

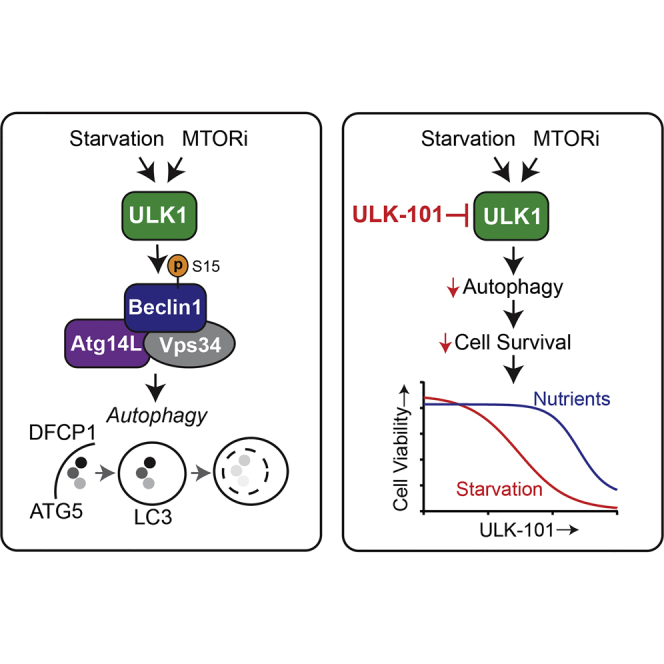
Summary
In response to stress, cancer cells generate nutrients and energy through a cellular recycling process called autophagy, which can promote survival and tumor progression. Accordingly, autophagy inhibition has emerged as a potential cancer treatment strategy. Inhibitors targeting ULK1, an essential and early autophagy regulator, have provided proof of concept for targeting this kinase to inhibit autophagy; however, these are limited individually in their potency, selectivity, or cellular activity. In this study, we report two small molecule ULK1 inhibitors, ULK-100 and ULK-101, and establish superior potency and selectivity over a noteworthy published inhibitor. Moreover, we show that ULK-101 suppresses autophagy induction and autophagic flux in response to different stimuli. Finally, we use ULK-101 to demonstrate that ULK1 inhibition sensitizes KRAS mutant lung cancer cells to nutrient stress. ULK-101 represents a powerful molecular tool to study the role of autophagy in cancer cells and to evaluate the therapeutic potential of autophagy inhibition.
Subject Areas: Therapeutics, Functional Aspects of Cell Biology, Cancer
Graphical Abstract
Highlights
-
•
ULK-101 has improved potency and selectivity when compared with SBI-0206965
-
•
ULK-101 inhibits both the nucleation of autophagic vesicles and turnover
-
•
ULK-101 sensitizes KRAS-driven lung cancer cells to nutrient restriction
-
•
ULK-101 is a valuable molecular tool to study the function of ULK1 and autophagy
Therapeutics; Functional Aspects of Cell Biology; Cancer
Introduction
Macroautophagy (hereafter autophagy) is an intracellular recycling pathway that generates biochemical building blocks through cytoplasmic breakdown (Klionsky, 2007). This process begins with nucleation of cup-shaped structures (phagophores) that grow into double-membrane autophagosomes as they sequester portions of cytosol. Fusion with lysosomes provides degradative enzymes to catabolize cargo into amino acids, lipids, and carbohydrates, which are then available for the cell to reuse. As an energy-efficient alternative to de novo synthesis, autophagy can promote cell survival during times of stress (Rabinowitz and White, 2010). For this reason, this process can contribute to the progression of certain cancers (e.g., by enabling survival in a nutrient-depleted tumor microenvironment) and also to therapeutic resistance. Autophagy appears to be particularly important in the survival and growth of KRAS-driven tumors, as evidenced in part by data from genetic mouse models (Eng et al., 2016, Guo et al., 2013, Guo et al., 2016, Karsli-Uzunbas et al., 2014, Rao et al., 2014). Autophagy inhibition is now being explored as a means to improve efficacy of existing cancer treatments, as well as a therapeutic strategy of its own (Chude and Amaravadi, 2017).
Autophagy induction is controlled primarily by the serine/threonine kinase, ULK1 (unc-51 like autophagy initiating kinase 1), a mammalian ortholog of yeast ATG1. ULK1 is part of a complex with binding partners ATG13 (autophagy related 13), RB1CC1 (RB1 inducible coiled-coil; also known as FIP200), and ATG101 (autophagy related 101) (Ganley et al., 2009, Mercer et al., 2009). Through this complex, ULK1 integrates upstream signals from both the mechanistic target of rapamycin complex 1 (mTORC1) nutrient-sensing and the AMP-activated protein kinase (AMPK) energy-sensing pathways to induce the production of early autophagic membranes (Ganley et al., 2009, Hosokawa et al., 2009, Jung et al., 2009, Kim et al., 2011). ULK1's essential role in autophagy has been shown in animals and mammalian cell culture systems where ULK1 depletion impairs autophagy (Chan et al., 2007, Cheong et al., 2011, Lee and Tournier, 2011). Although a second mammalian ATG1 ortholog, ULK2, also promotes autophagy (Lee and Tournier, 2011), loss of ULK1 alone is sufficient to abrogate autophagy in many cell types, underscoring its particularly important role (Chan et al., 2007, Zachari and Ganley, 2017).
