

Abstract
Smoking and major depression frequently co-occur, at least in part due to shared genetic risk. However, the nature of the shared genetic basis is poorly understood. To detect genetic risk variants for comorbid nicotine dependence (ND) and major depression (MD), we conducted genome-wide association study (GWAS) in two samples of African-American participants (Yale-Penn 1 and 2) using linear mixed model, followed by meta-analysis. 3724 nicotine-exposed subjects were analyzed: 2596 from Yale-Penn-1 and 1128 from Yale-Penn-2. Continuous measures (Fagerström Test for Nicotine Dependence (FTND) scores and DSM-IV MD criteria) rather than disorder status were used to maximize the power of the GWAS. Genotypes were ascertained using the Illumina HumanOmni1-Quad array (Yale-Penn-1 sample) or the Illumina HumanCore Exome array (Yale-Penn-2 sample), followed by imputation based on the 1000 Genomes reference panel. An intronic variant at the GRIA4 locus, rs68081839, was significantly associated with ND–MD comorbidity (β = 0.69 [95% CI, 0.43–0.89], P = 1.53 × 10−8). GRIA4 encodes an AMPA-sensitive glutamate receptor that mediates fast excitatory synaptic transmission and neuroplasticity. Conditional analyses revealed that the association was explained jointly by both traits. Enrichment analysis showed that the top risk genes and genes co-expressed with GRIA4 are enriched in cell adhesion, calcium ion binding, and synapses. They also have enriched expression in the brain and they have been implicated in the risk for other neuropsychiatric disorders. Further research is needed to determine the replicability of these findings and to identify the biological mechanisms through which genetic risk for each condition is conveyed.
Introduction
Substance use is highly associated with other psychiatric illnesses1–5. For instance, substance use disorders (SUDs) and major depression (MD) are highly comorbid in the general population2,3, and strong associations between alcohol misuse and other psychiatric disorders were observed in a U.S. Army cohort1. Clinical outcome is worse in the patients with comorbid psychiatric disorders and SUDs than in each disorder separately6. The causes of this comorbidity are poorly understood, and a better understanding of the causal relationship and etiology may provide opportunities for risk mitigation. In recent years, genetic associations (pleiotropy) between specific substance use and psychiatric disorders have been investigated by genome-wide approaches7–9 and some specific genome-wide significant (GWS) loci that affect SUD/psychiatric comorbidity have been identified in our previous study9.
The association between cigarette smoking and MD is a particularly well-studied comorbidity, with several epidemiological studies showing co-occurrence3,10–12. Smoking initiation, daily smoking, persistent daily smoking, and heavy smoking were significantly associated with increased risk of MD, and the association also applies to nicotine dependence (ND)13–15. Different hypotheses have been proposed to explain the association. It has been suggested that depression may result from the neuropharmacological effects of nicotine or nicotine withdrawal12,16,17, or alternatively, that depression may cause smoking as an attempt at self-medication of negative feelings18,19 or that there are bidirectional causal effects linking smoking and depression15. Genetic risk variants for ND (as well as smoking-related behaviors) and MD (as well as depressive symptoms) have been separately identified in large cohorts by genome-wide association study (GWAS)20–25. Common risk factors or shared etiology for smoking and depression have also been suggested26,27, and genetic factors that predispose to both smoking and MD were also suggested in a study of female twins28.
To detect shared genetic variants that predispose to comorbid ND and MD, we conducted GWAS and meta-analysis on criterion counts comprised of Fagerström Test for Nicotine Dependence (FTND) scores and DSM-IV MD criteria in two African-American samples. A variant in GRIA4, the gene that codes for the subunit 4 of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) glutamate receptor, showed genome-wide significance for association with ND–MD comorbidity.
Materials and methods
Participants and diagnostic procedures
A total of 4944 African American (AA) subjects were recruited for the Yale-Penn genetics of substance dependence study from 2000 to 2013, as previously described22,29. The subjects were grouped into two sets, Yale-Penn-1 (3227) and Yale-Penn-2 (1717), based on their epoch of recruitment and the genotyping platforms used. All subjects provided written informed consent, and certificates of confidentiality were obtained from National Institute on Drug Abuse (NIDA) and National Institute on Alcohol Abuse and Alcoholism (NIAAA). All subjects were interviewed using the Semi-Structured Assessment for Drug Dependence and Alcoholism (SSADDA)30. Lifetime FTND scores31 and criterion counts for MD from the DSM-IV (Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition)32 were derived. Six items were assessed for the FTND, generating scores from 0 to 10 (the higher the score, the more severe the nicotine use), and nine criteria were assessed for MD, generating scores from 0 to 9. We scaled the FTND scores uniformly using the same range as for MD criteria so as to weight them comparably for the GWAS9. Then, the comorbid (summed) criterion counts (ranging from 0 to 18) were treated as the outcomes, representing the overall severity of comorbidity. Subjects who were not exposed to tobacco (i.e., who answered “no” to the question, have you ever tried any form of tobacco?) were excluded from the 4994 participants, leaving 3724 eligible subjects, 2596 from Yale-Penn-1 and 1128 from Yale-Penn-2.
