

Abstract
Progressive attrition of telomeres triggers DNA damage response (DDR) and limits the regenerative capacity of adult stem cells during mammalian aging. Intriguingly, telomere integrity is not only determined by telomere length but also by the epigenetic status of telomeric/sub‐telomeric regions. However, the functional interplay between DDR induced by telomere shortening and epigenetic modifications in aging remains unclear. Here, we show that deletion of Gadd45a improves the maintenance and function of intestinal stem cells (ISCs) and prolongs lifespan of telomerase‐deficient mice (G3Terc −/−). Mechanistically, Gadd45a facilitates the generation of a permissive chromatin state for DDR signaling by inducing base excision repair‐dependent demethylation of CpG islands specifically at sub‐telomeric regions of short telomeres. Deletion of Gadd45a promotes chromatin compaction in sub‐telomeric regions and attenuates DDR initiation at short telomeres of G3Terc −/− ISCs. Treatment with a small molecule inhibitor of base excision repair reduces DDR and improves the maintenance and function of G3Terc −/− ISCs. Taken together, our study proposes a therapeutic approach to enhance stem cell function and prolong lifespan by targeting epigenetic modifiers.
Keywords: epigenetics, Gadd45a, stem cell aging, telomere
Subject Categories: Ageing; Chromatin, Epigenetics, Genomics & Functional Genomics; DNA Replication, Repair & Recombination
Introduction
Decline in stem cell regenerative capacity is one of the hallmarks in mammalian aging 1. Telomere attrition represents one of the essential molecular drivers in human aging and aging‐related diseases 2, 3, 4. Loss of telomeric DNA repeats leaves chromosome ends uncapped, triggers chronic DNA damage response (DDR), and results in cell death, cellular senescence, and organismal aging 5, 6, 7, 8, 9, 10. Late generation of telomerase RNA component knockout (G3Terc −/−) mice have been extensively utilized to probe the underlying mechanisms of telomere‐associated stem cell aging in vivo 11, 12, 13. Deletion of DDR components or restoration of the telomerase activity restores the function of adult stem cells, thereby improving tissue homeostasis and alleviating premature aging symptoms in G3Terc −/− mice 11, 12, 13, 14. One of the most significant aging phenotypes in G3Terc −/− mice is the dramatic loss of body weight with aging, which is strongly correlated with the deterioration of intestinal stem cells (ISCs) 5, 13. Inhibition of DDR initiation improved ISC function and tissue homeostasis, thereby alleviating premature aging symptoms of telomere dysfunctional mice 12.
Intriguingly, telomere integrity is not only determined by the length of telomeres but also regulated by the epigenetic dynamics of telomeric/sub‐telomeric regions 15, 16, 17, 18, 19. In wild‐type cells, sub‐telomeric regions are characterized by hyper‐methylation of CpG islands, enrichment of repressive histone marks (HP1a, H3K9me3, and H4K20me3), and lack of permissive histone modifications (H3K9ac and H4K20ac) 16, 17, 18, 20. Of note, primary cells from G3Terc −/− mice exhibit decondensed telomeric/sub‐telomeric chromatin, as indicated by marked decrease in CpG island methylation, loss of repressive histone modifications (H3K9me3 and H4K20me3), and increase in chromatin histone modifications (H3K9ac and H4K20ac) 16, 17, 18, 20. However, the regulation of telomeric/sub‐telomeric heterochromatin status in response to replicative aging remains unknown, and whether epigenetic dynamics at telomeric/sub‐telomeric regions contributes to organismal aging remains elusive.
Gadd45a (growth arrest and DNA damage‐inducible protein 45 alpha) plays multiple roles in cell cycle regulation, stress response, and DNA repair 21, 22, 23. As a stress response protein, Gadd45a is transcriptionally regulated by p53 24 and FoxO3a 25. Upon DNA damage, Gadd45a participates in DNA repair through direct interaction with PCNA or essential DNA repair complexes. Loss of Gadd45a results in significant impairment in both nucleotide excision repair (NER) 22 and base excision repair (BER) 26. In addition, Gadd45a participates in epigenetic gene activation through repair‐mediated active DNA demethylation 27, 28. Our previous study has shown that Gadd45a plays bilateral roles by repressing the cellular fitness of hematopoietic stem cells upon DNA lesions and preventing tumorigenesis in hematopoietic system 29. However, whether and how Gadd45a functions in replicative aging is unknown.
Here, we discover that Gadd45a deletion significantly attenuates the DDR, improves the maintenance and function of ISCs in G3Terc −/− mice, and consequently prolongs lifespan of G3Terc −/− mice. Mechanistically, Gadd45a facilitates telomeric heterochromatin remodeling via BER‐mediated active DNA demethylation in sub‐telomeric regions of G3Terc −/− cells, which generates a uncondensed chromatin structure to promote DDR signaling 30, 31. Together, our study suggests that stem cell function improvement and lifespan extension could be achieved by altering the site‐specific epigenetic dynamics, such as DNA methylation at telomeres/sub‐telomeres.
Results
Gadd45a deletion improves organ maintenance and extends lifespan of G3Terc −/− mice
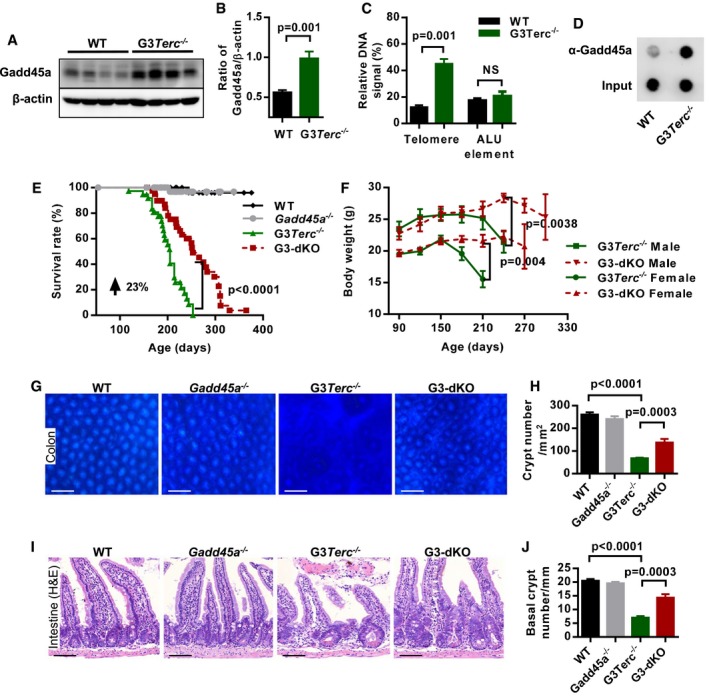
The protein level of Gadd45a was upregulated in cells from G3Terc −/− mice in comparison with wild‐type mice (Fig 1A and B). To test whether Gadd45a preferentially binds to short telomeres, we performed ChIP‐qPCR and dot‐blot analysis and found that Gadd45a was enriched in the telomeric/sub‐telomeric regions of G3Terc −/− cells as compared to wild‐type cells (Fig 1C and D). To understand the function of Gadd45a in the telomere shortening‐driven aging process, we generated G3Terc −/− Gadd45a −/− double knockout (G3‐dKO) mice (mice genotyping, see Fig EV1A). Gadd45a deficiency significantly prolonged the median lifespan of G3Terc −/− mice with a 23% increase (205 days in G3Terc −/− mice versus 253 days in G3‐dKO; Fig 1E). Detailed analysis showed that Gadd45a deletion prevented multiple organ atrophy observed in aged G3Terc −/− mice (weights of these organs are shown in Fig EV1B–F). Interestingly, Gadd45a deficiency prevented the loss of body weight in both male and female G3Terc −/− mice (Fig 1F). Given that the reduced body weight was tightly associated with the atrophy of intestine tissues in G3Terc −/− mice, we concentrated our analysis on the intestine tissues. Whole‐mount staining of large intestines showed around onefold increase in crypt numbers of G3‐dKO mice as compared to G3Terc −/− mice (Fig 1G and H). More strikingly, the villi length and numbers of basal crypts in small intestine were dramatically increased in G3‐dKO mice as compared to G3Terc −/− mice (Figs 1I and J, and EV1G).
Figure 1. Gadd45a deletion improves organ maintenance and prolongs the lifespan of G3Terc −/− mice.
-
A, BWestern blotting analysis of Gadd45a protein expression in mouse intestine crypt lysates from WT and G3Terc −/− mice (A). Quantification of Gadd45a protein expression, β‐actin protein level was used as the loading control (n = 7 mice per genotype), two independent experiments were carried out (B). P‐values were calculated with unpaired two‐tailed Student's t‐test.
-
CQuantification of Gadd45a‐ChIP values as percentage of total telomeric DNA and ALU elements in WT and G3Terc −/− MEF cells (n = 3 for each genotype). P‐values were calculated with unpaired two‐tailed Student's t‐test.
-
DDot‐blot assay following Gadd45a‐ChIP in WT and G3Terc −/− MEF cells.
-
EKaplan–Meier survival curves of the mice with indicated genotypes (WT, n = 44; Gadd45a −/−, n = 48; G3Terc −/−, n = 37; G3‐dKO, n = 41). P‐value comparing the survival of G3Terc −/− with G3‐dKO mice was calculated with the log‐rank test.
-
FThe average body weight of mice with indicated genotypes at indicated time points (G3‐dKO female (n = 19) versus G3Terc −/− female (n = 17), P = 0.004; G3‐dKO male (n = 14) versus G3Terc −/− male (n = 14), P = 0.0038). P‐values were calculated with log‐rank test.
-
G, HWhole‐mount images of colon samples from 7‐month‐old mice with indicated genotypes (G). Quantification on density of colon crypts (H) (n = 5 mice per group). P‐values were calculated with unpaired two‐tailed Student's t‐test.
-
I, JH&E‐stained small intestine sections from 7‐month‐old mice with indicated genotypes (I). Quantification of the number of small intestine basal crypts (J) (n = 5–6 mice per group). P‐values were calculated with unpaired two‐tailed Student's t‐test.