ULK1 is now considered a promising target for autophagy inhibition because of its central role in pathway activation, druggable nature, and apparent selectivity for autophagy over other cellular functions. Accordingly, several small molecule inhibitors of ULK1 have recently been reported (Egan et al., 2015, Lazarus et al., 2015, Lazarus and Shokat, 2015, Petherick et al., 2015, Wood et al., 2017). These compounds have provided insight into ULK1's structure and validated ULK1 inhibition as an effective means for suppressing autophagy, while also providing proof of concept for small molecule targeting of this enzyme. That being said, existing ULK1 inhibitors are limited individually in their potency, selectivity, or evidence of cellular activity.
Here, we describe two small molecule inhibitors, ULK-100 and ULK-101, which show strong activity toward ULK1 both in vitro and in cells. We establish improved potency and selectivity of ULK-101 compared with the published ULK1 inhibitor, SBI-0206965. Moreover, we demonstrate that ULK-101 blocks autophagy induction and suppresses autophagic flux both constitutively and in response to starvation and mTOR inhibition. Last, we discover that ULK-101 sensitizes KRAS-driven lung cancer cells to nutrient restriction. Taken together, ULK-101 is a valuable molecular tool to interrogate the cellular function of ULK1 and autophagy and to evaluate the therapeutic potential of autophagy inhibition at the level of the ULK1 complex and autophagy initiation.
Results
Small Molecules Potently Inhibit ULK1 In Vitro and in Cells
To identify small molecule ULK1 inhibitors, we searched existing pharmaceutical data for compounds that showed activity against ULK1 in selectivity screening. This process led us to two closely related molecules, ULK-100 and ULK-101, which share a common scaffold and differ in the two flanking R-groups (Figure 1A). We confirmed that both inhibit ULK1 directly in vitro with an IC50 of 1.6 nM (95% confidence interval [CI]: 1.5–1.8 nM) for ULK-100 and 8.3 nM (95% CI: 7.2–9.6 nM) for ULK-101 (Figure 1B; Table 1). For comparison, we determined the IC50 of a recently reported ULK1 inhibitor, SBI-0206965 (Egan et al., 2015), to be 38 nM (95% CI: 34–42 nM) (Figure 1B; Table 1).
Figure 1.

ULK-100 and ULK-101 Potently Inhibit ULK1 In Vitro and in Cells
(A) The chemical structures of ULK-100 and ULK-101 are shown with the shared scaffold shaded.
(B) ULK1 activity was measured in the presence of ULK-100 (blue), ULK-101 (red), or SBI-0206965 (green) at half-log dilutions from top concentrations of 10 μM. All concentrations were tested in 8 replicates with the exception of the two highest and lowest concentrations, which were performed in 4 replicates. Data are represented as mean activity normalized to control (0 μM inhibitor) ± SD. Solid lines represent IC50 curves fit by non-linear regression.
(C and D) 293FT lysates expressing HA-hULK1, Myc-mULK1 (wild-type or M92A), V5-Beclin 1, and/or EGFP-ATG14 were treated for 1 hr with full growth (F) or nutrient starvation (S) media and (C) probed by immunoblotting with the indicated antibodies. In (D), membranes were imaged by Odyssey (total and pS15 detected from same Beclin 1 band). S14 is the murine (mULK1) residue number corresponding to human (hULK1) S15.
(E–G) 293FT cells expressing HA-ULK1, V5-Beclin 1, and EGFP-ATG14 were treated with the indicated concentrations of SBI-0206965 (E), ULK-100 (F), or ULK-101 (G) for 1 hr. Lysates were probed with the indicated antibodies and imaged using an Odyssey imager. Representative blots from 3 biological replicates per inhibitor are shown.
(H) Relative pS15-BECLIN 1 was quantified as the pS15-Beclin 1 signal divided by total Beclin 1 signal, each normalized to the vehicle (0 μM inhibitor). Symbols represent means from 3 biological replicates ± SEM. EC50 curves (solid lines) were generated by non-linear regression using variable slopes.
Table 1.
