodimers with TLR1, and TLR1/TLR2 is involved in glial activation and the upregulation of inflammatory mediators that contribute to pathological conditions56,57. Further investigation into the functional roles of TLR1 and TLR2 in the SDH of diabetic monkeys is necessary. Being a well-characterized neuroinflammatory mediator19,39,40,54, it is surprising to find that although TLR4 was upregulated in the SDH of diabetic mice12, TLR4 expression did not change in the SDH and thalamus of diabetic monkeys.
Although some growth factors are known as inflammatory modulators23,26,40, all growth factors in the SDH and thalamus of diabetic monkeys examined in this study did not change. For example, BDNF plays a pivotal role in microglial activation and drives neuropathic pain in rodents58,59. In contrast, TGFβ1, an anti-inflammatory growth factor, improves spinal sensitization in rodents60,61. Moreover, cerebrospinal fluid levels of VEGF are correlated with pain in patients with failed back surgery syndrome62. Despite the abundance of these growth factors in the CNS, they are not significantly changed in diabetic monkeys. On the other hand, accumulating evidence suggests that CB1 and CB2 receptors could be the targets of anti-inflammatory therapeutics24,25,63. Activation of cannabinoid receptors also inhibits pain transmission in the SDH of rodents64. However, there was no change in CB1 and CB2 receptor expression in the SDH of diabetic monkeys. These findings together indicate that growth factors and cannabinoid receptors may play a minimal role in modulating neuroinflammation in primates with T2DM.
It is worth noting that the upregulation of TNFα mRNA correlated with an increase in TNFα protein levels in diabetic primates. This is in line with the results of a previous study. This study reported a similar correlation between changes in mRNA and protein levels13. Interestingly, the mRNA levels of pro-inflammatory mediators (i.e., IL-1β, TNFα, TLR1, and TLR2) in the SDH also correlate positively with plasma glucose levels in monkeys. It is also important to point out that the SDH is more susceptible to changes due to a variety of neuroinflammatory modulators in diabetic monkeys than the thalamus13. Pharmacological treatments via the intrathecal route may be a viable therapeutic option as these pro-inflammatory mediators play a critical role in neuroinflammation26,40,56. Diabetic monkeys used in this study were diagnosed with 3.1 to 6.8 years of T2DM. Monkeys with a similar duration of T2DM showed impaired nerve conduction29 and severe changes in the epidermis innervation similar to patients with diabetic neuropathy30, suggesting that the diabetic monkeys used in this study might have abnormal sensory processing. Future functional studies to determine if diabetic monkeys display mechanical hypersensitivity to noxious stimuli and to determine if these behavioral responses could be altered by pharmacological interventions are warranted.
Collectively, this study demonstrates the simultaneous upregulation of pro-inflammatory mediators (IL-1β, TNFα, TLR1, and TLR2) and downregulation of an anti-inflammatory mediator, IL-10, in the SDH of diabetic monkeys. Unlike rodents, no changes were observed in the expression of TLR4, growth factors, and cannabinoid receptors in the SDH of diabetic monkeys. Together with recent findings13, multiple ligands and receptors involved in neuroinflammation are simultaneously dysregulated (i.e., upregulation of pro-inflammatory cytokines, chemokines, and TLRs versus downregulation of opioid receptors and anti-inflammatory cytokines) in the spinal cord of diabetic monkeys. Drugs targeting several receptors instead of a single receptor may be more effective in managing neuroinflammatory symptoms in T2DM. Although it is challenging to recruit diabetic monkeys, this primate disease model not only helped demonstrate the pathological relevance of particular ligands and receptors involved in neuroinflammation, but may also facilitate the design of novel approaches for the treatment of neuroinflammation-driven adverse consequences in diabetic patients.
Supplementary Material
Highlight.
Whether neuroinflammatory modulators, such as cytokines, toll-like receptors (TLR), growth factors, and cannabinoid receptors, change differentially in type 2 diabetic primates is unknown. We found that mRNA expression levels of inflammatory cytokines (interleukin-1β and tumor necrosis factor-α), TLR1, and TLR2 were upregulated in the spinal cord of diabetic monkeys, and positively correlated with their plasma glucose levels. This study indicates that several ligands and receptors involved in neuroinflammation are simultaneously dysregulated in diabetic primates.