Genotyping, quality control, and imputation
The Yale-Penn-1 sample was genotyped using the Illumina HumanOmni1-Quad array containing ~988,000 SNPs. The Yale-Penn-2 sample was genotyped using the Illumina HumanCore Exome array containing ~266,000 exonic SNPs and ~240,000 tagging SNPs for genome-wide imputation. Individuals and SNPs with genotype call rates <98%, and SNPs with minor allele frequency (MAF) <1% were removed from downstream analyses. Yale-Penn-1 and Yale-Penn-2 data were analyzed separately.
To correct any misclassification from self-reported race, we conducted principal component (PC) analysis33 on SNPs common to both the two Yale-Penn genotype datasets and the 1000 Genomes phase 3 reference panel which contains African, American, Asian, and European populations34. SNPs were pruned based on LD (r2 < 0.2) using PLINK35. Yale-Penn subjects were clustered into different groups by the Euclidean distances to the reference populations (based on the first 3 PCs). For this study, subjects that clustered with non-African populations were removed from the downstream analyses. We then conducted a second PC analysis within the remaining Yale-Penn subjects and removed any outliers beyond three standard deviations from the mean. The first 10 PCs were used in all subsequent analyses to correct for residual population stratification.
We imputed additional single nucleotide variants (SNVs) using Minimac3 implemented in Michigan Imputation Server (https://imputationserver.sph.umich.edu/index.html)36 based on the 1000 Genomes phase 3 reference panel34. SNVs with Hardy–Weinberg equilibrium P values <10−5, imputation accuracy <0.8, or MAF <1% were excluded from downstream analyses. In the Yale-Penn-1 sample, 14,778,319 SNVs were included in the association analyses; in the Yale-Penn-2 sample, 9,658,251 SNVs were analyzed. 9,520,174 SNPs common in two samples were meta-analyzed.
Phenotype imputation
ND scores or the set of MD criteria were incomplete in a small proportion of the sample: 4.7% (121) of the Yale-Penn-1 and 4.6% (52) of the Yale-Penn-2 subjects. To address this without the power reduction that would result from simply excluding these subjects, we used PHENIX37, a variational Bayesian method fitting in a Bayesian multiple-phenotype mixed model, to impute the missing criteria. ND and MD were imputed separately in the two datasets, using the correlation matrix of the subjects derived from genome-wide efficient mixed model association (GEMMA)38.
Statistical analysis
We performed association tests for the ND+MD criterion counts (ranging from 0 to 18). All SNVs, both genotyped and imputed, were tested using a linear mixed model (GEMMA), adjusted by age, sex, and the first 10 PCs. Analyses were performed separately within each dataset. The association results were meta-analyzed across the two datasets, using the inverse variance method implemented in the program METAL39. Regional associations were plotted using LocusZoom40.
Functional annotation and enrichment analysis
Functional annotations for the top variants and genes were explored from the literature and from expression databases, including Gene-Tissue Expression (GTEx, https://www.gtexportal.org/home/) for gene-tissue expression41 and BrainSpan (http://www.brainspan.org/) for information regarding the transcriptome across human brain development42. Genes co-expressed with the target gene were identified using COXPRESdb v6.0 (http://coxpresdb.jp/)43, a coexpression database of DNA-microarray and RNAseq-based expression data. Disease enrichment among the co-expressed genes was assessed using WebGestalt (http://www.webgestalt.org/option.php)44, a functional enrichment analysis web tool. Gene ontology (GO) enrichment of the genes mapped to the top SNVs (P-value < 1 × 10−4 in either dataset or meta-analysis) was analyzed using the web-based gene set analysis tool Gorilla (http://cbl-gorilla.cs.technion.ac.il/)45. Terms with false discovery rate (FDR) <0.05 were considered to be significantly enriched.
Results
In total, 3724 AA subjects (mean age, 42 years [SD, 8.9]; 1523 women [40.9%]) were included in the analysis (2596 from Yale-Penn-1 and 1128 from Yale-Penn-2). There were 173 subjects (4.6%) with partially missing ND or MD criteria, which were imputed. Among subjects with imputed phenotype data, the average number of items that needed to be imputed was 1.2 for ND and 2.1 for MD. For ND, the imputation correlation was 0.74 for Yale-Penn-1 and 0.71 for Yale-Penn-2; for MD, the imputation correlation (between imputed phenotypes and their true hidden values) was 0.86 for both datasets. The median comorbid criterion count was 8.1 (interquartile range [IQR], 4.5–12.5) (Table 1). The distributions of comorbid criterion counts are shown in Figure S1.
Table 1.
