

Abstract
The Myc family of oncogenic transcription factors regulates myriad cellular functions. Myc proteins contain a basic region/helix‐loop‐helix/leucine zipper domain that mediates DNA binding and heterodimerization with its partner Max. Among the Myc proteins, c‐Myc is the most widely expressed and relevant in primary B lymphocytes. There is evidence suggesting that c‐Myc can perform some of its functions in the absence of Max in different cellular contexts. However, the functional in vivo interplay between c‐Myc and Max during B lymphocyte differentiation is not well understood. Using in vivo and ex vivo models, we show that while c‐Myc requires Max in primary B lymphocytes, several key biological processes, such as cell differentiation and DNA replication, can initially progress without the formation of c‐Myc/Max heterodimers. We also describe that B lymphocytes lacking Myc, Max, or both show upregulation of signaling pathways associated with the B‐cell receptor. These data suggest that c‐Myc/Max heterodimers are not essential for the initiation of a subset of important biological processes in B lymphocytes, but are required for fine‐tuning the initial response after activation.
Keywords: B lymphocytes, cell differentiation, c‐Myc, Max, replication
Subject Categories: Development & Differentiation, Immunology, Signal Transduction
Introduction
In bone marrow (BM), B lymphocyte progenitors progress through a series of well‐defined differentiation stages associated with rearrangements of the B‐cell receptor gene segments V(D)J. Several transcription factors have been shown to play critical roles during this process 1, 2. Among them, c‐Myc, a member of the Myc family (N‐, L‐, and c‐Myc), has a prominent role in many biological functions required for B lymphocyte development such as regulation of cell cycle, cell growth, metabolism, or apoptosis 3, 4. c‐Myc is necessary throughout B‐cell differentiation in vivo, and its deletion in early B‐cell progenitors prevents these cells from progressing beyond the pre‐B‐cell stage 5. At later stages in B‐cell maturation, c‐myc expression is induced by mitogenic stimulation and is required for cell proliferation 6, terminal differentiation, and germinal center (GC) formation 7, 8. Accordingly, deregulation of Myc has a major impact on human health. A large number of human cancers show enhanced expression of one of the three myc genes mediated by various mechanisms that include rearrangements, mutations, or alterations of the signaling pathways that control their expression 9, 10, 11. In Burkitt's lymphoma, a B‐cell lymphoma, c‐Myc is translocated to one of the three immunoglobulin loci and is overexpressed by regulatory elements of these loci 12, 13.
Myc proteins contain a basic region/helix‐loop‐helix/leucine zipper (bHLHZip) domain that mediates DNA binding and heterodimerization with the bHLHZip protein Max 14. To activate or repress target genes, c‐Myc/Max heterodimers bind to conserved DNA sequences called E‐boxes 15, 16, 17. Max can also heterodimerize with another group of bHLHZ proteins, the MXD family and MGA, which act as tumor suppressors and generally antagonize Myc functions 18, 19. Thus, Max has a central role in modulating the complex Myc protein network. Much of the scientific literature assumes that c‐Myc function relies on its ability to heterodimerize with Max, although several reports have shown that c‐Myc can perform some functions in its absence (reviewed in Ref. 20). For instance, c‐Myc has been shown to induce transcription from a reporter gene containing Myc/Max binding sites in Max‐deficient PC‐12 pheochromocytoma cells 21. Furthermore, c‐Myc‐induced apoptosis 22 or inhibition of Ras‐mediated cell differentiation 23 is Max‐independent in this cell line. A study of Max mutations in patients with hereditary pheochromocytoma, a rare neural crest tumor, suggest that loss of Max correlates with metastatic potential 24. Max inactivation is also observed in small cell lung carcinoma and is mutually exclusive with alterations in c‐Myc 25. Some in vitro studies point to the possibility of Max‐independent functions of c‐Myc in embryonic stem cells 26 and fibroblasts 27. Finally, in Drosophila, a myc mutant lacking the Max‐interaction domain retained partial activity 28. Interestingly, the onset of B lymphomas in eμ‐myc transgenic mice is attenuated by the overexpression of Max 29. Despite all these data, there are no definitive in vivo studies examining Myc/Max functional interrelationships, likely due in part to the embryonic lethality associated with germline deletions of Max 30. In this report, we examined the in vivo contribution of Max to c‐Myc function in B lymphocyte differentiation and in specific B‐cell functions. We observed that Max has an inhibitory effect in the absence of c‐Myc. However, the absence of both factors did not prevent the initiation of relevant biological functions in primary B lymphocytes.
Results and Discussion
Generation of Max and c‐Myc/Max conditional KO mice
To study Max function in B lymphocytes, we generated mice homozygous for the max flox conditional allele (max fl/fl mice) 31 and bred them to either mb1‐cre “knock‐in” mice 32 or cd19‐cre mice 33, to delete max in developing and mature B lymphocytes, respectively. Cre recombinase deletes exons 4 and 5 in max, which contain the HLHZip domain and the 3′untranslated region (3′UTR; Fig 1A). To monitor and isolate those cells with max deletion, we crossed the offspring with rosa26 gfp/gfp reporter mice 34 to generate homozygous max fl/fl ;mb1 cre/+ ;rosa26 gfp/gfp (MaxKO‐mb1), max fl/fl ;cd19 cre/+ ;rosa26 gfp/gfp (MaxKO‐cd19) and control mice. Flow cytometry analysis of green fluorescent protein (GFP) expression allowed the rapid identification and isolation of B lymphocytes that had deleted the max gene (Fig EV1A and B, and 35). Conditional double knock‐out c‐myc fl/fl ;max fl/fl ;mb1 cre/+ ;rosa26 gfp/gfp (DKO‐mb1) or c‐myc fl/fl ;max fl/fl ;cd19 cre/+ ;rosa26 gfp/gfp (DKO‐cd19) mice were generated by breeding MaxKO‐mb1 and MaxKO‐cd19 mice to myc fl/fl ;mb1 cre/+ ;rosa26 gfp/gfp (MycKO‐mb1) 5 or to c‐myc fl/fl ;cd19 cre/+ ;rosa26 gfp/gfp (MycKO‐cd19) mice, respectively 6, 35.
Figure 1. c‐Myc/Max in B lymphocyte differentiation.
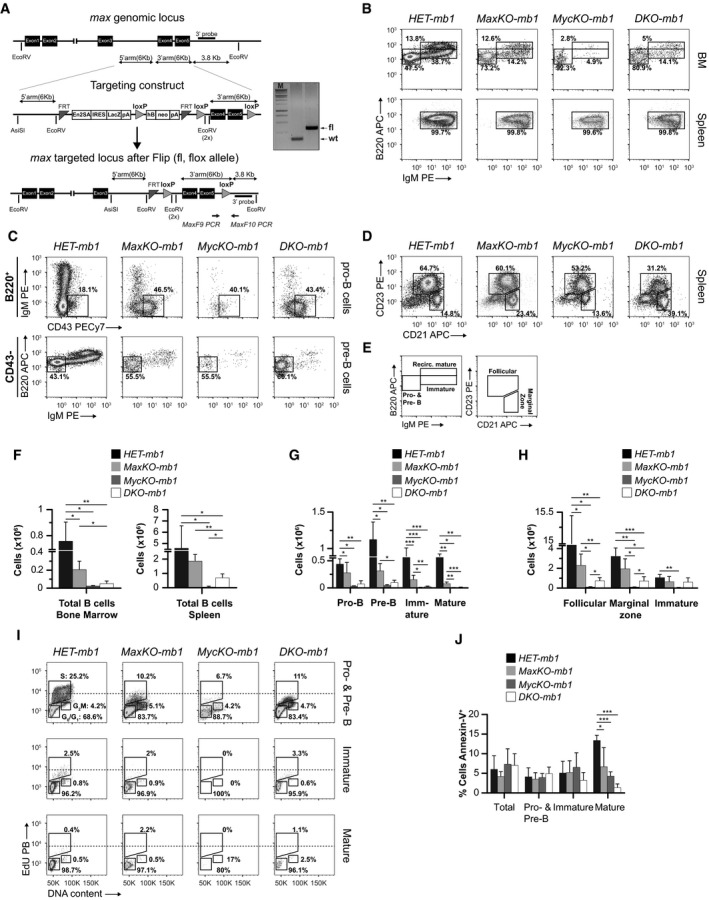
-
AGene targeting strategy. Left: max genomic locus, targeting construct and max targeted locus. In vitro Flipase (Flip) deletion of the selectable markers lacZ and neo results in a max targeted locus with exons 4 and 5 (coding for bHLHLZip domain) flanked by two loxP sites (fl, flox allele). Arrows represent PCR primers used for genotyping MaxF9 and MaxF10. Right: Genomic PCR analysis of tail DNA from F1 mice. PCR products of max wt (430 bp) and max flox (526 bp) alleles. M, 1 kb ladder.
-
B–DFlow cytometry analyses of B‐cell populations (GFP +) in bone marrow (BM) and spleen. Single‐cell suspensions were prepared from BM (B, C) or spleen (B, D) of MaxKO‐mb1, MycKO‐mb1, DKO‐mb1, or heterozygous HET‐mb1 control mice, stained with antibodies against the indicated markers, and analyzed by flow cytometry. For BM analysis, cells were isolated from femora and tibiae. Pro‐B cells (Ly‐6c− CD49b−B220+IgM− CD43+), pre‐B cells (Ly‐6c− CD49b−B220+IgM− CD43−), immature B cells (Ly‐6c− CD49b−B220loIgM+ CD43−), and mature B cells (Ly‐6c− CD49b−B220hiIgM+ CD43−), spleen follicular B cells (B220+ CD23+ CD21int), marginal zone B cells (B220+ CD23− CD21hi), and immature (newly formed/transitional stage 1) B cells (B220+ CD23− CD21lo). To identify KO cells (GFP +), all the plots show gated GFP + B lymphocytes.
-
ESchematic representation of the gated populations in the flow cytometry plots in (B and D).
-
F–HAbsolute numbers of B lymphocytes in bone marrow (F, G) and spleen (F, H) from MaxKO‐mb1 (n = 6), MycKO‐mb‐1 (n = 3), DKO‐mb1 (n = 5), and control HET‐mb1 (n = 4) mice. Columns represent mean values and error bars standard deviations. The different B‐cell populations were identified by flow cytometry as in (B–D). Experiment representative of at least three independent experiments.
-
ICell cycle analysis by flow cytometry of sorted GFP + pre‐ and pro‐B (B220+IgM−), immature (B220loIgM+), and mature (B220hiIgM+) bone marrow B cells from MaxKO‐mb1, MycKO‐mb1, DKO‐mb1, and HET‐mb1 control mice. Mice (n = 5) were injected (i.p.) with 200 μg of EdU and analyzed 4 h later. Single‐cell suspensions were prepared, stained, and sorted. Sorted cells were subsequently stained with propidium iodide for DNA content. Experiment representative of at least three independent experiments. Dotted line is shown for EdU MFI comparison.