Acknowledgments
We thank Drs. Janice Wagner, Mark Cline, and Li Zhang for their suggestions, and Ms. Jean Gardin, Christina Long, Diana Swaim, Melissa Ayers, Jade Lackey, and Kelsey Reynolds for their technical assistance regarding tissue collection and data analysis. We thank Editage for editing this manuscript. This work was supported by the National Institutes of Health, NIDA (R01-DA032568 to M.-C. K. and R21-DA040104 to M.-C. K.) and NIAMS (R01-AR059193 to M.-C. K. and R21-AR064456 to M.-C. K.).
Footnotes
Disclosure
None declared
References
- 1.Laiteerapong N, Karter AJ, Liu JY, et al. Correlates of quality of life in older adults with diabetes: the diabetes & aging study. Diabetes Care. 2011;34:1749–53. doi: 10.2337/dc10-2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nathan DM. Diabetes: Advances in Diagnosis and Treatment. JAMA. 2015;314:1052–62. doi: 10.1001/jama.2015.9536. [DOI] [PubMed] [Google Scholar]
- 3.Callaghan BC, Cheng HT, Stables CL, Smith AL, Feldman EL. Diabetic neuropathy: clinical manifestations and current treatments. Lancet Neurol. 2012;11:521–34. doi: 10.1016/S1474-4422(12)70065-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krein SL, Heisler M, Piette JD, Makki F, Kerr EA. The effect of chronic pain on diabetes patients’ self-management. Diabetes Care. 2005;28:65–70. doi: 10.2337/diacare.28.1.65. [DOI] [PubMed] [Google Scholar]
- 5.Sudore RL, Karter AJ, Huang ES, et al. Symptom burden of adults with type 2 diabetes across the disease course: diabetes & aging study. J Gen Intern Med. 2012;27:1674–81. doi: 10.1007/s11606-012-2132-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cai D. Neuroinflammation and neurodegeneration in overnutrition-induced diseases. Trends Endocrinol Metab. 2013;24:40–7. doi: 10.1016/j.tem.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Felice FG, Ferreira ST. Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease. Diabetes. 2014;63:2262–72. doi: 10.2337/db13-1954. [DOI] [PubMed] [Google Scholar]
- 8.Forbes JM, Cooper ME. Mechanisms of diabetic complications. Physiol Rev. 2013;93:137–88. doi: 10.1152/physrev.00045.2011. [DOI] [PubMed] [Google Scholar]
- 9.Feldman EL, Nave KA, Jensen TS, Bennett DL. New Horizons in Diabetic Neuropathy: Mechanisms, Bioenergetics, and Pain. Neuron. 2017;93:1296–313. doi: 10.1016/j.neuron.2017.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pop-Busui R, Ang L, Holmes C, Gallagher K, Feldman EL. Inflammation as a Therapeutic Target for Diabetic Neuropathies. Curr Diab Rep. 2016;16:29. doi: 10.1007/s11892-016-0727-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dauch JR, Yanik BM, Hsieh W, Oh SS, Cheng HT. Neuron-astrocyte signaling network in spinal cord dorsal horn mediates painful neuropathy of type 2 diabetes. Glia. 2012;60:1301–15. doi: 10.1002/glia.22349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang YP, Song CY, Yuan Y, et al. Diabetic neuropathic pain development in type 2 diabetic mouse model and the prophylactic and therapeutic effects of coenzyme Q10. Neurobiol Dis. 2013;58:169–78. doi: 10.1016/j.nbd.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 13.Kiguchi N, Ding H, Peters CM, et al. Altered expression of glial markers, chemokines, and opioid receptors in the spinal cord of type 2 diabetic monkeys. Biochim Biophys Acta. 2017;1863:274–83. doi: 10.1016/j.bbadis.2016.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–34. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ji RR, Xu ZZ, Gao YJ. Emerging targets in neuroinflammation-driven chronic pain. Nat Rev Drug Discov. 2014;13:533–48. doi: 10.1038/nrd4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nathan C, Ding A. Nonresolving inflammation. Cell. 2010;140:871–82. doi: 10.1016/j.cell.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 17.Prinz M, Priller J. The role of peripheral immune cells in the CNS in steady state and disease. Nat Neurosci. 2017;20:136–44. doi: 10.1038/nn.4475. [DOI] [PubMed] [Google Scholar]