Demographic characteristics of the samples
| Yale-Penn-1 | Yale-Penn-2 | Total | |
|---|---|---|---|
| Sample size (female %) | 3227 (47.1) | 1717 (41.7) | 4944 (45.2) |
| GWAS | |||
| Tobacco exposed (female %) | 2596 (44.3) | 1128 (33.2) | 3724 (40.9) |
| Age, mean (SD), years | 41.5 (8.2) | 42.0 (10.4) | 41.7 (8.9) |
| Subjects with partial missing (%) | 121 (4.7) | 52 (4.6) | 173 (4.6) |
| –with partial missing of ND | 20 | 11 | 31 |
| –with partial missing of MD | 101 | 41 | 142 |
| Imputation correlation of ND | 0.74 | 0.71 | |
| Imputation correlation of MD | 0.86 | 0.86 | |
| Median (IQR) of ND+MD | 7.9 (4.5–12.5) | 8.8 (3.8–12.4) | 8.1 (4.5–12.5) |
| Median (IQR) of ND | 5 (3–6) | 4 (3–6) | 5 (3–6) |
| Median (IQR) of MD | 5 (0–8) | 6 (0–8) | 5 (0–8) |
| Correlation between ND and MD | 0.15 | 0.20 | 0.16 |
ND nicotine dependence, MD major depression, IQR interquartile range
Genome-wide significant association
GWAS was performed in each dataset, followed by meta-analysis (Figure S2; SNVs with P-values < 1 × 10−4 (in either individual sample or the meta-analysis) are listed in Table S1). No GWS signals were detected in either sample analyzed individually. In the meta-analysis, a significant association was detected in GRIA4 (rs68081839, a single nucleotide deletion, −/T; the frequency of the risk allele (−) is 0.68, beta coefficient [β] = 0.69 [95% CI, 0.43–0.89], P = 1.53 × 10−8, Fig. 1). This variant was well imputed in both Yale-Penn-1 (INFO = 0.91) and Yale-Penn-2 (INFO = 0.87) samples. rs68081839 was nominally associated in both the Yale-Penn-1 (P = 1.17 × 10−5) and Yale-Penn-2 (P = 2.95 × 10−4) samples.
Fig. 1.
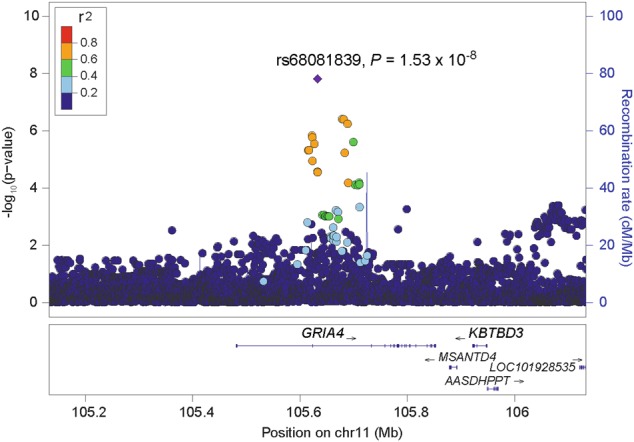
Regional Manhattan plot of rs68081839
Conditional analyses of rs68081839
We tested the association of rs68081839 with FTND scores (controlling for MD criterion counts) and MD criterion counts (controlling for FTND scores) to determine whether the association was being driven by a single disorder. Both traits were nominally associated with rs68081839 (P = 7.11 × 10−3 for ND and P = 7.34 × 10−6 for MD), indicating an additive or synergistic association for ND–MD comorbidity: i.e., the risk allele contributes to the risk of each trait taken individually. To test whether the association effect was age- or sex-related, we split the sample into older (>40 years old) and younger groups (≤40), adjusting for sex and 10 PCs, and into male and female groups, adjusting for age and 10 PCs. Similar associations between rs68081839 and ND+MD were observed with each of these approaches, indicating a consistent effect in all of the subgroups (Fig. 2). We also tested whether rs68081839 has pleiotropic effects with other substance dependence traits including alcohol, cocaine, marijuana, and opioids, and found no evidence for association (all P-values >0.5).
Fig. 2. Conditional analysis of rs68081839 and associations in different groups.
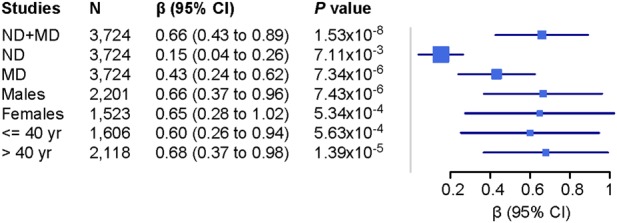
Association with ND was adjusted for MD; then, association with MD was adjusted for ND
Functional assessment of GRIA4
GRIA4 codes for subunit 4 of the AMPA glutamate receptor and it is implicated in glutamate signaling and neuroplasticity46,47. It is involved in several KEGG pathways (e.g., amphetamine addiction, nicotine addiction, the cAMP signaling pathway, neuroactive ligand–receptor interaction, glutamatergic synapses, dopaminergic synapses48). We explored the gene expression profiles of GRIA4 in different tissues from GTEx41, where it is shown to be widespread and primarily expressed in human brain (Figure S3). We then evaluated the spatio-temporal transcriptome of GRIA4 in human brain42. High expression of GRIA4 across several brain regions was observed in adulthood, increasing from the early fetal periods (Figure S4). The consistent high level of expression in brain supports the functional relevance of GRIA4 in psychiatric traits.