-
JPercentage of annexin‐V+ GFP + cells in the BM of MaxKO‐mb1 (n = 5), MycKO‐mb1 (n = 3), DKO‐mb1 (n = 3), and control (n = 4) mice. Pro‐B, pre‐B, immature, and mature B lymphocyte subpopulations were identified by flow cytometry as indicated in (E). Columns represent mean values and error bars standard deviations. Experiment representative of at least three independent experiments.
Figure EV1. Efficient deletion of c‐myc and max in B lymphocytes from bone marrow and spleen of conditional mice.
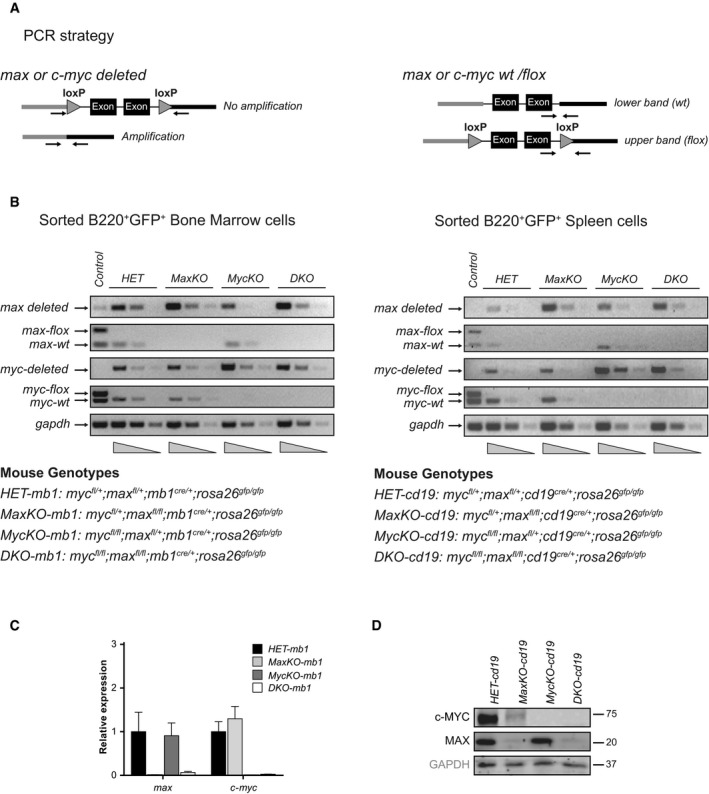
- PCR strategy to amplify flox, deleted, and wt alleles of c‐myc or max. For c‐myc, exons 2 and 3 were flanked by loxP sites and, for max, exons 4 and 5.
- Genomic PCR analysis of wt, deleted, and flox alleles of c‐myc and max from sorted B220+ GFP + bone marrow or spleen lymphocytes of mb1‐cre‐ or cd19‐cre‐bearing mice, respectively. gapdh was used for normalization. Experiment representative of three independent experiments.
- RNA expression of max and c‐myc genes from sorted bone marrow GFP +B220+ IgM+ cells of HET‐mb1, MaxKO‐mb1, MycKO‐mb, and DKO‐mb1 mice. n = 3. Error bars are standard deviations.
- Western blotting of c‐Myc and Max in sorted B220+IgM+ GFP + lymphocytes. Spleen cells from the indicated mice were sorted and activated with LPS and IL‐4 for 3 days before analysis. Experiment representative of three independent experiments.
Source data are available online for this figure.
Genomic PCR on sorted GFP+ B lymphocytes from BM or spleen of mice carrying mb1‐cre or cd19‐cre showed efficient deletion of c‐myc and max (Fig EV1A and B). Quantitative PCR analysis of c‐myc and max expression in these cells was consistent with the genomic status of the different alleles (Fig EV1C). Moreover, Western blotting confirmed the absence of Max protein in sorted mature GFP+ B lymphocytes from spleen of MaxKO‐cd19 and DKO‐cd19 mice; c‐Myc was also absent in B cells from spleen in MycKO‐cd19 and DKO‐cd19 mice (Fig EV1D). Surprisingly, the same analysis revealed a substantial decrease of c‐Myc expression in sorted GFP+ MaxKO B lymphocytes (Fig EV1D). This result suggests an indirect or direct regulatory function of Max on Myc stability or mRNA regulation.
MaxKO B lymphocytes differentiate into mature B cells
c‐Myc is expressed at the pro‐ to pre‐B‐cell stage in early B lymphocyte differentiation 36. Our previous results showed that c‐Myc‐deficient B lymphocytes fail to differentiate beyond the pre‐B‐cell stage in MycKO‐mb1 mice (5 and Fig 1B–H). To determine whether B lymphocyte differentiation was affected upon max or c‐myc/max deletion, we used flow cytometry to analyze the different B‐cell subpopulations in the BM and the spleen of MaxKO‐mb1, MycKO‐mb1, DKO‐mb1 and c‐myc fl/+ ;max fl/+ ;mb1 cre/+ ;rosa26 gfp/gfp heterozygous control mice (HET‐mb1). Max‐, c‐myc‐, and c‐myc/max‐deleted B lymphocytes (GFP+) were distinguished from non‐deleted (floxed, GFP−) cells based on their GFP expression. MaxKO and DKO B lymphocytes (GFP+) were capable of differentiating into pro‐B, pre‐B, immature, and mature B lymphocytes in the BM of MaxKO‐mb1 and DKO‐mb1 mice (Fig 1B–C), which was in contrast to the developmental block observed in MycKO‐mb1 mice (5 and Fig 1B–E). However, the absolute number of B cells in BM and spleen was significantly lower in MaxKO‐mb1 (BM 2.0 × 105, spleen 1.8 × 106) MycKO‐mb1 (BM 0.2 × 105, spleen 0.5 × 104) and DKO‐mb1 (BM 0.5 × 104, spleen 0.6 × 106) mice than in controls (BM 0.7 × 106, spleen 4.5 × 106; Fig 1F). As previously reported 5, the presence of mature B lymphocytes in the spleen of MycKO‐mb1 is likely due to cells that have escaped Cre‐mediated deletion. Accordingly, the absolute numbers of differentiating B lymphocytes were significantly lower in MaxKO‐mb1, MycKO‐mb1, and DKO‐mb1 mice than in control mice (Fig 1G). We also detected a significant decrease in the absolute numbers of follicular (HET 9.4 × 106 versus MaxKO 2.3 × 106, MycKO 6.3 × 103, DKO 6.6 × 104), immature (HET 1.0 × 106 versus MaxKO 6.6 × 104, MycKO 3.7 × 103, DKO 6.1 × 104), and marginal zone B cells (HET 3.2 × 106 versus MaxKO 1.9 × 106, MycKO 5.8 × 103, DKO 7.2 × 104) in the spleen of MaxKO‐mb1, MycKO‐mb1, and DKO‐mb1 mice as compared with control HET‐mb1 mice (Fig 1H).
To rule out the possibility that the presence of immature (B220loIgM+GFP+) and mature (B220hiIgM+GFP+) B cells in the BM of mutant mice was due to B lymphocytes that had escaped Cre‐mediated deletion, we performed differentiation assays in vitro with sorted B220+IgM−GFP+ cells from the BM of MaxKO‐mb1 and control HET‐mb1 mice. We observed that MaxKO B lymphocytes generated IgM+ cells after 4 days in culture in the presence of interleukin (IL)‐7 (Fig EV2). These results suggest that Max‐deficient B lymphocytes, unlike equivalent c‐Myc‐deficient cells 5, can progress through the differentiation program, albeit less efficiently than normal B cells.
Figure EV2. Max‐deficient cells generate B220+IgM+ lymphocytes in vitro .
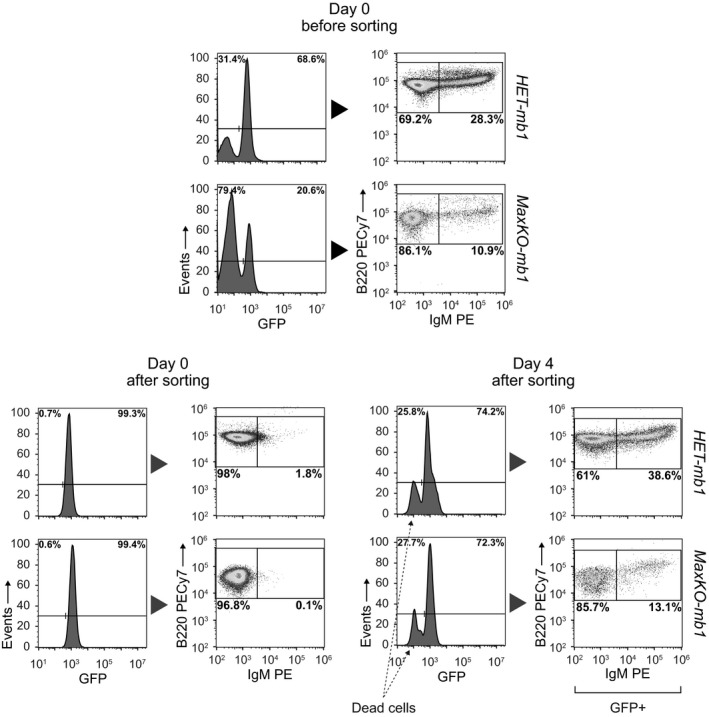
Sorted GFP +B220+IgM− BM cells from HET‐mb1 and MaxKO‐mb1 mice were cultured with IL‐7, rSCF, and rFlt3L and analyzed by flow cytometry 4 days later. For BM analysis, cells were isolated from femora and tibiae and sorted by flow cytometry (GFP +cells, purity > 99%) before culture. After 4 days, the generation of GFP +B220+IgM+ cells was analyzed. A GFP − population corresponding to dead cells is indicated. Experiment representative of at least three independent experiments.
During early differentiation in the BM, B lymphocytes undergo V(D)J recombination and proliferative expansion, mainly at the pro‐ to pre‐B‐cell stage 36, 37. To test whether proliferation or apoptosis could account for the observed differences in the absolute numbers, we performed EdU incorporation assays in vivo and analyzed annexin‐V staining in B lymphocytes from the BM of MaxKO‐mb1, MycKO‐mb1, DKO‐mb1, and control HET‐mb1 mice. Analysis of EdU incorporation in sorted GFP+ B lymphocytes revealed a decrease in the percentage of cells in S phase at the pro‐ and pre‐B‐cell stages and a decrease in EdU fluorescence intensity in MaxKO, MycKO, and DKO cells as compared with control cells [S‐phase mean fluorescence intensity (MFI): 19,886 HET versus 5,172 MaxKO, 4,017 MycKO, 4,416 DKO (Fig 1I)]. Interestingly, MaxKO and DKO B lymphocytes retained some capacity to proliferate when compared with c‐MycKO B cells. It is known that c‐MycKO mature B cells are more resistant to apoptosis than normal cells 35. Consistent with this, the level of apoptosis was significantly lower in mature B lymphocytes from mutant mice than from control mice (Fig 1J). Overall, these data show that a defect in proliferation could contribute to the decrease in absolute numbers of B lymphocytes in the BM and spleen of MaxKO‐mb1, MycKO‐mb1 5, and DKO‐mb1 mice.
