

Abstract
Familial hyperkalemic hypertension (FHHt) can be mainly attributed to increased activity of the renal Na+:Cl− cotransporter (NCC), which is caused by altered expression and regulation of the with-no-lysine (K) 1 (WNK1) or WNK4 kinases. The WNK1 gene gives rise to a kidney-specific isoform that lacks the kinase domain (KS-WNK1), the expression of which occurs primarily in the distal convoluted tubule. The role played by KS-WNK1 in the modulation of the WNK/STE20-proline-alanine rich kinase (SPAK)/NCC pathway remains elusive. In the present study, we assessed the effect of human KS-WNK1 on NCC activity and on the WNK4-SPAK pathway. Microinjection of oocytes with human KS-WNK1 cRNA induces remarkable activation and phosphorylation of SPAK and NCC. The effect of KS-WNK1 was abrogated by eliminating a WNK-WNK-interacting domain and by a specific WNK inhibitor, WNK463, indicating that the activation of SPAK/NCC by KS-WNK1 is due to interaction with another WNK kinase. Under control conditions in oocytes, the activating serine 335 of the WNK4 T loop is not phosphorylated. In contrast, this serine becomes phosphorylated when the intracellular chloride concentration ([Cl−]i) is reduced or when KS-WNK1 is coexpressed with WNK4. KS-WNK1-mediated activation of WNK4 is not due to a decrease of the [Cl−]i. Coimmunoprecipitation analysis revealed that KS-WNK1 and WNK4 interact with each other and that WNK4 becomes autophosphorylated at serine 335 when it is associated with KS-WNK1. Together, these observations suggest that WNK4 becomes active in the presence of KS-WNK1, despite a constant [Cl−]i.
Keywords: distal convoluted tubule, diuretics, Na+:Cl− cotransporter, salt transport, STE20-proline-alanine rich kinase
INTRODUCTION
Familiar hyperkalemic hypertension (FHHt) (OMIM 145260) is a monogenic disease caused by mutations in the with-no-lysine (K) 1 (WNK1) or WNK4 kinases or in the ubiquitin ligase complex proteins KLHL3 or Cul3 (14). The phenotype is mainly the result of increased activity of the renal thiazide-sensitive NaCl cotransporter (NCC) in the distal convoluted tubule (DCT) caused by WNKs. The effect of WNKs is transduced to NCC by an intermediary kinase known as STE20-proline-alanine rich kinase (SPAK) that is able to phosphorylate NCC only if it has been previously phosphorylated by WNKs at a key residue in the T-loop (34, 35).
In FHHt subtypes, deletions of the first intron of WNK1 upregulate the expression of the kinase (37), whereas mutations in any of the other genes result in decreased ubiquitination of WNKs and, in turn, increased expression of WNK kinases (22, 27), suggesting that the disease is caused by an increase in the number of WNK copies. However, studies have shown that WNK kinases, particularly WNK4, are very sensitive to serum potassium and decreased activity of the renin angiotensin system (4, 5, 8, 30). The mechanism by which the WNK kinases remain active despite the fact that FHHt phenotype exhibits physiological parameters expected to inhibit WNK kinases is not clear (14). Salt-sensitive arterial hypertension with volume expansion, which reduces the activity of the renin-angiotensin system, would be expected to reduce WNK4 activity attributable to the absence of angiotensin II (5, 8); additionally, there is a significant inverse relationship between serum potassium concentration and NCC activity (29, 30), and it has been shown that serum potassium acts as a powerful modulator of NCC activity via the WNK4-SPAK pathway (10). Thus hyperkalemia would also be expected to reduce WNK4 activity.
Initial studies of the WNK1 locus demonstrated that this gene gives rise to two major isoforms (9, 21), a long variant (L-WNK1) with a transcription site located in intron 1 and a shorter isoform starting at exon 4a that lacks a functional kinase domain and is only expressed in the kidney. This shorter variant is known as kidney-specific (KS-WNK1). Initial studies demonstrated that L-WNK1 transcripts are present at low abundance in many regions of the kidney, whereas KS-WNK1 is highly expressed but only in renal cortex (9, 21). Vidal-Pietot et al. (32) demonstrated that KS-WNK1 mRNA is almost exclusively present in the DCT, where it is 80 times more abundant than L-WNK1. Additionally, alternative splicing of exons 9, 11, 12, and/or 26 produces a diverse array of potential variants. The most abundant isoform in the kidney is one lacking exon 11 (32). We have shown that L-WNK1 lacking exon 11 (L-WNK1-Δ11) activates NCC (7), but the role of KS-WNK1 remains elusive.
A previous report proposed KS-WNK1 as a negative regulator of NCC (28). This was based, however, on a previous model of the effect of WNKs on NCC, in which WNK4 inhibits NCC and L-WNK1 has no direct effect on NCC but prevents the inhibitory effect of WNK4 (11). The recent cloning of human orthologs of WNK1 and WNK4 allowed us to demonstrate that both kinases are activators of NCC (7) and that this effect is modulated by intracellular Cl− concentration (1), resulting in the current model in which both WNKs activate NCC and the difference between them is their degree of sensitivity to chloride (14). Thus the previously suggested inhibitory role for KS-WNK1 does not fit the present model of NCC modulation by WNKs.
We therefore decided to analyze the effect of KS-WNK1 on NCC activity and phosphorylation using the kidney-enriched variant of human KS-WNK1-Δ11. Here, we present evidence that KS-WNK1 is an activator of NCC because it is able to interact with and activate the WNK4 kinase, apparently by decreasing the affinity of the resulting dimers to intracellular chloride concentration ([Cl−]i).
METHODS
Mutagenesis and constructs.