To investigate the functional relevance of GRIA4 further, the top 100 genes co-expressed with GRIA4 were derived from COXPRESSdb43 (Table S2). These include NLGN1, KCND2, ELAVL4, NXPH1, GRM5, and GABRB1. We assessed the disease enrichment of the co-expressed genes using web-based tool WebGestalt44 and found that mental disorders, depression, bipolar disorder, and anxiety disorder were significantly enriched (FDR < 0.05, Table S3).
Gene ontology enrichment analysis
The top SNVs (P < 1 × 10−4) were mapped to 223 genes (Table S1). GO enrichment analysis using the GOrilla web tool45 showed these genes to be enriched for cell adhesion, calcium ion binding, synapse, and plasma membrane (Table 2). We also tested the tissue expression enrichment using DAVID49,50; it showed significant enrichment in the brain (P = 5.44 × 10−6, FDR = 6.29 × 10−3). Disease enrichment analysis using WebGestalt showed that the top genes are enriched in various psychiatric disorders including bipolar disorder, anxiety disorder, depression, and substance-related disorders (Table S4). Taken together, the enrichments in signal transduction, synapse, and mental disorders support the interpretation that the polygenic risk of ND+MD is related to neural functions.
Table 2.
Gene ontology enrichment of the genes mapped to top SNVs (P < 1 × 10−4)
| Category | Term | P | FDR |
|---|---|---|---|
| Biological process | GO:0098742—cell–cell adhesion via plasma-membrane adhesion molecules | 1.81 × 10−12 | 2.67 × 10−8 |
| GO:0007156—homophilic cell adhesion via plasma membrane adhesion molecules | 6.14 × 10−11 | 4.52 × 10−7 | |
| GO:0098609—cell–cell adhesion | 4.14 × 10−8 | 2.03 × 10−4 | |
| GO:0007155—cell adhesion | 5.98 × 10−8 | 2.20 × 10−4 | |
| GO:0022610—biological adhesion | 6.52 × 10−8 | 1.92 × 10−4 | |
| Molecular function | GO:0005509—calcium ion binding | 1.43 × 10−6 | 6.29 × 10−3 |
| Cellular component | GO:0045202—synapse | 1.60 × 10−6 | 2.89 × 10−3 |
| GO:0044459—plasma membrane part | 2.67 × 10−6 | 2.42 × 10−3 | |
| GO:0005887—integral component of plasma membrane | 2.05 × 10−5 | 1.24 × 10−2 | |
| GO:0031226—intrinsic component of plasma membrane | 2.23 × 10−5 | 1.01 × 10−2 | |
| GO:0005886—plasma membrane | 8.50 × 10−5 | 3.08 × 10−2 | |
| GO:0031224—intrinsic component of membrane | 8.92 × 10−5 | 2.69 × 10−2 |
Discussion
ND and MD are among the most common psychiatric disorders worldwide and are associated with substantial morbidity and mortality51. The association between ND (as well as smoking) and MD has been well established, and both shared and distinct etiologies have been postulated. GWAS have identified risk or protective variants for ND and MD individually. To our knowledge, this is the first study of the shared genetic risks for ND and MD comorbidity. To accomplish this, we employed a dimensional approach using our phenotype data collected using the SSADDA. We found one SNP to be significantly associated with ND+MD comorbidity (β = 0.69 [95% CI, 0.43–0.89], P = 1.53 × 10−8, Fig. 1). rs68081839 is a single nucleotide deletion in the GRIA4 gene. Conditional analyses showed that the association was not driven by ND or MD alone; instead, there is an additive or synergistic effect of ND and MD. In our dataset, the contribution from MD is greater than that from ND (Fig. 2). There was no evidence of pleiotropy with other substance dependence traits (based on direct testing for association).
GRIA4 (glutamate ionotropic receptor AMPA type subunit 4)—also referred to as GluR-D or GluR4—is a member of the AMPA-selective glutamate receptor family (AMPARs). AMPARs are expressed ubiquitously in the central nervous system and are the predominant excitatory neurotransmitter receptors in the mammalian brain. They are localized at the postsynaptic membrane and are essential for synaptic plasticity52. The most thoroughly characterized examples of synaptic plasticity are long-term potentiation (LTP) and long-term depression (LTD), widely believed to be the cellular basis of learning and memory53. Further studies have shown that LTP and LTD participate in pathological processes such as Alzheimer’s disease, schizophrenia, and addiction52,54. GRIA4 functions as a ligand-gated ion channel in the central nervous system and plays an important role in fast excitatory synaptic transmission46. Expression of GRIA4 is sufficient to alter the signaling requirements for LTP during a critical period of synapse development47, and the membrane proximal region of GRIA4, needed for receptor trafficking and synaptic plasticity, is essential for long-term fear memory formation55.
