

Abstract
Histone acetylation influences protein interactions and chromatin accessibility and plays an important role in the regulation of transcription, replication, and DNA repair. Conversely, DNA damage affects these crucial cellular processes and induces changes in histone acetylation. However, a comprehensive overview of the effects of DNA damage on the histone acetylation landscape is currently lacking. To quantify changes in histone acetylation, we developed an unbiased quantitative mass spectrometry analysis on affinity‐purified acetylated histone peptides, generated by differential parallel proteolysis. We identify a large number of histone acetylation sites and observe an overall reduction of acetylated histone residues in response to DNA damage, indicative of a histone‐wide loss of acetyl modifications. This decrease is mainly caused by DNA damage‐induced replication stress coupled to specific proteasome‐dependent loss of acetylated histones. Strikingly, this degradation of acetylated histones is independent of ubiquitylation but requires the PA200‐proteasome activator, a complex that specifically targets acetylated histones for degradation. The uncovered replication stress‐induced degradation of acetylated histones represents an important chromatin‐modifying response to cope with replication stress.
Keywords: histone, DNA damage, replication stress, acetylation, PA200‐proteasome
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; DNA Replication, Repair & Recombination
Introduction
DNA transacting processes such as transcription, replication, and DNA repair take place in the context of chromatin. Chromatin is a highly organized structure in which DNA is wound around histone octamers to form nucleosomes. The histone octamer consists of a histone H3‐H4 heterotetramer flanked by two histone H2A‐H2B heterodimers. The linker histone H1 binds linker DNA entering and exiting nucleosomes, thereby regulating chromatin compaction 1. Histones are a target for many post‐translational modifications (PTMs), like methylation, phosphorylation, ubiquitylation, and acetylation that are predominantly located at histone tails protruding from the nucleosome 2. Histone PTMs can directly influence the strength of the histone interactions with each other and with the DNA 2. Furthermore, several PTMs provide a docking site for specific readers, like chromatin remodeling complexes containing bromodomains that have affinity for acetylated histones 3. Together, this interplay of histone PTMs and chromatin remodeling proteins controls the accessibility of the chromatin thereby playing an important role in transcription, replication, and DNA repair.
For example, histone PTMs play a crucial role during DNA repair and DNA damage signaling 2, 4, 5. Histone acetylation was one of the first histone PTMs, shown to be involved in DNA repair. More than 3 decades ago, it was found that after UV irradiation histones undergo a wave of rapid hyperacetylation followed by a hypoacetylation phase 6. UV light causes helix‐distorting lesions such as 6‐4 photoproducts (6‐4PP) and cyclobutane pyrimidine dimers (CPD), which block transcription and interfere with replication. These lesions can be removed by a specific DNA repair mechanism called nucleotide excision repair (NER) 7. The fact that hyperacetylated nucleosomes both increased chromatin accessibility in vivo and stimulated repair efficiency 8 led to the formulation of the access‐repair‐restore concept 9. This model proposed that chromatin is remodeled by ATP‐dependent chromatin remodelers, histone chaperones, and modifying enzymes to provide access of repair proteins to damaged sites. After repair, the chromatin conformation is restored to pre‐damage conditions to preserve epigenetic information and inhibit DNA damage signaling 9, 10. Over the years, several histone acetyltransferases (HATs) have been implicated in the response to UV damage 11, 12, 13, 14. For instance, p300 interacts with PCNA and is associated with newly synthesized DNA after UV irradiation 14. UV‐induced H3K9/K14 acetylation by GCN5 increases nucleosome accessibility at the repressed MFA2 locus in yeast 11, 15 through binding of the RSC remodeling complex, which stimulated CPD repair 16. In human cells, GCN5 is necessary for efficient recruitment of NER factors and repair 17. In addition to H3 acetylation, also histone H4 is rapidly acetylated after UV by ING2 leading to the recruitment of XPA to the lesion 13. Notably, besides acetylation also deacetylation plays an important role during the UV‐DDR. For instance, histone deacetylase enzymes (HDACs) 1 and 2 are recruited to damaged sites by the DNA damage recognition proteins, DDB1 and DDB2, resulting in H3K56 deacetylation 18.
Although these studies underscore the crucial interplay of histone acetylation and DNA repair, thus far a comprehensive overview of UV‐induced histone acetylation and deacetylation events during repair, but also caused by UV‐induced replication or transcription blocks is missing. Changes in histone modifications are often studied using modification‐specific antibodies 15, 19. However, these techniques rely on the specificity and availability of antibodies, and unknown modification sites can therefore not be identified. In particular, the acetylation status of histone H2A and H2B after UV irradiation remains largely unclear. In this study, we used a quantitative mass spectrometry approach to identify histone acetylation changes in response to UV irradiation in an unbiased manner. Surprisingly, we found that UV damage induces a histone‐wide reduction in acetylation levels. This loss of acetylated histones was not dependent on active transcription or NER. Instead, we show that it is the result of replication stress‐induced histone degradation by a specific type of proteasome, containing the PA200 subunit, which recognizes acetylated proteins.
Results
Isolation of acetylated histone peptides
To identify the effects of UV‐induced DNA damage on histone acetylation in an unbiased and quantitative manner, we used a stable isotope labeling by amino acids in cell culture (SILAC) MS‐based approach. Cells labeled with light‐isotope‐containing amino acids (K0R0) were mock‐treated, and cells labeled with heavy‐isotope‐containing amino acids (K6R10) were UV‐irradiated (16 J/m2) 1 h before harvesting. In order to increase detection and quantification of histone acetylation sites, including less abundant ones, we established an isolation procedure to enrich for acetylated histone peptides. To this end, we combined histone acid extraction 20, 21 with recently developed acetyl‐lysine immunoprecipitation (Ac‐IP) procedures 22, 23 (Fig 1A). Histones were isolated by a two‐step histone acid extraction protocol: extracting first linker histone H1 and the high mobility group proteins, followed by isolation of the core histones. Specificity of the histone extraction was confirmed by comparing the pellet fraction containing the precipitated non‐acid‐soluble proteins with the histone H1 and the core histone fractions using Coomassie staining (Fig 1B). Protein bands of the expected sizes of H1 (21 kDa) and the core histones (10–15 kDa) could be detected in the designated lanes. The presence of histone H1.2 in the histone H1 fraction and histone H2B in the core histone fraction was confirmed by Western blot analysis. Furthermore, the induction of γ‐H2AX following UV irradiation 24, 25 indicates that histone PTMs are preserved during the acid extraction procedure (Fig 1C). Prior to digestion, the isolated H1 and core histone fractions were pooled. To obtain peptide sizes that are compatible with MS analysis to ensure a high coverage of all histone proteins, we split our sample in fractions, each digested with a different protease; trypsin, pepsin, or GluC (Fig 1A). The trypsin fraction was further split into four and digested for different durations. After pooling these differential digested fractions, this approach led to the detection of many different unique and overlapping peptides, covering 74–98% of the core histone sequences and 27–62% of the different histone H1 variants (Fig 1D and Dataset EV1).
Figure 1. Isolation of acetylated histone peptides.
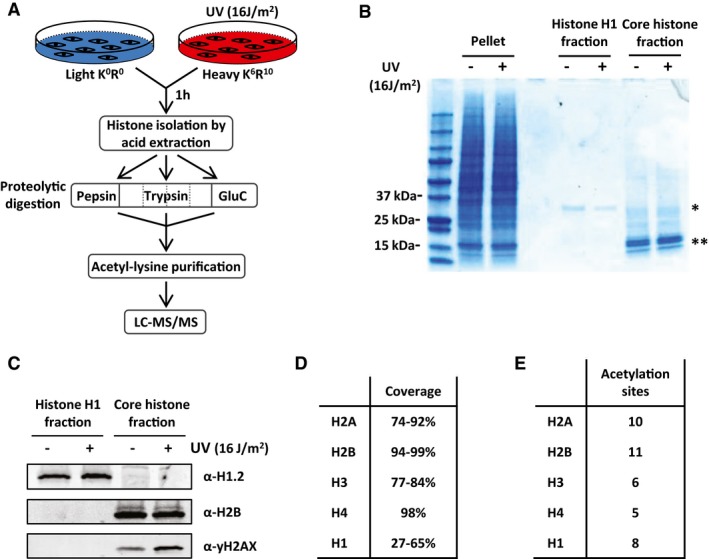
- Schematic overview of the experimental set‐up to isolate and identify acetylated histone peptides following UV irradiation. SILAC‐labeled cells are mock or UV (16 J/m2) treated 1 h before harvesting. Heavy and light labeled cells are mixed in a 1:1 ratio followed by acid extraction to isolate histones. Histones are separated in three fractions each digested with either pepsin, trypsin, or GluC. The limited digestion with trypsin is performed for 2, 10, 30, and 120 min. The different fractions of digested peptides are combined and followed by acetyl‐lysine immunopurification. The acetylated‐lysine‐enriched histone peptides are analyzed by LC‐MS/MS.
- Histones were isolated by acid extraction from HeLa cells 1 h after UV (16 J/m2) or mock treatment and were loaded on a 4–15% gradient SDS–PAGE gel and stained with Coomassie Brilliant Blue. The different fractions originate from the same amount of cells and contain the non‐acid‐soluble proteins (pellet), histone H1 and high mobility group proteins (H1 fraction), or the core histones (core histone fraction). *Indicates 21 kDa protein band, most likely representing histone H1. **Indicates several protein bands ranging between 10 and 15 kDa most likely representing core histones.
- Western blot of isolated histone fractions from HeLa cells 1 h after UV (16 J/m2) or mock treatment. Western blots were stained with α‐histone H1.2 (top panel), α‐histone H2B (middle panel), and α‐γ‐H2AX (bottom panel).
- Table listing the coverage of the histone sequences using our peptide digestion procedure by LC‐MS/MS analysis.
- Table listing the number of acetylation sites on core histones identified by MS after acetylated peptide enrichment.
Acetylated peptides were isolated by Ac‐IP and measured on LC‐MS/MS. While several acetylated histone peptides could already be identified without the specific isolation procedure (Dataset EV1), the Ac‐IP resulted in a fourfold increase in the number of identified unique acetylated histone peptides. We identified 301 different acetylated histone peptides in total, 75% of which carry more than one acetyl group (Dataset EV2). Of note, when we performed additional MaxQuant analysis for the presence of phosphorylation, methylation, and diGLY modifications, we found that about 19% of the peptides carried additional histone PTMs (Table EV1). This suggests that the affinity of the antibody used for the acetylation enrichments is not negatively affected by the presence of additional PTMs on the same peptide.