Biochemical IC50 and Cellular EC50 for ULK1 Inhibitors
| SBI-0206965 | ULK-100 | ULK-101 | |
|---|---|---|---|
| ULK1 IC50 (nM) | 38 | 1.6 | 8.3 |
| ULK2 IC50 (nM) | 212 | 2.6 | 30 |
| Cellular EC50 (nM) | 2,400 | 83 | 390 |
ULK1 promotes autophagy through phosphorylation of a protein complex that includes the lipid kinase, PIK3C3 (phosphatidylinositol 3-kinase catalytic subunit type 3), and its binding partners, ATG14 (autophagy related 14) and Beclin 1. Specifically, when Beclin 1 is phosphorylated at Ser15 by ULK1, it increases the activity of PIK3C3 to produce phosphoinositide-3-phosphate (PtdIns3P), a phospholipid required for the nucleation of autophagic membranes (Russell et al., 2013). To determine whether ULK-100 and ULK-101 inhibit ULK1 in cells, we evaluated the phosphorylation of Beclin 1 at Ser15. We exogenously expressed both Beclin 1 and ULK1 and found that Ser15 phosphorylation was substantially increased by nutrient starvation, a stimulus known to activate ULK1 and induce autophagy (Figure 1C) (Cheong et al., 2011). Furthermore, we found that co-expression of ATG14 caused phosphorylation of Ser15 that was no longer dependent on starvation (Figure 1C). Importantly, Beclin 1 Ser15 phosphorylation was ablated by the expression of a kinase-dead ULK1 mutant (Jung et al., 2009), confirming that this phosphorylation specifically required the catalytic activity of ULK1 and could serve as a cellular readout of ULK1 kinase activity (Figure 1D).
We treated cells expressing ULK1, Beclin 1, and ATG14 with concentration gradients of ULK-100, ULK-101, or SBI-0206965 for 1 hr and measured Ser15 phosphorylation by quantitative immunoblotting. All three ULK1 inhibitors reduced Ser15 phosphorylation in a concentration-dependent manner (Figures 1E–1G), with SBI-0206965 showing a cellular EC50 of 2.4 μM in this assay, compared with 390 nM for ULK-101 and just 83 nM for ULK-100 (Figure 1H; Table 1). Taken together, these data suggest that ULK-101 and ULK-100 are more potent ULK1 tool compounds than SBI-0206965 both in vitro and in cells.
ULK-101 Has a Desirable Kinome Selectivity Profile
ULK2 is the second mammalian ortholog of yeast ATG1 with a similar function in autophagy as ULK1 (Lee and Tournier, 2011); therefore, we reasoned that inhibiting ULK2 would be important for an ULK1 inhibitor intended to block autophagy. We tested ULK-100 and ULK-101, along with SBI-0206965, for the ability to inhibit ULK2 kinase activity in vitro and found that all three molecules show nanomolar ULK2 IC50 values (Figure 2A; Table 1). Similar to the results obtained for ULK1, ULK-100 (ULK2 IC50 2.6 nM) showed the greatest potency followed by ULK-101 (ULK2 IC50 30 nM) and SBI-0206965 (ULK2 IC50 212 nM).
Figure 2.

Selectivity Profiles of ULK1 Inhibitors
(A) ULK2 activity was measured in the presence of ULK-100 (blue), ULK-101 (red), or SBI-0206965 (green) at half-log dilutions from top concentrations of 10 μM. All concentrations were tested in 8 replicates with the exception of the two highest and lowest concentrations, which were performed in 4 replicates. Data are represented as activity normalized to control (0 μM inhibitor) ± SD. Solid lines represent IC50 curves fit by non-linear regression.
(B) The relative inhibition of human kinases by each compound is shown in a rainbow scale from low (red) to high (purple). Kinases are ordered by their relative inhibition within each compound.
(C) The number of non-ULK1 kinases (of 326 surveyed) inhibited by at least 50% (left), 75% (middle), or 100% (right) the level of ULK1 is plotted for SBI-0206965 (green), ULK-100 (blue), and ULK-101 (red).
(D–F) Kinases inhibited by at least 75% the level of ULK1 by SBI-0206965 (green; D), ULK-100 (blue; E), and ULK-101 (red; F) are identified on human kinome phylogenetic trees. Symbols are sized according to their relative inhibition, and major kinase groups identified. TK, tyrosine kinase; TKL, tyrosine kinase-like; STE, homologs of yeast Sterile 7, Sterile 11, Sterile 20 kinases; CK1, casein kinase 1; AGC, containing PKA, PKG, PKC families; CAMK, calcium/calmodulin-dependent protein kinase; CMGC, containing CDK, MAPK, GSK3, CLK families. Figures generated using KinMap Beta, and illustrations reproduced courtesy of Cell Signaling Technology.
See also Table S1.