Rat NCC, human WNK4, WNK4-L322F, the kinase death WNK4-KD, WNK4-S335A, human L-WNK1-Δ11, and its different splicing variants have been previously described (1, 6, 12, 16, 38). Human KS-WNK1-Δ11 was cloned from human kidney using the same strategy as previously reported (7). Other splicing variants of KS-WNK1 were generated by subcloning into the L-WNK1 constructs. The QuikChange mutagenesis system (Stratagene) and custom-made primers (Sigma) were used to prepare the human KS-WNK1-Δ11 HQ/AA mutant construct. Fast-cloning using GoTaq DNA Polymerase (Promega) and custom-made primers (Sigma) were used to generate the KS-WNK1 1-257 isoform. All mutations and deletions were confirmed by sequencing, and the mutant fragment was subcloned back into the appropriate expression constructs to avoid unwanted mutations.
Functional expression of NCC.
Adult female Xenopus laevis frogs were Tricaine (0.17%) anesthetized. Oocytes were surgically extracted and incubated in the following Ca2+-free ND-96 solution (in mM): 96.0 NaCl, 2.0 KCl, 1.0 MgCl2, and 5.0 HEPES/Tris, pH 7.4 with type B collagenase for 1.5 h. After four washes with ND-96 solution (in mM: 96.0 NaCl, 2.0 KCl, 1.8 CaCl2, 1.0 MgCl2, and 5.0 HEPES/Tris, pH 7.4), oocytes were incubated overnight at 16°C in ND-96 supplemented with 5 mg/100 ml gentamicin. The next day oocytes were injected with 50 nl of H2O alone or containing NCC alone, wild-type KS-WNK1-Δ11 or other splicing variants, mutant KS-WNK1-Δ11, wild-type L-WNK1-Δ11, wild-type WNK4, or WNK4-KD. All transcripts were injected at 0.2 μg/μl, unless another concentration is indicated for particular experiments. cRNA for injection was transcribed in vitro from cDNA linearized at the 3′ using the T7 RNA polymerase mMESSAGE kit (Ambion). Experiments were performed after 48 h of injection in oocytes that were maintained at 16°C with daily changes of the ND-96 with gentamicin medium.
All experimental data are based on a minimum of three different experiments. The committee on animal research of the Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán approved the use of Xenopus laevis frogs.
Transport assays.
The NCC activity was assessed following our protocol (23) utilizing the radioactive tracer [22Na+] (Perkin Elmer Life Sciences) uptake. For uptake in control conditions, oocytes were incubated the night before in ND-96. The next day, 10–15 oocytes per well were preincubated for 10 min in Cl−-free ND-96 medium (in mM: 96.0 Na+ isethionate, 2.0 K+ gluconate, 1.8 Ca2+ gluconate, 1.0 Mg2+gluconate, 5.0 mM HEPES, pH 7.4) containing 1.0 mM ouabain, 100.0 μM amiloride, 100.0 μM bumetanide, and the presence or absence of 100.0 μM metolazone. Oocytes were then transferred for uptake to a K+-free uptake medium (in mM: 40.0 NaCl, 56.0 sodium-gluconate, 4.0 CaCl2, 1.0 MgCl2, and 5.0 HEPES/Tris, pH 7.4), containing ouabain, amiloride, and bumetanide, with or without metolazone and with 1.0 μCi of 22Na+ for 60 min at 32°C. At the end of the uptake period, oocytes were transferred to ice-cold radioactive-free medium and washed five times to remove any excess tracer from oocyte membranes, placed in individual tubes. and lysed with SDS 1% solution, for tracer activity determination with the use of β-scintillation counting. For uptake in low-chloride hypotonic stress (LCHS), we followed our previously published protocol (23). Oocytes were incubated overnight in a Cl−-free solution with an osmolarity of 170 mOsm/Kg H2O (in mM: 70.0 Na+ isethionate, 1.8 Ca2+ gluconate, 1.0 Mg2+ gluconate, and 5.0 mM HEPES, pH 7.4). The next day, oocytes were incubated for 30 min in the same solution, supplemented with 1.0 mM ouabain, 100.0 μM amiloride, and 100.0 μM bumetanide, in the presence or absence of 100.0 μM metolazone. Uptake was then performed in hypotonic 170 mOsm/Kg H2O K+-free uptake medium (in mM: 40.0 NaCl, 30.0 sodium-gluconate, 4.0 CaCl2, 1.0 MgCl2, and 5.0 HEPES/Tris, pH 7.4) containing ouabain, amiloride, and bumetanide, with or without metolazone, and with 1.0 μCi of 22Na+ for 60 min at 32°C. At the end of uptake, oocytes were washed and treated as above for tracer activity determination.
Western blotting of Xenopus laevis oocyte proteins.