Changes in GRIA4 expression have been associated with both depression and stress. Postmortem studies showed GRIA4 upregulation in depressed patients. Expression of GRIA4 in Brodmann area 10 and amygdala was increased in subjects who died by suicide during an episode of MD compared to subjects who died by suicide without depression, or controls who died suddenly from other causes and had no history of suicidal behavior56. Higher expression of GRIA4 in the dorsolateral prefrontal cortex in female patients with MD than that in female controls has been reported57. In relation to stress, Gria4 was upregulated in the hippocampus in stressed rats, and this could be reversed by the antidepressant drug venlafaxine. Gria4 expression was also increased by chronic treatment with corticosterone, the major stress hormone58. An opposite effect was observed in the ventral (but not dorsal) hippocampus in rats which were treated by neonatal handling59.
Synaptic plasticity is known to play a key role in drug addiction. Indeed, addiction has been conceptualized as a pathological form of learning and memory, as they share synaptic plasticity mechanisms. Synaptic plasticity may contribute to different aspects of addiction, including craving, withdrawal, and relapse60,61. Altered expression of GRIA4 and other glutamatergic genes in postmortem hippocampus was observed after chronic exposure to alcohol or cocaine62, perhaps contributing to the development of craving63. Studies in mouse models showed that AMPARs and N-methyl-D-aspartate receptors (NMDAR) in the ventral tegmental area (VTA) are involved in behavioral sensitization, thus playing key roles in the development of addiction64,65. For example, a single exposure to cocaine in vivo can increase the AMPAR/NMDAR ratio in the VTA, which may be involved in an early stage of drug addiction66. Along with addictive substances such as cocaine64 and morphine65, nicotine activates nicotinic acetylcholine receptors (nAChRs) in the VTA to reinforce smoking behavior67,68. We therefore speculate that the synaptic plasticity effects of GRIA4 may explain its contribution to the risk of ND–MD comorbidity.
Besides the significant finding at GRIA4, we performed enrichment analyses taking the genes identified by top SNPs (P < 1 × 10−4) as a whole. The enriched GO terms include cell adhesion in biological processes, calcium ion binding in molecular function, and synapse and plasma membrane in cellular component (Table 2). A significant enrichment of tissue expression was reported in DAVID using the same list of genes. Disease-level enrichment is more informative than GO level enrichment, in this instance: the disease enrichment analyses for the top genes or top coexpressed genes with GRIA4 shows that the significant enriched disease traits are mainly related to mental disorders (Table S3 and S4). All the reported terms are significant after multiple testing correction (FDR < 0.05). In GWAS with limited sample size, it is very common that no significant enrichment can be detected or the enriched terms cannot be linked to the study trait in an obvious way. Here, despite the sample size limitation, we observed consistent GO or disease enrichments using different web-based tools or different gene lists, indicating that the nominally significant findings (P < 1 × 10−4) and the coexpressed genes with GRIA4 are robustly related to the ND+MD trait.
This study has important limitations including modest sample size and the lack of a replication sample. Further studies to understand the biological mechanisms of the genetic risk loci we identified are also warranted.
In conclusion, we identified variation at GRIA4, a gene that codes for an AMPA glutamate receptor subunit, as a genetic risk factor for ND and MD comorbidity. This provides initial evidence that variation in the glutamatergic system may underlie the common etiology of these highly comorbid disorders. Thus, the glutamatergic system may thus be a target for treatment of ND69, as it is already for MD70; especially so in clinical contexts where the two traits are comorbid.
Electronic supplementary material
Acknowledgements
We appreciate the work of recruitment and assessment by James Poling, PhD, at Yale University School of Medicine and the APT Foundation; by Roger Weiss, MD, at McLean Hospital; by Kathleen Brady, MD/PhD and Raymond Anton, MD, at the Medical University of South Carolina; and David Oslin, MD at the University of Pennsylvania. Genotyping services for a part of our GWAS were provided by the Center for Inherited Disease Research and Yale University (Center for Genome Analysis), which is fully funded by Federal contract N01-HG-65403 from the NIH to The Johns Hopkins University. We thank Ann Marie Lacobelle, MS, at the VA CT Healthcare Center, and Christa Robinson, BS, at the VA CT Healthcare Center, who provided technical assistance. We thank John Farrell, PhD, Section of Biomedical Genetics, Boston University School of Medicine, who provided database management assistance. This study was supported by National Institutes of Health grants RC2 DA028909, R01 DA12690, R01 DA12849, R01 DA18432, R01 AA11330, R01 AA017535, 2P50-AA012870, VA Connecticut Healthcare Center, Philadelphia VA MIRECCS, and National Center for Post Traumatic Stress Disorder. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of interest
Dr. Kranzler has been a consultant or advisory board member for Indivior and Lundbeck. He is also a member of the American Society of Clinical Psychopharmacology’s Alcohol Clinical Trials Initiative, which was supported for the last 3 years by AbbVie, Alkermes, Ethypharm, Indivior, Lilly, Lundbeck, Otsuka, Pfizer, Arbor, and Amygdala Neurosciences. Dr. Krystal has stock in ArRETT Neuroscience and Biohaven Pharmaceuticals Medical Sciences, and has stock options in Biohaven Pharmaceuticals Medical Sciences, Blackthorn Therapeutics, and Luc Therapeutics. Dr. Krystal also has patents and inventions 5,447,948, 8,778,979 B2, 14/197,767, 14/306,382, 61/973,961, Y0087.70116US00, and 62/444,552. The other authors have no biomedical financial interests or potential conflicts of interest.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary Information accompanies this paper at (10.1038/s41398-018-0258-8).