B‐cell function in MaxKO and DKO B lymphocytes
Previous results have shown that c‐Myc plays a major role in terminal B lymphocyte differentiation 7, 8. Specifically, c‐Myc‐deficient B cells cannot generate B220loCD138+ cells [antibody‐secreting cells (ASCs)] or undergo immunoglobulin class switch recombination (CSR) in vitro 8. To test the functional interplay between Max and c‐Myc in these processes, we used cd19‐cre mice, which provide higher absolute numbers of deleted mature B lymphocytes in the spleen for analysis 8 than mb1‐cre mice 5.
To generate B220lo CD138+ cells (plasmablasts) and induce CSR to the IgG1 isotype, we sorted B220+GFP+ mature B lymphocytes from the spleen of MaxKO‐cd19, MycKO‐cd19, DKO‐cd19, and heterozygous c‐myc fl/+ ;max fl/+ ;cd19 cre/+ ;rosa26 gfp/gfp (HET‐cd19) mice, and activated them with lipopolysaccharide (LPS) and IL‐4. We observed that the generation of B220loCD138+ cells from MaxKO (5.5 × 103), DKO (3.2 × 103), and c‐MycKO (1.1 × 103) B lymphocytes was lower than from control cells (4.8 × 104; Fig 2A and B, and 8). We analyzed the presence of B220+IgG1+GFP+ cells in the same cultures by flow cytometry, finding that MaxKO B lymphocytes generated significantly less IgG1+ cells than control cells (19.3 × 103 versus 22.1 × 104). Further, DKO B cells (5.4 × 103) presented a more dramatic defect in CSR, which was similar to that of c‐Myc‐deficient B cells (4.5 × 103; Fig 2C and D, and 8). The levels of apoptosis and the activation marker CD69 in these cells were similar for all the genotypes (Fig 2E–H). Based on these data, we conclude that both c‐Myc and Max are required to achieve normal levels of plasmablasts and CSR. However, in the absence of Max or c‐Myc/Max, B cells in cd19‐cre mice can still generate plasmablasts and perform CSR.
Figure 2. Role of c‐Myc/Max in B lymphocyte function and the immune response.
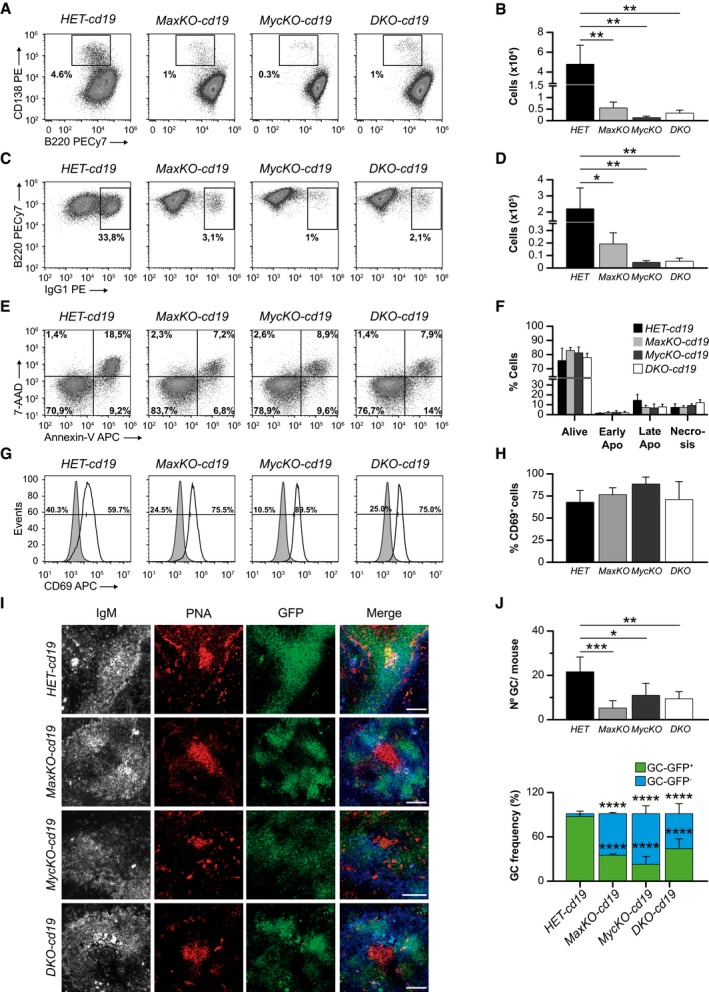
-
A–DFlow cytometry analysis and absolute numbers of B220lo CD138+ (plasmablasts) (A, B) and B220+IgG1+ switched B cells (C, D) from HET‐cd19 (n = 6), MaxKO‐cd19 (n = 8), MycKO‐cd19 (n = 4), and DKO‐cd19 (n = 5) mice. Spleen B220+ GFP + B lymphocytes were sorted and activated in vitro with LPS plus IL‐4 for 72 h. Cells were stained with monoclonal antibodies against the indicated markers and analyzed by flow cytometry. Columns represent mean values and error bars standard deviations. Experiment representative of at least three independent experiments.
-
ESpleen B220+ GFP + B lymphocytes from (A) or (C) were stained with 7AAD and annexin‐V to monitor apoptosis. 7‐AAD − annexin‐v− (alive), 7‐AAD − annexin‐v+ (early apoptotic), 7‐AAD + annexin‐v+ (late apoptotic), 7‐AAD + annexin‐v− (necrosis).
-
FRelative numbers of live, early apoptotic, late apoptotic, and necrotic cells from (E). Error bars are standard deviations (n = 4).
-
GCD69 surface expression in purified B220+ GFP + mature spleen cells. Sorted B cells were activated in vitro with LPS plus IL‐4 for 72 h, stained and analyzed by flow cytometry. The solid gray peak represents the expression levels of CD69 marker in non‐activated mature B lymphocytes at Day 0.
-
HRelative numbers of CD69+ B lymphocytes in the cultures from (G) (n = 4). Error bars represent standard deviations. Experiment representative of at least three independent experiments.
-
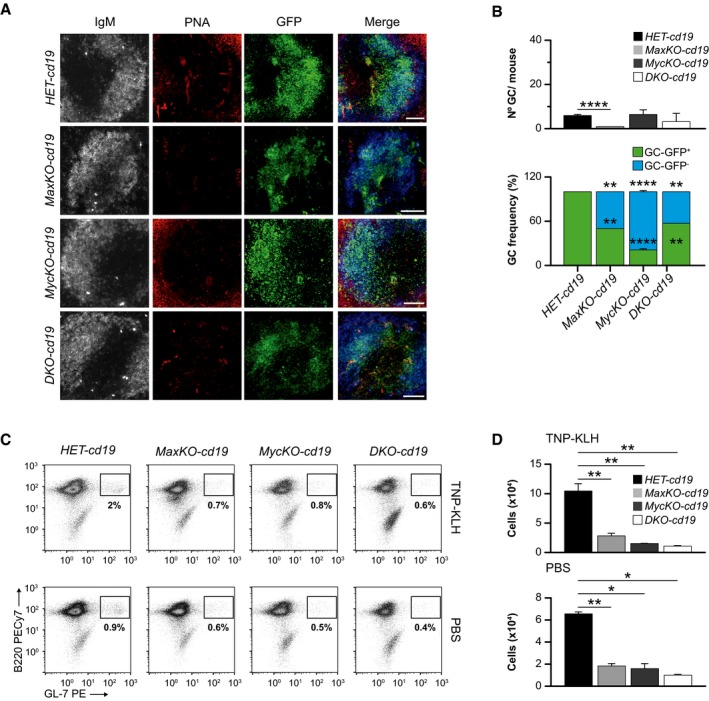
IAnalysis of germinal center (GC) formation in the spleen of MaxKO‐cd19, MycKO‐cd19,DKO‐cd19, and heterozygous control mice immunized with TNP‐KLH. Representative images of frozen spleen sections stained with IgM (gray/blue), PNA (GC marker; red), and GFP (Max‐, c‐Myc‐ or c‐Myc/Max‐deficient B cells; green). Scale bar, 80 μm.
-
JUpper, number of GCs per mouse in spleen sections of immunized MaxKO‐cd19 (n = 3 mice, n = 186 follicles analyzed), MycKO‐cd19 (n = 3 mice, n = 178 follicles analyzed), DKO‐cd19 (n = 3 mice, n = 191 follicles analyzed), and heterozygous control mice (n = 3 mice, n = 203 follicles analyzed). Lower, frequencies of GC (PNA +) GFP + (deleted cells) or GFP − (non‐deleted cells) in immunized MaxKO‐cd19 (n = 13 GCs), MycKO‐cd19 (n = 21 GCs), DKO‐cd19 (n = 27 GCs), and heterozygous control (n = 62 GCs) mice. Analysis was performed at 13 days post‐immunization. Error bars are standard deviations.
Max and c‐Myc are required for normal generation of germinal centers
The generation of GCs is a hallmark of a competent and robust T‐dependent immune response. c‐Myc has been previously shown to be necessary for the generation of these structures 7, 8. To test whether Max‐deficient B lymphocytes were capable of generating GCs, we immunized homozygous MaxKO‐cd19, MycKO‐cd19, DKO‐cd19, and HET‐cd19 control mice with the T‐dependent antigen 2,4,6‐trinitrophenyl hapten‐keyhole limpet hemocyanin (TNP‐KLH). After 12 days, we observed the presence of GCs (as indicated by peanut agglutinin staining, PNA) in the spleens of all immunized mice (Fig 2I), whereas mutant and control mice injected with phosphate‐buffered saline (PBS) did not generate GCs (Fig EV3A and B). We observed that the cell density of B cells (GFP+) in the spleen GCs in immunized MaxKO‐cd19, MycKO‐cd19, and DKO‐cd19 mice was lower than in equivalent HET‐cd19 control mice (Fig 2I). The number of GL‐7+ B cells was also decreased in all mutant mice (Fig EV3C and D). Moreover, the number of GCs per mouse in spleen sections of MaxKO‐cd19, MycKO‐cd19, and DKO‐cd19 mice was lower than in control mice (Fig 2J). An analysis of the presence of GC GFP+ (deleted) and GFP− (non‐deleted) cells showed that the majority of the cells present in the GCs of homozygous MaxKO‐cd19, MycKO‐cd19, and DKO‐cd19 mice were non‐deleted B cells (GFP−; Fig 2J). These results lead us to conclude that both Max and Myc are necessary for normal generation of the GC.
Figure EV3. Analysis of GC formation in the spleen of MaxKO‐cd19, MycKO‐cd19,DKO‐cd19, and heterozygous control mice upon TNP‐KLH and PBS immunization.