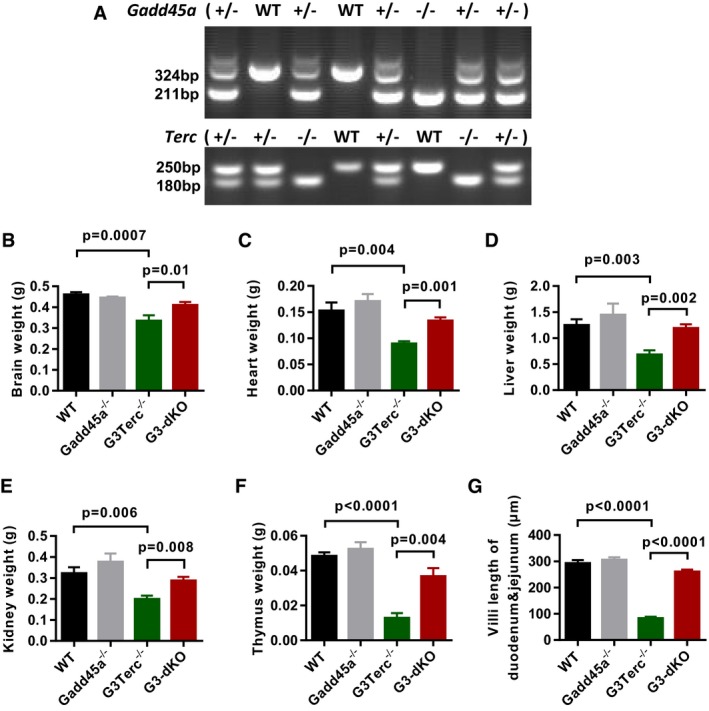
Figure EV1. Gadd45a deletion prevents overall organ atrophy of aged G3Terc −/− mice.
-
ARepresentative example of mice genotyping. Upper panel is genotyping of Gadd45a and lower panel is genotyping of Terc gene.
-
B–FThe average weights of brain (B), heart (C), liver (D), kidney (E), and thymus (F) from 6‐ to 7‐month‐old mice (n = 5–12 mice/group).
-
GQuantification of villi length in duodenum and jejunum of small intestines (n = 5–6 mice per genotype).
Gadd45a deletion improves intestine stem cell maintenance and function in G3Terc −/− mice
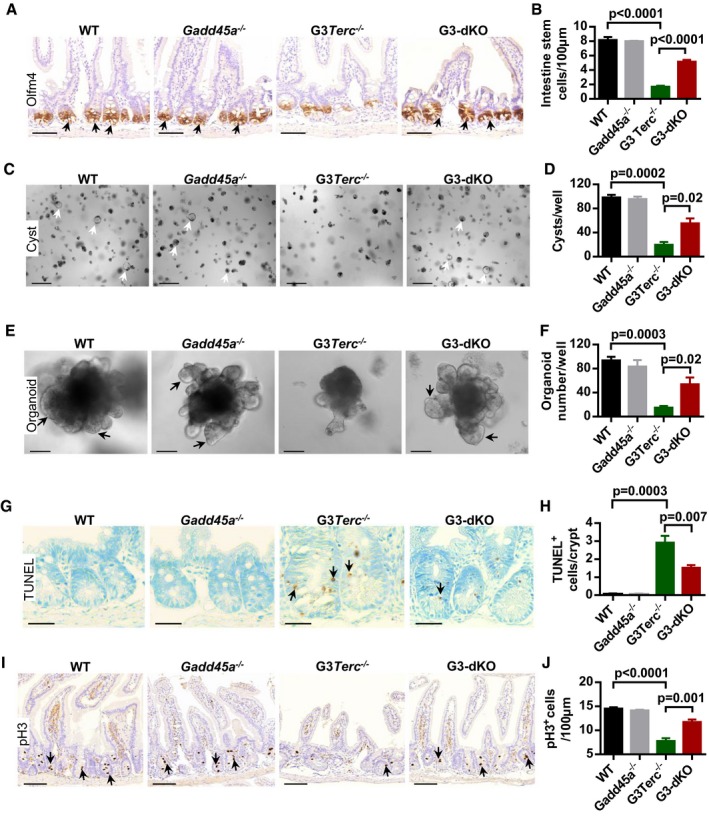
Loss of ISCs has been implicated in aged G3Terc −/− mice 13. In our experimental settings, the number of intestinal stem cells (represented by Olfm4+ cells in basal crypts 32 and PCNA+ cells next to paneth cells at the bottom of basal crypts) was dramatically increased in G3‐dKO mice as compared to G3Terc −/− mice (Fig 2A and B and Appendix Fig S1A and B). To test the functional capacity of ISCs, we isolated crypts from the 3‐month‐old mice of each genotype and performed in vitro crypt culture assay 13, 33. G3‐dKO crypts formed significantly more cysts at the early time point (day 2) (Fig 2C and D) and further developed into larger organoids at day 8 in culture as compared to G3Terc −/− crypts (Fig 2E and F and Appendix Fig S1C). Apoptosis and proliferation of ISCs were analyzed by TUNEL assay and pH3 antibody staining, respectively. Intestinal basal crypt cells from G3‐dKO mice exhibited significantly less cell death (Fig 2G and H) and more cell proliferation as compared to those from G3Terc −/− mice (Fig 2I and J). These data suggest that Gadd45a deletion improves the viability and proliferative capacity of ISCs with short telomeres.
Figure 2. Gadd45a deletion improves intestinal stem cell maintenance and function in G3Terc −/− mice.
-
A, BOlfm4‐positive intestinal stem cells (indicated by arrows) in small intestine sections (A). Quantification of intestinal stem cell (Olfm4+) number per 100 μm (n = 4–5 mice per group) of small intestine sections (B).
-
C, DRepresentative images of small intestine crypt culture (day 2). Arrows mark the surviving cysts (C). Quantification of numbers of cysts per culture well (D) (n = 4–5 mice/genotype).
-
E, FIn vitro crypt culture (day 8) from small intestine crypts isolated from mice with indicated genotypes. Arrows mark the crypt‐like structure (E). The number of organoids per culture wells (n = 4–5 mice per group). Two hundred crypts are seeded per well (F).
-
G, HRepresentative images of TUNEL staining in small intestine sections. Arrows indicate TUNEL‐positive cells (G). Quantification of TUNEL‐positive cells per crypt (n = 5 mice per group) (H).
-
I, JRepresentative images of phosphorylated histone H3 (pH3) antibody staining in mouse small intestine. Arrows indicate pH3‐positive cells (I). Quantification of pH3‐positive crypt cells per 100 μm of small intestines (n = 4–6 mice per group) (J).
Gadd45a deletion attenuates telomere dysfunction‐induced DDR initiation in G3Terc −/− mice
Given that Gadd45a deficiency did not extend the telomere length of G3Terc −/− basal crypt cells as shown by telomere Q‐FISH assay (Appendix Fig S2) 34, we suspected that loss of Gadd45a might affect DDR signaling transduction and thus improve ISC function and maintenance. Immunohistochemistry and Western blot analysis showed that p21 levels were drastically reduced upon Gadd45a deletion in G3Terc −/− mice (Fig EV2A–D). Gadd45a was reported as protein stabilizer of p53 35, and we also found that the frequency of p53‐positive cells was decreased in G3‐dKO crypts as compared to G3Terc −/− crypts (Fig 3A and B). Interestingly, the expression of other early DDR marks in G3‐dKO mice, such as γ‐H2AX, pS1981‐ATM, and 53BP1, was reduced almost to the wild‐type levels (Figs 3C–F and EV2E and F). These data suggest that, in addition to being a downstream target, Gadd45a also plays role upstream of p53 and functions in the early stage of DDR initiation. Shortened chromosome ends induce telomere–telomere fusions and repeated breakage–fusion–bridge cycles 36. To directly evaluate the telomere status, we examined chromosome anaphase bridges 13 and found that G3Terc −/− mice exhibited a dramatically increased frequency of cells manifesting anaphase bridges in the intestinal basal crypts as compared to wild‐type controls, which was significantly rescued after Gadd45a deletion (Fig 3G and H). In line with this, RNA‐Seq analysis of primary MEFs showed that Gadd45a deletion reversed the gene expression profiles of DDR, inflammation, p38‐MAPK pathways, cell cycle, and metabolic‐related genes in G3Terc −/− mice (Fig 3I and Appendix Fig S3). Thus, in addition to being a bona fide p53 downstream target 24, Gadd45a may exert its function upstream of p53 activation in response to telomere dysfunction. Together, these data suggest that Gadd45a contributes to the impaired stem cell function and maintenance by promoting DDR initiation at the short telomeres upstream of p53.
Figure EV2. Gadd45a deletion attenuated DDR signaling in the intestine basal crypts of G3‐dKO mice.
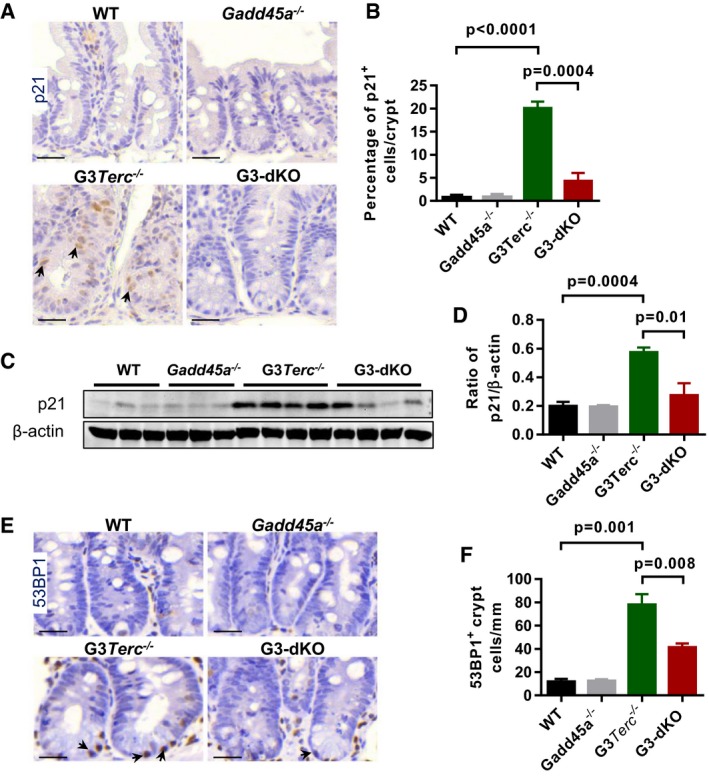
- Representative images of small intestine sections stained with p21 antibody. Arrows indicate the positive nuclear staining of p21 antibody.
- Quantification of the percentage of p21‐positive crypt cells (n = 4–6 of 7‐month‐old mice per genotype).
- Western blotting analysis of p21 protein expression in mouse intestine crypt lysates from WT, Gadd45a −/−, G3Terc −/−, and G3‐dKO mice.
- Quantification of p21 protein expression; β‐actin protein level was used as the loading control (n = 3–4 mice per genotype).
- Representative images of small intestine crypt sections stained with antibody against 53BP1. Arrows mark the positive nuclear staining of 53BP1 antibody.