Total protein samples were extracted from groups of 10 oocytes per condition using lysis buffer containing 50.00 mM Tris·HCl (pH 7.5), 1.00 mM EGTA, 1.00 mM EDTA, 50.00 mM sodium fluoride, 5.00 mM sodium pyrophosphate, 1.00 mM sodium orthovanadate, 1% (wt/vol) Nonidet P-40, 0.27 M sucrose, 0.10% (vol/vol) 2-mercaptoethanol, and protease inhibitors (Complete tablets; Roche). Proteins were quantified, and 60 µg of total protein was subjected to SDS-PAGE and transferred to a PVDF membrane. The membranes were blocked for 1.5 h in 5% (wt/vol) nonfat milk dissolved in Tris-buffered saline with Tween (TBS-T) solution (in mM: 2.0 Tris·HCl, 150.0 NaCl and 0.2% Tween-20, pH 7.5). The membranes were then immunoblotted with the indicated antibodies in TBS-T containing 5% (wt/vol) nonfat milk (blocking solution) for 16 h at 4°C. The following sheep antibodies were used at a concentration of 1–2 μg/ml: anti-NCC (recognizing residues 906–925 of human NCC, CHTKRFEDMIAPFRLNDGFKD), anti-phosphorylated NCC at threonine 46, threonine 50, and threonine 55 (T44, 48, and 53 in rats), and anti-phosphorylated WNK1 S382. This antibody recognizes S335 of WNK4 when phosphorylated, which is a conserved serine in the four WNKs. Incubation with the phospho-specific antibodies was performed with the addition of 10 μg/ml of the nonphosphopeptide antigen used to raise the antibody. These antibodies were produced at the Medical Research Council phosphorylation unit at Dundee University and were previously characterized (25, 31). The blots performed with sheep antibodies were washed four times with TBS-T and incubated for 1.5 h at room temperature with secondary anti-sheep horseradish peroxidase (HRP)-conjugated antibody diluted 1:5,000 (Santa Cruz Biotechnology) in blocking solution. The membranes were washed six times before signal detection by chemiluminescence using the Luminata Forte Western HRP substrate (Merck Millipore). The following commercial HRP-conjugated antibodies were used: anti-Flag 1:5,000 (Sigma), anti-Myc 1:1,000 (Sigma), and anti-β-actin 1:2,500 (Santa Cruz Biotechnology).
Band analysis.
Immunoblots were scanned using the C-Digit (Li-Cor) blot scanner. Densitometric analysis was performed using ImageStudioLite software. Figures were generated using Illustrator (Adobe).
Intracellular chloride concentration.
Ion-selective microelectrodes (ISMs) were fabricated using borosilicate glass (TW1450-4, WPI) pulled in one step by a laser device (P-2000, Sutter Instruments). The micrometric tip was filled by PVC membrane containing chloride ionophore II [4,5-dimethyl-3,6-dioctyloxy-O-phenylene bis(mercurytrifluoroacetate)] and tridodecylmethylammonium chloride. ISMs were dried and conditioned overnight and were then backfilled with an inner reference solution (0.1 M KCl). Microreference (μ-Ref) electrodes were prepared similar to ISMs but without a polymeric matrix and were backfilled with KCl (1 M). μ-Ref performance was evaluated by potentiometric titration, and its accuracy, instantaneous response, and stability were similar to bulk AgCl/Ag electrodes. A high-input impedance potentiometer was used to measure the potential. Oocytes were impaled with the help of a stereoscopic microscope (Stereozoom7, Bausch and Lomb) and xyz micromanipulators (MM33, Marzhauser). All measurements were performed in a Faraday cage to avoid electromagnetic interference. Each electrode was calibrated with 10−5 to 10−1 M KCl standard solutions exhibiting Nernstian behavior (20).
Biotinylation of membrane proteins in Xenopus laevis oocytes.
Groups of 20 oocytes per condition were incubated with 0.5 ml of EZ-Link sulfo-NHS L-C biotin (Thermo Scientific) containing 1.0 mg/mL ND-96-triethylamine (100.0 mM) at pH 8.0. After 1 h of incubation, each group was washed five times with 3 ml ice-cold ND96-triethylamine (100.0 mM) at pH 8.0. Oocytes were lysed with lysis buffer as described above and were centrifuged at 4,000 revolutions/min for 15 min at 4°C. The supernatants were collected, and protein quantification was performed by Bradford assay. Two hundred microliters of protein was incubated overnight with streptavidin agarose beads (Thermo Scientific). The mixture was then centrifuged, and supernatant was collected. Agarose beads were suspended in 100 µl Laemmli sample buffer and heated at 95°C for 10 min. The proteins were resolved on an SDS-PAGE, and immunodetection was performed by Western blotting as described above.
Immunoprecipitation.
Immunoprecipitation studies were performed using an Anti-c-Myc Immunoprecipitation Kit according to the manufacturer’s recommendations (Sigma-Aldrich). Four milligrams of protein from Xenopus laevis oocyte total protein lysate was incubated with 20 μl anti-c-Myc agarose suspension in a total volume of 700 μl attained using 1× immunoprecipitation buffer (catalog number I5779, Sigma-Aldrich). The mixture was incubated overnight as instructed by the manufacturer. The beads were washed one time with PBS buffer on spin columns, and the proteins were suspended in 80 μl Laemmli sample buffer and heated at 95°C for 5 min. Ten microliters of 2× Laemmli sample buffer was added to 20 μl flowthrough for a total volume of 30 μl. SDS-PAGE gels were then loaded with 30 µl of the immunoprecipitated fraction or 30 µl of the flowthrough fraction. Proteins were then transferred to PVDF membranes, and Western blotting was performed as detailed above.
Statistical analysis.
Statistical significance is defined as two-tailed P < 0.05, and the results are presented as means ± SE. The significance of the differences between two groups was tested with t-test and for three or more groups by one-way ANOVA with multiple comparisons with Bonferroni correction using GraphPad Prism version 6.00 for Mac (GraphPad Software).
RESULTS
The KS-WNK1 isoform is a powerful activator of NCC.
The most abundant variant of WNK1 produced in the kidney is the one lacking the exon 11 (32). Thus, in the present study, with the exception of those presented in Fig. 1B, all experiments were performed using L-WNK1 and/or KS-WNK1 variants that lack exon 11.
Fig. 1.