- Representative images of frozen spleen sections stained for IgM (gray/blue), PNA (GC marker; red), and GFP (deleted B cells; green) of mice immunized with PBS. Analysis was performed at 12 days post‐immunization. Scale bar, 80 μm.
- Upper, number of GCs per mouse in spleen sections of PBS‐immunized MaxKO‐cd19 (n = 2 mice, n = 84 follicles analyzed), MycKO‐cd19 (n = 2 mice, n = 163 follicles analyzed), DKO‐cd19 (n = 2 mice, n = 157 follicles analyzed), and heterozygous control mice (n = 2 mice, n = 182 follicles analyzed). Lower, frequencies of GC (PNA +) GFP + (deleted cells) or GFP − (non‐deleted cells) in PBS‐immunized MaxKO‐cd19 (n = 2 GCs), MycKO‐cd19 (n = 14 GCs), DKO‐cd19 (n = 7 GCs), and heterozygous control (n = 13 GCs) mice at 13 days post‐immunization. Error bars are standard deviations.
- Flow cytometry analyses of GC B lymphocytes (GFP +B220+ GL‐7+) in the spleen of TNP‐KLH or PBS‐immunized mice. Single‐cell suspensions from MaxKO‐cd19, MycKO‐cd19,DKO‐cd19, and heterozygous control mice were prepared, stained, and analyzed (n = 3).
- Absolute numbers of GC B cells (GFP +B220+ GL‐7+) in MaxKO‐cd19, MycKO‐cd19,DKO‐cd19, and heterozygous control mice from (C). Error bars are standard deviations.
MaxKO and DKO mature B lymphocytes undergo few cell divisions before ceasing proliferation
c‐Myc has been widely shown to control cell proliferation mainly through the regulation of the G1‐S transition of the cell cycle 38. c‐Myc‐deficient B lymphocytes cannot proliferate upon in vitro activation 6, 8. To evaluate the contribution of Max and c‐Myc to this process, we examined the capacity of MaxKO, c‐MycKO, and c‐Myc/Max DKO mature B lymphocytes to proliferate in vitro. With this aim, sorted B220+GFP+ cells from the spleens of MaxKO‐cd19, MycKO‐cd19, DKO‐cd19, and HET‐cd19 control mice were activated with LPS and IL‐4, and cell division was monitored with a cell tracking dye. Unexpectedly, MaxKO and DKO mature B lymphocytes showed some capacity to undergo cell division (2–4 cell divisions), whereas c‐MycKO cells did not divide (Fig 3A), as previously shown 8. Accordingly, we observed a similar absolute number of MaxKO (14.8 × 104) and DKO (15.5 × 104) B cells in 3‐day cultures, although no increase in B‐cell number was detected in c‐MycKO cultures (Fig 3B and 8). Interestingly, the smaller B‐cell size previously observed in c‐MycKO B lymphocytes 6 was compensated slightly in DKO B cells and was not seen in MaxKO cells (Fig 3C and D). These results show that c‐Myc and Max are both required for normal levels of cell proliferation; however, in the absence of c‐Myc, Max plays a strong inhibitory role, either alone or in collaboration with other proteins of the Myc network 18, as observed in c‐MycKO B cells. The capacity of B lymphocytes to undergo a limited number of cell divisions in the absence of Max or c‐Myc/Max suggests that c‐Myc itself is not capable of regulating proliferation, supporting the notion that c‐Myc requires Max for its normal proliferative function. In addition, this capacity could explain the better competence of MaxKO and DKO B cells to differentiate (Fig 1), in contrast to the early developmental block observed in c‐MycKO B lymphocytes 5.
Figure 3. Distinct roles of c‐Myc and Max in the proliferation in mature B lymphocytes.
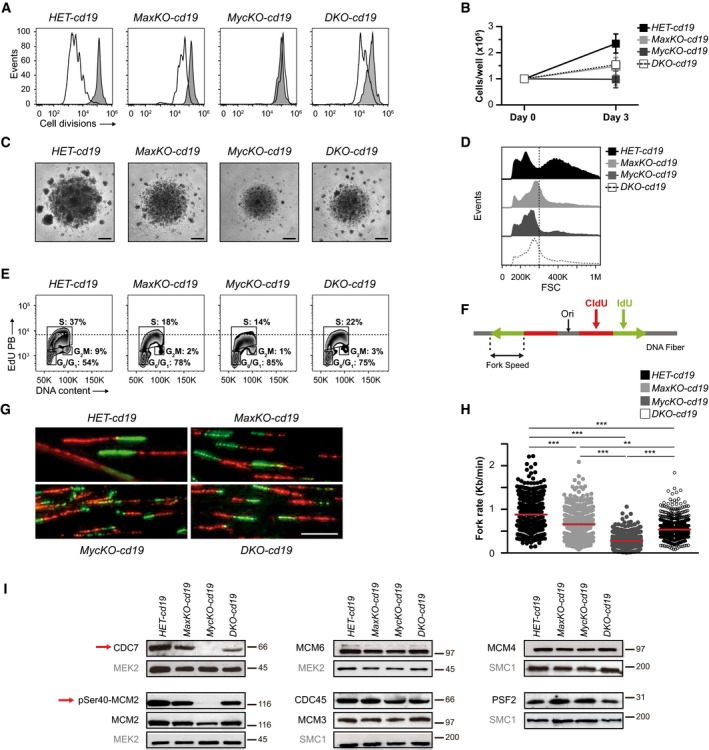
-
ANumber of cell divisions in activated mature B lymphocytes. Sorted B220+ GFP + spleen cells from HET‐cd19 (n = 5), MaxKO‐cd19 (n = 5), MycKO‐cd19 (n = 3), and DKO‐cd19 (n = 3) mice were stained with a cell tracer, activated in vitro with LPS plus IL‐4 for 72 h, and analyzed by flow cytometry. Experiment representative of at least three independent experiments.
-
BCell counts from the cultures in (A) (n = 5). Error bars are standard deviations.
-
CPhotographs of B‐cell cultures from (A). Scale bar, 500 μm. 4× magnification.
-
DForward scatter (FSC) histograms of activated B lymphocytes from (A). An arbitrary dashed line is shown for comparison.
-
ECell cycle analysis of sorted B220+ GFP + B lymphocytes activated in vitro. Cells were activated as in A and EdU was added 4 h before harvesting. Cells were stained with propidium iodide to measure DNA content. Experiment representative of at least three independent experiments. An arbitrary dashed line shows the difference of EdU MFI. S‐phase mean fluorescence intensity (MFI): 7,871 HET, 6,512 MaxKO, 5,860 MycKO, 6,420 DKO.
-
F, GRepresentation of labeling with nucleotide analogues CldU (red) and IdU (green) and photographs of DNA fibers from sorted B220+ GFP + B lymphocytes activated as in (A) for 48 h. Scale bar, 10 μm.
-
HFork rate of DNA fibers from primary B lymphocytes sorted and activated as in (G). Data are pooled from three replicate experiments. Horizontal bars represent median values. 150–250 forks were measured per condition. ***, P < 0.001; **, P < 0.01. Statistical significance was assessed using non‐parametric, one‐way ANOVA Kruskal–Wallis test followed by Dunn's post‐test.
-
IWestern blotting of DNA replication proteins in activated B lymphocytes from (F, G). Sorted B220+ GFP + spleen cells were activated in vitro with LPS plus IL‐4 for 72 h. MEK2, SMC1 are shown as loading controls. Red arrows indicate the absence of Cdc7 and pSer40‐MCM2 in MycKO B cells. The blots shown are representative of three independent experiments.
Source data are available online for this figure.
To better understand the proliferative defect observed in the mutant cells, we monitored the cell cycle in vitro by EdU incorporation and propidium iodide staining. We observed that the percentage of B lymphocytes in S and G2/M phases was appreciably lower in MaxKO, c‐MycKO, and DKO cells than in control cells (Fig 3E). This effect was less dramatic than in pro‐and pre‐B cells (Fig 1I), probably due to the prominent proliferative expansion at this stage in vivo and the different cell contexts (in vitro versus in vivo). The observed differences in EdU incorporation and the cell cycle between mutant and control cells prompted us to next investigate the replication machinery of these cells.
The role of Max in the regulation of DNA replication
Replication origins are frequently located at or around gene promoters, underscoring the link between replication and transcription 39. c‐Myc influences DNA replication through transcription‐dependent and transcription‐independent mechanisms. In addition to regulating the activity of cyclin‐dependent kinases at the G1/S transition, c‐Myc may create a chromatin environment that favors the assembly of pre‐replication complexes (pre‐RCs) at origins. Indeed, c‐Myc has been reported to physically interact with the pre‐RC proteins CDC6 and MCM and to promote the association of CDC45 to chromatin, a key step in the activation of the CMG (Cdc45‐MCM‐GINS) helicase 40.
We assessed the participation of c‐Myc and Max in DNA replication in activated mature B lymphocytes using stretched DNA fibers. Fork speed was strikingly lower in c‐MycKO cells (median 0.28 kb/min) than in control cells (0.88 bb/min) and was decreased to a lesser extent in MaxKO and DKO cells (0.66 and 0.54 kb/min, respectively; Fig 3F–H).
No significant differences were found in the amount of MCM3, MCM4, MCM6, CDC45, and PSF2 proteins between control, MaxKO‐, c‐MycKO‐, and DKO‐activated B lymphocytes (Fig 3I and Appendix Fig S1). However, we detected a substantial decrease in the level of CDC7 in c‐Myc‐deficient B lymphocytes (Fig 3I). CDC7 is the catalytic component of the Dbf4‐dependent kinase (DDK), responsible for MCM phosphorylation and activation. Indeed, DDK‐mediated MCM2 phosphorylation at Ser40 was absent in c‐MycKO B cells and the total levels of MCM2 protein were also low (Fig 3I and Appendix Fig S1). The loss of CDC7 provides a plausible explanation for the severe decrease in DNA replication in c‐MycKO mature B lymphocytes. RNA‐Seq did not reveal differences in cdc7 expression between c‐MycKO B cells and control cells (Dataset EV1), despite the presence of an E‐box located at 328 nt from the transcription start site of the cdc7 gene 41. Thus, the dramatic decrease in CDC7 protein might be attributed to post‐transcriptional mechanisms. The unexpected finding of reduced levels of c‐Myc in MaxKO B cells provides a new context to explain this result. We speculate that, in the absence of c‐Myc and Max, B lymphocytes are capable of initiating different functions, such as the regulation of CDC7; however, both factors are required to fine‐tune this function. We hypothesize that, in the absence of c‐Myc, Max alone or with other members of the Myc network downregulates CDC7 levels.