- Quantification of frequencies of 53BP1‐positive crypt cells (n = 4–6 of 7‐month‐old mice per genotype).
Figure 3. Gadd45a deletion attenuates telomere dysfunction‐induced DDR in the basal crypts of G3Terc −/− mice.
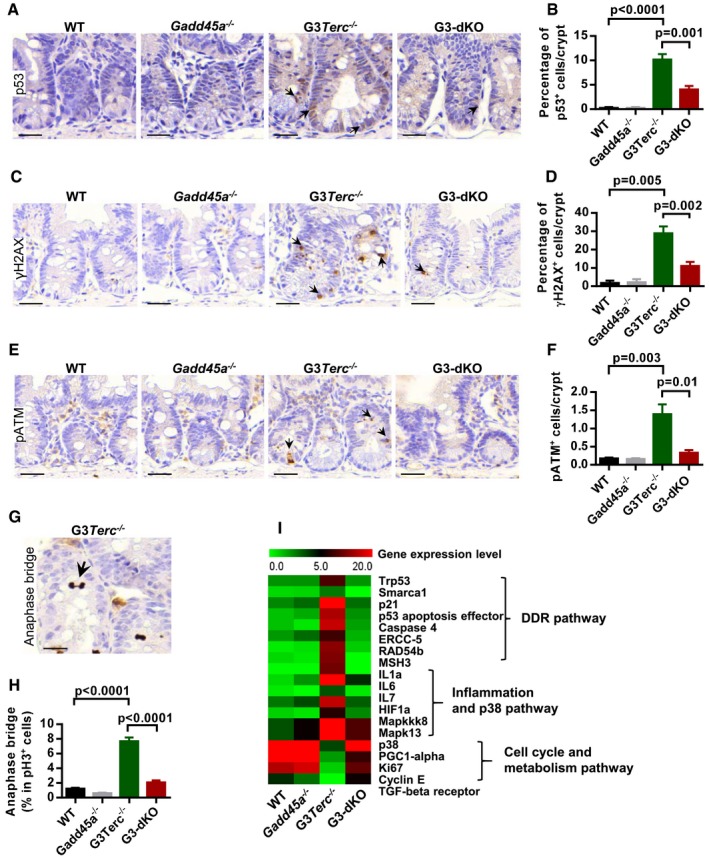
-
A, BRepresentative images of immunostaining for p53 on small intestine sections. Arrows indicate p53‐positive cells (A). Quantification of the percentage of p53‐positive cells per crypt (n = 5–6 mice per group) (B).
-
C, DRepresentative images of γH2AX antibody staining on small intestine paraffin sections from WT, Gadd45a −/−, G3Terc −/−, and G3‐dKO mice (7 months old). Arrows indicate γH2AX‐positive nuclei (C). Quantification of the percentage of γH2AX‐positive cells per small intestine crypt (n = 4–6 mice per group) (D).
-
E, FRepresentative images of small intestine crypt sections stained with antibody against pATM (Ser1981) (E). Quantification of frequencies of pATM (Ser1981)‐positive crypt cells (indicated with arrows in E) (n = 4–6 of 7‐month‐old mice per genotype) (F).
-
G, HRepresentative image of the anaphase bridge (indicated with arrows) in the basal crypt of G3Terc −/− mice (G). The percentage of cells with anaphase bridges in pH3‐positive basal crypt cell population (n = 4–6 mice per genotype) (H).
-
IHeat map indicating the relative expression levels of DDR and cell cycle‐related gene transcripts in MEFs from indicated genotypes (data are extracted from RNA‐Seq datasets).
Gadd45a deletion restores telomeric heterochromatin marks in G3Terc −/− cells
In addition to being a direct player in DDR, Gadd45a participates in transcriptional regulation of gene expression by active DNA demethylation 27, 28. We proposed that Gadd45a‐mediated DNA demethylation mechanism could contribute to G3Terc −/− mice aging. To test this hypothesis, we conducted the whole‐genome methylation sequencing on CpG islands in wild‐type, Gadd45a −/−, G3Terc −/−, and G3‐dKO MEFs, and found that Gadd45a loss does not change the global CpG island methylation status in all genotypes analyzed. Furthermore, methylation levels at the promoter regions of genes involving in DDR, cell death, or cell proliferation were not altered (Appendix Fig S4A). Since Gadd45a has been implicated in DNA demethylation around DNA lesions 27, 28, we speculated that Gadd45a may exert its DDR promoting function by altering the chromatin status at critically short telomeres. Given that murine chromosomes 1, 2, 16 are more vulnerable to telomere erosion in telomerase knockout cells 17, we utilized a PCR‐based bisulfite sequencing method to determine DNA methylation levels at sub‐telomeric regions of chromosomes 1. Consistent with previous study 17, G3Terc −/− cells showed marked reduction in CpG methylation at sub‐telomeric regions compared to wild‐type cells with long telomere reserves. Gadd45 deficiency largely rescued the loss of CpG demethylation in G3Terc −/− cells, and G3‐dKO MEFs showed similar DNA methylation patterns as shown in wild‐type and Gadd45 −/− MEFs (Fig 4A and B). However, the methylation status of other non‐telomeric repetitive elements does not change (Appendix Fig S4B), and furthermore, knockdown of DDR gene (p21) does not change the DNA methylation in sub‐telomeric region of G3Terc −/− cells (Appendix Fig S5A and B). DNA demethylation could remodel the heterochromatin and trigger further histone modification to open the chromatin of telomeric/sub‐telomeric regions 17. ChIP assays using H3K9me3 (a mark for heterochromatin) and H3K9ac (a uncondensed chromatin mark) antibodies showed that G3Terc −/− telomeric/sub‐telomeric regions were reduced with H3K9me3 and enriched with H3K9ac, while Gadd45a deletion in G3Terc −/− background efficiently restored these histone modifications to the levels as shown in wild‐type and Gadd45a −/− MEFs (Fig 4C and D). Furthermore, ChIP with antibody against HP1a, a heterochromatin mark, further showed that Gadd45a deficiency restored heterochromatic status of telomeric/sub‐telomeric regions of G3Terc −/− cells (Fig 4E). Our data suggested that Gadd45a deletion ameliorated DDR initiation at short telomeres by suppressing telomeric/sub‐telomeric chromatin accessibility.
Figure 4. Gadd45a deletion restores sub‐telomeric DNA methylation and telomeric heterochromatin marks in G3Terc −/− mice.
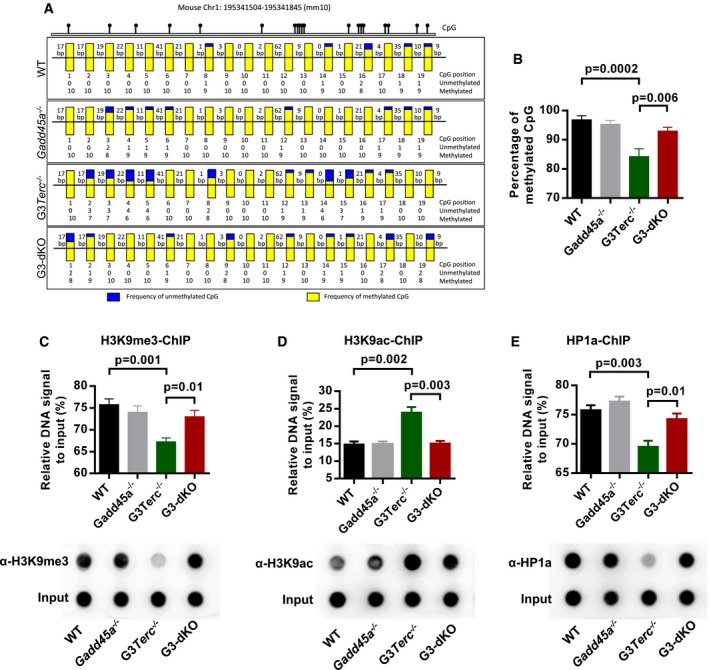
-
A, BCpG island methylation status of sub‐telomeric regions on chromosome 1q. gDNAs from MEFs with indicated genotypes were PCR‐amplified and analyzed by bisulfite sequencing. Yellow and blue bars denote the frequencies of methylated and unmethylated CpG islands, respectively, at each position (A). Quantification of sub‐telomeric CpG island methylation (B).
-
C–ETelomeric quantification and dot‐blot assay of tri‐methyl H3K9 (H3K9me3)‐ChIP in indicated genotypes (C, upper and lower panel). Telomeric quantification and dot‐blot assay of acetylated H3K9 (H3K9ac)‐ChIP in indicated genotypes (D, upper and lower panel). Telomeric quantification and dot‐blot assay of HP1a‐ChIP in indicated genotypes (E, upper and lower panel).
APE1 inhibition restores telomeric heterochromatin compactness and retains G3Terc −/− ISC function
Gadd45a functions in the active demethylation process through interaction with the components of BER (base excision repair) or NER (nucleotide excision repair) pathways 37. To test the potential pathway that may be involved in telomere dysfunction‐induced defects in ISCs, we used the APE1 (a key endonuclease in BER pathway) inhibitor CRT0044876 38 to treat freshly isolated intestine crypts in cultures. In our analysis, APE1 inhibitor CRT0044876 treatment could rescue the impaired organoid formation (in vitro) of G3Terc −/− crypts (Fig 5A–C and Appendix Fig S6A–C). The CRT0044876 treatment reduced the formation of DNA damage foci (γ‐H2AX, 53BP1, etc.) in the G3Terc −/− organoid‐derived cells (Fig 5D and E and Appendix Fig S6D–G). Furthermore, the CRT0044876 treatment largely restored heterochromatin compactness of G3Terc −/− cells, as shown with enrichment of H3K9me3 and HP1a and decrease in H3K9ac in telomeric/sub‐telomeric regions (Fig 5F–H and Appendix Fig S6H–J). However, APE1 inhibitor treatment does not show any visible effect in G3‐dKO crypt (Fig EV3A and B). In vivo experiments also showed that the treatment of G3Terc −/− mice with CRT0044876 largely restored the numbers of Olfm4+ intestine stem cell (Fig 5I and J) and basal crypts (Fig EV3C and D). Meanwhile, the formation of DNA damage foci (γ‐H2AX) was reduced in G3Terc −/− intestine crypt after CRT0044876 treatment (Fig EV3E and F). The increase in AP site accumulation in G3Terc −/− cells and mice after APE1 inhibitor CRT0044876 treatment indicates that the APE1 enzyme activity was inhibited (Fig EV3G). APE1 knockdown in G3Terc −/− MEF cells also shows reduced DNA demethylation in sub‐telomeric region (Fig EV4A–C). These data reinforced our assumption that Gadd45a‐mediated BER pathway facilitates the DDR signaling activation at short telomeres and thereby promotes organismal aging.