Within this set of peptides, 40 unique histone acetylation sites were identified, including many previously described sites, like histone H3 acetylation on lysine 9, 14, 18, 23, 27, and 64 26, 27. Interestingly, we found H2B to have most unique acetylation sites (Fig 1E). As expected, acetylated sites of core histones were mainly found in the tails 28, whereas lysine 64 of histone H3 was the only acetylated residue found within the globular domains (Dataset EV3). In contrast to the core histones, the majority of acetyl‐modified lysines of the linker histone H1 were located in the globular domain 29.
Histone acetylation levels are decreased after UV irradiation
After validating our approach to identify histone acetylation sites efficiently, we analyzed differences in the extent of histone acetylation 1 h after UV‐induced DNA damage. To visualize UV‐induced changes in the acetylation status, all unique acetyl‐modified histone peptides were plotted against their SILAC‐based UV/mock log2 ratio (Fig 2A). Surprisingly, the vast majority of the acetylated histone peptides had a negative normalized SILAC ratio following UV treatment (61% had a UV/mock log2 ratio < −0.5), indicative of an overall reduction in acetylation level of histones 1 h after UV‐induced DNA damage (Fig 2B and Dataset EV2). This is in contrast to previously observed UV‐induced acetylation at chromatin locations nearby DNA lesions, like H3K9/14Ac, which was observed using site‐specific antibodies combined with a ChIP procedure 11, 15. Our mass spectrometry approach, which quantifies all histones in the cell, showed a loss of histone H3 peptides that were acetylated at lysine 9 or 14 (Dataset EV3). In addition to these two acetylation sites on histone H3, we found several other H3 acetylation sites to be reduced after UV damage. All core histones displayed lower acetylation levels for most quantified acetyl modifications after UV‐induced DNA damage; however, histone H2B and H2A were affected the most (Figs 2B and EV1A, and Datasets EV2 and EV3). Most acetylated peptides we identified showed multiple acetyl modifications on different residues, which were all reduced following DNA damage. Together, these data indicate a non‐site‐specific, general loss of acetylated histones. This loss can be either caused by deacetylation of histones, reduced activity of histone acetylases or by degradation of acetylated histones. Of note, only for histone H2A variant H2AZ sites with increased acetylation after UV irradiation (UV/mock log2 ratio > 0.5) were observed, while its unmodified peptides are not changed (Datasets EV2 and EV3).
Figure 2. UV‐induced decrease in acetylated histones.
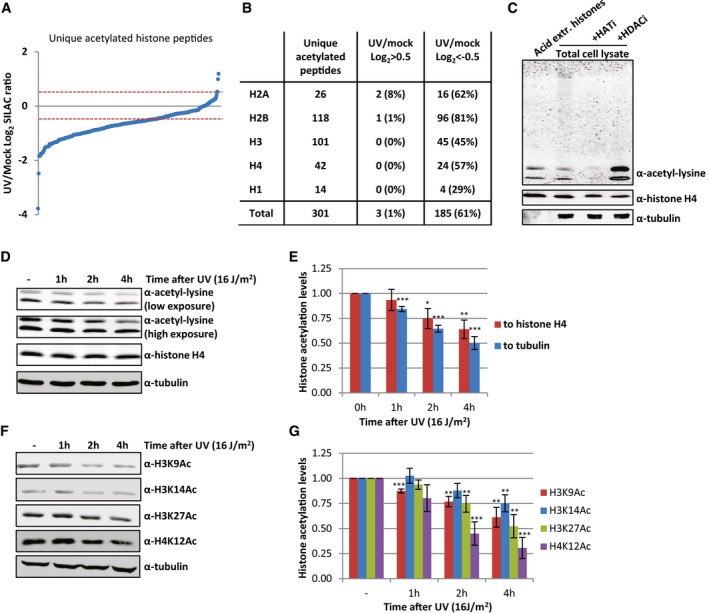
- Identified acetylated histone peptides plotted against their log2 SILAC ratio (1 h after 16 J/m2 UV/mock), ranked by SILAC ratio.
- Table listing the number of identified peptides per histone using MS following enrichment of acetylated peptides, and the number of peptides identified that are decreased (UV/mock log2 < −0.5) or increased (UV/mock log2 > 0.5) 1 h after UV irradiation (16 J/m2).
- Western blot of acid extracted histones and WCE from HeLa cells, treated with HATi (CTK7A, 100 μM and CPTH2, 50 μM) or HDACi (TSA, 1 μM) for 4 h or mock‐treated as indicated.
- A representative Western blot of histone acetylation levels of HeLa cells lysed at indicated time points after UV irradiation (16 J/m2). Blots were stained with α‐acetyl‐lysine (top panels, high and low exposure), α‐histone H4 (middle panel), and α‐tubulin (bottom panel).
- Quantification of histone acetylation levels, normalized against either the histone H4 levels (red bars) or tubulin levels (blue bars). Error bars represent SEM. N = 5 independent experiments. Significant differences between UV‐treated and mock‐treated conditions were calculated using a t‐test and are indicated with *P < 0.1, **P < 0.05, and ***P < 0.01.
- Representative Western blots of HeLa WCE from different time points after UV‐C irradiation (16 J/m2) using the indicated histone modification‐specific antibodies. Histone H4 and tubulin were used as loading controls.
- Quantification of specific histone acetylation marks normalized to tubulin levels. Significant differences were calculated using a t‐test and are indicated with **P < 0.05 and ***P < 0.01. N = 3, error bars represent SEM.
Figure EV1. UV‐induced decrease in acetylated histones.
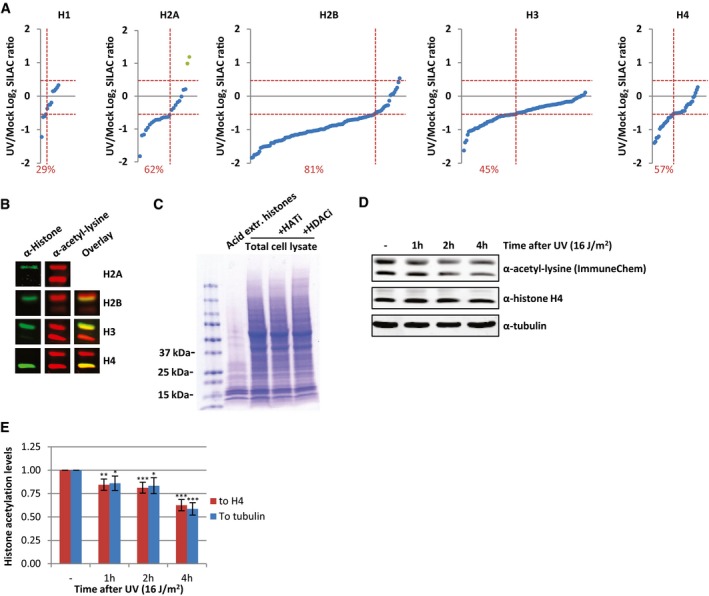
- Identified acetylated peptides per histone plotted against their log2 SILAC ratio (1 h after 16 J/m2 UV/mock), ranked by SILAC ratio. Green dots represent the identified peptides from histone variant H2AZ. The percentage of peptides with a SILAC ratio < −0.5 are indicated in red.
- Western blots of WCE from HeLa cells stained with α‐histone H2A, α‐histone H2B, α‐histone H3, and α‐histone H4 (depicted in green) and α‐acetyl‐lysine (depicted in red) indicating that the strong acetyl‐lysine signal of the low molecular weight bands originates from acetylated core histones. H2A and acetyl‐lysine antibodies were raised in the same species and therefore did not allow an overlay.
- Coomassie staining of extracted histones and WCE from the same number of HeLa cells, treated with HATi (CTK7A, 100 μM and CPTH2, 50 μM) or HDACi (TSA, 1 μM) or mock‐treated.
- Western blots of HeLa cells lysed at different time points after irradiation with UV (16 J/m2). Blots were stained with α‐acetyl‐lysine (Immunechem, top panel), α‐histone H4 (middle panel), and α‐tubulin (bottom panel).
- Quantification of the α‐acetyl‐lysine signal normalized against either the histone H4 levels (red bars) or tubulin levels (blue bars). Error bars represent SEM. N = 7. Significant differences between UV‐treated and mock‐treated conditions are calculated with t‐test and indicated with *P < 0.1, **P < 0.05, and ***P < 0.01.
To confirm this striking histone‐wide reduction of acetylation detected by MS, we quantified overall histone acetylation levels on Western blot using the α‐acetyl‐lysine antibody. Western blotting of sonicated whole cell extract (WCE), obtained by lysing HeLa cells directly in Laemmli buffer, showed that the vast majority of the signal from the α‐acetyl‐lysine antibody is confined in two bands around 10–15 kDa (Fig 2C). This α‐acetyl‐lysine signal fully overlapped with bands obtained by staining against the different core histones, the lower band mainly representing acetylated H4, while the upper band represent acetylated histones H2A, H2B, and H3 (Fig EV1B). A similar α‐acetyl‐lysine signal was obtained from an acid extracted histone fraction (Figs 2C and EV1C). This strongly suggests that these low molecular weight acetyl‐lysine signals represent acetylated histones and that the majority of lysine modifications with acetyl within the cell take place at the highly expressed and heavily modified core histones. This was further corroborated by the reduction in acetylation signal following incubation with HAT inhibitors and the increase in signal after HDACs were inhibited (Fig 2C). Together, this shows that the overall histone acetylation status can be assessed in a quantitative manner by Western blot using α‐acetyl‐lysine staining. In line with our MS results, we observed a UV‐induced decrease in acetylated histones 1 h after UV (Fig 2D and E). Interestingly, the histone acetylation levels decreased even further over time. Quantification of the histone acetylation levels normalized to either histone H4 or tubulin showed a 30–50% reduction of acetylated histones 4 h following UV exposure (Fig 2D and E). Our MS results were further confirmed using Western blot in combination with antibodies recognizing specific histone acetylation marks (H3K9, H3K14, H3K27, and H4K12) that were identified to be decreased in our MS experiments (Fig 2F and G). In addition, using another acetyl‐lysine antibody 22, we confirmed the UV‐induced reduction in overall histone acetylation levels by both MS (Dataset EV4) and Western blotting experiments (Fig EV1D and E), excluding the possibility that our results were due to preferred recognition motifs or other biases of the used antibodies.