Gene expression profiles in MaxKO, c‐MycKO, and DKO B lymphocytes
In an effort to determine the genes regulated by c‐Myc, Max, or both, we performed transcriptome profiling in mature B lymphocytes from MaxKO‐cd19, MycKO‐cd19, DKO‐cd19, and control HET‐cd19 mice. Accordingly, RNA‐Seq was performed in sorted primary B lymphocytes (B220+GFP+) isolated from the spleen of these mice and activated with LPS and IL‐4 (Fig 4). We identified 2,604, 3,313, and 2,559 differentially expressed genes (DEGs; P < 0.01) in B cells from MaxKO‐cd19, MycKO‐cd19, and DKO‐cd19 mice, respectively, when independently compared with HET‐cd19 B cells (Fig 4A and Dataset EV1). Venn diagram analysis revealed 1,921 genes that were simultaneously deregulated in MaxKO, c‐MycKO, and DKO B lymphocytes (Fig 4B). We observed a similar gene expression profile of previously reported genes 8 affected in MycKO‐cd19 mice (Fig EV4A). Hierarchical clustering analysis of all DEGs for the three experimental conditions showed that MaxKO and DKO B lymphocytes clustered closer to each other than to c‐MycKO (Fig 4C). To enhance the biological relevance, we calculated the number of overlapping DEGs with ≥ 1.5‐ and ≤ −1.5‐fold change among the 1,921 genes. In total, 1,445 DEGs were common for all three conditions (797 up‐, 648 downregulated), suggesting a high degree of overlap between c‐Myc and Max regulatory networks (Fig 4D and Dataset EV2).
Figure 4. RNA‐sequencing analyses in mature B lymphocytes.
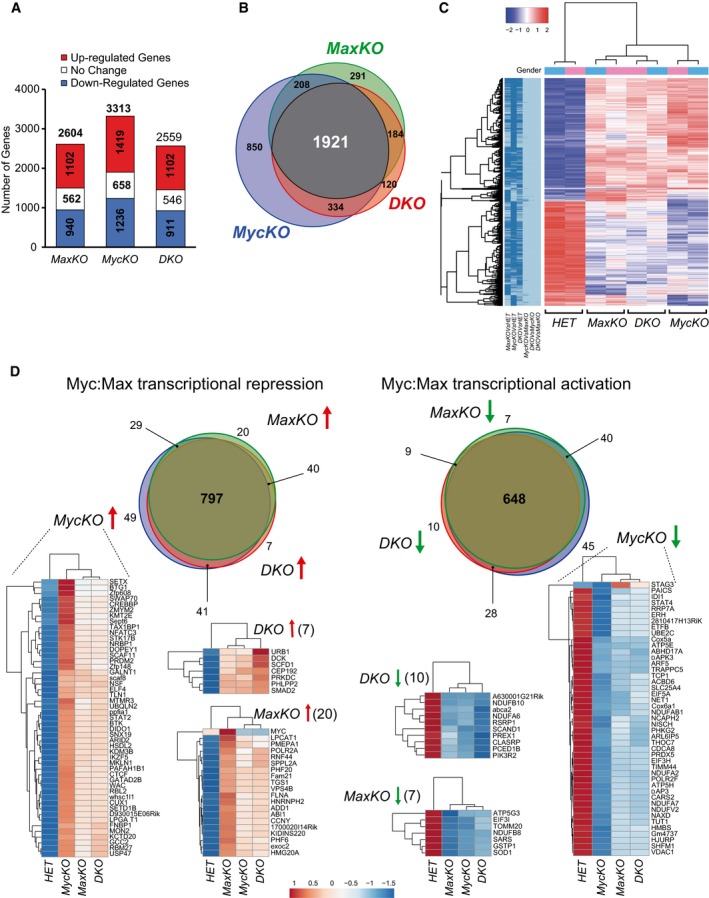
- Bar plot showing the number of DEGs (adj. P‐value < 0.01, FC +1.5 in red, 1.5 < FC > −1.5 in white, FC < −1.5 in blue). Sorted B220+ GFP + spleen B cells from MaxKO‐cd19, MycKO‐cd19, DKO‐cd19, and control HET‐cd19 mice were activated with LPS plus IL‐4 for 72 h and analyzed. Two samples for each condition were used.
- Venn diagrams showing the number of common DEGs in all conditions.
- Hierarchical clustering heatmap of DEGs in any comparison (two samples per condition). Dark blue in left panel indicates significance for that contrast column. Color code: Red represents upregulation; white, unchanged; and blue, downregulation of expression. Gender: color code for genetic condition. Pink are female and blue color male mice. Colour key (−2 to 2): raw Z‐score.
- Venn diagrams of up‐ (left) and downregulated genes (right) for all three conditions (±1.5‐FC and P < 0.01). Heatmaps of genes only deregulated in MaxKO‐cd19, MycKO‐cd19, or DKO‐cd19 mice are shown. Color code is the same as (C).
Figure EV4. RNA‐sequencing analyses.
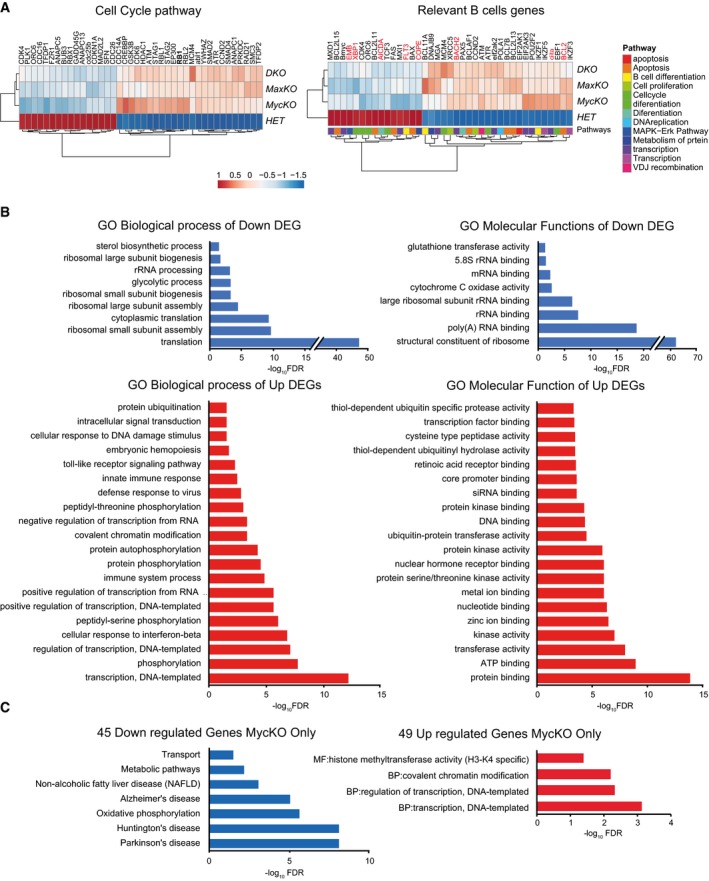
- Heatmaps of DEGs (adj. P‐value < 0.01) of cell cycle pathways and genes involved in B lymphocyte differentiation. Genes highlighted in red were previously tested by qPCR 22.
- GO classification of biological process and molecular functions of downregulated (blue) and upregulated (red) genes for all three conditions (±1.5‐FC and P < 0.01). The 20 most significantly (FDR < 0.05, Dataset EV1) affected GO categories are represented.
- GO and pathways enrichment analysis of 45 downregulated and 49 upregulated genes in MycKO only indicated in Fig 4D. Statistically significant GO categories and KEGG pathways (FDR < 0.05) are shown.
We next carried out functional classification analyses of altered genes (±1.5‐fold and P < 0.01) using the DAVID Bioinformatics Resources 42 through classification into Gene Ontology (GO) categories [false discovery rate (FDR) ≤ 0.05], based on biological processes and molecular functions, as well as KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways 43 (Figs 5A and B, and EV4B, Dataset EV3). RNA‐Seq data were validated by qPCR analysis of randomly chosen up‐ or downregulated genes. Noteworthy, the same analyses did not reveal differences in cdc7 expression between c‐MycKO B cells and control cells (Fig 5C). We observed that downregulated genes in all three conditions were involved in several functional categories, with ribosome pathways, representing the largest groups with a high significance (FDR 2.72 × 10−81; Fig 5B). All of these genes were more affected in c‐MycKO B lymphocytes than in equivalent MaxKO or DKO cells. Other pathways, such as oxidative phosphorylation or metabolic pathways, were also identified in the pool of downregulated genes (Figs 5B and EV4B).
Figure 5. Major up‐ and downregulated pathways in mature B lymphocytes.
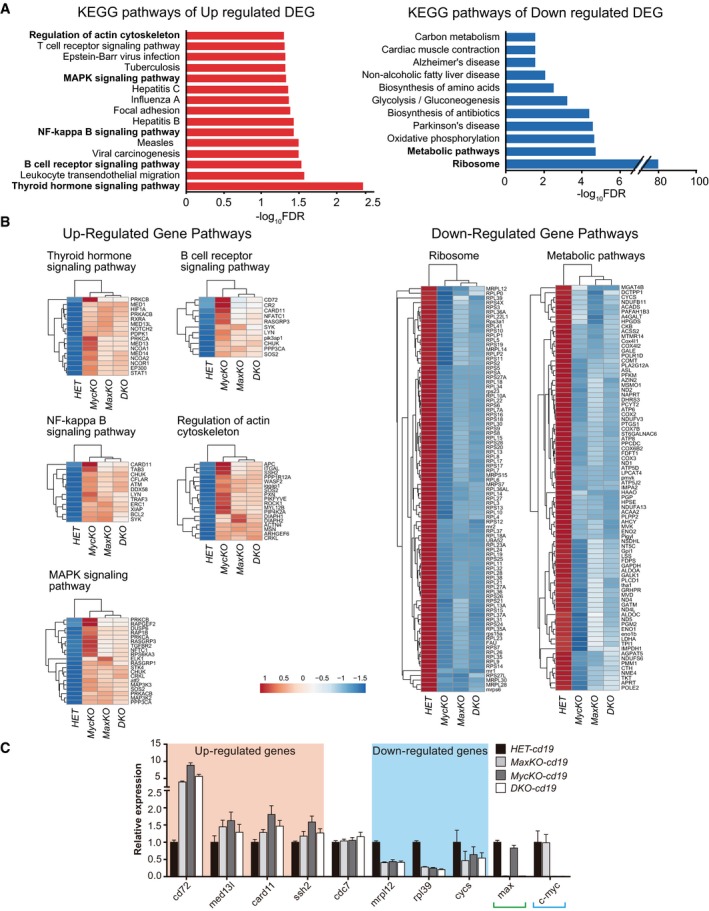
- Enrichment analysis of KEGG pathways (FDR < 0.05) of up‐ or downregulated genes common for all three conditions (±1.5‐FC and P < 0.01). Relevant pathways are labeled in bold.
- Heatmaps of genes involved in the relevant pathways indicated in (A). Color code: Red represents upregulation; white, unchanged; and blue, downregulation of expression.
- qPCR analysis of up‐ and downregulated genes from (B). Pooled RNA was prepared from sorted B220+ GFP + spleen B cells of MaxKO‐cd19 (n = 4), MycKO‐cd19 (n = 4), or DKO‐cd19 (n = 4) and control HET‐cd19 (n = 4) mice activated as in Fig 4A. Genes were randomly selected, and qPCR was performed using specific primers for cd72, med13l, card11, ssh2, cdc7, mrpl12, rpl39, and cysc. max and c‐myc were included as controls, and actin was used for normalization. Error bars are standard deviations.