Next, we wanted to understand the kinome-wide selectivity profiles of each inhibitor. For this, we determined their ability to inhibit 327 recombinant human kinases in vitro using radiometric activity assays. Each kinase was screened in duplicate with a single concentration of compound that achieved almost identical ULK1 inhibition for SBI-0206965 and ULK-101 (74% and 73% inhibition, respectively), and slightly better ULK1 inhibition for ULK-100 (88% inhibition). To compare the selectivity of the three compounds, we normalized the inhibition of each kinase to that of ULK1 (Table S1). Overall, ULK-101 showed the cleanest kinome-wide profile (Figure 2B). SBI-0206965 and ULK-100 inhibited 17 or 18 kinases at least 75% as well as they inhibited ULK1, respectively, whereas ULK-101 inhibited just four other kinases at this level (Figure 2C). We visualized these on the human kinase phylogenetic tree and found that the off-targets of both SBI-0206965 and ULK-100 were diversely distributed (Figures 2D and 2E), whereas three of the four off-targets of ULK-101 were CAMK family members (Figure 2F). To fully understand the relative inhibition of the closest ULK-101 off-targets, we determined the IC50 for DRAK1 to be 14 nM and that of MNK2 to be 22 nM (compared with 8.3 nM for ULK1). From this study, we concluded that ULK-101 offers superior selectively properties than both SBI-0206965 and ULK-100, and therefore we prioritized this inhibitor for further investigation.
ULK-101 Reduces the Nucleation of Autophagic Vesicles
When ULK1 phosphorylates Beclin 1 at Ser15, it increases the formation of PtdIns3P by the autophagic PIK3C3 complex (Russell et al., 2013). This PtdIns3P production occurs in rough endoplasmic reticulum microdomains called omegasomes, which serve as platforms for the formation of early autophagic membranes (Axe et al., 2008). To determine whether ULK-101 suppressed autophagy induction, we stably expressed the omegasome marker, EGFP-DFCP1, in U2OS cells and monitored omegasome formation by live-cell fluorescent microscopy. We treated cells for 2.5 hr with AZD8055, a catalytic mTOR inhibitor that strongly induces autophagy in U2OS cells (Martin et al., 2013) and observed that the abundance of DFCP1-positive puncta increased over 3-fold (Figures 3A and 3B). Co-treatment of cells with 5 μM ULK-101 completely abrogated the formation of DFCP1-positive structures induced by AZD8055 treatment, consistent with the loss of Beclin 1 phosphorylation (Figures 3A and 3B).
Figure 3.

ULK-101 Suppresses the Induction of Early Autophagy
(A) U2OS cells stably expressing EGFP-DFCP1 were treated for 2.5 hr with vehicle control or 100 nM AZD8055 with or without 5 μM ULK-101 and imaged live by fluorescent microscopy. Representative images are shown (grayscale of green fluorescein isothiocyanate channel).
(B) EGFP-DFCP1 objects per cell were quantified from 20 to 25 cells per condition. Data are represented as mean ± SEM. Two-tailed, unpaired t test; ***p < 0.001.
(C) U2OS cells were treated for 2.5 hr with vehicle control or 100 nM AZD8055 with or without 5 μM ULK-101 and then immunostained for endogenous ATG12 (green). Nuclei (blue) were counterstained. Representative images are shown. Lower panels are 3.3× magnifications of upper panel insets.
Omegasomes support the formation of phagophores, cup-shaped autophagic membranes marked specifically by the ubiquitin-like ATG12-ATG5-ATG16L complex (Axe et al., 2008, Mizushima et al., 2003). To determine whether ULK-101 also reduced the formation of phagophores, we immunostained U2OS cells for endogenous ATG12. Indeed, we found that AZD8055 treatment resulted in distinct ATG12-positive puncta and that co-treatment with ULK-101 strongly suppressed this (Figure 3C). From these experiments, we concluded that ULK-101 limits AZD8055-induced omegasome and phagophore formation.
Autophagic Turnover Is Suppressed by ULK-101 in Response to Multiple Stimuli
Autophagy is a dynamic membrane process in which vesicles form, grow, enclose, and fuse with lysosomes for degradation. MAP1LC3B (microtubule-associated proteins 1A/1B light chain 3B; hereafter LC3B) is a ubiquitin-like protein that becomes conjugated to the earliest autophagic membranes and remains on vesicles in the lysosome, where it is degraded along with sequestered cargo (Tanida et al., 2005). As such, its turnover can be measured to indicate the overall rate of autophagy (Klionsky et al., 2016). To do this, we blocked lysosomal function with bafilomycin A1 (BafA1), a proton pump inhibitor, for a short period. We then measured the accumulation of membrane-bound LC3B (detected as LC3B-II by immunoblotting) and compared with the amount observed in the absence of BafA1. We found that a 3-hr treatment with ULK-101 reduced BafA1-induced LC3B-II accumulation in U2OS cells in a concentration-dependent manner (Figure 4A). The EC50 value for this inhibition was approximately 700 nM (Figure 4B). To validate this phenotype, we quantified autophagic puncta in EGFP-LC3B-expressing U2OS cells using fluorescent microscopy (Figure 4C). Again, ULK-101 blocked the BafA1-induced accumulation of LC3B-positive vesicles (a 6-fold reduction from the vehicle control), suggesting a nearly complete inhibition of autophagic activity (Figure 4D).