Recovery of acetylation levels 16 h after UV is dependent on NER
To study the temporal behavior of histone acetylation levels after UV irradiation in more detail, Western blot experiments were performed with WCE obtained at later time points after UV irradiation. While the acetylation signal was still reduced 8 h after UV, it recovered to levels similar as mock‐treated cells 16 h after irradiation (Fig 3A (left panel) and B), a time point when most DNA repair is finished 30, 31. This suggests that repair of UV‐induced damage by NER might be necessary for the recovery of histone acetylation levels. Indeed, the recovery of the histone acetylation signal is abolished in cell lines deficient for NER proteins XPC or XPA, indicating that repair of UV‐induced DNA damage is crucial for recovery of histone acetylation levels and that the persistent presence of DNA damage prevents this (Fig 3). Interestingly, in NER‐deficient cells, a similar loss of acetylated histones was observed compared to NER‐proficient cells in the first 8 h after UV, showing that the UV‐induced decrease in acetylated histones is not dependent on DNA damage recognition or on repair by NER (Fig 3). Together, these data suggest that the trigger for the observed decrease in histone acetylation levels occurs upstream or in parallel to the damage recognition step of NER.
Figure 3. The recovery of histone acetylation levels at later time points after UV is dependent on NER .
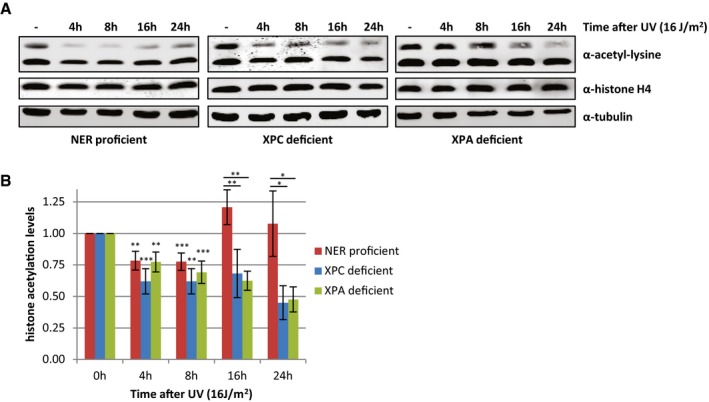
- Representative Western blots of WCE from NER‐proficient HeLa cells and NER‐deficient XP4PA (XP‐C) and XP2OS (XP‐A) cells obtained at indicated time points after UV irradiation (16 J/m2) and stained with the indicated antibodies.
- Quantification of histone acetylation levels of NER‐proficient cells (HeLa) and NER‐deficient (XP‐A and XP‐C) patient cells at the indicated time points after UV irradiation (16 J/m2). Histone acetylation levels are normalized against histone H4 levels. Average of at least five independent experiments and error bars represent SEM. Significant differences (t‐test) are indicated with *P < 0.1, **P < 0.05, and ***P < 0.01.
Transcription does not influence the decrease in acetylated histones after UV irradiation
Persistent UV‐induced DNA lesions severely impede transcription and replication 32, 33. As active transcription is highly associated with increased levels of histone acetylation 2, we tested whether transcription inhibition, in the absence of DNA damage, affects the acetylation status of histones. Transcription was impeded by THZ1 and flavopiridol, which both inhibit transcription preceding productive elongation, and α‐amanitin that blocks elongating RNAPII 34. In contrast to UV, the histone acetylation levels remain rather stable after transcription inhibition, indicating that transcription inhibition is not the main cause of the UV‐induced loss of acetylated histones (Fig 4A and B). However, even though chemical transcription inhibition did not result in a decrease in acetylated histones, we could not exclude that active transcription or lesion stalled RNAPII might initiate this UV‐induced process. To test this, cells were pre‐treated with the transcription inhibitors THZ1 or flavopiridol to deplete cells of elongating RNAPII before UV irradiation. This, however, did not affect the decrease in histone acetylation levels after UV irradiation (Fig 4C and D), indicating that the UV‐induced loss of acetylated histones is a process independent of transcription.
Figure 4. Loss of acetylated histones is independent of transcription inhibition.
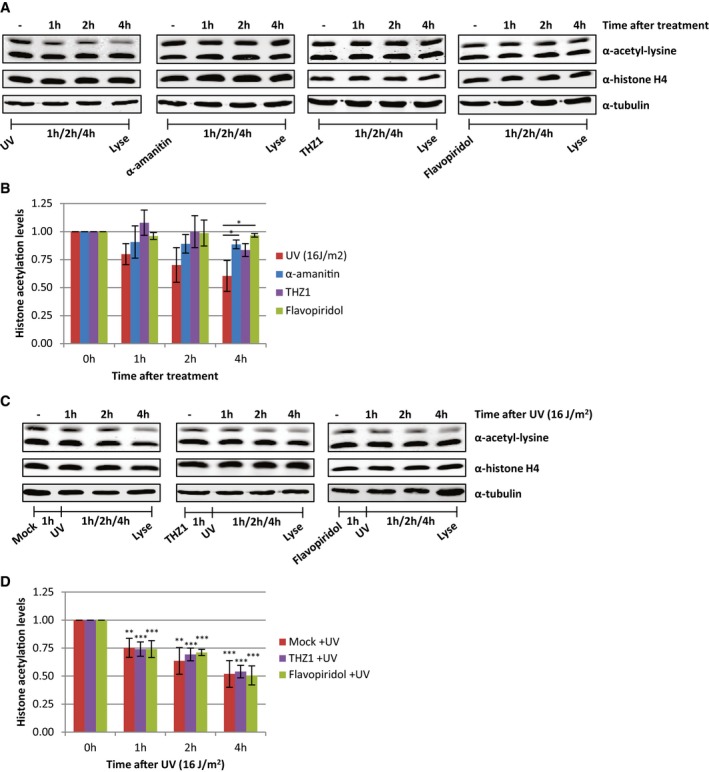
- Representative Western blots of histone acetylation levels of HeLa cells lysed at indicated time points after UV irradiation (16 J/m2, left panel), α‐amanitin (second panel, 100 μg/ml), THZ1 (third panel, 1 μM), or flavopiridol (right panel, 1 μM) treatment. Blots were stained with α‐acetyl‐lysine (top panel), α‐histone H4 (middle panel), and α‐tubulin (bottom panel).
- Quantified histone acetylation levels, normalized against histone H4 levels for quantification. Average of at least three experiments. Error bars represent SEM. Significant differences, calculated by t‐test, between UV‐irradiated and inhibitor‐treated conditions are indicated with *P < 0.1.
- Representative Western blots of histone acetylation levels of HeLa cells pre‐treated with transcription inhibitors [mock (left), THZ1 (middle, 1 μM), or flavopiridol (right, 1 μM)] an hour before UV irradiation and lysed at the indicated time points after UV (16 J/m2). Blots were stained with the indicated antibodies.
- Quantification of α‐acetyl‐lysine signal, normalized against histone H4 levels. Average of five experiments. Error bars represent SEM. Significant differences, calculated by t‐test, between UV‐treated and mock‐treated conditions are indicated with **P < 0.05 and ***P < 0.01.
Decrease in histone acetylation levels is the result of UV‐induced replication stress
In addition to transcription inhibition, UV‐induced DNA lesions also cause replication stress, by slowing down or stalling replication forks. To test whether the loss of acetylated histones is caused by UV‐induced replication stress, we also induced replication stress by exposing cells to a combination of hydroxyurea (HU) and arabinofuranosyl cytidine (AraC) or by mitomycin C (MMC). These experiments showed that induction of replication stress results in a similar loss of acetylated histones as after UV irradiation (Fig 5A and B). This indicates that replication stress itself can indeed induce a general decrease in histone acetylation levels and suggests that the loss of acetylated histones following DNA damage might be the direct consequence of UV‐induced replication stress. To test whether the loss of acetylated histones following UV exposure could be attributed to replication stress, we studied UV‐induced effects on histone acetylation in non‐replicating cells. To this end, HeLa cells were blocked in S‐phase using the Cdc7/CDK9 inhibitor PHA 767491 hydrochloride (Fig EV2A). These cells displayed lower histone acetylation levels under unperturbed conditions (Figs 5C and EV2B), which suggests that the high histone acetylation levels in cycling cells are mainly induced by replication‐related events. However, no additional UV‐induced decrease was observed in these cells (Fig 5C and D). Similar results were obtained in contact‐inhibited non‐replicating VH10 cells, which showed less reduction in histone acetylation levels compared to cycling VH10 cells following UV exposure (Fig EV2C–E). Together, these results indicate that the loss of acetylated histones after UV irradiation is mainly the result of replication stress.
Figure 5. Replication stress induces loss of acetylated histones.
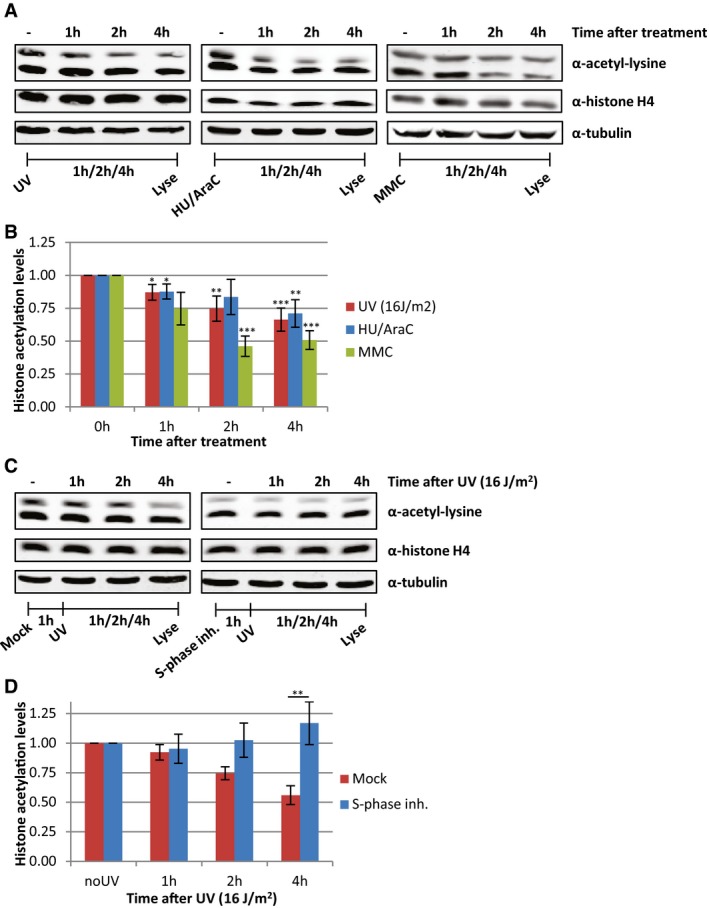
- Representative Western blots of histone acetylation levels of HeLa cells obtained at the indicated time points after UV irradiation (16 J/m2), HU/AraC treatment (100 mM/10 μM), or MMC treatment (10 μg/ml). Blots were stained with α‐acetyl‐lysine (top panel), α‐histone H4 (middle panel), and α‐tubulin (bottom panel).