- 18.Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science. 2016;353:777–83. doi: 10.1126/science.aag2590. [DOI] [PubMed] [Google Scholar]
- 19.Hanke ML, Kielian T. Toll-like receptors in health and disease in the brain: mechanisms and therapeutic potential. Clin Sci (Lond) 2011;121:367–87. doi: 10.1042/CS20110164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kopf M, Bachmann MF, Marsland BJ. Averting inflammation by targeting the cytokine environment. Nat Rev Drug Discov. 2010;9:703–18. doi: 10.1038/nrd2805. [DOI] [PubMed] [Google Scholar]
- 21.Liu X, Fang L, Guo TB, Mei H, Zhang JZ. Drug targets in the cytokine universe for autoimmune disease. Trends Immunol. 2013;34:120–8. doi: 10.1016/j.it.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 22.O’Neill LA, Golenbock D, Bowie AG. The history of Toll-like receptors - redefining innate immunity. Nat Rev Immunol. 2013;13:453–60. doi: 10.1038/nri3446. [DOI] [PubMed] [Google Scholar]
- 23.Xanthos DN, Sandkuhler J. Neurogenic neuroinflammation: inflammatory CNS reactions in response to neuronal activity. Nat Rev Neurosci. 2014;15:43–53. doi: 10.1038/nrn3617. [DOI] [PubMed] [Google Scholar]
- 24.Cassano T, Calcagnini S, Pace L, De Marco F, Romano A, Gaetani S. Cannabinoid Receptor 2 Signaling in Neurodegenerative Disorders: From Pathogenesis to a Promising Therapeutic Target. Front Neurosci. 2017;11:30. doi: 10.3389/fnins.2017.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Navarro G, Morales P, Rodriguez-Cueto C, Fernandez-Ruiz J, Jagerovic N, Franco R. Targeting Cannabinoid CB2 Receptors in the Central Nervous System. Medicinal Chemistry Approaches with Focus on Neurodegenerative Disorders. Front Neurosci. 2016;10:406. doi: 10.3389/fnins.2016.00406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ji RR, Chamessian A, Zhang YQ. Pain regulation by non-neuronal cells and inflammation. Science. 2016;354:572–77. doi: 10.1126/science.aaf8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kiguchi N, Kobayashi Y, Kishioka S. Chemokines and cytokines in neuroinflammation leading to neuropathic pain. Curr Opin Pharmacol. 2012;12:55–61. doi: 10.1016/j.coph.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 28.McMahon SB, La Russa F, Bennett DL. Crosstalk between the nociceptive and immune systems in host defence and disease. Nat Rev Neurosci. 2015;16:389–402. doi: 10.1038/nrn3946. [DOI] [PubMed] [Google Scholar]
- 29.Cornblath DR, Hillman MA, Striffler JS, Herman CN, Hansen BC. Peripheral neuropathy in diabetic monkeys. Diabetes. 1989;38:1365–70. doi: 10.2337/diab.38.11.1365. [DOI] [PubMed] [Google Scholar]
- 30.Pare M, Albrecht PJ, Noto CJ, et al. Differential hypertrophy and atrophy among all types of cutaneous innervation in the glabrous skin of the monkey hand during aging and naturally occurring type 2 diabetes. J Comp Neurol. 2007;501:543–67. doi: 10.1002/cne.21262. [DOI] [PubMed] [Google Scholar]
- 31.Gong L, Zeng W, Yang Z, et al. Comparison of the clinical manifestations of type 2 diabetes mellitus between rhesus monkey (Macaca mulatta lasiotis) and human being. Pancreas. 2013;42:537–42. doi: 10.1097/MPA.0b013e3182732501. [DOI] [PubMed] [Google Scholar]