Kidney-specific with no lysine kinase 1 (KS-WNK1) is a powerful activator of Na+:Cl− cotransporter (NCC). A: functional expression assay reveals NCC activity in groups of oocytes injected with NCC cRNA alone or together with human KS-WNK1 lacking exon 11 (KS-WNK1-Δ11) cRNA, as stated. *P < 0.00001 vs. no KS-WNK1; n = 30. B: NCC activity in oocytes injected with NCC cRNA (solid bar), NCC + KS-WNK1 Full, KS-WNK1-Δ11, KS-WNK1-Δ11-12, Δ-9-12, KS-WNK1-Δ11-HQ/AA, or KS-WNK1-Δ11-Δ4a cRNA (open bars), as stated. *P < 0.001 vs. NCC; n = 3. For both figures, thiazide-sensitive 22Na+ uptake in oocytes injected with NCC alone was set to 100%, and the other groups were normalized accordingly.
We previously demonstrated that the injection of human L-WNK1-Δ11 cRNA into Xenopus laevis oocytes induces significant increases in NCC activity and phosphorylation (7). In the present study, we first assessed the effect of KS-WNK1-Δ11 on the functional activity of NCC using the same system. As shown in Fig. 1A, the coinjection of NCC with KS-WNK1-Δ11 resulted in a remarkable increase in NCC activity. This positive effect of KS-WNK1 on NCC was consistently observed in more than 30 independent experiments. Because a previous study described the KS-WNK1 isoform lacking exons 11 and 12 as having no effect on NCC (28), we assessed whether potential variants of KS-WNK1, including full-length KS-WNK1 (containing all exons from 4 to 26) and isoforms lacking exon 11, exons 11 and 12, or exons 9 and 11, exerted the same effect. We found that all of the KS-WNK1 variants tested increased the activity of NCC (Fig. 1B), indicating that the effect of KS-WNK1 on NCC prevails regardless of variation at the carboxyl-terminal domain. Note in the last two bars of Fig. 1B that two mutant variants lost the positive effect on NCC. One is the mutant in which we eliminated the so-called HQ motif within the coiled-coil domain at the COOH terminus by substituting H and Q for alanine residues. The HQ motif is known to be required for WNK-WNK interaction and activation (7, 31). In addition, elimination of the initial 30 amino-terminal domain residues that are unique to KS-WNK1 (i.e., not present in L-WNK1) prevented the effect of KS-WNK1 on NCC, suggesting that this segment is critical for the proper folding and function of KS-WNK1.
The effect of the KS-WNK1 and L-WNK1 isoforms on NCC is synergistic.
Because L-WNK1 also activates NCC (7), we compared the dose-dependent effects of L-WNK1 and KS-WNK1 and found that the positive effect of KS-WNK1-Δ11 on NCC was observed with significantly lower amounts of injected cRNA, compared with L-WNK1-Δ11 (Fig. 2A). Microinjection of low concentrations of L-WNK1 (0.025 and 0.050 μg/μl) had no effect on NCC activity, whereas the same concentrations of KS-WNK1 induced NCC activation that was similar to that observed with 0.200 μg/μl of L-WNNK1. Note in Fig. 2B that the expression of L-WNK1 and KS-WNK1 in oocytes was similar across the different quantities of cRNA injected.
Fig. 2.
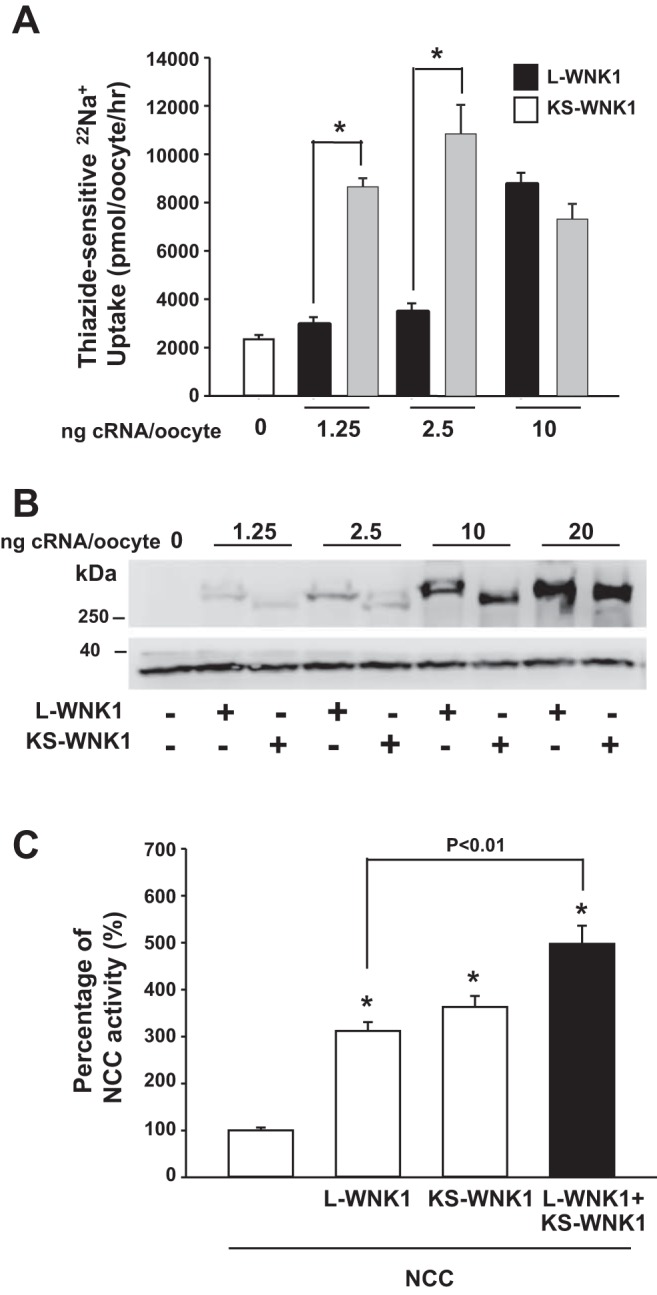
Kidney-specific with no lysine kinase 1 (KS-WNK1) is more potent than long-variant (L)-WNK1. A: thiazide-sensitive 22Na+ uptake in oocytes injected with Na+:Cl− cotransporter (NCC) alone or together with human L-WNK1 lacking exon 11 (L-WNK1-Δ11) (solid bars) or KS-WNK1-Δ11 (shaded bars) cRNA. cRNA of L-WNK1-Δ11 and KS-WNK1-Δ11 was injected at a concentration of 0, 1.25, 2.50, or 10.00 ng per oocyte. *P < 0.001; n = 5. B: immunodetection of L-WNK1 and KS-WNK1 (upper blot) and actin (lower blot) from total protein lysate of oocytes injected with 0, 1.25, 2.50, 10.00, or 20.00 ng L-WNK1 or KS-WNK1 cRNA per oocyte. C: functional expression assay shows NCC activity in groups of oocytes injected with NCC cRNA, alone or together with L-WNK1-Δ11 cRNA, KS-WNK1-Δ11 cRNA, or both. For this figure, the level of thiazide-sensitive 22Na+ uptake in the oocytes injected with NCC alone was set to 100%, and the rest of the groups were normalized accordingly. *P < 0.001 vs. NCC; n = 3.