- Quantification of average histone acetylation signal, normalized to loading control. N ≥ 3. Error bars represent SEM. Significant differences, calculated with t‐test, between treated and mock conditions are indicated with *P < 0.1, **P < 0.05, and ***P < 0.01.
- Representative Western blots, stained with the indicated antibodies of HeLa cells pre‐treated o/n with an S‐phase inhibitor (PHA 767491 hydrochloride, 10 μM) and lysed at indicated time points after UV (16 J/m2).
- Quantification of the α‐acetyl‐lysine signals, normalized against histone H4 levels, N = 5, error bars represent SEM. Significant differences, calculated with t‐test, between UV‐irradiated and inhibitor‐treated conditions are indicated with **P < 0.05.
Figure EV2. Loss of acetylated histones is caused by replication stress.
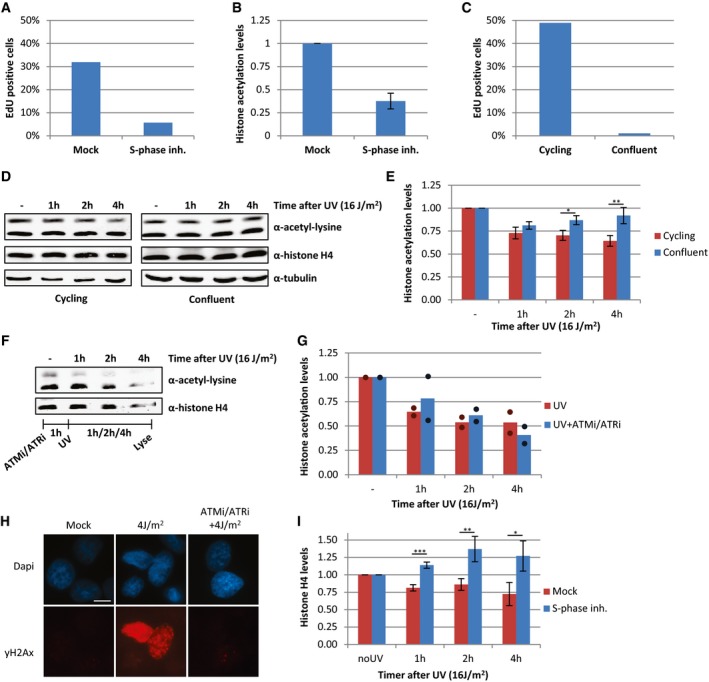
- Relative amount of HeLa cells positively stained for EdU incorporation (2 h, 20 μM), quantified from immunofluorescence images (> 120 cells analyzed).
- Quantification of histone acetylation levels, normalized against histone H4 levels of HeLa cells that are cultured with S‐phase inhibitor (10 μM, o/n) or mock‐treated. Error bars represent SEM, N = 5.
- Relative amount of EdU‐positive, cycling, or contact‐inhibited (confluent) VH10 cells, quantified from immunofluorescence images (> 100 cells analyzed).
- Western blots of WCE from confluent (right panel) or cycling (left panel) VH10 cells lysed at different time points after UV (16 J/m2). Blots were stained with α‐acetyl‐lysine (top panel), α‐histone H4 (middle panel), and α‐tubulin (bottom panel).
- Quantification of histone acetylation levels in confluent or cycling VH10 cells at different time points in which the histone acetylation levels are normalized against histone H4 levels after UV (16 J/m2). N = 3, error bars represent SEM. Significance of differences between cycling and confluent cells is calculated by t‐test and indicated with *P < 0.1 and **P < 0.05.
- Western blot of WCE, made at different time points after UV irradiation (16 J/m2) from HeLa cells pre‐treated for 1 h with a combination of ATM (Ku55933, 10 μM) and ATR (VE‐821, 10 μM) inhibitors.
- Quantification of histone acetylation levels in cells treated with ATM and ATR inhibitors an hour before UV irradiation (16 J/m2). Bar graph represents mean, and dots represent individual data points. N = 2
- Representative immunofluorescence images of γ‐H2AX staining following, mock treatment, UV‐induced DNA damage (4 J/m2) with or without ATM and ATR inhibitors as indicated. Scale bar 10 μm.
- Quantification of histone H4 levels in HeLa cells pre‐treated o/n with an S‐phase inhibitor (PHA 767491 hydrochloride, 10 μM) and lysed at indicated time points after UV (16 J/m2). N = 5, error bars represent SEM, and data are normalized against tubulin levels. Significant differences, calculated with t‐test, between UV‐irradiated and inhibitor‐treated conditions are indicated with *P < 0.1, **P < 0.05, and ***P < 0.01.
Although ATR and ATM signaling pathways play an important role following DNA damage and replication stress 35, 36, the replication stress‐induced loss of acetylated histones was not dependent on the activity of these kinases as histone acetylation was still reduced in the presence of both ATM and ATR inhibitors (Fig EV2F and G).
Chromatin‐bound acetylated histones are degraded
Besides the reduction in histone acetylation levels, we observed a concomitant subtle decrease in the histone H4 levels after UV irradiation, which was more pronounced at later time points (Fig 6A and B), however, to a lesser extent than the loss of acetylated histones (Fig 6C). This suggests that the observed UV‐induced decrease in histone acetylation levels could be caused by degradation of a specific subset of acetylated histones. Accordingly, we observed that the decrease in histone acetylation after UV was larger when normalized to tubulin compared to normalization to H4 levels (Fig 2E). To test this hypothesis, cells were pre‐treated with the proteasome inhibitor MG132 1 h before UV irradiation. Proteasome inhibition completely rescued the UV‐induced loss of both acetylated histones and histone H4 (Fig 6A–C). Of note, this loss of H4 is, like the loss of histone acetylation, dependent on replication (Fig EV2I).
Figure 6. Proteasomal degradation of acetylated histones following replication stress.
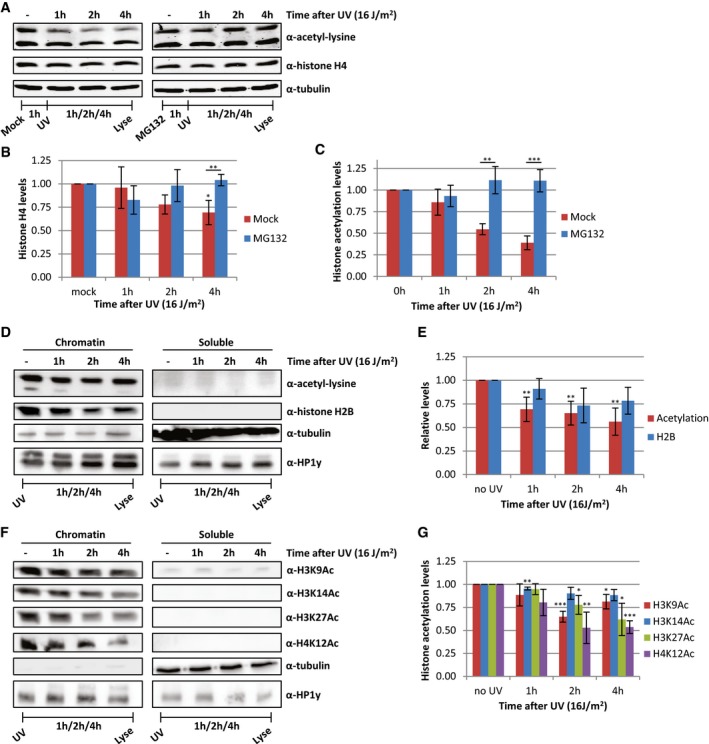
-
A–C(A) Western blots of HeLa cells treated with proteasome inhibitor MG132 1 h before UV irradiation and lysed at different time points after UV (16 J/m2). Blots were stained with α‐acetyl‐lysine (top panel), α‐histone H4 (middle panel), and α‐tubulin (bottom panel). Quantifications of (B) histone H4 levels and (C) acetylation levels, normalized against tubulin levels. Average of six experiments, and error bars represent SEM. Representative blots are shown in panel (A).
-
DFractionation experiments comparing histone and acetylation levels in chromatin and soluble fractions at the indicated time points after UV irradiation (16 J/m2). HP1y was used as loading control for chromatin fraction, tubulin for soluble fraction.
-
EQuantification of histone acetylation and H2B levels in chromatin fraction. Non‐irradiated samples were set as 1. N ≥ 4, error bars indicate SEM.
-
FRepresentative Western blots of fractionated cell extracts of HeLa cells at indicated time after UV irradiation (16 J/m2) using histone modification‐specific antibodies. Loading controls for H3K9 and H3K27 are depicted in Figure 6D.
-
GQuantification of the signal of the indicated histone modification‐specific antibodies stainings in the chromatin fraction. N ≥ 4 and error bars represent SEM.
During replication, the levels of soluble histones are increased. In addition, it has previously been shown that newly synthesized histone H3/H4 dimers are acetylated to stimulate incorporation into chromatin 37, 38, 39. Therefore, we hypothesized that this pool of newly synthesized, yet unincorporated acetylated histones might be degraded upon replication stress, thereby preventing an excess of non‐incorporated histones. To test this, we performed cell fractionation experiments to separate newly synthesized, free histones from chromatin‐bound histones. Compared to chromatin‐bound histones, the levels of soluble histones are, as expected, very low and often hardly detectable on blots (Fig 6D). Most importantly, the acetylation signal is almost completely restricted to the chromatin fraction. This suggests that the observed 30–50% decrease in acetylation levels can only be explained by degradation of chromatin‐bound histones. Indeed, after UV irradiation, the acetylation levels in the chromatin fraction are decreasing over time, to a similar extent as was observed in whole cell extract (Fig 6D and E). This replication stress‐induced decrease in acetylation levels of chromatin‐bound histones was confirmed by additional stainings using acetylation site‐specific histone antibodies (Fig 6D, F and G).