Figure 4.

ULK-101 Reduces Autophagic Turnover
(A) U2OS cells were treated for 3 hr with the indicated concentrations of ULK-101, with (+) or without (−) 100 nM BafA1 for the final 1.5 hr. Lysates were probed for LC3B and β-actin as a loading control and imaged. A representative from 3 biological replicates is shown.
(B) Relative LC3B-II signal was determined by dividing the LC3B-II signal by the β-actin signal. The difference between (+) and (−) BafA1 bands was calculated for each ULK-101 concentration, and this value was plotted (normalized to 1.0 for the vehicle control). Symbols represent the mean of 3 biological experiments ± SEM. The red line is an EC50 curve fit by non-linear regression.
(C) EGFP-LC3B (green) was imaged in U2OS cells treated for 3 hr with vehicle or 5 μM ULK-101 with (+) or without (−) 100 nM BafA1 added for the final 1.5 hr. Green, EGFP-LC3B; blue, Hoechst-33342 (nuclei). Right panels are 5× magnifications of insets (boxes).
(D) The number of EGFP-LC3B puncta per cell was quantified from images in (C) (≥70 cells per condition). Data are represented as mean ± SEM. Two-tailed, unpaired t test; ***p < 0.001.
(E) U2OS cells were treated with vehicle control (gray bars) or 100 nM AZD8055 (blue bars) supplemented with or without 5 μM ULK-101 for 3 hr. Vehicle control or 100 nM BafA1 was added for the last 1.5 hr, and the relative amount of BafA1-induced LC3B-II (normalized to β-actin) was quantified as in (B). Data are represented as mean of 3 biological replicates ±SEM. Two-tailed, unpaired t test; **p < 0.01.
(F) U2OS cells were treated with full growth media or an Hank's balanced salt solution (HBSS)-based starvation media for 3 hr. ULK-101 was added at the indicated concentrations with (+) or without (−) 100 nM BafA1 for the final 1.5 hr. Lysates were probed for LC3B and β-actin as a loading control and imaged.
Having established that ULK-101 suppressed basal autophagic turnover, we next wanted to determine whether it also reduced stimulus-induced autophagy. To test this, we again treated cells with AZD8055 and found that BafA1-induced LC3B-II accumulation increased over 2-fold within 1 hr, an effect that was negated by ULK-101 (Figure 4E). We then stimulated autophagy by starving cells of nutrients, including amino acids and growth factors, for 3 hr in the presence or absence of ULK-101. Again, ULK-101 reduced BafA1-induced LC3B-II accumulation in a concentration-dependent manner (Figure 4F). Taken together, ULK-101 suppresses both basal and induced autophagy, including formation of early autophagic structures and autophagic flux.
Nutrient Stress Sensitizes Cells to ULK-101
Finally, we wanted to explore the therapeutic potential of ULK-101 by determining its effects on cell viability. Specifically, we hypothesized that ULK-101 would suppress cell survival during nutrient withdrawal when cells are most dependent on autophagy. To test this hypothesis, we optimized a starvation media that induced autophagy on a longer timescale without compromising cell viability (Figure S1). This media, which we called Optistarve (OS), contained reduced (but not ablated) levels of amino acids, serum, glucose, glutamine, and vitamins compared with full growth media (FM) (see Transparent Methods). To determine the effectiveness of ULK-101 in cells subjected to these different nutrient conditions, we performed a clonogenic survival experiment in which we treated cells with a concentration gradient of ULK-101 (or vehicle control) for 2 days during culture with either FM or OS. Following this 2-day treatment, media was washed and replaced with drug-free FM in a 5-day outgrowth (recovery) phase (Figure 5A). We then measured cell viability and generated relative cell viability curves. Consistent with our hypothesis, we found that ULK-101 was nearly four times as effective in reducing survival of osteosarcoma U2OS cells when cultured in OS (EC50 6.3 μM) when compared with FM (EC50 22.3 μM) (Figure 5B).
Figure 5.

ULK-101 Reduces Viability of Nutrient-Restricted Cancer Cells
(A) Clonogenic survival experimental design. Cells were seeded for 24 hr before a 2-day treatment with FM or OS media in the presence or absence of ULK-101. Thereafter, media were washed and cells treated with drug-free FM for a 5-day recovery period before cell viability was measured.