Gene Ontology analysis of significantly upregulated DEGs revealed an enrichment of immune‐related and transcriptional processes (Fig EV4B). The major pathways for upregulated genes in all three conditions included regulation of actin cytoskeleton 44, B‐cell receptor 45, NF‐kB 46, and MAPK signaling 47, among others (Fig 5B). These data suggest that the absence of c‐Myc, Max, or both, in B cells results in the upregulation of genes involved in different signaling pathways. B‐cell receptor signaling 48 or LPS activation of Toll‐like receptor 4 49 induces the canonical NF‐κB pathway 46 and MAPK signaling 47. These pathways promote reorganization of the actin cytoskeleton 44 and upregulate the transcription of c‐myc, among other genes 48, 50. It is possible that mutant B cells are not capable of processing the activation signal and/or that the absence of c‐Myc/Max might prevent these factors from inducing a negative feedback loop to modulate the response, as seen in immature B cells 51. We postulate that the presence of Max only (c‐MycKO B cells) enhances this phenotype by occupying E‐boxes and preventing normal regulation of the activation signal. In addition, major defects in ribosome components and function likely contribute to hamper the ability of B lymphocytes to perform their biological functions.
We next compared the results of our gene analysis (Fig 5B) with previously reported c‐myc expression data in primary mouse B cells 52 and T lymphocytes 53 (Fig EV5). Despite the differences in cell type, genetic background, nature of stimulus, kinetics, and methodology, we found striking similarities in the upregulated and downregulated pathways between our data and those of c‐Myc‐deficient T cells 53. We observed that metabolic and ribosome pathways were downregulated, whereas signaling pathways such as NF‐κB and MAPK were clearly upregulated in T lymphocytes lacking c‐Myc (Fig EV5). In addition, LPS‐activated primary B lymphocytes 52 showed the opposite pattern of pathway expression when compared with our findings in c‐MycKO B lymphocytes stimulated with LPS plus IL‐4 (Fig EV5A). These results suggest that c‐Myc function follows a similar pattern in B and T lymphocytes.
Figure EV5. Comparison of c‐Myc transcriptome.
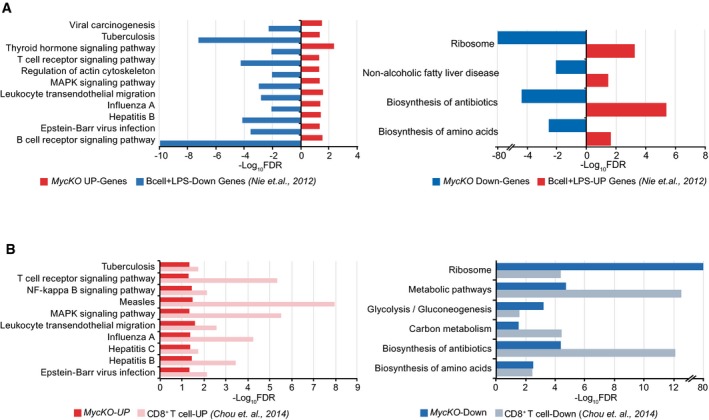
We identified 45 down‐ and 49 upregulated genes that were altered only in B lymphocytes from MycKO‐cd19 mice and would theoretically correspond to induced or repressed genes, respectively, by c‐Myc only (Fig 4D). Downregulated genes corresponded to a variety of molecular pathways (Fig EV4C), whereas upregulated genes were involved in the regulation of transcription, covalent chromatin modification, and histone methyltransferase activity (H3‐K4‐specific; Fig EV4C). The up‐ or downregulated gene sets in MaxKO‐cd19 or DKO‐cd19 mice only did not show statistical significance in the enrichment analysis.
RNA‐Seq data revealed a more similar gene expression profile between MaxKO and DKO than between either and c‐MycKO B cells (Figs 4 and 5). This finding correlated with the similar phenotype observed in MaxKO and DKO B lymphocytes for cell differentiation, proliferation, and DNA replication (Figs 1 and 3). Functional compensation among the different Myc family members is unlikely since we did not observe significant differences in n‐ and l‐myc expression between all the mutants and the heterozygous controls (Dataset EV1).
Considering all the RNA‐Seq data, the number of pathways regulated by c‐Myc and Max is limited and similar in different cell types. c‐Myc requires Max to regulate a selected number of genes that can subsequently, indirectly, amplify the signal, as described 17.
The generation of c‐MycKO, MaxKO, and DKO mouse models allowed us to individually assess the role of c‐Myc and Max in different biological processes in B lymphocytes. It should be noted that the mixed background of these models can play an important role in the observed differences in phenotype and gene expression. The unexpected reduction of Myc protein in MaxKO mature B cells, despite normal levels of c‐myc RNA (Fig 5C), reveals potential new mechanisms of Myc regulation that will be examined in a future study. Nevertheless, our results show that c‐Myc needs Max not only as heterodimer to perform its normal function in mature B lymphocytes, but also for mRNA translation and/or protein stability. This might provide an explanation for why MaxKO and DKO mature B cells are phenotypically closer when compared against c‐MycKO B cells. c‐MycKO B lymphocytes present the most severe phenotype of all the mutant phenotypes analyzed in this report, which is consistent with other studies 5, 8. c‐MycKO B lymphocytes seem to uncover the antagonistic role of Max by binding to E‐boxes, likely in association with proteins of the MXD family or MGA 18, 19. Our data suggest that c‐Myc/Max heterodimers are the main effectors of Myc functions in primary B lymphocytes. Interestingly, MaxKO and DKO mature B lymphocytes can still initiate relevant biological functions such as proliferation or cell differentiation. We propose that the first stages of these processes are not as strictly dependent on c‐Myc/Max heterodimers, but these factors are required to maintain, amplify, or fine‐tune these functions after the initial activation.
Materials and Methods
Generation of max fl mice and genotyping
The targeting construct was generated by the European Mouse Mutagenesis Consortium (Fig 1). The max gene contains five exons encoding a protein of 160 amino acids. Cre recombinase deletes exons 4 and 5 (aa: 57–160) containing the HLHZip domain and the 3′UTR. A total of 107 G4 (129S6/SvEv × C57Bl/6NTac) F1 ES cells 54 were transfected with 30 μg of linear DNA and plated on 106 mitomycin‐C treated neo‐resistant mouse embryonic fibroblasts/plate in KO‐DMEM (GIBCO) containing 15% HyClone serum, murine embryonic tested (Thermo), GlutaMAX (GIBCO), 2‐mercaptoethanol (GIBCO), MEM NEAA (GIBCO), penicillin/streptomycin (GIBCO), and 103 U/ml LIF (Millipore). At 24 h post‐electroporation, the medium was replenished and 200 μg/ml G418 was added. After 8 days, 96 clones were picked and correctly targeted mutations were genotyped by long‐range PCR with specific primers for 3′ and 5′ homology arms. Clones were tested for mycoplasma (MycoAlert Kit, Lonza) and the correct euploid karyotype. Embryonic day 2.5 embryos were collected from superovulated C57Bl/6N female mice. Microinjected embryos were transferred into pseudopregnant CD‐1 females. Chimeras (95%) were identified based on coat pigmentation and two were confirmed positive after genotyping. Mice were genotyped by PCR analysis of tail genomic DNA. Primers MaxF9 and MaxF10 were used to amplify max floxed (526 bp) and wt (430 bp) alleles. Genomic DNA of sorted B220+GFP+ B cells from spleen or BM was isolated and floxed, and deleted max alleles (589 bp) were amplified using MaxNull_2F and MaxFlox_2R. Primers GAPDH 5′and GAPDH 3′ were used for DNA normalization. Primer sequences are detailed in Appendix Table S1. myc fl/fl ;cd19 cre/+ ; rosa26 gfp/gfp (MycKO‐cd19) mice were previously reported 35. MycKO‐cd19, MaxKO‐cd19, DKO‐cd19, MycKO‐mb1, MaxKO‐mb1, and DKO‐mb1 mice are on a 129 × C57Bl6 mixed background. For all experiments, 8‐ 10‐week‐old mice were used.
Cell sorting and cell culture conditions
Freshly isolated cells from BM and spleen were incubated in NH4Cl buffer to lyse erythrocytes and were then stained with anti‐B220‐PE‐Cy7 (Biolegend) and anti‐IgM‐PE (Southern Biotech) antibodies or anti‐B220‐PE‐Cy7 only, in PBS with 2% fetal bovine serum (FBS). Cells were sorted (> 97% purity) based on the expression of GFP and different combinations of the aforementioned antibodies on a FACSAria Ilu sorter (BD Biosciences). For in vitro B‐cell differentiation assays, sorted BM GFP+B220+IgM− B lymphocytes were plated (4 × 105 cells/well) in RPMI 1,640 medium (GIBCO) with 15% FBS, 2‐mercaptoethanol, penicillin/streptomycin and supplemented with recombinant murine IL‐7 (10 ng/ml), murine stem cell factor (10 ng/ml), and rFlt3 ligand (10 ng/ml; all from Peprotech) for 4 days. For in vitro activation of mature B lymphocytes, sorted spleen GFP+B220+ B lymphocytes were cultured (105 cells/well) in RPMI 1,640 medium with 15% FBS, 2‐mercaptoethanol, penicillin/streptomycin and were activated with LPS (20 μg/ml; Sigma‐Aldrich) and IL‐4 (20 ng/ml; R&D Systems). Activated cells and supernatants were harvested after 72 h.
Flow cytometry
Single‐cell suspensions were incubated in NH4Cl buffer to lyse erythrocytes and were antibody‐stained in PBS with 2% FBS. Antibodies used were from Biolegend (B220‐PE‐Cy7, IgD‐Alexa Fluor 647), eBioscience (CD49b‐biotin, IgG1‐PE, IgM‐APC, GL‐7‐biotin), Beckman Coulter (B220‐APC, Ly‐6C‐biotin,), BD Pharmingen [CD21‐APC, CD23‐PE, CD43‐PE‐Cy7, CD69‐biotin, CD138 (Syndecan‐1)‐PE, IgG1‐PE], and Southern Biotech (IgM‐PE). Biotinylated antibodies were developed with streptavidin‐eFluor 450 (eBioscience) and streptavidin‐PE or streptavidin‐APC (Beckman Coulter). BM B‐cell populations were characterized as pro‐B cells (Ly‐6c−CD49b−B220+IgM−CD43+), pre‐B cells (Ly‐6c−CD49b−B220+IgM−CD43−), immature B cells (Ly‐6c−CD49b−B220loIgM+CD43−), and mature B cells (Ly‐6c−CD49b−B220hiIgM+CD43−). Spleen B lymphocytes were identified as follicular B cells (B220+CD23+CD21int), marginal zone B cells (B220+CD23−CD21hi), and immature (newly formed/transitional stage 1) B cells (B220+CD23−CD21lo). Cell death was monitored using annexin‐V‐eFluor 450 or annexin‐V‐APC (Immunostep) and 7‐aminoactinomycin D (7‐AAD; Beckman Coulter) and distinguished as live (7AAD− annexin‐V−), early apoptotic (7AAD− annexin‐V+), late apoptotic (7AAD+ annexin‐V+), and necrotic (7AAD+ annexin‐V−) cells. Cell cycle parameters were assessed directly by measuring DNA synthesis via EdU incorporation 4 h after intraperitoneal (i.p.) injection (200 μg) using the Click‐iT Plus EdU Pacific Blue (picolyl azide) Flow Cytometry Kit (ThermoFisher Scientific) for in vivo assays or after 4 h (at 10 μM) for in vitro studies on activated mature B lymphocytes. Cells were then stained with propidium iodide (Beckman Coulter) to measure DNA content. Cell proliferation was monitored with the CellTrace Violet Cell Proliferation Kit (ThermoFisher Scientific).