Acetylated histones are degraded by PA200‐proteasome complexes
Interestingly, inhibition of the ubiquitin‐activating enzyme (E1) using PYR‐41 did not rescue the decrease in acetylated histones (Fig 7A and B). This indicates, in contrast to most proteasome‐dependent protein degradation, that the proteasomal degradation of acetylated histones is independent of protein ubiquitylation. It has been shown that acetylated histones can be recognized and degraded by a specific proteasome complex independent of protein ubiquitylation 40. This specific proteasome complex consists of the 20S core complex and the nuclear proteasome activator PA200 40. In contrast to the 26S proteasome, in which the 19S regulatory cap recognizes ubiquitylated proteins, the PA200‐proteasome complex does not recognize poly‐ubiquitin chains but binds to acetylated proteins and targets them for degradation 40. Therefore, we tested the involvement of PA200 in the UV‐induced decrease in histone H4 and histone acetylation levels. siRNA‐mediated knockdown of PA200 (Fig EV3A) inhibited the UV‐induced proteasomal degradation of acetylated histones (Fig 7C–E). Together, these results show that following replication stress, for example, induced by UV‐induced DNA damage, acetylated histones are degraded by the PA200‐20S proteasome complex in an ubiquitin‐independent manner. To address the biological relevance of the degradation of acetylated histones, we performed cellular survival assays following UV‐induced DNA damage. Interestingly, PA200‐depleted cells were more sensitive to UV‐induced DNA damage compared to control‐transfected cells (Figs 7F and EV3C), suggesting that the degradation of acetylated histones plays a crucial role during the cellular responses to replication stress. Of note, this UV‐sensitivity following PA200 depletion is not caused by an effect on NER‐mediated repair rate, as knockdown of PA200 did not affect NER efficiency as shown by unscheduled DNA synthesis (UDS) experiments, which quantifies the gap‐filling DNA synthesis step NER (Figs 7G and EV3B). Together, these data indicate that the replication stress‐induced degradation of acetylated histones is important for cells to cope with replication stress.
Figure 7. PA200‐proteasome degrades acetylated histones.
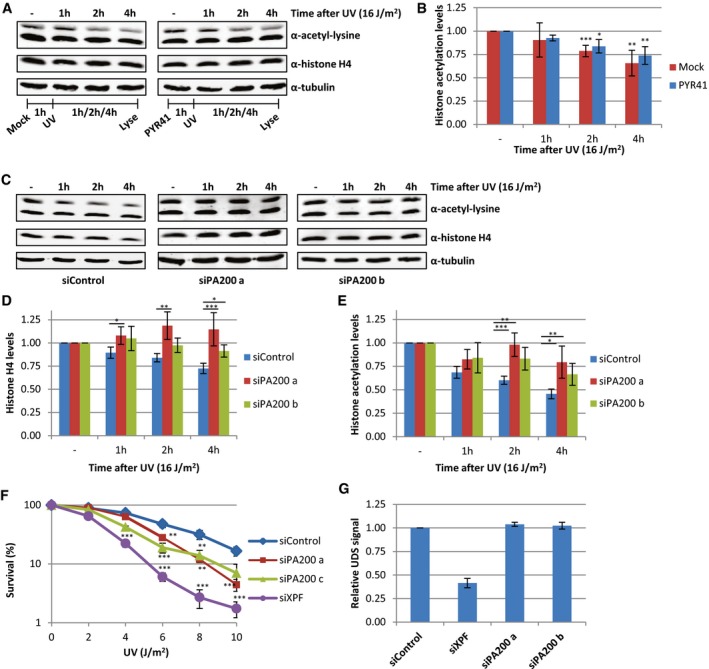
-
AWestern blots of HeLa cells pre‐treated with the E1 enzyme inhibitor PYR‐41 (10 μM) 1 h before UV irradiation and lysed at different time points after UV (16 J/m2). Blots were stained with the indicated antibodies, and representative blots are shown.
-
BQuantification of histone acetylation levels, normalized against histone H4 levels, average of five experiments. Error bars represent SEM.
-
CRepresentative Western blots of siControl or siPA200 (two independent siRNAs, a and b) transfected cells, lysed at indicated time points after UV irradiation (16 J/m2).
-
D, E(D) Quantification of histone H4 and (E) histone acetylation levels, normalized against tubulin and non‐treated levels are set as 1. The average of at least nine independent experiments and SEM is shown.
-
FClonogenic survival of HeLa cells treated with different doses of UV‐C. Relative survival (in percentage) is plotted against UV‐C dose. N ≥ 2, error bars represent SEM.
-
GUnscheduled DNA synthesis in non‐cycling C5RO cells measured by the incorporation of EdU (20 μM) after UV irradiation (16 J/m2) visualized by ATTO594 labeling using Click‐chemistry. Levels in siControl cells were set as 1. N = 2, > 200 cells analyzed, error bars represent SEM.
Figure EV3. Histone degradation does not affect repair of UV lesions.
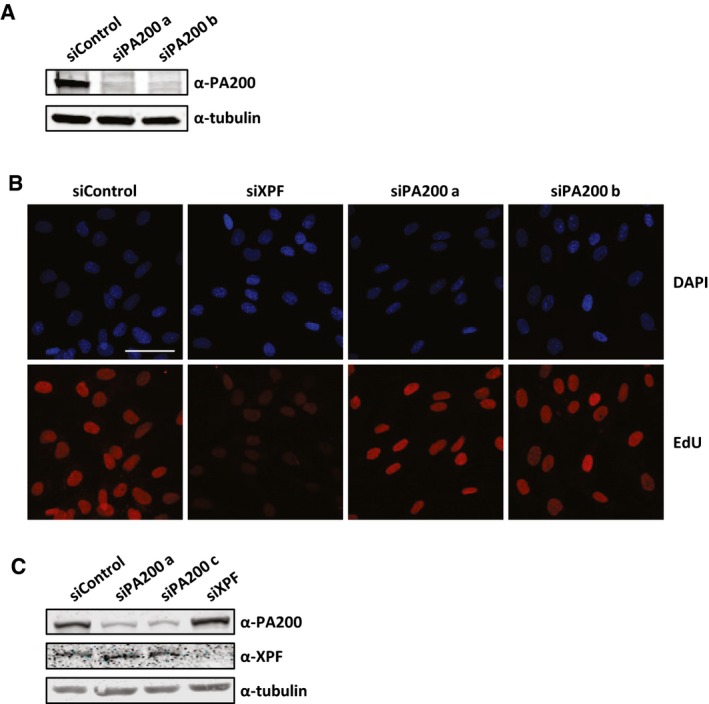
- Western blot showing the knockdown efficiency of the siRNAs targeting PA200.
- Representative images of non‐cycling C5RO cells treated with EdU (20 μM) for 3 h after UV irradiation (16 J/m2) visualized by ATTO594 labeling using Click‐chemistry. Scale bar 50 μm.
- Western blot showing the knockdown efficiency of the siRNAs targeting PA200 and XPF.
Discussion
To study the effects of UV‐induced DNA damage on histone acetylation in an unbiased manner, we established a protocol to efficiently isolate acetyl‐modified histone peptides. This stepwise purification approach, consisting of an acid extraction of histones, combined with proteolytic digestion with different proteases in parallel and followed by immunopurification of acetylated peptides and analysis by mass spectrometry, resulted in the identification of 40 histone acetylation sites originating from over 300 unique acetylated peptides. Using this procedure, we uncovered a striking UV‐induced reduction of the vast majority of detected acetylated residues originating from all histones, except for variant H2AZ. Western blot experiments confirmed this UV‐induced overall loss of acetylated histones and showed a loss of overall histone acetylation levels up to 40% 4 h after UV exposure. Thus, far most research on histone acetylation was focused on H3 and H4, for example, on the role of H4K16Ac in chromatin compaction 41 and H3K9/14Ac in transcription 42 and during the UV‐DDR 15, 16, 17. Our data show that acetylation levels of H2A and H2B are more reduced in response to UV than H3 and H4 acetylation levels (Figs 2B and EV1A, and Datasets EV2 and EV3), suggesting an important role for the regulation of H2A and H2B acetylation levels in the UV‐DDR or in the extensive chromatin remodeling processes observed following DNA damage 43, 44. In striking contrast to the here described global decrease of histone acetylation, multiple other studies have observed an increase in acetylation in response to UV irradiation 11, 12, 13, 15, 16, 17, 45. For example, the UV‐induced histone acetylation on H3K9/14 was shown to stimulate efficient NER 17 by opening the chromatin structure as a result of recruitment of the chromatin remodeling complex RSC 16. This UV‐induced acetylation is mediated by histone acetyltransferases GCN5 and p300, which are both found at UV lesions and interact with the UV‐DDB complex 11, 12, 15, 17, 46, 47. Our MS results show a reduction in H3K9 and K14 acetylation an hour after UV damage. In addition, Western blot analysis using antibodies specifically recognizing H3K9 and K14 acetylation shows a further decrease in the levels of these acetylation marks at later time points after damage (Fig 2F and G). This apparent discrepancy may be explained by the fact that the observed UV‐induced increase of these marks may specifically happen in close vicinity of UV‐induced DNA lesions 17. It is thus likely that this local increase of acetylated histones is masked by the overall replication stress‐derived loss of acetylated histones. In addition, in our MS dataset, we do not find any peptides that only carry H3K9Ac or only H3K14Ac, but all peptides containing these modifications carry at least one other additional acetylation mark. Additionally, this discrepancy could also be due to differences in cell cycle or caused by the fact that following replication stress a specific subset of hyperacetylated histones is degraded, while histones in the vicinity of UV‐induced lesions carry a single H3K9 or H3K14 acetylation mark.
We propose that our observed overall loss of histone acetylation is a separate event that acts independent of the reported histone acetylation events during NER 11, 12, 13, 15, 16, 17, 45. First, the large 40% decrease in total histone acetylation levels occurs on all core histones and includes multiple lysines, suggesting a chromatin‐wide effect in contrast to the reported histone acetylation events that are often lysine‐specific and are mainly locally observed centered around the DNA lesion 11, 15, 17. Furthermore, NER‐stimulating acetylation was proposed to be facilitated by the recruitment of HATs to the site of DNA damage 14, 17, 46, 47, 48, while the here described replication stress‐associated loss of acetylated histones is due to proteasomal degradation rather than caused by changes in HATs or HDACs activity. In addition, the degradation of acetylated histones is only observed in cycling cells (Fig 5C and D), while NER and the previously described histone acetylation events are expected to take place throughout the cell cycle. Finally, we found that the loss of acetylated histones after UV irradiation is completely independent on NER; it is still observed in NER‐deficient cells (Fig 3) and the degradation of acetylated histones does not affect NER efficiency (Fig 7G). These observations suggest that the overall decrease in histone acetylation in response to UV is not directly associated with DNA repair of UV lesions, but more likely a consequence of UV‐induced replication stress. Indeed, induction of replication stress by other mechanism, treatment with MMC or HU/AraC, resulted in a similar loss of acetylated histones (Fig 5A and B). In line with this, it was previously observed, using acetyl histone‐specific antibodies, that H3K9Ac and H3K56Ac are reduced after hydroxyurea treatment 19. The fact that mainly an overall loss of histone acetylation was observed, despite the existence of independent and opposing UV‐induced histone acetylation influencing events, indicates that in dividing cells the replication stress‐induced loss of acetylated histones is much more abundant than the DNA damage‐localized, site‐specific acetylation that regulates NER. To specifically investigate acetylation events that are induced by UV lesions or associated with NER, it is thus important to execute experiments in non‐replicating cells.