- 32.Wagner JE, Kavanagh K, Ward GM, Auerbach BJ, Harwood HJ, Jr, Kaplan JR. Old world nonhuman primate models of type 2 diabetes mellitus. ILAR J. 2006;47:259–71. doi: 10.1093/ilar.47.3.259. [DOI] [PubMed] [Google Scholar]
- 33.Anderson WS, O’Hara S, Lawson HC, Treede RD, Lenz FA. Plasticity of pain-related neuronal activity in the human thalamus. Prog Brain Res. 2006;157:353–64. doi: 10.1016/S0079-6123(06)57021-9. [DOI] [PubMed] [Google Scholar]
- 34.Peirs C, Seal RP. Neural circuits for pain: Recent advances and current views. Science. 2016;354:578–84. doi: 10.1126/science.aaf8933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McTighe MS, Hansen BC, Ely JJ, Lee DR. Determination of hemoglobin A1c and fasting blood glucose reference intervals in captive chimpanzees (Pan troglodytes) J Am Assoc Lab Anim Sci. 2011;50:165–70. [PMC free article] [PubMed] [Google Scholar]
- 36.Wang X, Wang B, Sun G, Wu J, Liu Y, Wang Y, Xiao YF. Dysglycemia and Dyslipidemia Models in Nonhuman Primates: Part I. Models of Naturally Occurring Diabetes. J Diabetes Metab. 2015;S13:010. [Google Scholar]
- 37.Saleem KS, Logothetis NK. A Combined MRI and Histology Atlas of the Rhesus Monkey Brain in Stereotaxic Coordinates. Second. Academic Press; 2012. [Google Scholar]
- 38.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–8. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 39.Grace PM, Hutchinson MR, Maier SF, Watkins LR. Pathological pain and the neuroimmune interface. Nat Rev Immunol. 2014;14:217–31. doi: 10.1038/nri3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci. 2007;10:1361–8. doi: 10.1038/nn1992. [DOI] [PubMed] [Google Scholar]
- 41.Samad TA, Moore KA, Sapirstein A, et al. Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410:471–5. doi: 10.1038/35068566. [DOI] [PubMed] [Google Scholar]
- 42.Wolf G, Gabay E, Tal M, Yirmiya R, Shavit Y. Genetic impairment of interleukin-1 signaling attenuates neuropathic pain, autotomy, and spontaneous ectopic neuronal activity, following nerve injury in mice. Pain. 2006;120:315–24. doi: 10.1016/j.pain.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 43.Xu JT, Xin WJ, Zang Y, Wu CY, Liu XG. The role of tumor necrosis factor-alpha in the neuropathic pain induced by Lumbar 5 ventral root transection in rat. Pain. 2006;123:306–21. doi: 10.1016/j.pain.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 44.O’Garra A, Vieira P. T(H)1 cells control themselves by producing interleukin-10. Nat Rev Immunol. 2007;7:425–8. doi: 10.1038/nri2097. [DOI] [PubMed] [Google Scholar]
- 45.Wynn TA. Type 2 cytokines: mechanisms and therapeutic strategies. Nat Rev Immunol. 2015;15:271–82. doi: 10.1038/nri3831. [DOI] [PubMed] [Google Scholar]
- 46.Plunkett JA, Yu CG, Easton JM, Bethea JR, Yezierski RP. Effects of interleukin-10 (IL-10) on pain behavior and gene expression following excitotoxic spinal cord injury in the rat. Exp Neurol. 2001;168:144–54. doi: 10.1006/exnr.2000.7604. [DOI] [PubMed] [Google Scholar]
- 47.Milligan ED, Penzkover KR, Soderquist RG, Mahoney MJ. Spinal interleukin-10 therapy to treat peripheral neuropathic pain. Neuromodulation. 2012;15:520–6. doi: 10.1111/j.1525-1403.2012.00462.x. discussion 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.George A, Marziniak M, Schafers M, Toyka KV, Sommer C. Thalidomide treatment in chronic constrictive neuropathy decreases endoneurial tumor necrosis factor-alpha, increases interleukin-10 and has long-term effects on spinal cord dorsal horn met-enkephalin. Pain. 2000;88:267–75. doi: 10.1016/S0304-3959(00)00333-X. [DOI] [PubMed] [Google Scholar]