To elucidate the mechanism by which KS-WNK1 increases NCC activity, we assessed whether the effect of L-WNK1 and KS-WNK1 is additive. As shown in Fig. 2C, when KS-WNK1 cRNA was coinjected with L-WNK1 cRNA, we observed a significant increase in the effect on NCC.
The observations presented in Fig. 2 suggest that L-WNK1 and KS-WNK1 may differ in their mechanisms of activation of NCC. WNKs must dimerize to be active and thus to activate SPAK (31). It is therefore likely that L-WNK1 forms homodimers that activate SPAK and then NCC and that KS-WNK1 likely forms the heterodimers with endogenously expressed WNKs in the oocyte, thereby increasing its activity toward SPAK and NCC. This could explain why the effect of KS-WNK1 is observed with a much lower quantity of injected cRNA; interaction with an endogenous WNK requires very few copies of KS-WNK1. In this case, the synergistic effect of L-WNK1 and KS-WNK1 could be explained because KS-WNK1 activates an endogenous pathway that is not affected by L-WNK1, thereby adding to the activation of SPAK and NCC.
The effect of KS-WNK1 on NCC is associated with SPAK phosphorylation and increased accumulation of NCC in the plasma membrane.
Western blot analysis of oocytes injected with NCC alone or together with L-WNK1 or KS-WNK1 revealed that the positive effect of L-WNK1-Δ11 or KS-WNK1-Δ11 on NCC activity is associated with increased NCC and SPAK phosphorylation (Fig. 3, A and B) and with increased accumulation of NCC at the plasma membrane (Fig. 3, C and D). Thus, although the major difference between L-WNK1 and KS-WNK1 is the lack of a kinase domain in the latter, they are both able to induce an increase in NCC and SPAK phosphorylation.
Fig. 3.

Long-variant with no lysine kinase 1 (L-WNK1) and kidney-specific (KS)-WNK1 increase Na+:Cl− cotransporter (NCC) phosphorylation and membrane protein abundance. A: representative Western blot showing the effect of L-WNK1 lacking exon 11 (L-WNK1-Δ11) or KS-WNK1-Δ11, as stated, on NCC and protein odd-skipped related 1 (OSR1) expression and phosphorylation. The blot shows L-WNK1/KS-WNK1, pNCC, NCC, phosphorylated STE20-proline-alanine rich kinase (pSPAK) OSR1, and actin expression, as stated. B: densitometric analysis of 4 different experiments to show pNCC/NCC ratio and pSPAK/SPAK ratio. Gray bars KS-WNK1 and black bars L-WNK1. *P < 0.05 vs. control groups. C: KS-WNK1 increases the abundance of NCC in a manner similar to L-WNK1. Representative Western blot performed using samples extracted from a biotinylation assay (membrane fraction) and in total lysates, as stated. L-WNK1-Δ11 or KS-WNK1-Δ11 increases the translocation of NCC to the membrane. D: densitometric analysis of 5 different experiments to show the membrane NCC/total NCC ratio. *P < 0.05 vs. control groups.
Because KS-WNK1 lacks the kinase domain, it cannot phosphorylate and activate the intermediary kinases SPAK and OSR1 directly. Instead, NCC activation might result from interaction with, and activation of, endogenous WNK in the oocyte. This is supported by the observation that elimination of the HQ motif blocked the effect of KS-WNK1 on NCC (Fig. 2B). To test this possibility, we assessed the ability of a specific inhibitor of WNK kinases, WNK463 (40), to modulate the effect of L-WNK1 and KS-WNK1 on NCC. WNK463 exhibits selectivity for the WNK kinase family because it binds to a particular ATP-binding site in WNKs that is the result of the unusual placement of a catalytic lysine in subdomain I (Lys233 of WNK1), which is a unique feature of the WNK kinase family. In most kinases, the catalytic lysine is located in subdomain II, thereby forming a completely different pocket. Thus WNK463 completely inhibits all four WNKs with an IC50 of ~4 nM. In contrast, at 10 μM (2,500-fold greater than 4 nM), WNK463 inhibits 50% of the activity of only 2 out of 442 human kinases tested (40). In addition, WNK463 is not expected to bind to KS-WNK1, which lacks the kinase domain. Thus the inhibitory effect of WNK463 would occur only if KS-WNK1 induces the activation of another WNK kinase.