Of note, unlike the loss of acetylated histones, the recovery of histone acetylation levels after UV‐induced replication stress is dependent on NER. As NER is the mammalian DNA repair system capable of removing UV‐induced lesions 7, its absence will prohibit repair of replication stress‐inducing UV lesions. The recovery of histone acetylation levels is likely the result of synthesis of new histones required for the resumption of replication, which are subsequently acetylated, to replace the acetylated histones that were degraded in response to DNA damage.
The decrease in histone acetylation levels following replication stress is proteasome‐dependent and is likely explained by the degradation of a specific subset of histones, i.e. acetylated histones, as the histone acetylation levels are more reduced than the histone levels in response to UV irradiation (Fig 6A–C). Surprisingly, the observed histone degradation is not dependent on ubiquitylation, but instead, it depends on the PA200‐proteasome that specifically targets acetylated proteins for degradation (Fig 7A–E) 40. It is not known whether or how PA200 can discriminate between differentially acetylated histones. For example, it is not expected that histones in transcriptionally active regions, which also carry acetylation marks 49, are constantly targeted by the PA200‐proteasome. We observed a reduction in almost all acetylated peptides, many of which carried multiple acetyl groups (Dataset EV2). This might suggest that PA200 may not recognize specific histone marks, but rather targets hyperacetylated histones for degradation. Future studies should uncover if and how PA200 can discriminate between different subsets of acetylated histones, or that, for example, the activity of the PA200‐complex itself is regulated to control histone degradation following replication stress. It is also not clear yet whether incorporated histones can be degraded by the PA200‐proteasome or whether histones need to be evicted by a specific histone chaperone from chromatin prior to degradation.
An important question that remains to be answered is why histones are degraded in response to replication stress. In response to DNA damage, chromatin is remodeled to a more accessible conformation to enable efficient repair and this might be facilitated by the degradation of histones. In line with this, histone degradation in response to zeocin treatment was recently observed in yeast. This degradation was shown to lead to enhanced chromatin dynamics and recombination rates 50. We have shown that PA200‐dependent histone degradation does not affect nucleotide excision repair; however, it might be necessary to efficiently overcome stalled replication forks. In accordance with this hypothesis, PA200 was found to localize to chromatin in response to DNA damage 51, which suggests that it might degrade nucleosomal histones. Previously, it was shown that an excess of histones interferes with homologous recombination in yeast 52. It is thus plausible that the observed histone degradation in response to replication stress is required to prevent an excess of histones to resolve stalled replication forks efficiently. Degradation of chromatin‐bound histones around stalled forks might make space for the proteins involved in resolving stalled forks, for example, by facilitating reversal of the replication fork followed by template switching to allow resumption of replication 53. Additionally, this mechanism might be involved in the repair of DNA breaks, caused by the collapse of stalled replication forks. As degradation of acetylated histones is much more abundant as the UV‐induced histone acetylation, this suggests that chromatin remodeling by histone degradation around stalled replication forks possibly involves longer stretches of chromatin than remodeling around UV‐induced lesions that are resolved by NER. However, it is currently not known whether histones are indeed hyperacetylated near stalled replication forks. Interestingly, in yeast, all core histones, except H2AZ, are degraded in response to DNA damage 50. The only two sites that in our study were identified to be more acetylated in response to UV irradiation are both located on this histone variant (Dataset EV3), which was previously described to be important for UV survival and CPD repair in yeast 16. This might imply that also in human cells H2AZ might not be degraded in response to replication stress. However, the function of this UV‐induced H2AZ acetylation 16 and why the PA200‐proteasome would not recognize this specific histone variant remains unclear.
In summary, our data indicate that in response to replication stress acetylated histones are specifically degraded by the PA200‐proteasome in an ubiquitin‐independent manner. This degradation most likely represents an important chromatin‐modifying mechanism for cells to cope with stalled replication forks.
Materials and Methods
Cell culture and treatments
HeLa and VH10, SV40 or hTert immortalized, cells were cultured in DMEM/F10 (Lonza) supplemented with 10% fetal calf serum (FCS) and 1% penicillin‐streptomycin (PS, P0781 Sigma) at 37°C in a humidified incubator containing 5% CO2. For stable isotope labeling by amino acids in cell culture (SILAC), HeLa cells were cultured for at least 10 cell doublings in lysine, arginine, and l‐glutamine (PAA)‐deficient DMEM with 10% dialyzed FCS (Invitrogen), 1% PS, and 1% ultraglutamine (200 mM Lonza), supplemented with either 73 μg/ml light [12C6]lysine and 42 μg/ml light [12C6, 14N4]arginine (Sigma) or similar concentrations of heavy [13C6]lysine and heavy [13C6, 15N4]arginine (Cambridge Isotope Laboratories). If not stated otherwise in the figure legends, cells were at 70–80% confluency when treated. For UV treatments, cells were washed with phosphate‐buffered saline (PBS) and irradiated with 16 J/m2 (254 nm, Philips TUV lamp). To induce replication stress, a combination of hydroxyurea (100 mM) and AraC (10 μM) or mitomycin C (10 μg/ml) was used. Transcription was inhibited using α‐amanitin (0.1 mg/ml), THZ1 (1 μM), and flavopiridol (1 μM). To inhibit cell cycle, PHA 767491 hydrochloride (10 μM) was added o/n to the culture medium. MG132 (50 μM) and PYR41 (10 μM) were added to inhibit the proteasome or ubiquitylation, respectively. DNA damage signaling was inhibited by the addition of ATM (Ku55933, 10 μM) and ATR (VE‐821, 10 μM) inhibitors. Sequences of siRNAs used siControl UGGUUUACAUGUCGACUAA, siXPF (Dharmacon SMARTpool), siPA200 a GAAAAGAGAUGCAAAGUUA, siPA200 b AGAAAUAAGGCUCAGCAAA, and siPA200 c GCUUCAACUUAGUAAAGAA.
Histone isolation and protein digestion
Cells were washed twice with cold PBS, scraped, and mixed in a 1:1 ratio based on cell pellet size (UV:mock). The cell pellet was resuspended in 1 volume 5% perchloric acid. After incubating for 10 min, the solution was centrifuged for 10 min at 16,100 g. The supernatant containing histone H1 was collected, and the procedure was repeated twice. The pellet was then resuspended in 2 volumes 0.4 N hydrochloric acid (HCl) and incubated for 15 min before spinning down (10 min 16,100 g). This procedure was repeated twice, and all supernatants containing the core histones were collected. The histone containing supernatants were precipitated by the addition of trichloric acid to 25% and incubated on ice for 30 min and centrifuged for 20 min at 16,100 g. Pellets were washed in acetone with 0.006% HCl and subsequently in acetone, and finally, the histone pellets were dissolved in 50 mM ammonium bicarbonate. Digestions were performed with proteomics‐grade trypsin (Roche, 1:100, 30°C, 2/10/30/120 min; 50% of sample), GluC [100×, o/n, room temperature (RT)] (25% of sample), or on an immobilized pepsin (Pierce) column [in 0.5% trifluoroacetic acid (TFA)] (25% of sample). Tryptic and GluC digestion was stopped by acidification to 0.5% TFA. The digested peptides were mixed and subsequently purified using 200 mg tC18 SEP‐PAK SPE cartridges (Waters) and eluted with 40% acetonitrile (ACN) containing 0.1% TFA. The peptides were then lyophilized for 48 h (Scanvac CoolSafe 110‐4, Scala Scientific).
Ac‐K peptide enrichment
Lyophilized peptides were dissolved in 1.4 ml of IAP buffer (PTMscan, Cell Signaling) and incubated with anti‐Ac‐K antibody beads (PTMscan, Cell Signaling) for 2 h at 4°C on a rotating unit. Beads were washed three times in IAP buffer followed by two washes in H2O, and subsequently, the peptides were eluted using 0.1% of TFA in H2O. The enrichments with the anti‐Ac‐K antibody from Immunechem were performed as described previously 22. In short, peptides were dissolved in 1.4 ml IP buffer (50 mM MOPS pH7.2, 10 mM sodium phosphate, 50 mM NaCl) and incubated o/n with 62.5 μg anti‐Ac‐K antibody conjugated to 12 μl protein A beads (GE Healthcare). Beads were washed three times in IP buffer and two times with water before peptide elution with 0.15% TFA. Eluted peptides were purified with C18 stage tips (Millipore).
Mass spectrometry
Digested histone peptides, not enriched for acetyl‐lysines, were analyzed with an Orbitrap Fusion Tribrid mass spectrometer (Thermo Fisher Scientific) or a quadrupole Orbitrap (Q‐Exactive, Thermo Fisher Scientific) and samples enriched for acetylated peptides were analyzed with the Orbitrap Fusion Tribrid mass spectrometer according to protocols below.
Mass spectra were acquired on an Orbitrap Fusion Tribrid mass spectrometer (Thermo Fisher Scientific) coupled to an EASY‐nLC 1000 system (Thermo Fisher Scientific). Peptide samples were loaded onto a ReproSil C18 reversed phase column (20 cm × 75 μm ID) and eluted with a gradient of 5–80% (acetonitrile containing 0.1% formic acid) over 90 min at 300 nl/min. For all experiments, the instrument was operated in data‐dependent acquisition (DDA) mode. MS1 spectra were collected at a resolution of 120,000, with an automated gain control (AGC) target of 2E5 and a max injection time of 50 ms in the scan range from 375 to 1,500 m/z. The most intense ions were selected for MS/MS, top speed method with a 3‐s cycle time. The normalized collision energy was optimized at 30% for HCD. Precursors were filtered according to charge state (2–7 z) and monoisotopic peak assignment. Previously interrogated precursors were dynamically excluded for 70 s. Peptide precursors were isolated with a quadrupole mass filter set to a width of 0.7 Th.