- 49.Milligan ED, Sloane EM, Langer SJ, et al. Repeated intrathecal injections of plasmid DNA encoding interleukin-10 produce prolonged reversal of neuropathic pain. Pain. 2006;126:294–308. doi: 10.1016/j.pain.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 50.Ledeboer A, Jekich BM, Sloane EM, et al. Intrathecal interleukin-10 gene therapy attenuates paclitaxel-induced mechanical allodynia and proinflammatory cytokine expression in dorsal root ganglia in rats. Brain Behav Immun. 2007;21:686–98. doi: 10.1016/j.bbi.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miyoshi K, Obata K, Kondo T, Okamura H, Noguchi K. Interleukin-18-mediated microglia/astrocyte interaction in the spinal cord enhances neuropathic pain processing after nerve injury. J Neurosci. 2008;28:12775–87. doi: 10.1523/JNEUROSCI.3512-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dinarello CA, Novick D, Kim S, Kaplanski G. Interleukin-18 and IL-18 binding protein. Front Immunol. 2013;4:289. doi: 10.3389/fimmu.2013.00289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim D, Kim MA, Cho IH, et al. A critical role of toll-like receptor 2 in nerve injury-induced spinal cord glial cell activation and pain hypersensitivity. J Biol Chem. 2007;282:14975–83. doi: 10.1074/jbc.M607277200. [DOI] [PubMed] [Google Scholar]
- 54.Liu T, Gao YJ, Ji RR. Emerging role of Toll-like receptors in the control of pain and itch. Neurosci Bull. 2012;28:131–44. doi: 10.1007/s12264-012-1219-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Casula M, Iyer AM, Spliet WG, et al. Toll-like receptor signaling in amyotrophic lateral sclerosis spinal cord tissue. Neuroscience. 2011;179:233–43. doi: 10.1016/j.neuroscience.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 56.Kielian T. Toll-like receptors in central nervous system glial inflammation and homeostasis. J Neurosci Res. 2006;83:711–30. doi: 10.1002/jnr.20767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Okun E, Griffioen KJ, Mattson MP. Toll-like receptor signaling in neural plasticity and disease. Trends Neurosci. 2011;34:269–81. doi: 10.1016/j.tins.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Coull JA, Beggs S, Boudreau D, et al. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–21. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- 59.Vanelderen P, Rouwette T, Kozicz T, et al. The role of brain-derived neurotrophic factor in different animal models of neuropathic pain. Eur J Pain. 2010;14:473e1–9. doi: 10.1016/j.ejpain.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 60.Echeverry S, Shi XQ, Haw A, Liu H, Zhang ZW, Zhang J. Transforming growth factor-beta1 impairs neuropathic pain through pleiotropic effects. Mol Pain. 2009;5:16. doi: 10.1186/1744-8069-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lantero A, Tramullas M, Diaz A, Hurle MA. Transforming growth factor-beta in normal nociceptive processing and pathological pain models. Mol Neurobiol. 2012;45:76–86. doi: 10.1007/s12035-011-8221-1. [DOI] [PubMed] [Google Scholar]
- 62.McCarthy KF, Connor TJ, McCrory C. Cerebrospinal fluid levels of vascular endothelial growth factor correlate with reported pain and are reduced by spinal cord stimulation in patients with failed back surgery syndrome. Neuromodulation. 2013;16:519–22. doi: 10.1111/j.1525-1403.2012.00527.x. discussion 22. [DOI] [PubMed] [Google Scholar]
- 63.Klein TW. Cannabinoid-based drugs as anti-inflammatory therapeutics. Nat Rev Immunol. 2005;5:400–11. doi: 10.1038/nri1602. [DOI] [PubMed] [Google Scholar]
- 64.Woodhams SG, Sagar DR, Burston JJ, Chapman V. The role of the endocannabinoid system in pain. Handb Exp Pharmacol. 2015;227:119–43. doi: 10.1007/978-3-662-46450-2_7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.