Immunization
T‐cell‐dependent immunization was performed in 6‐ to 8‐week‐old mice using 200 μg of TNP‐KLH (Biosearch Technologies) in Imject Alum (ThermoFisher Scientific) at a 1:1 ratio in 0.2 ml of PBS by i.p. injection. Mice injected with PBS were used as controls. Mice were analyzed 13 days after immunization.
Tissue immunofluorescence
Freshly isolated spleens were immersed in OCT and frozen with liquid nitrogen. Cryostat sections (10 μm) were fixed in 4% paraformaldehyde [10 min, room temperature (RT)]; blocked with PBS containing 2% fetal calf serum, 2% bovine serum albumin, and 10% goat serum (30 min, RT); and stained with FITC‐conjugated PNA (Vector Laboratories), Cy3‐conjugated Fab fragment goat anti‐mouse IgM (μ heavy chain‐specific; Jackson ImmunoResearch Laboratories), or a rabbit anti‐GFP antibody (A‐11122, ThermoFisher Scientific) followed by Alexa647‐conjugated goat anti‐rabbit IgG (Southern Biotech) at RT. Sections were then mounted in Fluoromount (Southern Biotech) and imaged on a Zeiss Axiovert LSM 510‐META inverted microscope with 20×/air objective. Images were analyzed using LSM 510 software (Zeiss). GC analysis and quantification were done using FiJi software (ImageJ, NIH). For each mouse model, GCs were identified as PNA+ in a comparable number of follicles on spleen sections; colocalization analysis of PNA/GFP was used to determine the presence of GFP+ cells at GCs in the tissue sections. The frequency of GC GFP+ and GC GFP− cells was calculated as (number of GC GFP+/total number of GC) × 100 and (number of GC GFP−/total number of GC) × 100, respectively.
Analysis of replication fork rate in stretched DNA fibers
Sorted spleen B220+GFP+ B lymphocytes were isolated by flow cytometry and stimulated to proliferate with LPS and IL‐4. After 48 h, cells were sequentially pulse‐labeled with the BrdU analogs CldU and IdU for 20 min. DNA fibers were spread in buffer containing 0.5% SDS, 200 mM Tris pH 7.4 and 50 mM EDTA, as described 55. CldU and IdU tracks were detected by immunofluorescence with anti‐BrdU antibodies (Abcam ab1791 for CldU; Beckton‐Dickinson 347580 for IdU). Fiber integrity was assessed with an anti‐ssDNA antibody (Millipore Mab3034). Slides were examined with a Leica DM6000 B microscope.
Preparation of whole‐cell extracts and immunoblots
Whole‐cell extracts were prepared by resuspending cells in Laemmli sample buffer followed by sonication (2 × 30 s in a Branson Digital sonifier set to 20% amplitude). SDS–PAGE and immunoblotting were performed following standard protocols. Primary antibodies were sourced as follows: MCM2 and MCM3 56; MCM4 and MCM6 57; pS40‐MCM2 (Abcam ab133243), CDC6 (Millipore 05‐550); CDC7 (Abcam ab108332); CDC45 and PSF2 58; MEK2 (BD Biosciences Pharmingen 610236); and SMC1 59.
Gene expression analysis
Sorted spleen B220+GFP+ B lymphocytes were isolated by flow cytometry and stimulated with LPS and IL‐4 for 3 days. Total RNA was extracted using the RNeasy Micro Kit (Cat No./ID: 74004, Qiagen), and cDNA was generated with avian myeloblastosis virus reverse transcriptase using random hexamers (SuperScript™ III First‐Strand Synthesis System, Cat 18080051, ThermoFisher). cDNA was used for real‐time PCR with specific primers (Appendix Fig S2). qPCR was performed in an ABI Prism 7700 using SYBR‐green core reagents and results were analyzed with SDS software v1.9. Relative gene expression was normalized to that of β‐actin. For BM B cells, total RNA was extracted from sorted B220+IgM+GFP+ B lymphocytes.
RNA‐Seq library preparation, sequencing, and generation of FastQ files
Total RNA (200 ng) was used to generate barcoded RNA‐Seq libraries using the NEB Next Ultra RNA library Prep Kit for Illumina (New England Biolabs) after poly A+ RNA selection using oligo‐d(T)25 magnetic beads. The size of the libraries was checked using the Agilent 2100 Bioanalyzer DNA 1000 chip, and their concentration was determined using the Qubit® fluorometer (Life Technologies). Libraries were sequenced on a HiSeq2500 (Illumina) to generate 60‐base single reads. FastQ files for each sample were obtained using CASAVA v1.8 software (Illumina).
RNA‐Seq analysis
Sequencing adaptor contaminations were removed from reads using cutadapt software, and the resulting reads were mapped and quantified on the transcriptome (Ensembl GRCm38, gene‐build 82) using RSEM v1.2.3 60. Genes with at least 1 count per million in at least two samples were considered for statistical analysis. Data were TMM normalized and transformed with function voom for differential expression test using the bioconductor packages edgeR 61 and limma 62. We considered as differentially expressed those genes with a Benjamini–Hochberg‐adjusted P‐value ≤ 0.01. Batch effects were accounted for by using a random variable in the model. A functional analysis on differentially expressed genes was performed using DAVID GO 42 and R package GOplot 63.
Statistical analyses
All the statistic analyses were performed using Student's two‐tailed unpaired t‐test (Prism 6.0; GraphPad) except for fiber analyses (Fig 3H) where statistical significance was assessed using non‐parametric, one‐way ANOVA Kruskal–Wallis test followed by Dunn's post‐test.
Data availability
RNA‐sequencing data are deposited in Gene Expression Omnibus database GSE115915.
Author contributions
MP‐O and AT designed and performed the experiments and analyzed and interpreted data. DF‐A performed the initial characterization experiments. SR‐G and YRC advised and performed the immunochemistry experiments. DM and TL‐B advised and performed the cell sorting. CT designed and performed statistical analysis of RNA‐Seq. DG‐A, SR‐A, and JM designed and executed the DNA replication experiments, with help from AT. IMA designed experiments, interpreted data, wrote the manuscript, and supervised the project.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Dataset EV1
Dataset EV2
Dataset EV3
Source Data for Figure 3
Acknowledgements
We thank the CNB Animal and Flow Cytometry facilities and the Transgenic CNB‐CBMSO UAM/CSIC Unit for the generation of max flox mice. Next‐generation sequencing experiments were performed in the Genomics Unit of the CNIC. We thank K McCreath for editing the manuscript. This work was supported by a grant of the Spanish Ministry of Economy and Competitiveness (SAF2014‐52398, AEI/FEDER, UE).
EMBO Reports (2018) 19: e45770
References
- 1. Busslinger M (2004) Transcriptional control of early B cell development. Annu Rev Immunol 22: 55–79 [DOI] [PubMed] [Google Scholar]
- 2. Pang SH, Carotta S, Nutt SL (2014) Transcriptional control of pre‐B cell development and leukemia prevention. Curr Top Microbiol Immunol 381: 189–213 [DOI] [PubMed] [Google Scholar]
- 3. Meyer N, Penn LZ (2008) Reflecting on 25 years with MYC. Nat Rev Cancer 8: 976–990 [DOI] [PubMed] [Google Scholar]
- 4. Kress TR, Sabo A, Amati B (2015) MYC: connecting selective transcriptional control to global RNA production. Nat Rev Cancer 15: 593–607 [DOI] [PubMed] [Google Scholar]
- 5. Vallespinos M, Fernandez D, Rodriguez L, Alvaro‐Blanco J, Baena E, Ortiz M, Dukovska D, Martinez D, Rojas A, Campanero MR et al (2011) B Lymphocyte commitment program is driven by the proto‐oncogene c‐Myc. J Immunol 186: 6726–6736 [DOI] [PubMed] [Google Scholar]
- 6. de Alboran IM, O'Hagan RC, Gartner F, Malynn B, Davidson L, Rickert R, Rajewsky K, DePinho RA, Alt FW (2001) Analysis of C‐MYC function in normal cells via conditional gene‐targeted mutation. Immunity 14: 45–55 [DOI] [PubMed] [Google Scholar]
- 7. Calado DP, Sasaki Y, Godinho SA, Pellerin A, Kochert K, Sleckman BP, de Alboran IM, Janz M, Rodig S, Rajewsky K (2012) The cell‐cycle regulator c‐Myc is essential for the formation and maintenance of germinal centers. Nat Immunol 13: 1092–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fernandez D, Ortiz M, Rodriguez L, Garcia A, Martinez D, Moreno de Alboran I (2013) The proto‐oncogene c‐myc regulates antibody secretion and Ig class switch recombination. J Immunol 190: 6135–6144 [DOI] [PubMed] [Google Scholar]
- 9. Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, Karnezis AN, Swigart LB, Nasi S, Evan GI (2008) Modelling Myc inhibition as a cancer therapy. Nature 455: 679–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dang CV (2012) MYC on the path to cancer. Cell 149: 22–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Whitfield JR, Beaulieu ME, Soucek L (2017) Strategies to inhibit Myc and their clinical applicability. Front Cell Dev Biol 5: 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schmitz R, Ceribelli M, Pittaluga S, Wright G, Staudt LM (2014) Oncogenic mechanisms in Burkitt lymphoma. Cold Spring Harb Perspect Med 4: a014282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gelmann EP, Psallidopoulos MC, Papas TS, Dalla‐Favera R (1983) Identification of reciprocal translocation sites within the c‐myc oncogene and immunoglobulin mu locus in a Burkitt lymphoma. Nature 306: 799–803 [DOI] [PubMed] [Google Scholar]
- 14. Blackwood EM, Eisenman RN (1991) Max: a helix‐loop‐helix zipper protein that forms a sequence‐specific DNA‐binding complex with Myc. Science 251: 1211–1217 [DOI] [PubMed] [Google Scholar]
- 15. Blackwell TK, Kretzner L, Blackwood EM, Eisenman RN, Weintraub H (1990) Sequence‐specific DNA binding by the c‐Myc protein. Science 250: 1149–1151 [DOI] [PubMed] [Google Scholar]
- 16. Desbarats L, Gaubatz S, Eilers M (1996) Discrimination between different E‐box‐binding proteins at an endogenous target gene of c‐myc. Genes Dev 10: 447–460 [DOI] [PubMed] [Google Scholar]