For ETD and EThcD fragmentation experiments, a decision tree was applied in the Orbitrap Tune software for instrument control. For precursor ions with charges 2–3, EThcD was used for fragmentation, while for precursors with charges 4–8 ETD was used. Calibrated charge‐dependent ETD parameters were applied, and fragment ions were measured in the ion trap. Alternatively, peptides were analyzed on a quadrupole Orbitrap (Q‐Exactive, Thermo Fisher Scientific) mass spectrometer equipped with an EASY‐nLC 1000 (Thermo Fisher Scientific). Peptide samples were loaded onto a ReproSil C18 reversed phase column (20 cm × 75 μm ID) and eluted with a gradient of 5–80% (acetonitrile containing 0.1% formic acid) over 70 min at 300 nl/min. Fragmentation of the peptides was performed in DDA mode. MS1 spectra were collected at a resolution of 70,000, with an automated gain control (AGC) target of 1E6 and a max injection time of 50 ms. The 10 most intense ions were selected for MS/MS. Precursors were filtered according to charge state (2–7 z) and monoisotopic peak assignment. Previously interrogated precursors were dynamically excluded for 30 s, and peptide precursors were isolated with a quadrupole mass filter set to a width of 2.0 Th.
Peptide identification
Raw data files were analyzed using MaxQuant software (version 1.5.2.8). MS/MS spectra were searched against a histone database, containing all UniProt entries of which the protein name contains the string “histone” and organism “Homo sapiens”, using the Andromeda search engine. The protease specificity was set to non‐specific cleavage. Cysteine carbamidomethylation was included as a fixed modification, whereas methionine oxidation, N‐terminal protein acetylation, and lysine acetylation were set as variable modifications. To check for the presence of additional histone modifications, as shown in Table EV1, dedicated MaxQuant searches were performed in which also ubiquitylation, phosphorylation, methylation, dimethylation, and trimethylation were set as variable modifications. A false discovery rate of 0.05 for peptides and a minimum peptide length of 7 were set. Before further data analysis, known contaminants and reverse hits were removed from the modification‐specific peptides list.
Cell fractionations
To separate chromatin‐bound proteins from soluble proteins, cells (80% confluent, 3 cm2 dishes) were washed 2× with PBS and scraped from the plates. Cells were lysed for 1 h in buffer A (30 mM HEPES pH7.5, 250 mM NaCl, 1 mM MgCl2, 0.5% Triton X‐100, protease inhibitor) on ice and centrifuged for 15 min at full speed at 4°C. Supernatants containing soluble proteins were taken, and pellets, containing chromatin, were washed 1× with 1 ml buffer A. To both the soluble and chromatin fractions, 2× SDS sample buffer was added, and samples were boiled for 5 min at 95°C prior to loading on 14% SDS–PAGE gels.
Western blotting
Cell lysates were made in 2× Laemmli buffer. Lysates were boiled and sonicated with a Diagenode Bioruptor (30 s on; 30 s off for 10 min) to shear the DNA. Lysates were separated on a 6 or 14% SDS–PAGE acrylamide gel and transferred to a PVDF membrane (0.45 μm, Merck Millipore ltd). Membranes were blocked with 5% milk in PBS for 1 h at RT and incubated with primary antibody for 2 h at RT or overnight at 4°C. Secondary Alexa Fluor 795 donkey anti‐mouse antibodies and Alexa Fluor 680 donkey anti‐rabbit antibodies (Sigma) were used to visualize the proteins using an infrared imaging system (Odyssey; LI‐COR Biosciences). Primary antibodies were as follows: rabbit‐α‐histone H2B (1:2,000, Cat # ab64039 Abcam), mouse‐α‐phospho‐H2A.X (SER139) (1:1,000, Cat # 05‐636, Millipore), rabbit‐α‐histone H1.2 (1:1,000, Abcam), rabbit‐α‐histone H2A (1:200, ab13923, Abcam), goat‐α‐histone H2B (1:100, Cat # SC‐8650, Santacruz), goat‐α‐histone H3 (1:1,000, Cat # CS‐8654, Santacruz), mouse‐α‐histone H4 (1:1,000, Cat # ab31830, Abcam), rabbit‐α‐acetyl‐lysine (1:2,000, Ac‐K‐100, Cat # 9814S, PTMscan), rabbit‐α‐acetyl‐lysine (1:1,000, Cat # ICP0380, Immunechem), rabbit‐α‐acetyl histone H3 Lys9 (1:1,000, Cat # 06‐942, Millipore), rabbit‐α‐acetyl histone H3 Lys14 (1:2,000, Cat # 07‐353, Millipore), rabbit‐α‐acetyl histone H3 Lys27 (1:2,000, Cat # ab4729, Abcam), rabbit‐α‐acetyl histone H4 Lys12 (1:2,000, Cat # ab46983, Abcam), mouse anti‐HP1y (1:1,000, Cat # ab56978, Abcam), and mouse‐α‐tubulin (1:5,000, Cat # T5168, Sigma‐Aldrich).
For Western blots, the rabbit‐α‐acetyl‐lysine from PTMscan was used, except when stated otherwise. Quantifications were performed using the Odyssey software (LI‐COR Biosciences). The intensity of the acetyl‐lysine signal was normalized against the histone H4 or tubulin signals, and the mock‐treated time point was set as 1. Each experiment was performed at least three times, and mean values and standard error of the means (SEM) are shown. A two‐tailed t‐test was performed, and P‐values < 0.1 (*), < 0.05 (**), and < 0.005 (***) are depicted.
EdU incorporation
Cells were incubated with 20 μM 5‐ethynyl‐2′‐deoxyuridine (EdU, Invitrogen) and 1 μM 5‐fluoro‐2′‐deoxyuridine for 2 h. Cells were fixed in 3.6% formaldehyde in PBS and permeabilized in 0.5% Triton X in PBS. EdU was visualized with a click‐it reaction using Alexa Fluorophore 488 nm according to manufacturer's protocol (Invitrogen). For unscheduled DNA synthesis experiments, cells were labeled with 20 μM EdU in medium containing 20 mM Hepes buffer (Lonza) directly after UV irradiation (16 J/m2). After 3 h of labeling, a chase with 10 μM thymidine was performed after which the cells were fixed in 3.7% formaldehyde and 0.5% Triton in PBS. Cells were shortly washed twice in PBS and blocked 2× in 3% BSA in PBS for 10 min. Permeabilization was performed in 0.5% Triton in PBS for 20 min. Click‐it reaction was performed using a 594 nm azide (Atto Tec) for 30 min. Images were obtained using a LSM700 microscope (Carl Zeiss Microimaging, Inc.) and analyzed using ImageJ software 54.
Clonogenic survival assays
HeLa cells were seeded in 6‐well plates (400 cells/well) a day before treatment with a single doses of UV‐C. Each experiment was performed in triplicate. After 6 days, the colonies were fixed and stained with 50% methanol, 43% H2O, 7% acetic acid, and 0.1% Brilliant blue R (Sigma). Number of colonies was counted using a GelCount™ (Oxford Optronix, version 1.1.2.0). The survival was plotted as the relative amount of colonies after treatment compared to the non‐treated samples.
Immunofluorescence
Cells were grown on coverslips until 70–80% confluency, washed in PBS, and fixed in 2% paraformaldehyde in PBS containing 0.1% Triton X. Cells were permeabilized for 20 min in 0.5% Triton X in PBS and washed in PBS containing 0.5% bovine serum albumin (BSA) and 0.15% glycine prior to primary antibody mouse‐α‐phospho‐H2A.X (SER139; 1:1,000, Cat # 05‐636, Millipore) staining for 1 h at RT. Coverslips were washed three times short and twice for 10 min in 0.1% Triton X in PBS and once in PBS with BSA and glycine and subsequently stained with secondary antibodies labeled with Alexa Fluorochrome 555 (Invitrogen) and DAPI (0.1 μg/ml) for 1 h at RT. Images were obtained using a Leica DM4000B microscope.
Data availability
The mass spectrometry data from this publication have been deposited to the PeptideAtlas database (http://www.peptideatlas.org) and assigned the identifier PASS01209 (http://www.peptideatlas.org/PASS/PASS01209).
Author contribution
IKM performed the majority of the experiments. MEG, IK, and RCJ performed Western blots. AR performed UDS experiments. KB, ER, and JAAD performed MS experiments and data analysis. IKM, JHJH, HL, WV, and JAM designed the experiments. IKM and JAM analyzed the data and wrote the manuscript. All authors reviewed the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Review Process File
Acknowledgements
This work is dedicated to the memory of Iris Kik. This work was supported by the Dutch Organization for Scientific Research (NWO) TOP Grants of Earth and Life Sciences and ZonMW (854.11.002 and 912.12.132), European Research Council Advanced Grant (340988‐ERC‐ID), Dutch Organization for Scientific Research Earth and Life Sciences VIDI grant (846.13.004), and Cancer Genomics Netherlands. This work is part of the Oncode Institute which is partly financed by the Dutch Cancer Society.