(B) Data from clonogenic survival assay with U2OS cells. Symbols represent means of 3 technical replicates ± SD. Solid lines are EC50 curves fit by non-linear regression. Blue, FM; red, OS. Cell viability normalized to the 0 μM ULK-101 concentration.
(C–F) Clonogenic survival assay data for non-small-cell lung cancer lines (cell line and KRAS mutation indicated above each graph). Symbols represent means of 3 technical replicates ± SD. Solid lines are EC50 curves fit by non-linear regression. Blue, FM; red, OS. Cell viability normalized to the 0 μM ULK-101 concentration.
To determine whether this phenotype was common among other cell lines, we examined non-small-cell lung cancer (NSCLC) cell lines for ULK-101 sensitivity. We chose this model because oncogenic KRAS, which is found in 15%–25% of NSCLC cell lines, has been suggested to promote autophagy-dependent cell survival, and autophagy contributes to lung cancer progression in animal models (Guo et al., 2011, Guo et al., 2013, Lock et al., 2011, Lock et al., 2014). Indeed, we found that KRAS mutant or amplified NSCLC lines showed at least a 5-fold increase in ULK-101 sensitivity when cultured in OS compared with FM (Figures 5C–5F; Table S2).
Discussion
Autophagy is a conserved recycling process that has emerged as a critical effector of both oncogenes and tumor suppressors and a potent regulator of cancer cell fate (Liu and Ryan, 2012, Rosenfeldt and Ryan, 2009). Although autophagy is carried out by the coordinated activity of more than 30 proteins, just a few are enzymes with clear drug-targeting potential. Among these is ULK1, which has garnered interest as a small molecule target given its essential and early role in the pathway. Here, we have presented ULK-101 as a potent and selective ULK1 inhibitor and demonstrated its ability to suppress autophagy in human cells.
ULK-101 joins at least six other ULK1 inhibitors reported since 2015. The Shokat laboratory has developed a series of ULK1-targeted compounds that have provided valuable insights into the structure of ULK1, despite limited selectivity and potency in cells (Lazarus et al., 2015, Lazarus and Shokat, 2015). Two other notable inhibitors were found by mining pharmaceutical data for compounds with activity against ULK1, analogous to our approach. SBI-0206965 was developed from a FAK inhibitor and shown to reduce Beclin 1 Ser15 phosphorylation in cells (Egan et al., 2015). This compound was reported as selective, based primarily on a large-scale competition binding assay; however, our direct comparison using in vitro kinase assays found ULK-101 to be considerably more selective than SBI-0206965. MRT68921, derived from a TBK1 inhibitor, inhibited ULK1 potently in vitro and strongly suppressed autophagy in cells, with 1 μM shown to block BafA1-induced LC3-II accumulation in nutrient-starved murine embryonic fibroblasts (Petherick et al., 2015). Although the authors screened 80 other kinases for inhibition by MRT68921, it is difficult to compare the selectivity profiles of MRT68921 and ULK-101. ULK-101 was screened against 327 kinases. Of interest, MRT68921 cross-reacts with AMPK, which may represent a therapeutic liability given the broad tumor suppressive functions of AMPK signaling. Interestingly, whereas AMPK was also inhibited by ULK-100 in vitro, it was spared by ULK-101 (Table S1). Finally, a study employing in silico screening and structure-activity relationship analyses identified potent indazole-derived ULK1 inhibitors, although their selectivity and activity in cells remains to be determined (Wood et al., 2017).
A major unresolved issue in the autophagy field concerns the genetic and environmental contexts in which autophagy promotes tumor growth and represents a therapeutic target. Here, we have used ULK-101 to show that nutrient-stressed cells may be particularly susceptible to ULK1 inhibition. SBI-0206965 was similarly found to increase cell death in nutrient-starved cells or in those with chemical mTORC1 inhibition (Egan et al., 2015). These findings are consistent with other studies in which autophagy inhibition was particularly effective in cells deprived of nutrients (Eng et al., 2016, Guo et al., 2016). Together, this suggests that nutrient depletion caused by rapid tumor growth may create a unique vulnerability to autophagy inhibition. Finally, although we found that several lung cancer cell lines with oncogenic KRAS were sensitive to ULK-101, future work is required to fully define the genetic backgrounds in which targeting ULK1 and autophagy will be effective.