References
- 1.Stein MB, et al. Alcohol misuse and co-occurring mental disorders among new soldiers in the U.S. Army. Alcohol. Clin. Exp. Res. 2017;41:139–148. doi: 10.1111/acer.13269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Edwards AC, et al. Adolescent alcohol use is positively associated with later depression in a population-based U.K. cohort. J. Stud. Alcohol Drugs. 2014;75:758–765. doi: 10.15288/jsad.2014.75.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grant BF. Comorbidity between DSM-IV drug use disorders and major depression: results of a national survey of adults. J. Subst. Abuse. 1995;7:481–497. doi: 10.1016/0899-3289(95)90017-9. [DOI] [PubMed] [Google Scholar]
- 4.Regier DA, et al. Comorbidity of mental disorders with alcohol and other drug abuse. Results from the Epidemiologic Catchment Area (ECA) Study. JAMA. 1990;264:2511–2518. doi: 10.1001/jama.1990.03450190043026. [DOI] [PubMed] [Google Scholar]
- 5.Kessler RC, et al. Lifetime and 12-month prevalence of DSM-III-R psychiatric disorders in the United States. Results from the National Comorbidity Survey. Arch. Gen. Psychiatry. 1994;51:8–19. doi: 10.1001/archpsyc.1994.03950010008002. [DOI] [PubMed] [Google Scholar]
- 6.Volkow ND. Substance use disorders in schizophrenia—clinical implications of comorbidity. Schizophr. Bull. 2009;35:469–472. doi: 10.1093/schbul/sbp016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edwards AC, et al. Genome-wide association study of comorbid depressive syndrome and alcohol dependence. Psychiatr. Genet. 2012;22:31–41. doi: 10.1097/YPG.0b013e32834acd07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andersen Allan M., Pietrzak Robert H., Kranzler Henry R., Ma Li, Zhou Hang, Liu Xiaoming, Kramer John, Kuperman Samuel, Edenberg Howard J., Nurnberger John I., Rice John P., Tischfield Jay A., Goate Alison, Foroud Tatiana M., Meyers Jacquelyn L., Porjesz Bernice, Dick Danielle M., Hesselbrock Victor, Boerwinkle Eric, Southwick Steven M., Krystal John H., Weissman Myrna M., Levinson Douglas F., Potash James B., Gelernter Joel, Han Shizhong. Polygenic Scores for Major Depressive Disorder and Risk of Alcohol Dependence. JAMA Psychiatry. 2017;74(11):1153. doi: 10.1001/jamapsychiatry.2017.2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou H, et al. Genetic risk variants associated with comorbid alcohol dependence and major depression. JAMA Psychiatry. 2017;74:1234–1241. doi: 10.1001/jamapsychiatry.2017.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bakhshaie J, Zvolensky MJ, Goodwin RD. Cigarette smoking and the onset and persistence of depression among adults in the United States: 1994–2005. Compr. Psychiatry. 2015;60:142–148. doi: 10.1016/j.comppsych.2014.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steuber TL, Danner F. Adolescent smoking and depression: which comes first? Addict. Behav. 2006;31:133–136. doi: 10.1016/j.addbeh.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 12.Klungsoyr O, Nygard JF, Sorensen T, Sandanger I. Cigarette smoking and incidence of first depressive episode: an 11-year, population-based follow-up study. Am. J. Epidemiol. 2006;163:421–432. doi: 10.1093/aje/kwj058. [DOI] [PubMed] [Google Scholar]
- 13.Hu MC, Davies M, Kandel DB. Epidemiology and correlates of daily smoking and nicotine dependence among young adults in the United States. Am. J. Public Health. 2006;96:299–308. doi: 10.2105/AJPH.2004.057232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grant BF, Hasin DS, Chou SP, Stinson FS, Dawson DA. Nicotine dependence and psychiatric disorders in the United States: results from the national epidemiologic survey on alcohol and related conditions. Arch. Gen. Psychiatry. 2004;61:1107–1115. doi: 10.1001/archpsyc.61.11.1107. [DOI] [PubMed] [Google Scholar]
- 15.Breslau N, Kilbey MM, Andreski P. Nicotine dependence and major depression. New evidence from a prospective investigation. Arch. Gen. Psychiatry. 1993;50:31–35. doi: 10.1001/archpsyc.1993.01820130033006. [DOI] [PubMed] [Google Scholar]
- 16.Pasco JA, et al. Tobacco smoking as a risk factor for major depressive disorder: population-based study. Br. J. Psychiatry. 2008;193:322–326. doi: 10.1192/bjp.bp.107.046706. [DOI] [PubMed] [Google Scholar]
- 17.Covey LS, Glassman AH, Stetner F. Major depression following smoking cessation. Am. J. Psychiatry. 1997;154:263–265. doi: 10.1176/ajp.154.2.263. [DOI] [PubMed] [Google Scholar]