- 17. Sabo A, Kress TR, Pelizzola M, de Pretis S, Gorski MM, Tesi A, Morelli MJ, Bora P, Doni M, Verrecchia A et al (2014) Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature 511: 488–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Link JM, Hurlin PJ (2015) The activities of MYC, MNT and the MAX‐interactome in lymphocyte proliferation and oncogenesis. Biochim Biophys Acta 1849: 554–562 [DOI] [PubMed] [Google Scholar]
- 19. Conacci‐Sorrell M, McFerrin L, Eisenman RN (2014) An overview of MYC and its interactome. Cold Spring Harb Perspect Med 4: a014357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gallant P, Steiger D (2009) Myc's secret life without Max. Cell Cycle 8: 3848–3853 [DOI] [PubMed] [Google Scholar]
- 21. Ribon V, Leff T, Saltiel AR (1994) c‐Myc does not require max for transcriptional activity in PC‐12 cells. Mol Cell Neurosci 5: 277–282 [DOI] [PubMed] [Google Scholar]
- 22. DuHadaway JB, Sakamuro D, Ewert DL, Prendergast GC (2001) Bin1 mediates apoptosis by c‐Myc in transformed primary cells. Cancer Res 61: 3151–3156 [PubMed] [Google Scholar]
- 23. Vaque JP, Fernandez‐Garcia B, Garcia‐Sanz P, Ferrandiz N, Bretones G, Calvo F, Crespo P, Marin MC, Leon J (2008) c‐Myc inhibits Ras‐mediated differentiation of pheochromocytoma cells by blocking c‐Jun up‐regulation. Mol Cancer Res 6: 325–339 [DOI] [PubMed] [Google Scholar]
- 24. Comino‐Mendez I, Gracia‐Aznarez FJ, Schiavi F, Landa I, Leandro‐Garcia LJ, Leton R, Honrado E, Ramos‐Medina R, Caronia D, Pita G et al (2011) Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat Genet 43: 663–667 [DOI] [PubMed] [Google Scholar]
- 25. Romero OA, Torres‐Diz M, Pros E, Savola S, Gomez A, Moran S, Saez C, Iwakawa R, Villanueva A, Montuenga LM et al (2014) MAX inactivation in small cell lung cancer disrupts MYC‐SWI/SNF programs and is synthetic lethal with BRG1. Cancer Discov 4: 292–303 [DOI] [PubMed] [Google Scholar]
- 26. Hishida T, Nozaki Y, Nakachi Y, Mizuno Y, Okazaki Y, Ema M, Takahashi S, Nishimoto M, Okuda A (2011) Indefinite self‐renewal of ESCs through Myc/Max transcriptional complex‐independent mechanisms. Cell Stem Cell 9: 37–49 [DOI] [PubMed] [Google Scholar]
- 27. Cowling VH, Cole MD (2007) The Myc transactivation domain promotes global phosphorylation of the RNA polymerase II carboxy‐terminal domain independently of direct DNA binding. Mol Cell Biol 27: 2059–2073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Steiger D, Furrer M, Schwinkendorf D, Gallant P (2008) Max‐independent functions of Myc in Drosophila melanogaster . Nat Genet 40: 1084–1091 [DOI] [PubMed] [Google Scholar]
- 29. Lindeman GJ, Harris AW, Bath ML, Eisenman RN, Adams JM (1995) Overexpressed max is not oncogenic and attenuates myc‐induced lymphoproliferation and lymphomagenesis in transgenic mice. Oncogene 10: 1013–1017 [PubMed] [Google Scholar]
- 30. Shen‐Li H, O'Hagan RC, Hou H Jr, Horner JW II, Lee HW, DePinho, RA (2000) Essential role for Max in early embryonic growth and development. Genes Dev 14: 17–22 [PMC free article] [PubMed] [Google Scholar]
- 31. Gu H, Zou YR, Rajewsky K (1993) Independent control of immunoglobulin switch recombination at individual switch regions evidenced through Cre‐loxP‐mediated gene targeting. Cell 73: 1155–1164 [DOI] [PubMed] [Google Scholar]
- 32. Hobeika E, Thiemann S, Storch B, Jumaa H, Nielsen PJ, Pelanda R, Reth M (2006) Testing gene function early in the B cell lineage in mb1‐cre mice. Proc Natl Acad Sci USA 103: 13789–13794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rickert RC, Rajewsky K, Roes J (1995) Impairment of T‐cell‐dependent B‐cell responses and B‐1 cell development in CD19‐deficient mice. Nature 376: 352–355 [DOI] [PubMed] [Google Scholar]
- 34. Mao X, Fujiwara Y, Orkin SH (1999) Improved reporter strain for monitoring Cre recombinase‐mediated DNA excisions in mice. Proc Natl Acad Sci USA 96: 5037–5042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. de Alboran IM, Baena E, Martinez AC (2004) c‐Myc‐deficient B lymphocytes are resistant to spontaneous and induced cell death. Cell Death Differ 11: 61–68 [DOI] [PubMed] [Google Scholar]
- 36. Huang CY, Bredemeyer AL, Walker LM, Bassing CH, Sleckman BP (2008) Dynamic regulation of c‐Myc proto‐oncogene expression during lymphocyte development revealed by a GFP‐c‐Myc knock‐in mouse. Eur J Immunol 38: 342–349 [DOI] [PubMed] [Google Scholar]
- 37. Jung D, Giallourakis C, Mostoslavsky R, Alt FW (2006) Mechanism and control of V(D)J recombination at the immunoglobulin heavy chain locus. Annu Rev Immunol 24: 541–570 [DOI] [PubMed] [Google Scholar]
- 38. Bretones G, Delgado MD, Leon J (2015) Myc and cell cycle control. Biochim Biophys Acta 1849: 506–516 [DOI] [PubMed] [Google Scholar]
- 39. Aladjem MI, Redon CE (2017) Order from clutter: selective interactions at mammalian replication origins. Nat Rev Genet 18: 101–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dominguez‐Sola D, Gautier J (2014) MYC and the control of DNA replication. Cold Spring Harb Perspect Med 4: a014423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yevshin I, Sharipov R, Valeev T, Kel A, Kolpakov F (2017) GTRD: a database of transcription factor binding sites identified by ChIP‐seq experiments. Nucleic Acids Res 45: D61–D67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. da Huang W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57 [DOI] [PubMed] [Google Scholar]
- 43. Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M (1999) KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 27: 29–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tolar P (2017) Cytoskeletal control of B cell responses to antigens. Nat Rev Immunol 17: 621–634 [DOI] [PubMed] [Google Scholar]
- 45. Schweighoffer E, Tybulewicz VL (2017) Signalling for B cell survival. Curr Opin Cell Biol 51: 8–14 [DOI] [PubMed] [Google Scholar]
- 46. Kaileh M, Sen R (2012) NF‐kappaB function in B lymphocytes. Immunol Rev 246: 254–271 [DOI] [PubMed] [Google Scholar]
- 47. Yasuda T (2016) MAP kinase cascades in antigen receptor signaling and physiology. Curr Top Microbiol Immunol 393: 211–231 [DOI] [PubMed] [Google Scholar]
- 48. Kurosaki T, Wienands J (2016) Preface. Regulatory signal networks of the B cell antigen receptor. Curr Top Microbiol Immunol 393: v–vi. [PubMed] [Google Scholar]
- 49. Kremlitzka M, Macsik‐Valent B, Erdei A (2016) Regulation of B cell functions by Toll‐like receptors and complement. Immunol Lett 178: 37–44 [DOI] [PubMed] [Google Scholar]
- 50. Luo W, Weisel F, Shlomchik MJ (2018) B cell receptor and CD40 signaling are rewired for synergistic induction of the c‐Myc transcription factor in germinal center B cells. Immunity 48(313–326): e315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Benhamou D, Labi V, Novak R, Dai I, Shafir‐Alon S, Weiss A, Gaujoux R, Arnold R, Shen‐Orr SS, Rajewsky K et al (2016) A c‐Myc/miR17‐92/Pten axis controls PI3K‐mediated positive and negative selection in B cell development and reconstitutes CD19 deficiency. Cell Rep 16: 419–431 [DOI] [PubMed] [Google Scholar]
- 52. Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R et al (2012) c‐Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 151: 68–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chou C, Pinto AK, Curtis JD, Persaud SP, Cella M, Lin CC, Edelson BT, Allen PM, Colonna M, Pearce EL et al (2014) c‐Myc‐induced transcription factor AP4 is required for host protection mediated by CD8+ T cells. Nat Immunol 15: 884–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. George SH, Gertsenstein M, Vintersten K, Korets‐Smith E, Murphy J, Stevens ME, Haigh JJ, Nagy A (2007) Developmental and adult phenotyping directly from mutant embryonic stem cells. Proc Natl Acad Sci USA 104: 4455–4460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mouron S, Rodriguez‐Acebes S, Martinez‐Jimenez MI, Garcia‐Gomez S, Chocron S, Blanco L, Mendez J (2013) Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat Struct Mol Biol 20: 1383–1389 [DOI] [PubMed] [Google Scholar]
- 56. Alvarez S, Diaz M, Flach J, Rodriguez‐Acebes S, Lopez‐Contreras AJ, Martinez D, Canamero M, Fernandez‐Capetillo O, Isern J, Passegue E et al (2015) Replication stress caused by low MCM expression limits fetal erythropoiesis and hematopoietic stem cell functionality. Nat Commun 6: 8548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bua S, Sotiropoulou P, Sgarlata C, Borlado LR, Eguren M, Dominguez O, Ortega S, Malumbres M, Blanpain C, Mendez J (2015) Deregulated expression of Cdc6 in the skin facilitates papilloma formation and affects the hair growth cycle. Cell Cycle 14: 3897–3907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Aparicio T, Guillou E, Coloma J, Montoya G, Mendez J (2009) The human GINS complex associates with Cdc45 and MCM and is essential for DNA replication. Nucleic Acids Res 37: 2087–2095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Remeseiro S, Cuadrado A, Gomez‐Lopez G, Pisano DG, Losada A (2012) A unique role of cohesin‐SA1 in gene regulation and development. EMBO J 31: 2090–2102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA‐Seq data with or without a reference genome. BMC Bioinformatics 12: 323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK (2015) limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res 43: e47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Walter W, Sanchez‐Cabo F, Ricote M (2015) GOplot: an R package for visually combining expression data with functional analysis. Bioinformatics 31: 2912–2914 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Dataset EV1
Dataset EV2
Dataset EV3
Source Data for Figure 3
Data Availability Statement
RNA‐sequencing data are deposited in Gene Expression Omnibus database GSE115915.