Figure 5. APE1 inhibition restores telomeric heterochromatin marks and promotes the function of intestinal stem cells in G3Terc −/− mice.
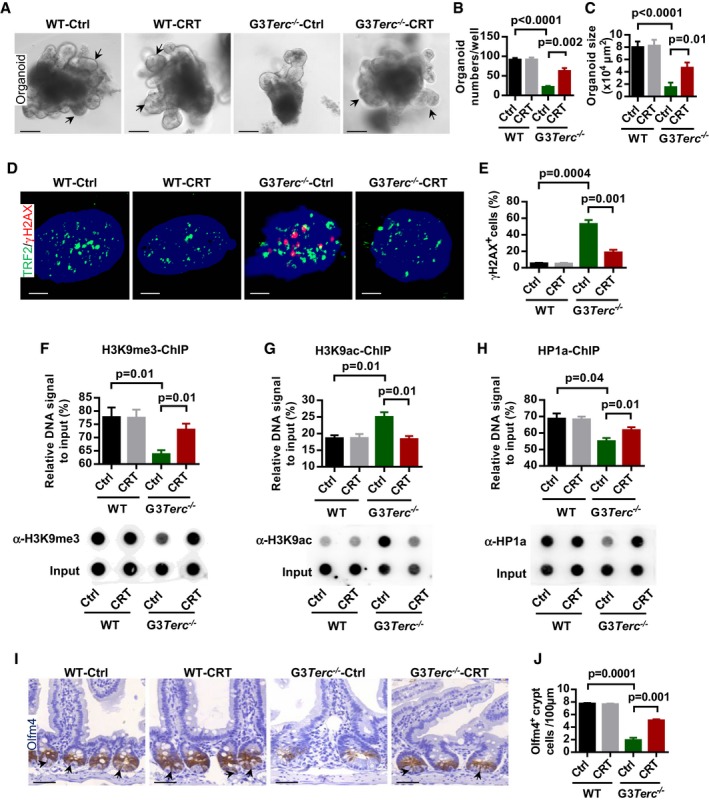
-
A–CRepresentative images of small intestine organoids from WT and G3Terc −/− mice incubated with or without APE1 inhibitor (CRT0044876, 10 μM) treatment. Arrows indicate the crypt‐like structure in organoid cultures (A). Quantification of organoid number (per well) and size (in 104 μm2) (n = 3–4 mice per genotype/treatment) (B, C).
-
D, ERepresentative images of γH2AX antibody staining on dissociated organoid cells from WT and G3Terc −/− organoid cultures with or without APE1 inhibitor (CRT0044876, 10 μM) (DNAs are stained with DAPI) (D). Quantification of γH2AX‐positive cells (n = 3–4 mice per group) (E).
-
F–HTelomeric DNA quantification and dot‐blot assay of H3K9me3‐ChIP (F, upper and lower panel), H3K9ac‐ChIP (G, upper and lower panel) and HP1a‐ChIP (H, upper and lower panel).
-
I, JRepresentative images of Olfm4 staining in small intestine sections from mice treated with or without APE1 inhibitor (CRT0044876, CRT). Arrows indicate Olfm4‐positive intestine stem cells (I). Quantification of Olfm4‐positive intestine stem cells (n = 4 mice per group) (J).
Figure EV3. APE1 inhibition rescues intestine maintenance in G3Terc −/− mice.
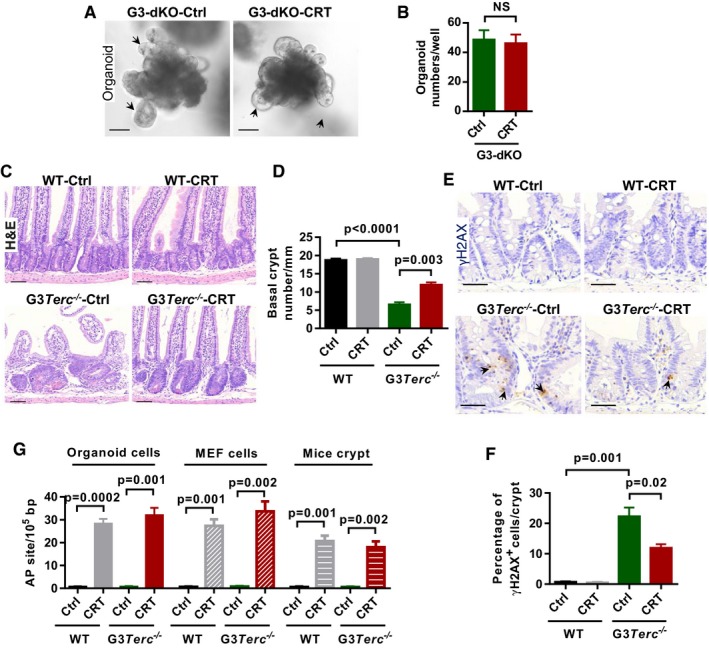
- Representative images of small intestine organoids from G3‐dKO mice incubated with or without APE1 inhibitor CRT0044876 (CRT, 10 μM) treatment. Arrows indicate the crypt‐like structure in organoid cultures.
- Quantification of organoid number (per well) from crypt cultures (n = 3–4 mice per treatment).
- H&E‐stained small intestine sections from 7‐month‐old mice treated with or without APE1 inhibitor CRT0044876 (CRT).
- Quantification of the number of small intestine basal crypts (n = 4 mice per group).
- Representative images of γH2AX antibody staining on small intestine sections from WT and G3Terc −/− mice treated with or without APE1 inhibitor CRT0044876 (CRT). Arrows indicate γH2AX‐positive nuclei.
- Quantification of the percentage of γH2AX‐positive cells per small intestine crypt (n = 4 mice per group).
- Inhibition efficiency of enzyme activity by using APE1 inhibitor CRT0044876 (CRT). Relative AP site accumulation was used as an indicator for enzyme activity inhibition. Organoid and MEF cells were treated with and without CRT0044876. Crypt isolated from WT and G3Terc −/− mice treated with and without CRT0044876 (n = 3 replicates per group).
Figure EV4. APE1 knockdown reduces DNA demethylation in the sub‐telomeric region of G3Terc −/− mice.
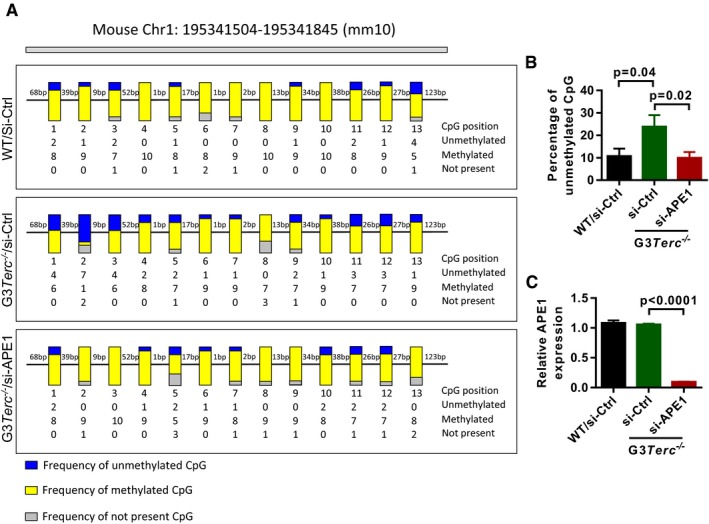
- The methylation status of CpG island in sub‐telomeric regions of chromosome 1q. gDNAs were isolated from MEF cells treated with and without APE1 siRNA and analyzed by bisulfite sequencing. Yellow and blue bars indicate the frequencies of methylated and unmethylated CpG islands, respectively.
- Quantification of the percentage of unmethylated CpG islands in (A) (Data was quantified from 13 CpG islands and each for 10 colony replicates).
- Quantification of APE1 knockdown efficiency in G3Terc −/− cells (n = 3 replicates per group).
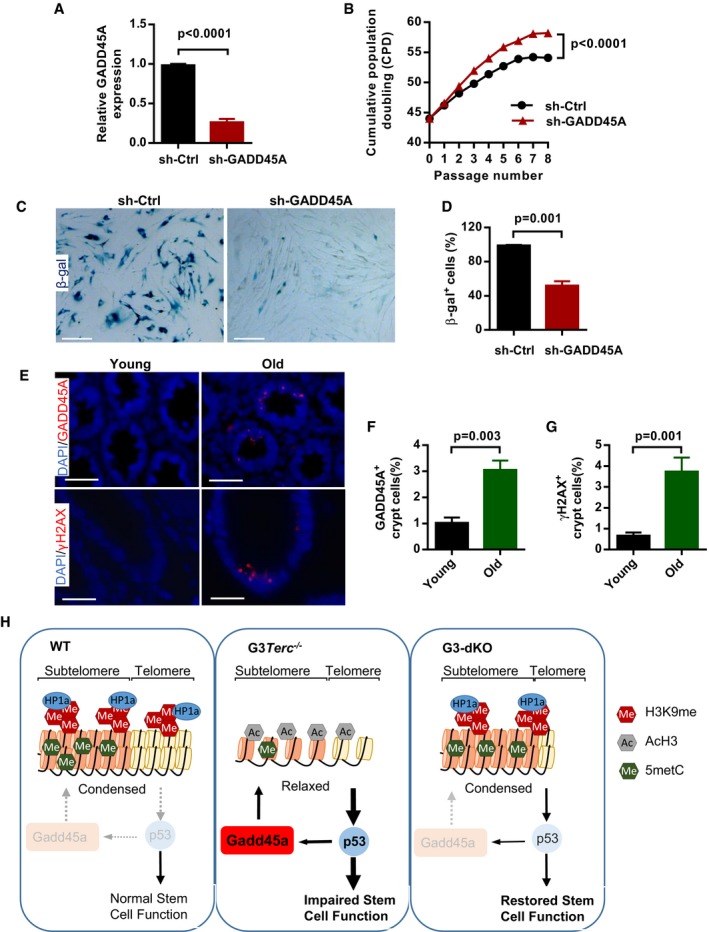
GADD45A knockdown or APE1 inhibition delays replicative senescence of human fibroblasts
Telomere shortening is one of the key drivers of human aging 7, 8. To further substantiate our finding on GADD45A in human aging, we investigated whether GADD45A participates in replicative aging of human fibroblasts. Experiments using human fibroblast cell line WI38 showed that GADD45A knockdown enhanced cell proliferation and importantly prolonged its replicative lifespan (Fig 6A and B), as characterized by increased population doubling and reduced senescence‐associated β‐galactosidase staining (Fig 6C and D). APE1 inhibitor (CRT0044876) treatment and knockdown of APE1 reduced the formation of DNA damage foci (γ‐H2AX) and senescence‐associated β‐galactosidase staining in human WI38 cells (Fig EV5A–E). The knockdown of APE1 by siRNA increased the proliferation capacity of WI38 cells and also reduced the mRNA expression of senescence‐related genes (e.g., p16, p21) (Fig EV5F and G). Furthermore, colon samples from old people showed significantly higher percentage of GADD45A and γ‐H2AX‐positive cells as compared to young ones (Fig 6E–G and Appendix Table S1), and the upregulation of GADD45A is related to inflammation in human colon 39. These data indicate that GADD45A and BER pathways participate in the replicative aging of human cells.