EMBO Reports (2018) 19: e45566
References
- 1. Zhu P, Li G (2016) Structural insights of nucleosome and the 30‐nm chromatin fiber. Curr Opin Struct Biol 36: 106–115 [DOI] [PubMed] [Google Scholar]
- 2. Kouzarides T (2007) Chromatin modifications and their function. Cell 128: 693–705 [DOI] [PubMed] [Google Scholar]
- 3. Langst G, Manelyte L (2015) Chromatin remodelers: from function to dysfunction. Genes (Basel) 6: 299–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Escargueil AE, Soares DG, Salvador M, Larsen AK, Henriques JA (2008) What histone code for DNA repair? Mutat Res 658: 259–270 [DOI] [PubMed] [Google Scholar]
- 5. van Attikum H, Gasser SM (2009) Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol 19: 207–217 [DOI] [PubMed] [Google Scholar]
- 6. Ramanathan B, Smerdon MJ (1986) Changes in nuclear protein acetylation in u.v.‐damaged human cells. Carcinogenesis 7: 1087–1094 [DOI] [PubMed] [Google Scholar]
- 7. Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JH (2014) Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol 15: 465–481 [DOI] [PubMed] [Google Scholar]
- 8. Ramanathan B, Smerdon MJ (1989) Enhanced DNA repair synthesis in hyperacetylated nucleosomes. J Biol Chem 264: 11026–11034 [PubMed] [Google Scholar]
- 9. Smerdon MJ (1991) DNA repair and the role of chromatin structure. Curr Opin Cell Biol 3: 422–428 [DOI] [PubMed] [Google Scholar]
- 10. Green CM, Almouzni G (2002) When repair meets chromatin. First in series on chromatin dynamics. EMBO Rep 3: 28–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Teng Y, Yu Y, Waters R (2002) The Saccharomyces cerevisiae histone acetyltransferase Gcn5 has a role in the photoreactivation and nucleotide excision repair of UV‐induced cyclobutane pyrimidine dimers in the MFA2 gene. J Mol Biol 316: 489–499 [DOI] [PubMed] [Google Scholar]
- 12. Kim MK, Shin JM, Eun HC, Chung JH (2009) The role of p300 histone acetyltransferase in UV‐induced histone modifications and MMP‐1 gene transcription. PLoS One 4: e4864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang J, Chin MY, Li G (2006) The novel tumor suppressor p33ING2 enhances nucleotide excision repair via inducement of histone H4 acetylation and chromatin relaxation. Cancer Res 66: 1906–1911 [DOI] [PubMed] [Google Scholar]
- 14. Hasan S, Hassa PO, Imhof R, Hottiger MO (2001) Transcription coactivator p300 binds PCNA and may have a role in DNA repair synthesis. Nature 410: 387–391 [DOI] [PubMed] [Google Scholar]
- 15. Yu Y, Teng Y, Liu H, Reed SH, Waters R (2005) UV irradiation stimulates histone acetylation and chromatin remodeling at a repressed yeast locus. Proc Natl Acad Sci USA 102: 8650–8655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Duan MR, Smerdon MJ (2014) Histone H3 lysine 14 (H3K14) acetylation facilitates DNA repair in a positioned nucleosome by stabilizing the binding of the chromatin Remodeler RSC (Remodels Structure of Chromatin). J Biol Chem 289: 8353–8363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guo R, Chen J, Mitchell DL, Johnson DG (2011) GCN5 and E2F1 stimulate nucleotide excision repair by promoting H3K9 acetylation at sites of damage. Nucleic Acids Res 39: 1390–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhu Q, Battu A, Ray A, Wani G, Qian J, He J, Wang QE, Wani AA (2015) Damaged DNA‐binding protein down‐regulates epigenetic mark H3K56Ac through histone deacetylase 1 and 2. Mutat Res 776: 16–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tjeertes JV, Miller KM, Jackson SP (2009) Screen for DNA‐damage‐responsive histone modifications identifies H3K9Ac and H3K56Ac in human cells. EMBO J 28: 1878–1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bergink S, Salomons FA, Hoogstraten D, Groothuis TA, de Waard H, Wu J, Yuan L, Citterio E, Houtsmuller AB, Neefjes J et al (2006) DNA damage triggers nucleotide excision repair‐dependent monoubiquitylation of histone H2A. Genes Dev 20: 1343–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Citterio E, Van Den Boom V, Schnitzler G, Kanaar R, Bonte E, Kingston RE, Hoeijmakers JHJ, Vermeulen W (2000) ATP‐dependent chromatin remodeling by the Cockayne syndrome B DNA repair‐transcription‐coupling factor. Mol Cell Biol 20: 7643–7653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Elia AE, Boardman AP, Wang DC, Huttlin EL, Everley RA, Dephoure N, Zhou C, Koren I, Gygi SP, Elledge SJ (2015) Quantitative proteomic atlas of ubiquitination and acetylation in the DNA damage response. Mol Cell 59: 867–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Svinkina T, Gu H, Silva JC, Mertins P, Qiao J, Fereshetian S, Jaffe JD, Kuhn E, Udeshi ND, Carr SA (2015) Deep, quantitative coverage of the lysine acetylome using novel anti‐acetyl‐lysine antibodies and an optimized proteomic workflow. Mol Cell Proteomics 14: 2429–2440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Marteijn JA, Bekker‐Jensen S, Mailand N, Lans H, Schwertman P, Gourdin AM, Dantuma NP, Lukas J, Vermeulen W (2009) Nucleotide excision repair‐induced H2A ubiquitination is dependent on MDC1 and RNF8 and reveals a universal DNA damage response. J Cell Biol 186: 835–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hanasoge S, Ljungman M (2007) H2AX phosphorylation after UV irradiation is triggered by DNA repair intermediates and is mediated by the ATR kinase. Carcinogenesis 28: 2298–2304 [DOI] [PubMed] [Google Scholar]
- 26. Turner BM (2002) Cellular memory and the histone code. Cell 111: 285–291 [DOI] [PubMed] [Google Scholar]
- 27. Kallin EZY (2004) Chromatin remodeling In Encyclopedia of biological chemistry, Lennarz WJLM. (ed), pp 456–462. London: Academic Press; [Google Scholar]
- 28. Sadakierska‐Chudy A, Filip M (2015) A comprehensive view of the epigenetic landscape. Part II: histone post‐translational modification, nucleosome level, and chromatin regulation by ncRNAs. Neurotox Res 27: 172–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wisniewski JR, Zougman A, Kruger S, Mann M (2007) Mass spectrometric mapping of linker histone H1 variants reveals multiple acetylations, methylations, and phosphorylation as well as differences between cell culture and tissue. Mol Cell Proteomics 6: 72–87 [DOI] [PubMed] [Google Scholar]
- 30. van Cuijk L, van Belle GJ, Turkyilmaz Y, Poulsen SL, Janssens RC, Theil AF, Sabatella M, Lans H, Mailand N, Houtsmuller AB et al (2015) SUMO and ubiquitin‐dependent XPC exchange drives nucleotide excision repair. Nat Commun 6: 7499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Choi JH, Gaddameedhi S, Kim SY, Hu J, Kemp MG, Sancar A (2014) Highly specific and sensitive method for measuring nucleotide excision repair kinetics of ultraviolet photoproducts in human cells. Nucleic Acids Res 42: e29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Orren DK, Petersen LN, Bohr VA (1997) Persistent DNA damage inhibits S‐phase and G2 progression, and results in apoptosis. Mol Biol Cell 8: 1129–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Steurer B, Marteijn JA (2016) Traveling rocky roads: the consequences of transcription‐blocking DNA lesions on RNA polymerase II. J Mol Biol 429: 3146–3155 [DOI] [PubMed] [Google Scholar]
- 34. Bensaude O (2011) Inhibiting eukaryotic transcription: which compound to choose? How to evaluate its activity? Transcription 2: 103–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bensimon A, Aebersold R, Shiloh Y (2011) Beyond ATM: the protein kinase landscape of the DNA damage response. FEBS Lett 585: 1625–1639 [DOI] [PubMed] [Google Scholar]
- 36. Nam EA, Cortez D (2011) ATR signalling: more than meeting at the fork. Biochem J 436: 527–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sobel RE, Cook RG, Perry CA, Annunziato AT, Allis CD (1995) Conservation of deposition‐related acetylation sites in newly synthesized histones H3 and H4. Proc Natl Acad Sci USA 92: 1237–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li Q, Zhou H, Wurtele H, Davies B, Horazdovsky B, Verreault A, Zhang Z (2008) Acetylation of histone H3 lysine 56 regulates replication‐coupled nucleosome assembly. Cell 134: 244–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Verreault A (2000) De novo nucleosome assembly: new pieces in an old puzzle. Genes Dev 14: 1430–1438 [PubMed] [Google Scholar]
- 40. Qian MX, Pang Y, Liu CH, Haratake K, Du BY, Ji DY, Wang GF, Zhu QQ, Song W, Yu Y et al (2013) Acetylation‐mediated proteasomal degradation of core histones during DNA repair and spermatogenesis. Cell 153: 1012–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shogren‐Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL (2006) Histone H4‐K16 acetylation controls chromatin structure and protein interactions. Science 311: 844–847 [DOI] [PubMed] [Google Scholar]
- 42. Agalioti T, Chen G, Thanos D (2002) Deciphering the transcriptional histone acetylation code for a human gene. Cell 111: 381–392 [DOI] [PubMed] [Google Scholar]
- 43. Lans H, Marteijn JA, Vermeulen W (2012) ATP‐dependent chromatin remodeling in the DNA‐damage response. Epigenetics Chromatin 5: 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Soria G, Polo SE, Almouzni G (2012) Prime, repair, restore: the active role of chromatin in the DNA damage response. Mol Cell 46: 722–734 [DOI] [PubMed] [Google Scholar]
- 45. Brand M, Moggs JG, Oulad‐Abdelghani M, Lejeune F, Dilworth FJ, Stevenin J, Almouzni G, Tora L (2001) UV‐damaged DNA‐binding protein in the TFTC complex links DNA damage recognition to nucleosome acetylation. EMBO J 20: 3187–3196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rapic‐Otrin V, McLenigan MP, Bisi DC, Gonzalez M, Levine AS (2002) Sequential binding of UV DNA damage binding factor and degradation of the p48 subunit as early events after UV irradiation. Nucleic Acids Res 30: 2588–2598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Datta A, Bagchi S, Nag A, Shiyanov P, Adami GR, Yoon T, Raychaudhuri P (2001) The p48 subunit of the damaged‐DNA binding protein DDB associates with the CBP/p300 family of histone acetyltransferase. Mutat Res 486: 89–97 [DOI] [PubMed] [Google Scholar]
- 48. Yang X, Li L, Liang J, Shi L, Yang J, Yi X, Zhang D, Han X, Yu N, Shang Y (2013) Histone acetyltransferase 1 promotes homologous recombination in DNA repair by facilitating histone turnover. J Biol Chem 288: 18271–18282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kurdistani SK, Tavazoie S, Grunstein M (2004) Mapping global histone acetylation patterns to gene expression. Cell 117: 721–733 [DOI] [PubMed] [Google Scholar]
- 50. Hauer MH, Seeber A, Singh V, Thierry R, Sack R, Amitai A, Kryzhanovska M, Eglinger J, Holcman D, Owen‐Hughes T et al (2017) Histone degradation in response to DNA damage enhances chromatin dynamics and recombination rates. Nat Struct Mol Biol 24: 99–107 [DOI] [PubMed] [Google Scholar]
- 51. Blickwedehl J, Agarwal M, Seong C, Pandita RK, Melendy T, Sung P, Pandita TK, Bangia N (2008) Role for proteasome activator PA200 and postglutamyl proteasome activity in genomic stability. Proc Natl Acad Sci USA 105: 16165–16170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liang D, Burkhart SL, Singh RK, Kabbaj MH, Gunjan A (2012) Histone dosage regulates DNA damage sensitivity in a checkpoint‐independent manner by the homologous recombination pathway. Nucleic Acids Res 40: 9604–9620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mailand N, Gibbs‐Seymour I, Bekker‐Jensen S (2013) Regulation of PCNA‐protein interactions for genome stability. Nat Rev Mol Cell Biol 14: 269–282 [DOI] [PubMed] [Google Scholar]
- 54. Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Review Process File
Data Availability Statement
The mass spectrometry data from this publication have been deposited to the PeptideAtlas database (http://www.peptideatlas.org) and assigned the identifier PASS01209 (http://www.peptideatlas.org/PASS/PASS01209).