As shown in Fig. 4, WNK463 prevented the stimulation of NCC by both L-WNK1 and KS-WNK1-Δ11. Although the inhibitory effect was significant in all groups, it was more marked in oocytes injected with water or KS-WNK1 that in those injected with L-WNK1. This is probably because in the water- or KS-WNK1-injected oocytes the WNK463 effect is due to inhibition of the endogenous WNK that should be less abundant than the L-WNK1 that was overexpressed by the injection of L-WNK1 cRNA. This observation, together with the loss of the effect of KS-WNK1-Δ11 upon ablation of the WNK-WNK interaction by HQ motif mutation (Fig. 1B), strongly suggests that KS-WNK1 induces SPAK/NCC activation by interacting with and activating an endogenous WNK. This could explain why the effect of KS-WNK1 is observed upon coinjection of very low concentrations of KS-WNK1 cRNA (Fig. 2A); to activate an endogenous WNK, only a miniscule amount of KS-WNK1 protein would be required.
Fig. 4.
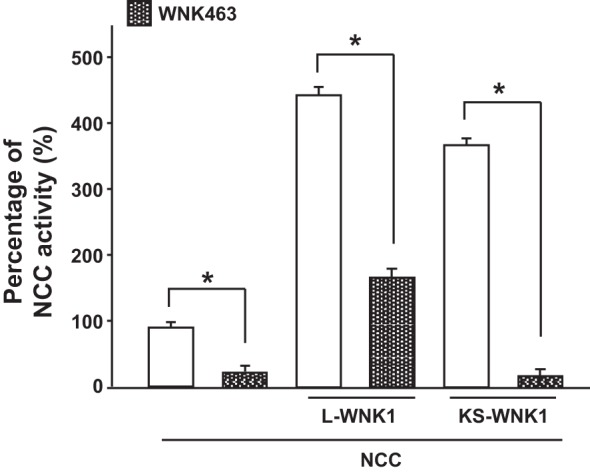
The effect of long-variant with no lysine kinase 1 (L-WNK1) and kidney-specific (KS)-WNK1 on Na+:Cl− cotransporter (NCC) is abrogated by the WNK inhibitor WNK463. NCC activity in oocytes injected with NCC cRNA, NCC + L-WNK1 lacking exon 11 (L-WNK1-Δ11), or NCC + KS-WNK1-Δ11is shown. Na+ uptake was measured in the presence (hashed bars) or absence (open bars) of the small molecule inhibitor WNK463 (1 μM). Thiazide-sensitive 22Na+ uptake in oocytes injected with NCC alone was set to 100%, and the other groups were normalized accordingly. *P < 0.001; n = 3.
We have previously shown that Xenopus oocytes express transcripts of all four WNKs (26). Thus KS-WNK1 could be interacting with any of them. It is most likely that the effect on the endogenous WNK could be in the paralogs of WNK1 or WNK4, which are the most chloride-sensitive WNKs (2). Supporting this, analysis of the genome project of Xenopus (Xenbase; http://www.xenbase.org) shows that at stages V–VI of Xenopus laevis oocytes the most abundant RNA corresponds to WNK1.
KS-WNK1 induces WNK4 activation.
We and others have previously reported that exposing cells to LCHS, which promotes chloride efflux and thus decreases [Cl−]i, results in the activation of NCC in oocytes and mammalian cells (2, 23, 25). This phenomenon occurs because WNKs are sensitive to [Cl−]i; the higher the [Cl−]i, the lower the activity of WNKs. This is due to the presence of a chloride-binding site within the kinase domain of WNKs that when occupied by chloride prevents the autophosphorylation of the kinase in a key serine of the T-loop that is required for activation (24). The chloride-binding site has been localized to lysine residue 379 in WNK1 and lysine 322 in WNK4. Substitution of this lysine by phenylalanine renders the kinase constitutively active (1, 24).
We have suggested that the sensitivity of WNKs to [Cl−]i ranks as follows: WNK4 > WNK1 > WNK3. This is based on the following observations: 1) functional experiments involving NCC under control conditions show that relative NCC activation by WNK3 is approximately threefold, by WNK1 is approximately half-fold, and by WNK4 is near zero. In the same experiment, however, after the decrease of [Cl−]i using LCHS, activation by either WNK3, WNK1, or WNK4 was threefold, suggesting that, at the [Cl−]i in control conditions, WNK3 is fully active, WNK1 is partially active, and WNK4 is not active, whereas, after the decrease in [Cl−]i, all three kinases become fully activated (2). 2) Under control conditions, WNK3 is phosphorylated at the activating serine in the T-loop and does not change upon decreasing [Cl−]i, whereas WNK4 under control conditions is not phosphorylated at the homologous serine and becomes phosphorylated under conditions of decreasing [Cl−]i (1). 3) In vitro assessment of the effect of WNK on its target kinase SPAK reveals that WNK4 activity is affected by chloride concentration, with an IC50 that is significantly lower than that of WNK1 or WNK3 (29).
We thus reasoned that KS-WNK1 activates NCC because this isoform likely interacts with endogenous WNK, thereby reducing its affinity for chloride and activating the kinase without changing [Cl−]i. In support of this possibility, note in Fig. 3A that SPAK phosphorylation is higher in oocytes injected with KS-WNK1 than in those injected with L-WNK1. Because both isoforms activate NCC to promote chloride influx into the oocyte, it is possible that L-WNK1 is partially inhibited by the influx of chloride, whereas KS-WNK1 is not, and that this is reflected in their effect on SPAK phosphorylation. If KS-WNK1 activates NCC by changing the chloride affinity of endogenous WNK, we hypothesized that this could also occur with WNK4, which is the WNK kinase with the highest affinity for chloride (1, 2, 29).