Figure 6. GADD45A is induced in old human colon and its knockdown delays replicative senescence of human cells.
-
AThe knockdown efficiency of shRNA‐GADD45A in human WI38 fibroblasts (n = 3 replicates per group).
-
BThe proliferation curves of human WI38 fibroblasts treated with control shRNA or shRNA against GADD45A (n = 3 replicates per group).
-
C, DRepresentative images of β‐gal staining in human fibroblast cultures with control and shRNA‐GADD45A treatments at passage 54 (C). Quantification of the percentage of β‐gal‐positive cells in human fibroblasts with control and shRNA‐GADD45A treatments (duplicates per group) (D).
-
E–GRepresentative images of GADD45A and γH2AX staining in colon sections from young (35–50 y/o) and old (75–85 y/o) people (E). Quantification of the percentage of GADD45A‐positive cells in young and old human colon tissues (n = 8 per age group) (F). Quantification of the percentage of γH2AX‐positive cells in young and old human colon tissues (n = 6 per age group) (G).
-
HModel for the role of Gadd45a in regulation of DNA damage response initiation. In G3Terc −/− mice, Gadd45a promotes the accessibility of telomeric heterochromatin and enhances the DNA damage response initiation (e.g., pATM activation) through a BER‐dependent pathway. The initiated DNA damage response further causes upregulation of p53 and p21, which leads to impairment of intestine stem cell maintenance and function. In G3‐dKO mice, the telomeric heterochromatin is condensed and damage response initiation is reduced upon Gadd45a deletion, which further leads to decreased expression of p53 and p21. Therefore, the maintenance and function of intestine stem cell are improved.
Figure EV5. APE1 inhibition and knockdown reduce DNA damage response and replicative senescence in human cells.
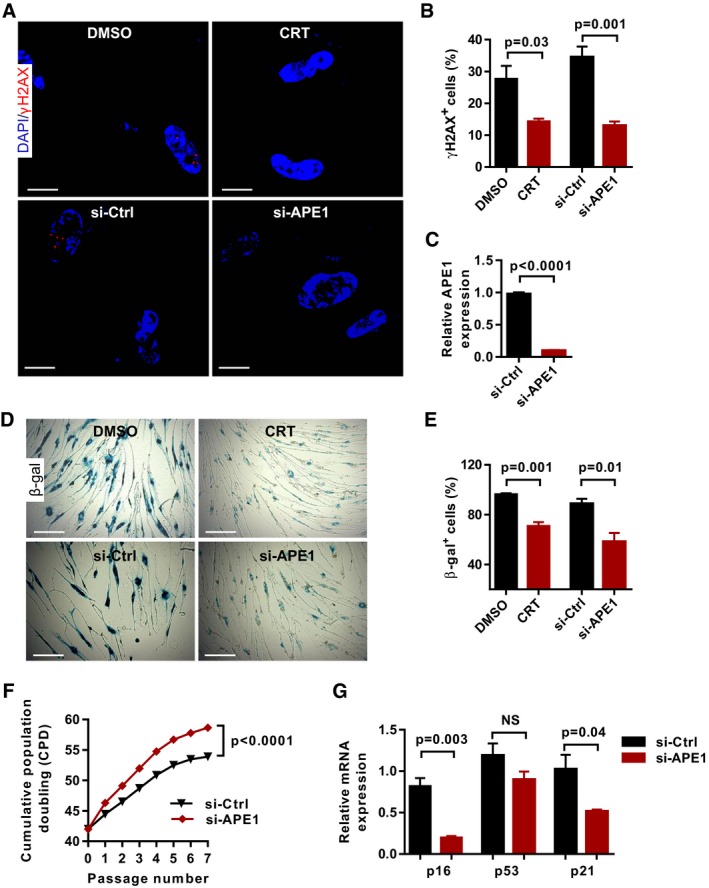
- Representative images of γH2AX antibody staining on human fibroblasts WI38 treated with APE1 inhibitor CRT0044876 (CRT, 100 μM) and APE1 siRNA (si‐APE1) at passage 48 (DNA is stained with DAPI).
- Quantification of γH2AX‐positive cells in (A) (triplicates per group).
- The knockdown efficiency of APE1 in human fibroblast WI38 cells (n = 3 replicates per group).
- Representative images of β‐gal staining in human fibroblasts treated with APE1 inhibitor CRT0044876 (CRT, 100 μM) and APE1 siRNA at passage 48.
- Quantification of β‐gal‐positive cells in human fibroblasts treated with APE1 inhibitor CRT0044876 (CRT) and APE1 siRNA treatments (triplicates per group).
- The growth curves of human WI38 fibroblasts treated with control and APE1 siRNA (n = 3 replicates per group).
- The relative mRNA expression of senescence‐related genes (p16, p53, and p21) in WI38 fibroblasts after APE1 siRNA treatment (n = 3 replicates per group).
Discussion
In mammals, loss of telomere integrity triggered by genetic mutations in telomerase components or telomere maintenance factors generates pathological symptoms in tissues with high turnover rate, such as skin, bone marrow, and intestine 5, 40, 41. It has been suggested that, in these high turnover tissues, the impairment of maintenance and function of stem cell compartments are mainly responsible for the clinical manifestations of telomere‐associated premature aging syndromes 41. Laboratory mice have longer telomeres, while human cells have comparatively shorter telomeres. To probe the significance of stem cell maintenance and function in the telomere shortening‐related aging process in vivo, G3Terc −/− mice were engineered to experience telomere dysfunction as humans do 5, 42. G3Terc −/− mice manifest many of the pathological conditions of human aging and aging‐related diseases, including loss of ISC homeostasis. As reported previously, the impairment of adult stem cell function and loss of tissue homeostasis in G3Terc −/− mice were mainly induced by chronic DNA damage signaling at telomere dysfunction 10, 11, 12, 13. Preventing the initiation of DDR at short telomeres seems to be a feasible approach to ameliorate stem cell aging and to rescue the premature aging phenotypes in G3Terc −/− mice 12, 13. Here, we show that Gadd45a‐mediated epigenetic response participates in the initiation of DDR signaling cascade and plays an essential role in the initiation of DDR at short telomeres (Fig 3A–F).
Gadd45a facilitates DDR initiation at short telomeres
Our previous study shows that loss of Gadd45a enhances hematopoietic stem cell (HSC) self‐renewal and protects HSC from IR‐induced DNA damage 29. In this study, we discovered that Gadd45a plays a critical role in the maintenance and function of intestinal stem cells in the context of telomere dysfunction‐driven aging. Gadd45a loss rescued largely the ISC defects in G3Terc −/− mice and subsequently improved intestine homeostasis and lifespan. In addition to its function as a downstream effector of p53 activation 24, 29, Gadd45a seems to play a role in the epigenetic regulation of heterochromatin status at short telomeres in G3Terc −/− ISCs. Gadd45a deletion restored the compactness of telomeric heterochromatin and thereby attenuating DDR initiation in response to telomere shortening. This finding adds a new layer of understanding of Gadd45a‐mediated DDR on top of its function as a downstream target of p53, which directly interacts with p21 or PCNA to induce cell cycle arrest 12, 23. Nevertheless, Gadd45a still could be transcriptionally upregulated by p53 in response to telomere dysfunction, since we noticed a higher level of Gadd45a expression in G3Terc −/− mice (Fig 1A and B). Taken together with previous findings from our group and others, we propose a model that Gadd45a functions as a positive feedback loop to enhance the DDR signaling initiation at short telomeres, i.e., p53 activation upregulates Gadd45a to further open the telomeric heterochromatin and thus creates a more permissive chromatin structure for the binding of DDR initiation proteins (such as 53BP1, γH2AX, pATM) in G3Terc −/− cells, while Gadd45a deletion abolishes the vicious cycle of DDR signaling and thus improves stem cell function and extends lifespan of G3Terc −/− mice (see Model in Fig 6H).
Gadd45a functions as a site‐specific chromatin modulator in response to telomere dysfunction
Recent study showed that Gadd45a was a global chromatin modulator to promote cellular reprogramming through directly interacting with core histones 43. In the current replicative aging model, Gadd45a participated in the CpG island demethylation specifically at sub‐telomeric regions and regulated mammalian aging by altering the telomeric/sub‐telomeric heterochromatin dynamics. Of note, Gadd45a itself did not possess DNA demethylase activity. Gadd45a functioned in DNA demethylation through its association with BER factor thymine DNA glycosylase (TDG) or NER factor xeroderma pigmentosum group G (XPG) 28, 44. Our study revealed that BER inhibition could improve the ISC maintenance and function by restoration of epigenetic status of telomeric/sub‐telomeric regions in G3Terc −/− mice. This finding is intriguing since, up to date, no study has reported a role of BER‐mediated DNA demethylation in the regulation of stem cell aging induced by short telomeres. Although the exact molecular mechanism on Gadd45a‐BER recruitment to short telomeres needs to be further elucidated, the current study reveals Gadd45a‐BER may function as a chromatin modification machinery to promote the DNA damage response signaling on critically short telomeres.
Gadd45a/APE1 inhibition delays stem cell aging through epigenetic modification
Accumulating evidences suggest an association of epigenetic alterations with mammalian aging 45, 46, 47, 48. Recently, studies in adult hematopoietic stem cells (HSCs) showed that aged HSCs exhibited distinct genome‐wide epigenetic landscapes as compared to young HSCs 49, 50, 51. In addition, deficiencies in epigenetic modulators, such as DNMTs, compromised the HSC homeostasis and function 52, 53, 54. Furthermore, locus‐specific DNA hyper‐methylations in gene regions were associated with aging process 43, 47, 48, 55. Interestingly, the heterochromatin condensation has been linked to cellular aging in Werner syndrome patient‐derived mesenchymal stem cells 56. In line with this, telomere dysfunctional cells exhibited hypo‐methylation in the telomeric/sub‐telomeric regions 17. In this study, we provided the first evidence that manipulation of an epigenetic factor could improve organ maintenance and prolong the lifespan of mice with short telomeres. Although Gadd45a has been suggested to be functioned in transcriptional regulation, Gadd45a loss does not globally change the methylation profile in our experimental setting. The current study discloses a transcription‐independent role of Gadd45a‐mediated DNA demethylation event, regulating stem cell aging by altering the heterochromatin status specifically at telomeric/sub‐telomeric regions 57.