- 18.Patton GC, et al. Is smoking associated with depression and anxiety in teenagers? Am. J. Public Health. 1996;86:225–230. doi: 10.2105/AJPH.86.2.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crone MR, Reijneveld SA. The association of behavioural and emotional problems with tobacco use in adolescence. Addict. Behav. 2007;32:1692–1698. doi: 10.1016/j.addbeh.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 20.Thorgeirsson TE, et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008;452:638–642. doi: 10.1038/nature06846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tobacco and Genetics Consortium. Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nat. Genet.42, 441–447 (2010). [DOI] [PMC free article] [PubMed]
- 22.Gelernter J, et al. Genome-wide association study of nicotine dependence in American populations: identification of novel risk loci in both African-Americans and European-Americans. Biol. Psychiatry. 2015;77:493–503. doi: 10.1016/j.biopsych.2014.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Converge Consortium Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature. 2015;523:588–591. doi: 10.1038/nature14659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hyde CL, et al. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat. Genet. 2016;48:1031–1036. doi: 10.1038/ng.3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okbay A, et al. Genetic variants associated with subjective well-being, depressive symptoms, and neuroticism identified through genome-wide analyses. Nat. Genet. 2016;48:624–633. doi: 10.1038/ng.3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fergusson DM, Lynskey MT, Horwood LJ. Comorbidity between depressive disorders and nicotine dependence in a cohort of 16-year-olds. Arch. Gen. Psychiatry. 1996;53:1043–1047. doi: 10.1001/archpsyc.1996.01830110081010. [DOI] [PubMed] [Google Scholar]
- 27.Breslau N, Peterson EL, Schultz LR, Chilcoat HD, Andreski P. Major depression and stages of smoking. A longitudinal investigation. Arch. Gen. Psychiatry. 1998;55:161–166. doi: 10.1001/archpsyc.55.2.161. [DOI] [PubMed] [Google Scholar]
- 28.Kendler KS, et al. Smoking and major depression. A causal analysis. Arch. Gen. Psychiatry. 1993;50:36–43. doi: 10.1001/archpsyc.1993.01820130038007. [DOI] [PubMed] [Google Scholar]
- 29.Sherva R, et al. Genome-wide association study of Cannabis dependence severity, novel risk variants, and shared genetic risks. JAMA Psychiatry. 2016;73:472–480. doi: 10.1001/jamapsychiatry.2016.0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pierucci-Lagha A, et al. Diagnostic reliability of the Semi-structured Assessment for Drug Dependence and Alcoholism (SSADDA) Drug Alcohol Depend. 2005;80:303–312. doi: 10.1016/j.drugalcdep.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 31.Heatherton TF, Kozlowski LT, Frecker RC, Fagerstrom KO. The Fagerstrom test for nicotine dependence: a revision of the Fagerstrom Tolerance Questionnaire. Br. J. Addict. 1991;86:1119–1127. doi: 10.1111/j.1360-0443.1991.tb01879.x. [DOI] [PubMed] [Google Scholar]
- 32.American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders. 4th edn. Washington, DC, USA: American Psychiatric Press; 1994. [Google Scholar]
- 33.Price AL, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 34.Auton A, et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Das S, et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016;48:1284–1287. doi: 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dahl A, et al. A multiple-phenotype imputation method for genetic studies. Nat. Genet. 2016;48:466–472. doi: 10.1038/ng.3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou X, Stephens M. Efficient multivariate linear mixed model algorithms for genome-wide association studies. Nat. Methods. 2014;11:407–409. doi: 10.1038/nmeth.2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pruim RJ, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–2337. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.GTEx Consortium Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kang HJ, et al. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okamura Y, et al. COXPRESdb in 2015: coexpression database for animal species by DNA-microarray and RNAseq-based expression data with multiple quality assessment systems. Nucleic Acids Res. 2015;43:D82–D86. doi: 10.1093/nar/gku1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang J, Duncan D, Shi Z, Zhang B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res. 2013;41:W77–W83. doi: 10.1093/nar/gkt439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu JJ, Esteban JA, Hayashi Y, Malinow R. Postnatal synaptic potentiation: delivery of GluR4-containing AMPA receptors by spontaneous activity. Nat. Neurosci. 2000;3:1098–1106. doi: 10.1038/80614. [DOI] [PubMed] [Google Scholar]