WNK4 is the dominant activator of NCC in vivo; its genetic inactivation leads to a strong reduction in the expression and phosphorylation of the cotransporter (5). WNK4 and NCC are coexpressed in DCT1 (17, 18, 41), in which KS-WNK1 is heavily expressed (32) and WNK4 is highly sensitive to [Cl−]i. We therefore tested whether KS-WNK1-Δ11 activates WNK4. As previously shown (1, 7), under control conditions in oocytes ([Cl−]i 45 mM), WNK4 remains inactive, as evidenced here by the absence of T-loop phosphorylation (Fig. 5A). We showed previously that, under this condition, WNK4 has no effect on NCC and can even behave as an inhibitory kinase, most likely by reducing the effect of endogenous WNK on SPAK/NCC (7). As shown in Fig. 5A, coinjection of WNK4 with KS-WNK1-Δ11 resulted in increased WNK4 T-loop phosphorylation. We consistently observed this result in six independent experiments (Fig. 5B). Thus the presence of KS-WNK1 induces the T-loop phosphorylation and activation of WNK4. These observations suggest that KS-WNK1-Δ11 either reduces [Cl−]i or facilitates WNK4 activation, despite the lack of change in [Cl−]i. Figure 5C demonstrates that [Cl−]i was not modified in oocytes injected with KS-WNK1-Δ11 cRNA, strongly supporting the model that WNK4 activation by KS-WNK1-Δ11 is not secondary to a reduction in [Cl−]i.
Fig. 5.
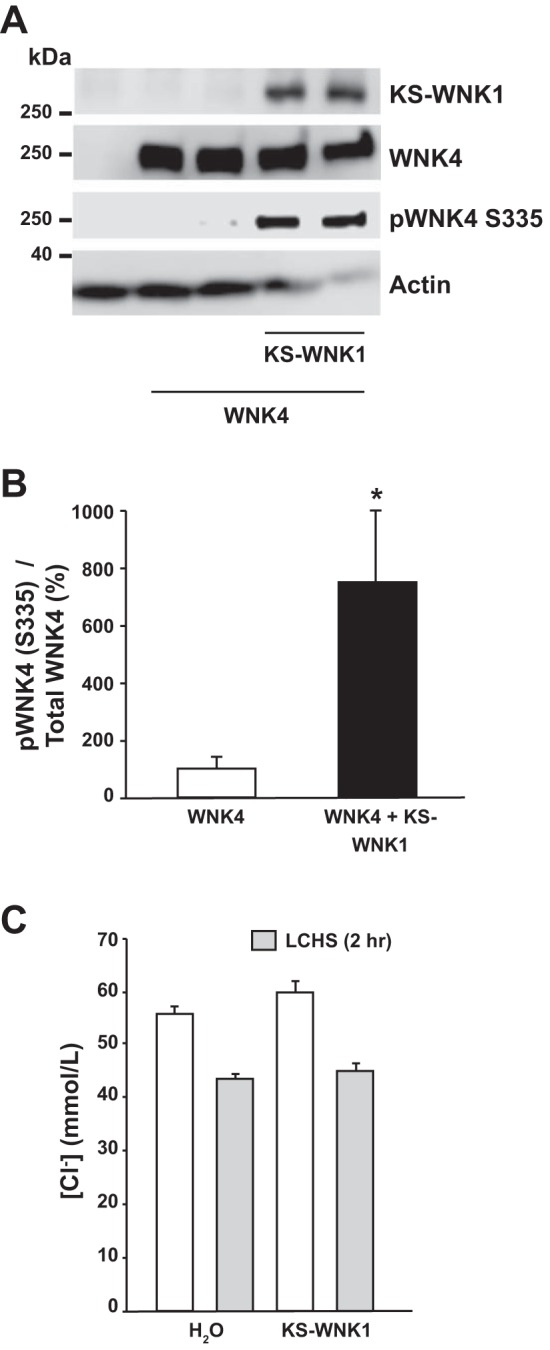
Kidney-specific with no lysine kinase 1 (KS-WNK1) induces WNK4 T-loop phosphorylation despite no change in [Cl−]i. A: representative Western blot showing the effect of KS-WNK1 lacking exon 11 (KS-WNK1-Δ11) and WNK4 coexpression on WNK4 T-loop phosphorylation. The blot shows total WNK4 expression and pWNK4 S335, in the absence or presence of KS-WNK1-Δ11. B: densitometric analysis of results compiled from 6 different experiments. The ratio of phosphorylated WNK4/total WNK4 in the absence of KS-WNK1-Δ11 was arbitrarily set to 100%, and the ratio of phosphorylated WNK4/total WNK4 in the presence of KS-WNK1-Δ11 was normalized accordingly. *P < 0.01 vs. control. C: [Cl−]i in oocytes was assessed with custom-made glass capillary ion-selective microelectrodes 48 h after the injection of water, Na+:Cl− cotransporter (NCC), or NCC + KS-WNK1-Δ11 cRNA, as stated, under control conditions (open bars) or after 2 h of low-chloride hypotonic stress (LCHS) (shaded bars); n = 3 (6 oocytes per group, per experiment). No statistically significant difference was found between the groups.
Figure 6A shows a representative Western blot of the effect of L-WNK1 or KS-WNK1 on WNK4 and NCC phosphorylation in oocytes under control conditions and after LCHS exposure. Densitometric analysis from four different experiments is shown in Fig. 6B. Under control conditions, the presence of L-WNK1 had no effect on WNK4 phosphorylation and induced a slight increase in NCC phosphorylation, as reported previously (and in Fig. 2A), which results in NCC activation (7). Under these conditions, KS-WNK1 induced both WNK4 and NCC phosphorylation. In contrast, in oocytes exposed to LCHS, a condition in which [Cl−]i is reduced (1), WNK4 phosphorylation and NCC phosphorylation were observed in all groups. Thus the presence of KS-WNK1-Δ11 mimics the effect of reducing [Cl−]i on WNK4. Interestingly, in LCHS WNK4 phosphorylation was significantly increased in the presence of L-WNK1, supporting previous work that suggested trans-phosphorylation between WNKs (31), which is probably promoted by intracellular chloride depletion.