Taken together, our finding adds a new layer of understanding on Gadd45a‐mediated DNA demethylation in response to telomere dysfunction‐induced DDR, and further extends our knowledge that lifespan extension and tissue homeostasis improvement could be achieved by altering site‐specific epigenetic changes, such as heterochromatin at telomeres/sub‐telomeres. According to our data, the APE1 inhibition represents a potential anti‐aging strategy for human physiological aging. Of note, a global change in epigenetic profile is deleterious for stem cell maintenance 52, 53, 54 and practically not applicable. Since Gadd45a functions in both DDR and epigenetic regulation, Gadd45a‐APE1 could be an ideal candidate for the intervention of epi‐aging and therefore serves as a potential target to specifically manipulate the DNA damage ‐related epigenetic modifications.
Materials and Methods
Mice
Gadd45a −/− mice 58 were gifted from Professor Albert J. Fornace Jr. and were maintained in C57BL/6 background. For the mice used in this study, Gadd45a −/− mice and their wild‐type littermate controls were produced from the intercrossing of Gadd45a +/− mice. Heterozygous telomerase RNA component Terc knockout mice 5, 42 (Terc +/−) were crossed with Gadd45a +/− to produce G1Terc −/− Gadd45a +/− mice, which were subsequently intercrossed to generate G2Terc −/− Gadd45a +/− mice. For the final analysis, G3Terc −/− Gadd45a +/+ (G3Terc −/−) and G3Terc −/− Gadd45a −/− (G3‐dKO) mice were produced from intercrossings of G2Terc −/− Gadd45a +/− mice. The animal breeding and experiments were conducted at the animal facility of Jinan University with the approval of the Animal Care and Ethics Committee.
Histology
Intestines were opened longitudinally, fixed in 4% buffered formalin overnight, and rolled up carefully. Colon whole mounts were prepared as previously described 59. Hematoxylin and eosin (H&E) sections were prepared according to standard protocol.
Immunohistochemistry (IHC) and immunofluorescence (IF) analysis of paraffin sections
Five‐micrometer (μm) intestine tissue sections were de‐paraffinized and rehydrated before antigen retrieval in citrate buffer (pH 6.0) in microwave. Sections were further incubated with primary antibodies overnight at 4°C: anti‐γH2AX (Millipore, 1:200), anti‐53BP1 (Abcam, 1:100), anti‐ATM (phospho‐Ser1981) (Millipore, 1:200), anti‐p53 (Santa Cruz, 1:50), anti‐p21 (Santa Cruz, 1:50), anti‐phospho‐histone H3 (Ser10) (Cell Signaling, 1:200), and anti‐olfm4 (Cell Signaling, 1:400). For IHC, the sections were developed with a Vectastain ABC Elite kit (Vector Laboratories). The slides were scanned with 3DHISTECH Digital Pathology System (Pannoramic MIDI). For each genotype, at least 100 crypts were quantified. For IF analysis, tissue sections were blocked with 1% BSA for 1 h and then incubated with primary antibodies in 1% BSA overnight at 4°C. The slides were mounted in a mounting medium with DAPI (Vector Laboratories) and photographed.
In vitro crypt culture
Crypt culture was conducted as previously described 33. In brief, 200 crypts isolated from WT, Gadd45a −/−, G3Terc −/−, and G3‐dKO mice (4 months old) were mixed with 20 μl of Matrigel (BD) and plated in 48‐well plates. The crypts were cultured in 200 μl of Advanced DMEM/F12 supplemented with B27 and N2 supplements (Invitrogen), epidermal growth factor (Peprotech), Noggin (Peprotech), R‐spondin‐1 (R&D), and N‐acetylcysteine (Sigma). On day 2, the number of cysts was quantified, and on day 8, the number and morphology of organoids were quantified. Alternatively, the organoids were disassociated and cytospinned onto coverslips for immunofluorescence analysis. For APE1 inhibitor treatment, crypt cultures from WT and G3Terc −/− mice (3 months old) were treated with high (100 μM) and low (10 μM) concentration of APE1 inhibitors (CRT0044876 60) for 8 days. A same volume of DMSO was used as a control treatment.
DNA methylation assay
Sub‐telomeric DNA methylation analysis on mouse chromosome 1 was performed by Generay Biotech Co., Ltd (Shanghai, China) following the previously published protocol 17. Briefly, around 10 colonies were sequenced from WT, Gadd45a −/−, G3Terc −/−, and G3‐dKO MEF cells. The bisulfite genomic sequencing primers for mouse chromosome 1 are as follows: p1: 5′‐TTACCAATACCACCATTCCTCCA‐3′ and p2: 5′‐GAGAGTAGTTAATTAGATGAGGAATA‐3′. A BiQ Analyzer was used to analyze the data collected in sequencing. The two primers used are located spanning Chr1: 195,341,504–195,341,845 (annotated mouse genome version mm10), where 19 CpGs are used for the methylation analysis.
ChIP‐qPCR
MEF cells from WT, Gadd45a −/−, G3Terc −/−, G3‐dKO genotypes were used for the ChIP assay. ChIP was carried out according to the protocol from Chromatin Immunoprecipitation Kit (EZ‐CHIP; Millipore, 17‐371). Briefly, for each ChIP experiment, ten million (1 × 107) cells was used and crosslinked with 1% formaldehyde. The DNA was sheared into 200–1,000 base pair fragments by sonication. Ten micrograms of following antibodies was used: anti‐H3K9me3 (Millipore, 07‐442), anti‐H3K9ac (Sigma‐Aldrich, H9286), anti‐HP1a (Cell Signaling, CST‐2616), and anti‐Gadd45a (Santa Cruz, sc‐797). For each ChIP and input, 30 μl elution buffer was used to elute the DNA. Two microliters of DNA eluent was used for qPCR with the following primers:
1. Telomere primers:
Fw: 5′‐CGGTTTGTTTGGGTTTGGGTTTGGGTTTGGGTTTGGGTT‐3′.
Rev5′‐GGCTTGCCTTACCCTTACCCTTACCCTTACCCTTACCCT‐3′.
2. ALU element primer 61:
Fw: 5′‐GAGGCAGGCGGATTTCTGA‐3′.
Rev: 5′‐CCCTGGTGTCCTGGAACTCA‐3′.
ChIP‐Dot‐blot
For ChIP‐Dot‐blot, 2 × 107 MEF cells were used and 10% of DNA was used as input after sonication. The ChIP assay was done as described above. DNA samples were diluted in 100 μl 2× SSC and then heated at 95°C for 10 min and followed by cooling on ice, centrifuged for 2 min at 15,800 g, 4°C. Denatured DNA samples were spotted on a nylon membrane (pre‐soaked in 2× SSC for 10 min and dried on tissue paper) in an assembled Bio‐Dot apparatus (Bio‐Rad) according to the manufacturer's instructions. The membrane was denatured in denaturing buffer (1.5 M NaCl, 0.5 M NaOH) for 10 min and neutralized in neutralization buffer (3 M NaCl, 0.5 M Tris–HCl, pH 7.0) for 10 min. The membrane was air‐dried and ultraviolet‐crosslinked for 5 min. The following procedure was done according to the “Telo TAGGG Telomere Length Assay” kit (Roche Cat. No. 12209136001). Then, the membrane was pre‐hybridized in hybridization buffer 1 h at 42°C and hybridized in hybridization buffer containing telomere probe overnight. The membrane was washed with stringent wash buffer I for two times, 5 min each; stringent wash buffer II for two times at 50°C, 20 min each time, and wash buffer once, 5 min. The membrane was blocked in blocking buffer for 30 min RT, incubated in anti‐DIG‐AP for 30 min RT, and washed with wash buffer for two times, 15 min each. The membrane was treated with detection buffer 5 min and exposed in sealed bag with substrate buffer under VersaDoc image device.
Apoptosis assay
TUNEL assay (in situ cell death detection kit, Millipore, S7101) was used to detect the apoptotic cells in intestine paraffin sections from mice of different genotypes according to the manufacturer's instruction. The frequency of apoptotic cells was quantified by counting TUNEL‐positive cells from at least 100 crypts/mouse/genotype.
Western blotting
Freshly isolated intestine crypt cells were lysed with RIPA buffer. Fifty micrograms of protein was resolved in 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and further transferred to PVDF membrane and detected using antibodies against p21 (Santa Cruz, sc‐6246, 1:1,000), Gadd45a (Abnova, H00001647‐M01, 1:1,000), and β‐actin (Sigma‐Aldrich, A5316, 1:5,000).
Telomere length measurement (Q‐FISH)
Q‐FISH using the PNA probes (PANAGENE) was conducted on paraffin sections of WT, Gadd45a −/−, G3Terc −/−, and G3‐dKO intestine tissues following the manufacturer's instruction. The FISH images were captured with a confocal fluorescence microscope (LSM 710, Carl Zeiss International). The telomere length is quantified at least 20 crypts/mouse following the protocol described previously 34.
Immunofluorescence
MEF cells or crypt cells dissociated from cultured organoids were cultured or cytospinned onto poly‐D‐lysine‐coated coverslips, respectively, fixed with 4% formaldehyde, and permeabilized with 0.3% Triton X‐100. The cells were further incubated for 1 h with blocking buffer (2% BSA in 1× PBS) before application of the primary antibodies. The γH2AX antibody (Millipore) and/or 53BP1 antibody (Abcam) was used at 1:200 in the blocking solution. The slides were mounted in a mounting medium with DAPI (Vector Laboratories) and imaged using a confocal fluorescence microscope (LSM 710, Carl Zeiss International).