- 47.Luchkina NV, et al. Developmental switch in the kinase dependency of long-term potentiation depends on expression of GluA4 subunit-containing AMPA receptors. Proc. Natl. Acad. Sci. USA. 2014;111:4321–4326. doi: 10.1073/pnas.1315769111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ogata H, et al. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999;27:29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.da Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 51.Murray CJ, Lopez AD. Measuring the global burden of disease. N. Engl. J. Med. 2013;369:448–457. doi: 10.1056/NEJMra1201534. [DOI] [PubMed] [Google Scholar]
- 52.Henley JM, Wilkinson KA. Synaptic AMPA receptor composition in development, plasticity and disease. Nat. Rev. Neurosci. 2016;17:337–350. doi: 10.1038/nrn.2016.37. [DOI] [PubMed] [Google Scholar]
- 53.Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 54.Huganir RL, Nicoll RA. AMPARs and synaptic plasticity: the last 25 years. Neuron. 2013;80:704–717. doi: 10.1016/j.neuron.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ganea DA, Dines M, Basu S, Lamprecht R. The membrane proximal region of AMPA receptors in lateral amygdala is essential for fear memory formation. Neuropsychopharmacology. 2015;40:2727–2735. doi: 10.1038/npp.2015.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sequeira A, et al. Global brain gene expression analysis links glutamatergic and GABAergic alterations to suicide and major depression. PLoS ONE. 2009;4:e6585. doi: 10.1371/journal.pone.0006585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gray AL, Hyde TM, Deep-Soboslay A, Kleinman JE, Sodhi MS. Sex differences in glutamate receptor gene expression in major depression and suicide. Mol. Psychiatry. 2015;20:1057–1068. doi: 10.1038/mp.2015.91. [DOI] [PubMed] [Google Scholar]
- 58.Martisova E, et al. Long lasting effects of early-life stress on glutamatergic/GABAergic circuitry in the rat hippocampus. Neuropharmacology. 2012;62:1944–1953. doi: 10.1016/j.neuropharm.2011.12.019. [DOI] [PubMed] [Google Scholar]
- 59.Katsouli S, et al. Sexually dimorphic long-term effects of an early life experience on AMPA receptor subunit expression in rat brain. Neuroscience. 2014;257:49–64. doi: 10.1016/j.neuroscience.2013.10.073. [DOI] [PubMed] [Google Scholar]
- 60.Dani JA, Ji D, Zhou FM. Synaptic plasticity and nicotine addiction. Neuron. 2001;31:349–352. doi: 10.1016/S0896-6273(01)00379-8. [DOI] [PubMed] [Google Scholar]
- 61.Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nat. Rev. Neurosci. 2007;8:844–858. doi: 10.1038/nrn2234. [DOI] [PubMed] [Google Scholar]
- 62.Enoch MA, et al. Expression of glutamatergic genes in healthy humans across 16 brain regions; altered expression in the hippocampus after chronic exposure to alcohol or cocaine. Genes Brain Behav. 2014;13:758–768. doi: 10.1111/gbb.12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Robinson TE, Berridge KC. The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res. Brain Res. Rev. 1993;18:247–291. doi: 10.1016/0165-0173(93)90013-P. [DOI] [PubMed] [Google Scholar]
- 64.Harris GC, Aston-Jones G. Critical role for ventral tegmental glutamate in preference for a cocaine-conditioned environment. Neuropsychopharmacology. 2003;28:73–76. doi: 10.1038/sj.npp.1300011. [DOI] [PubMed] [Google Scholar]
- 65.Harris GC, Wimmer M, Byrne R, Aston-Jones G. Glutamate-associated plasticity in the ventral tegmental area is necessary for conditioning environmental stimuli with morphine. Neuroscience. 2004;129:841–847. doi: 10.1016/j.neuroscience.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 66.Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- 67.Mansvelder HD, McGehee DS. Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron. 2000;27:349–357. doi: 10.1016/S0896-6273(00)00042-8. [DOI] [PubMed] [Google Scholar]
- 68.Pidoplichko VI, DeBiasi M, Williams JT, Dani JA. Nicotine activates and desensitizes midbrain dopamine neurons. Nature. 1997;390:401–404. doi: 10.1038/37120. [DOI] [PubMed] [Google Scholar]
- 69.Liechti ME, Markou A. Role of the glutamatergic system in nicotine dependence: implications for the discovery and development of new pharmacological smoking cessation therapies. CNS Drugs. 2008;22:705–724. doi: 10.2165/00023210-200822090-00001. [DOI] [PubMed] [Google Scholar]
- 70.Krystal JH, et al. Glutamate and GABA systems as targets for novel antidepressant and mood-stabilizing treatments. Mol. Psychiatry. 2002;7(Suppl. 1):S71–S80. doi: 10.1038/sj.mp.4001021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.