Limitations of the Study
Interest is mounting in developing novel therapeutics that can modulate the fundamental mechanisms of human disease, including autophagy. Despite encouraging research progress, only a limited number of compounds that target autophagy are developed beyond basic research. Accordingly, we aim to move these autophagy inhibitors through preclinical development. ULK-100 and ULK-101 have performed well in vitro, but these compounds require further validation in vivo to proceed with preclinical testing. In addition, ULK1 targeting as a therapeutic mechanism may not be effective in all genetic or environmental contexts, and further research is needed to identify when this strategy would be most effective.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank members of the MacKeigan laboratory for critical discussions and feedback. J.P.M. has support from the National Cancer Institute of the National Institutes of Health under Award Number R01CA197398.
Author Contributions
Conceptualization, K.R.M., R.C.O., and J.P.M.; Methodology, K.R.M., S.L.C., A.R.S., H.G., M.S., S.D.S., P.F., and J.P.M.; Validation, K.R.M., S.L.C., and A.R.S.; Formal Analysis, K.R.M., S.L.C., A.R.S., and J.P.M.; Investigation, K.R.M., S.L.C., and J.P.M; Resources, K.R.M., S.L.C., S.D.S., P.F., and J.P.M.; Writing – Original Draft, K.R.M., S.L.C., and J.P.M.; Writing – Review & Editing, K.R.M., S.L.C., A.R.S., R.C.O., S.D.S., P.F., and J.P.M.; Visualization, K.R.M., S.L.C., and J.P.M.; Supervision, K.R.M., S.D.S., P.F., and J.P.M.; Project Administration, R.C.O., S.D.S., P.F., and J.P.M.; Funding Acquisition, R.C.O. and J.P.M.
Declaration of Interests
The authors declare no competing interests.
Published: October 26, 2018
Footnotes
Supplemental Information includes Transparent Methods, one figure, and two tables and can be found with this article online at https://doi.org/10.1016/j.isci.2018.09.012.
Supplemental Information
The enzymatic activity of human recombinant kinases was measured after treatment with ULK1 inhibitors at the indicated concentrations using radiometric assays. The percent activity remaining (compared with vehicle control) is indicated, as is the percent inhibition, calculated by subtracting the % activity remaining from 100%. The relative inhibition compared with ULK1 is also shown (where relative inhibition [%] = [kinase % inhibition/ULK1 % inhibition] * 100) within each compound. ULK1 data are indicated in red, and all other kinases are alphabetized.
References
- Axe E.L., Walker S.A., Manifava M., Chandra P., Roderick H.L., Habermann A., Griffiths G., Ktistakis N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008;182:685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan E.Y., Kir S., Tooze S.A. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J. Biol. Chem. 2007;282:25464–25474. doi: 10.1074/jbc.M703663200. [DOI] [PubMed] [Google Scholar]
- Cheong H., Lindsten T., Wu J., Lu C., Thompson C.B. Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc. Natl. Acad. Sci. USA. 2011;108:11121–11126. doi: 10.1073/pnas.1107969108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chude C.I., Amaravadi R.K. Targeting autophagy in cancer: update on clinical trials and novel inhibitors. Int. J. Mol. Sci. 2017;18:1–11. doi: 10.3390/ijms18061279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan D.F., Chun M.G., Vamos M., Zou H., Rong J., Miller C.J., Lou H.J., Raveendra-Panickar D., Yang C.C., Sheffler D.J. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol. Cell. 2015;59:285–297. doi: 10.1016/j.molcel.2015.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng C.H., Wang Z., Tkach D., Toral-Barza L., Ugwonali S., Liu S., Fitzgerald S.L., George E., Frias E., Cochran N. Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc. Natl. Acad. Sci. USA. 2016;113:182–187. doi: 10.1073/pnas.1515617113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganley I.G., Lam du H., Wang J., Ding X., Chen S., Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009;284:12297–12305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J.Y., Chen H.Y., Mathew R., Fan J., Strohecker A.M., Karsli-Uzunbas G., Kamphorst J.J., Chen G., Lemons J.M., Karantza V. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25:460–470. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J.Y., Karsli-Uzunbas G., Mathew R., Aisner S.C., Kamphorst J.J., Strohecker A.M., Chen G., Price S., Lu W., Teng X. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013;27:1447–1461. doi: 10.1101/gad.219642.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J.Y., Teng X., Laddha S.V., Ma S., Van Nostrand S.C., Yang Y., Khor S., Chan C.S., Rabinowitz J.D., White E. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 2016;30:1704–1717. doi: 10.1101/gad.283416.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa N., Hara T., Kaizuka T., Kishi C., Takamura A., Miura Y., Iemura S., Natsume T., Takehana K., Yamada N. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell. 2009;20:1981–1991. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung C.H., Jun C.B., Ro S.H., Kim Y.M., Otto N.M., Cao J., Kundu M., Kim D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsli-Uzunbas G., Guo J.Y., Price S., Teng X., Laddha S.V., Khor S., Kalaany N.Y., Jacks T., Chan C.S., Rabinowitz J.D. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014;4:914–927. doi: 10.1158/2159-8290.CD-14-0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Kundu M., Viollet B., Guan K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky D.J. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007;8:931–937. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- Klionsky D.J., Abdelmohsen K., Abe A., Abedin M.J., Abeliovich H., Acevedo Arozena A., Adachi H., Adams C.M., Adams P.D., Adeli K. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) Autophagy. 2016;12:1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarus M.B., Novotny C.J., Shokat K.M. Structure of the human autophagy initiating kinase ULK1 in complex with potent inhibitors. ACS Chem. Biol. 2015;10:257–261. doi: 10.1021/cb500835z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarus M.B., Shokat K.M. Discovery and structure of a new inhibitor scaffold of the autophagy initiating kinase ULK1. Bioorg. Med. Chem. 2015;23:5483–5488. doi: 10.1016/j.bmc.2015.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E.J., Tournier C. The requirement of uncoordinated 51-like kinase 1 (ULK1) and ULK2 in the regulation of autophagy. Autophagy. 2011;7:689–695. doi: 10.4161/auto.7.7.15450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu E.Y., Ryan K.M. Autophagy and cancer–issues we need to digest. J. Cell Sci. 2012;125:2349–2358. doi: 10.1242/jcs.093708. [DOI] [PubMed] [Google Scholar]
- Lock R., Kenific C.M., Leidal A.M., Salas E., Debnath J. Autophagy-dependent production of secreted factors facilitates oncogenic RAS-driven invasion. Cancer Discov. 2014;4:466–479. doi: 10.1158/2159-8290.CD-13-0841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock R., Roy S., Kenific C.M., Su J.S., Salas E., Ronen S.M., Debnath J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Biol. Cell. 2011;22:165–178. doi: 10.1091/mbc.E10-06-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin K.R., Barua D., Kauffman A.L., Westrate L.M., Posner R.G., Hlavacek W.S., Mackeigan J.P. Computational model for autophagic vesicle dynamics in single cells. Autophagy. 2013;9:74–92. doi: 10.4161/auto.22532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer C.A., Kaliappan A., Dennis P.B. A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy. 2009;5:649–662. doi: 10.4161/auto.5.5.8249. [DOI] [PubMed] [Google Scholar]
- Mizushima N., Kuma A., Kobayashi Y., Yamamoto A., Matsubae M., Takao T., Natsume T., Ohsumi Y., Yoshimori T. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J. Cell Sci. 2003;116:1679–1688. doi: 10.1242/jcs.00381. [DOI] [PubMed] [Google Scholar]
- Petherick K.J., Conway O.J., Mpamhanga C., Osborne S.A., Kamal A., Saxty B., Ganley I.G. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J. Biol. Chem. 2015;290:28726. doi: 10.1074/jbc.A114.627778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowitz J.D., White E. Autophagy and metabolism. Science. 2010;330:1344–1348. doi: 10.1126/science.1193497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S., Tortola L., Perlot T., Wirnsberger G., Novatchkova M., Nitsch R., Sykacek P., Frank L., Schramek D., Komnenovic V. A dual role for autophagy in a murine model of lung cancer. Nat. Commun. 2014;5:3056. doi: 10.1038/ncomms4056. [DOI] [PubMed] [Google Scholar]
- Rosenfeldt M.T., Ryan K.M. The role of autophagy in tumour development and cancer therapy. Expert Rev. Mol. Med. 2009;11:e36. doi: 10.1017/S1462399409001306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell R.C., Tian Y., Yuan H., Park H.W., Chang Y.Y., Kim J., Kim H., Neufeld T.P., Dillin A., Guan K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013;15:741–750. doi: 10.1038/ncb2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I., Minematsu-Ikeguchi N., Ueno T., Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005;1:84–91. doi: 10.4161/auto.1.2.1697. [DOI] [PubMed] [Google Scholar]
- Wood S.D., Grant W., Adrados I., Choi J.Y., Alburger J.M., Duckett D.R., Roush W.R. Silico HTS and structure based optimization of indazole-derived ULK1 inhibitors. ACS Med. Chem. Lett. 2017;8:1258–1263. doi: 10.1021/acsmedchemlett.7b00344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachari M., Ganley I.G. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017;61:585–596. doi: 10.1042/EBC20170021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The enzymatic activity of human recombinant kinases was measured after treatment with ULK1 inhibitors at the indicated concentrations using radiometric assays. The percent activity remaining (compared with vehicle control) is indicated, as is the percent inhibition, calculated by subtracting the % activity remaining from 100%. The relative inhibition compared with ULK1 is also shown (where relative inhibition [%] = [kinase % inhibition/ULK1 % inhibition] * 100) within each compound. ULK1 data are indicated in red, and all other kinases are alphabetized.