Transcriptome analysis
The RNAs were isolated from WT, Gadd45a −/−, G3Terc −/−, and G3‐dKO MEF cells using TRI® reagent (Sigma‐Aldrich). The library construction and sequencing were performed in BGI‐Shenzhen, China. Briefly, RNA samples for transcriptome analysis was pre‐treated with DNase and processed following Illumina manufacturer's instructions where magnetic beads with oligo (dT) were used to isolate polyadenylated mRNA (polyA+ mRNA) from the total RNA. Fragmentation buffer consisting of divalent cations was added for shearing mRNA to short fragments of 200–700 nucleotides in length. These short fragments were used as templates to synthesize the first‐strand cDNA using random hexamer‐primer. The second‐strand cDNA was synthesized using buffer including dNTPs, RNase H, and DNA polymerase I. The products were purified and resolved with QIAquick PCR Purification Kit (Qiagen, California, USA) and Elution buffer for end preparation and tailing A, respectively. Purified cDNA fragments were connected with sequencing adapters and gel electrophoresed to select suitable fragments for PCR amplification. Agilent 2100 Bioanalyzer and Applied Biosystems StepOnePlus™ Real‐Time PCR System were used in quantification and qualification of the sample library for quality control. A paired‐end cDNA library was constructed and sequenced using Illumina HiSeq™ 4000 at BGI‐Shenzhen, China. The sequencing reads were mapped to the mouse reference genome (mm9) using HISAT 62. Differentially expressed genes (DEGs) between each genotype were calculated by standard bioinformatic analysis package, and the hierarchical clustering for DEGs between samples was generated based on DEGs.
APE1 inhibitor (CRT0044876) treatment
Human fibroblasts and MEF cells were treated with APE1 inhibitor (CRT0044876 1, 100 μM and 10 μM) for 4 and 2 weeks, respectively, and DMSO was used as control. For the mice treatment, 6‐month‐old WT and G3Terc −/− mice were treated with APE1 inhibitor (CRT0044876) every other day for 4 weeks, at the dosage of 15 mg/kg/mouse by intraperitoneal injection.
AP site accumulation assay
The number of AP sites was used as an indicator for APE1 enzyme activity inhibition. The increase in AP site accumulation represents the inhibition of enzyme activity 38. The genomic DNA was isolated from organoid cells, MEF cells, and crypt cells of mice by using “Universal Genomic DNA Extraction Kit” (Takara Bio Inc.). DNA was eluted with TE buffer and diluted to a final concentration of 100 μg/ml. AP site determinations were performed by using “DNA Damage Quantification Kit‐AP Site Counting” kit and according to the protocol provided by the manufacturer (Dojindo Molecular Technologies, Inc.). All experiments were performed in triplicate.
Immunofluorescence (IF) on human colon samples
The study on GADD45A expression in colon samples from young and old humans was approved by the Ethics Committee of Sir Run Run Shaw Hospital, Zhejiang University. Before the human colon specimen collections, written informed consents were obtained from all patients. The dissected colon tissues were fixed in 4% buffered formalin overnight before embedded into paraffin. IF staining with Gadd45a antibody was conducted on 5‐μm‐thick human colon tissue sections following the same protocol described before.
Knockdown GADD45A in human WI‐38 fibroblasts
The shRNA sequence (5′‐AACGTCGACCCCGATAACGTG‐3′) targeting human GADD45A was cloned into lentiviral vector SFLV‐mir30a‐EV 63. sh‐GADD45A lentiviral particles were produced by transfecting 293T cells with three plasmids‐pMD2.G, psPAX2, and Lenti‐sh‐GADD45A or Lenti‐sh‐Luciferase (sh‐Control) vectors. Virus was collected 36 h post‐transfection and applied onto WI‐38 cells in 60‐mm dish with 80% confluence. The efficiency of Gadd45a knockdown was confirmed by qRT–PCR 48 h after virus infection.
Knockdown of p21 and Ape1 in human FB WI‐38 cells and mouse MEF cells
p21 and APE1 were knocked down in WT and G3Terc −/− MEF cells by using Lipofectamine® RNAiMAX Reagent (Life Technologies™). The transfections were carried out according to the typical RNAiMAX transfection procedure. The cells were collected after 3 days of incubation at 37°C. Knockdown of APE1 in human fibroblast was continued for 4 weeks, and siRNAs were refreshed every 3–4 days when the culture medium was changed. siRNAs were synthesized from Sigma‐Aldrich, and two siRNAs were used for each gene. siRNAs IDs for p21 were SASI_Mm01_00086762 and SASI_Mm01_00086763, and for APE1 were SASI_Mm01_00152564 and SASI_Mm01_00152565.
SA‐β‐gal staining of cellular senescence
SA‐β‐gal staining was conducted with Senescence β‐Galactosidase Staining Kit (Cell Signaling, #9860) following the company manual. Briefly, cultured human WI‐38 cells at indicated passages were washed in 1× PBS twice and then fixed at RT for 3 min in 2% formaldehyde and 0.2% glutaraldehyde. Fixed cells were stained with fresh β‐gal staining solution at 37°C overnight. The percentage of cells positive for SA‐β‐gal staining were quantified and statistically analyzed.
Quantification and statistical analysis
GraphPad Prism 6.0 software is used for the statistics presented in the manuscript. Unless specified, unpaired two‐tailed Student's t‐test was conducted between each group. All results are presented as mean ± SEM.
Data availability
The RNA‐Seq data from this publication have been deposited to GEO (Gene Expression Omnibus) and with the accession number: GSE117845.
Author contributions
DD designed and performed experiments, analyzed data, and wrote comments on the manuscript; HW designed and performed experiments, analyzed data and wrote comments on the manuscript; TL designed experiments and wrote the manuscript; ZS performed experiments; XJ wrote comments on the manuscript; TS designed experiments and wrote comments on the manuscript; XZ wrote comments on the manuscript; MZ performed experiments; FY wrote comments on the manuscript; YC wrote comments on the manuscript; LS analyzed data and wrote comments on the manuscript; QZ designed experiments and wrote comments on the manuscript; JY wrote comments on the manuscript; ZS provided human colon sample and wrote comments on the manuscript; ZJ designed experiments, wrote comments on the manuscript, and provided funding.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
This work is supported by the National Key Research and Development Program of China (Stem Cell and Translational Research, grant #2016YFA0100600, 2017YFA0103302) and the National Natural Science Foundation of China (grants #81420108017 and 81525010) to Z.J., the National Natural Science Foundation of China for youth scientists (grant #81300263) to H.W., the National Natural Science Foundation of China (grant #81571380), the Natural Science Foundation of Zhejiang Province (grant #LY16H080009) to T.L. Z.S. was supported by a grant from the National Natural Science Foundation of China (grant #81370461).
EMBO Reports (2018) 19: e45494
Contributor Information
Daojun Diao, Email: diaodaojun@hotmail.com.
Zhenyu Ju, Email: zhenyuju@163.com.
References
- 1. López‐Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013) The hallmarks of aging. Cell 153: 1194–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Atzmon G, Cho M, Cawthon RM, Budagov T, Katz M, Yang X, Siegel G, Bergman A, Huffman DM, Schechter CB et al (2010) GatGenetic variation in human telomerase is associated with telomere length in Ashkenazi centenarians. Proc Natl Acad Sci USA 107: 1710–1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Njajou OT, Hsueh WC, Blackburn EH, Newman AB, Wu SH, Li R, Simonsick EM, Harris TM, Cummings SR, Cawthon RM et al (2009) Association between telomere length, specific causes of death, and years of healthy life in health, aging, and body composition, a population‐based cohort study. J Gerontol A 64: 860–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shaw AC, Joshi S, Greenwood H, Panda A, Lord JM (2010) Aging of the innate immune system. Curr Opin Immunol 22: 507–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rudolph KL, Chang S, Lee HW, Blasco M, Gottlieb GJ, Greider C, DePinho RA (1999) Longevity, stress response, and cancer in aging telomerase‐deficient mice. Cell 96: 701–712 [DOI] [PubMed] [Google Scholar]
- 6. Deng Y, Chan SS, Chang S (2008) Telomere dysfunction and tumour suppression: the senescence connection. Nat Rev Cancer 8: 450–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Newgard CB, Sharpless NE (2013) Coming of age: molecular drivers of aging and therapeutic opportunities. J Clin Invest 123: 946–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Blackburn EH, Epel ES, Lin J (2015) Human telomere biology: a contributory and interactive factor in aging, disease risks, and protection. Science 350: 1193–1198 [DOI] [PubMed] [Google Scholar]
- 9. Shay JW (2016) Role of telomeres and telomerase in aging and cancer. Cancer Discov 6: 584–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sperka T, Wang J, Rudolph KL (2012b) DNA damage checkpoints in stem cells, ageing and cancer. Nat Rev Mol Cell Biol 13: 579–590 [DOI] [PubMed] [Google Scholar]
- 11. Choudhury AR, Ju Z, Djojosubroto MW, Schienke A, Lechel A, Schaetzlein S, Jiang H, Stepczynska A, Wang C, Buer J et al (2007) Cdkn1a deletion improves stem cell function and lifespan of mice with dysfunctional telomeres without accelerating cancer formation. Nat Genet 39: 99–105 [DOI] [PubMed] [Google Scholar]
- 12. Schaetzlein S, Kodandaramireddy NR, Ju Z, Lechel A, Stepczynska A, Lilli DR, Clark AB, Rudolph C, Kuhnel F, Wei K et al (2007) Exonuclease‐1 deletion impairs DNA damage signaling and prolongs lifespan of telomere‐dysfunctional mice. Cell 130: 863–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sperka T, Song Z, Morita Y, Nalapareddy K, Guachalla LM, Lechel A, Begus‐Nahrmann Y, Burkhalter MD, Mach M, Schlaudraff F et al (2012a) Puma and p21 represent cooperating checkpoints limiting self‐renewal and chromosomal instability of somatic stem cells in response to telomere dysfunction. Nat Cell Biol 14: 73–79 [DOI] [PubMed] [Google Scholar]
- 14. Jaskelioff M, Muller FL, Paik JH, Thomas E, Jiang S, Adams AC, Sahin E, Kost‐Alimova M, Protopopov A, Cadiñanos J et al (2011) Telomerase reactivation reverses tissue degeneration in aged telomerase‐deficient mice. Nature 469: 102–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gonzalo S, Jaco I, Fraga MF, Chen T, Li E, Esteller M, Blasco MA (2006) DNA methyltransferases control telomere length and telomere recombination in mammalian cells. Nat Cell Biol 8: 416–424 [DOI] [PubMed] [Google Scholar]
- 16. Benetti R, Gonzalo S, Jaco I, Schotta G, Klatt P, Jenuwein T, Blasco M (2007a) Suv4‐20 h deficiency results in telomere elongation and derepression of telomere recombination. J Cell Biol 178: 925–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Benetti R, Garcia‐Cao M, Blasco MA (2007b) Telomere length regulates the epigenetic status of mammalian telomeres and subtelomeres. Nat Genet 39: 243–250 [DOI] [PubMed] [Google Scholar]
- 18. Blasco MA (2007) The epigenetic regulation of mammalian telomeres. Nat Rev Genet 8: 299–309 [DOI] [PubMed] [Google Scholar]
- 19. He Q, Kim H, Huang R, Lu W, Tang M, Shi F, Yang D, Zhang X, Huang J, Liu D et al (2015) The Daxx/Atrx complex protects tandem repetitive elements during DNA hypomethylation by promoting H3K9 trimethylation. Cell Stem Cell 17: 273–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Garcia‐Cao M, O'Sullivan R, Peters AH, Jenuwein T, Blasco MA (2004) Epigenetic regulation of telomere length in mammalian cells by the Suv39h1 and Suv39h2 histone methyltransferases. Nat Genet 36: 94–99 [DOI] [PubMed] [Google Scholar]
- 21. Amanullah A, Azam N, Balliet A, Hollander C, Hoffman B, Fornace A, Liebermann D (2003) Cell signalling: cell survival and a Gadd45‐factor deficiency. Nature 424: 741–742 [DOI] [PubMed] [Google Scholar]
- 22. Gao M, Guo N, Huang C, Song L (2009) Diverse roles of GADD45alpha in stress signaling. Curr Protein Pept Sci 10: 388–394 [DOI] [PubMed] [Google Scholar]
- 23. Smith ML, Chen IT, Zhan Q, Bae I, Chen CY, Gilmer TM, Kastan MB, O'Connor PM, Fornace AJ (1994) Interaction of the p53‐regulated protein Gadd45 with proliferating cell nuclear antigen. Science 266: 1376–1380 [DOI] [PubMed] [Google Scholar]
- 24. Zhan Q (2005) Gadd45a, a p53‐ and BRCA1‐regulated stress protein, in cellular response to DNA damage. Mutat Res 569: 133–143 [DOI] [PubMed] [Google Scholar]
- 25. Tran H, Brunet A, Grenier JM, Datta SR, Fornace AJ, DiStefano PS, Chiang LW, Greenberg ME (2002) DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science 296: 530–534 [DOI] [PubMed] [Google Scholar]
- 26. Dan AL, Hoffman B (2013) Gadd45 stress sensor genes, Vol. 793 New York, NY: Springer New York; [Google Scholar]
- 27. Schafer A (2013) Gadd45 proteins: key players of repair‐mediated DNA demethylation. Adv Exp Med Biol 793: 35–50 [DOI] [PubMed] [Google Scholar]
- 28. Barreto G, Schäfer A, Marhold J, Stach D, Swaminathan S, Handa V, Döderlein G, Maltry N, Wu W, Lyko F et al (2007) Gadd45a promotes epigenetic gene activation by repair‐mediated DNA demethylation. Nature 445: 671–675 [DOI] [PubMed] [Google Scholar]
- 29. Chen Y, Ma X, Zhang M, Wang X, Wang C, Wang H, Guo P, Yuan W, Rudolph KL, Zhan Q et al (2014) Gadd45a regulates hematopoietic stem cell stress responses in mice. Blood 123: 851–862 [DOI] [PubMed] [Google Scholar]
- 30. Saidi A, Li T, Weih F, Concannon P, Wang ZQ (2010) Dual functions of Nbs1 in the repair of DNA breaks and proliferation ensure proper V(D)J recombination and T‐cell development. Mol Cell Biol 30: 5572–5581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dabin J, Fortuny A, Polo SE (2016) Epigenome maintenance in response to DNA damage. Mol Cell 62: 712–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Muñoz J, Stange DE, Schepers AG, van de Wetering M, Koo BK, Itzkovitz S, Volckmann R, Kung K, Koster J, Radulescu S et al (2012) The Lgr5 intestinal stem cell signature: robust expression of proposed quiescent “+4” cell markers. EMBO J 31: 3079–3091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van EJ, Abo A, Kujala P, Peters PJ et al (2009) Single Lgr5 stem cells build crypt‐villus structures in vitro without a mesenchymal niche. Nature 459: 262–265 [DOI] [PubMed] [Google Scholar]
- 34. Ourliac‐Garnier I, Londono‐Vallejo A (2011) Telomere length analysis by quantitative fluorescent in situ hybridization (Q‐FISH). Methods Mol Biol 735: 21–31 [DOI] [PubMed] [Google Scholar]
- 35. Jin S, Mazzacurati L, Zhu X, Tong T, Song Y, Shujuan S, Petrik KL, Rajasekaran B, Wu M, Zhan Q (2003) Gadd45a contributes to p53 stabilization in response to DNA damage. Oncogene 22: 8536–8540 [DOI] [PubMed] [Google Scholar]
- 36. Artandi SE, Chang S, Lee SL, Alson S, Gottlieb GJ, Chin L, DePinho RA (2000) Telomere dysfunction promotes non‐reciprocal translocations and epithelial cancers in mice. Nature 406: 641–645 [DOI] [PubMed] [Google Scholar]
- 37. Ma DK, Guo JU, Ming GL, Song H (2009) DNA excision repair proteins and Gadd45 as molecular players for active DNA demethylation. Cell Cycle 8: 1526–1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Madhusudan S, Smart F, Shrimpton P, Parsons JL, Gardiner L, Houlbrook S, Talbot DC, Hammonds T, Freemont PA, Sternberg MJ et al (2005) Isolation of a small molecule inhibitor of DNA base excision repair. Nucleic Acids Res 33: 4711–4724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lembo‐Fazio L, Nigro G, Noël G, Rossi G, Chiara F, Tsilingiri K, Rescigno M, Rasola A, Bernardini ML (2011) Gadd45α activity is the principal effector of Shigella mitochondria‐dependent epithelial cell death in vitro and ex vivo . Cell Death Dis 2: e122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jonassaint NL, Guo N, Califano JA, Montgomery EA, Armanios M (2013) The gastrointestinal manifestations of telomere‐mediated disease. Aging Cell 12: 319–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Armanios M (2009) Syndromes of telomere shortening. Annu Rev Genomics Hum Genet 10: 45–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Blasco MA, Lee HW, Hande MP, Samper E, Lansdorp PM, DePinho RA, Greider CW (1997) Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 91: 25–34 [DOI] [PubMed] [Google Scholar]
- 43. Chen K, Long Q, Wang T, Zhao D, Zhou Y, Qi J, Wu Y, Li S, Chen C, Zeng X et al (2016) Gadd45a is a heterochromatin relaxer that enhances iPS cell generation. EMBO Rep 17: 1641–1656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li Z, Gu TP, Weber AR, Shen JZ, Li BZ, Xie ZG, Yin R, Guo F, Liu X, Tang F et al (2015) Gadd45a promotes DNA demethylation through TDG. Nucleic Acids Res 43: 3986–3997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shumaker DK, Dechat T, Kohlmaier A, Adam SA, Bozovsky MR, Erdos MR, Eriksson M, Goldman AE, Khuon S, Collins FS et al (2006) Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci USA 103: 8703–8708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sen P, Shah PP, Nativio R, Berger SL (2016) Epigenetic mechanisms of longevity and aging. Cell 166: 822–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Heyn H, Li N, Ferreira HJ, Moran S, Pisano DG, Gomez A, Diez J, Sanchez‐Mut JV, Setien F, Carmona FJ et al (2012) Distinct DNA methylomes of newborns and centenarians. Proc Natl Acad Sci USA 109: 10522–10527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gonzalo S (2010) Epigenetic alterations in aging. J Appl Physiol 109: 586–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sun D, Luo M, Jeong M, Rodriguez B, Xia Z, Hannah R, Wang H, Le T, Faull KF, Chen R et al (2014) Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self‐renewal. Cell Stem Cell 14: 673–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chambers SM, Shaw CA, Gatza C, Fisk CJ, Donehower LA, Goodell MA (2007) Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol 5: e201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Beerman I, Rossi DJ (2015) Epigenetic control of stem cell potential during homeostasis, aging, and disease. Cell Stem Cell 16: 613–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang X, Su J, Jeong M, Ko M, Huang Y, Park HJ, Guzman A, Lei Y, Huang YH, Rao A et al (2016) DNMT3A and TET2 compete and cooperate to repress lineage‐specific transcription factors in hematopoietic stem cells. Nat Genet 48: 1014–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Challen GA, Sun D, Mayle A, Jeong M, Luo M, Rodriguez B, Mallaney C, Celik H, Yang L, Xia Z et al (2014) Dnmt3a and Dnmt3b have overlapping and distinct functions in hematopoietic stem cells. Cell Stem Cell 15: 350–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Trowbridge JJ, Snow JW, Kim J, Orkin SH (2009) DNA methyltransferase 1 is essential for and uniquely regulates hematopoietic stem and progenitor cells. Cell Stem Cell 5: 442–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bollati V, Schwartz J, Wright R, Litonjua A, Tarantini L, Suh H, Sparrow D, Vokonas P, Baccarelli A (2009) Decline in genomic DNA methylation through aging in a cohort of elderly subjects. Mech Ageing Dev 130: 234–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhang W, Li J, Suzuki K, Qu J, Wang P, Zhou J, Liu X, Ren R, Xu X, Ocampo A et al (2015) Aging stem cells. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science 348: 1160–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Engel N, Tront JS, Erinle T, Nguyen N, Latham KE, Sapienza C, Hoffman B, Liebermann DA (2009) Conserved DNA methylation in Gadd45a(−/−) mice. Epigenetics 4: 98–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hildesheim J, Bulavin DV, Anver MR, Alvord WG, Hollander MC, Vardanian L, Fornace AJ (2002) Gadd45a protects against UV irradiation‐induced skin tumors, and promotes apoptosis and stress signaling via MAPK and p53. Cancer Res 62: 7305–7315 [PubMed] [Google Scholar]
- 59. Pretlow TP, Barrow BJ, Ashton WS, O'Riordan MA, Pretlow TG, Jurcisek JA, Stellato TA (1991) Aberrant crypts: putative preneoplastic foci in human colonic mucosa. Cancer Res 51: 1564–1567 [PubMed] [Google Scholar]
- 60. Wilson DM III, Simeonov A (2010) Small molecule inhibitors of DNA repair nuclease activities of APE1. Cell Mol Life Sci 67: 3621–3631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhang W, Wu M, Menesale E, Lu T, Magliola A, Bergelson S (2014) Development and qualification of a high sensitivity, high throughput Q‐PCR assay for quantitation of residual host cell DNA in purification process intermediate and drug substance samples. J Pharm Biomed Anal 100: 145–149 [DOI] [PubMed] [Google Scholar]
- 62. Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12: 357–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang J, Sun Q, Morita Y, Jiang H, Gross A, Lechel A, Hildner K, Guachalla LM, Gompf A, Hartmann D et al (2012) A differentiation checkpoint limits hematopoietic stem cell self‐renewal in response to DNA damage. Cell 148: 1001–1014 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Data Availability Statement
The RNA‐Seq data from this publication have been deposited to GEO (Gene Expression Omnibus) and with the accession number: GSE117845.