Fig. 6.

Kidney-specific with no lysine kinase 1 (KS-WNK1) and WNK4 coexpression results in WNK4 phosphorylation. A: representative Western blot showing the effect of long-variant-WNK1 lacking exon 11 (L-WNK1-Δ11) or KS-WNK1-Δ11, as stated, on Na+:Cl− cotransporter (NCC) and WNK4 expression and phosphorylation under control conditions. The blot shows L-WNK1/KS-WNK1, WNK4, pWNK4 S335, pNCC, and NCC, as indicated. The experiment was conducted under both control and low-chloride hypotonic stress (LCHS) conditions, as stated. B: densitometric analysis of the phosphoWNK4/total WNK4 ratio from 4 different experiments. *P < 0.05 vs. H2O-coinjected oocytes. C: effect of wild-type WNK4 (shaded bars) or WNK4 kinase death (KD) (solid bars) on NCC activity in oocytes injected with NCC cRNA, NCC + L-WNK1-Δ11, or NCC + KS-WNK1-Δ11. Thiazide-sensitive 22Na+ uptake in oocytes injected with NCC alone was set to 100%, and the other groups were normalized accordingly. *P < 0.01; n = 3. D: effect of KS-WNK1 on NCC activity in the absence or presence of WNK4 (shaded bars). Thiazide-sensitive 22Na+ uptake in oocytes injected with NCC alone was set to 100%, and the oocytes injected with KS-WNK1 were normalized accordingly (open bars); thiazide-sensitive 22Na+ uptake in oocytes injected with NCC + WNK4 was set to 100%, and the oocytes injected with KS-WNK1 group were normalized accordingly. *P < 0.0001; n = 3.
WNK4 activation in the presence of KS-WNK1-Δ11 is also supported by the following observation. As previously shown (7), under control conditions, wild-type WNK4 or inactive WNK4-KD inhibits the effect of L-WNK1 overexpression on NCC (Fig. 6B). This can be secondary to a WNK4-induced dominant-negative effect resulting from either the formation of inactive WNK4 dimers that sequester SPAK and NCC or by the formation of heterodimers between inactive WNK4 and L-WNK1 that are inactive at the oocyte [Cl−]i. Figure 6B shows that, in similar circumstances, wild-type WNK4 did not inhibit the KS-WNK1-Δ11-induced activation of NCC, suggesting that WNK4-KS-WNK1 dimers are active. Indeed, in contrast to what was observed with wild-type WNK4, catalytically inactive WNK4-KD inhibited the effect of KS-WNK1 on NCC (Fig. 6B). If KS-WNK1 induces the activation of WNK4, the coinjection of KS-WNK1 and WNK4 would result in the synergic activation of NCC, further increasing the activity of the cotransporter. As shown in Fig. 6C, this effect was not evident. However, under control conditions, WNK4 inhibits the activity of NCC; we therefore analyzed the effect of KS-WNK1 compared with the NCC alone or NCC + WNK4 conditions. As shown in Fig. 6D, coinjection with KS-WNK1 increased the activity by 253 ± 14% compared with the NCC baseline (P < 0.05). In contrast, when NCC + WNK4 was taken as 100%, the activation induced by adding KS-WNK1 increased by 595 ± 53%. Thus, in the presence of WNK4, KS-WNK1 exerts a more powerful effect on NCC activity.
WNK4 activation by KS-WNK1 requires interaction between the variants.
To explore whether WNK4 activation by KS-WNK1-Δ11 is dependent on physical interaction, we used coimmunoprecipitation to show that KS-WNK1-Δ11 and WNK4 interact (Fig. 7A). We also observed that fraction of WNK4 in the flowthrough (Fig. 7A), indicating that some of the WNK4 peptides interact with KS-WNK1-Δ11, whereas others do not. Importantly, phosphorylated WNK4 was only detected in the immunoprecipitated fraction, suggesting that only the copies of WNK4 that interact with KS-WNK1 become phosphorylated. In addition, coinjection of WNK4 with KS-WNK1-Δ11-HQ/AA did not result in WNK4 immunoprecipitation or phosphorylation (Fig. 7A). These data suggest that the interaction of KS-WNK1-Δ11 with WNK4 is responsible for the increased WNK4 T-loop phosphorylation. However, it remains possible that, in the presence of KS-WNK1-Δ11, WNK4 becomes indirectly phosphorylated via an unknown mechanism and that KS-WNK1-Δ11 binds only phosphorylated WNK4. To eliminate this possibility, we performed a similar immunoprecipitation experiment using WNK4-S335A cDNA, which cannot be phosphorylated, and WNK4-L322F cDNA, which is constitutively phosphorylated. As shown in Fig. 7B, both clones bound KS-WNK1-Δ11, but only the L322F clone displayed increased phosphorylation. These results suggest that WNK4 phosphorylation status does not modulate its binding to KS-WNK1-Δ11 and support our proposal that binding to KS-WNK1-Δ11 is required for WNK4 phosphorylation. Furthermore, we tested the effect of KS-WNK1-Δ11-Δ4a. As shown in Fig. 1B, KS-WNK1-Δ11-Δ4a lacks a positive effect on NCC. Consistent with this, KS-WNK1-Δ11-Δ4a did not induce WNK4 phosphorylation, even though both proteins interacted as evidenced by coprecipitation (Fig. 7C).
Fig. 7.
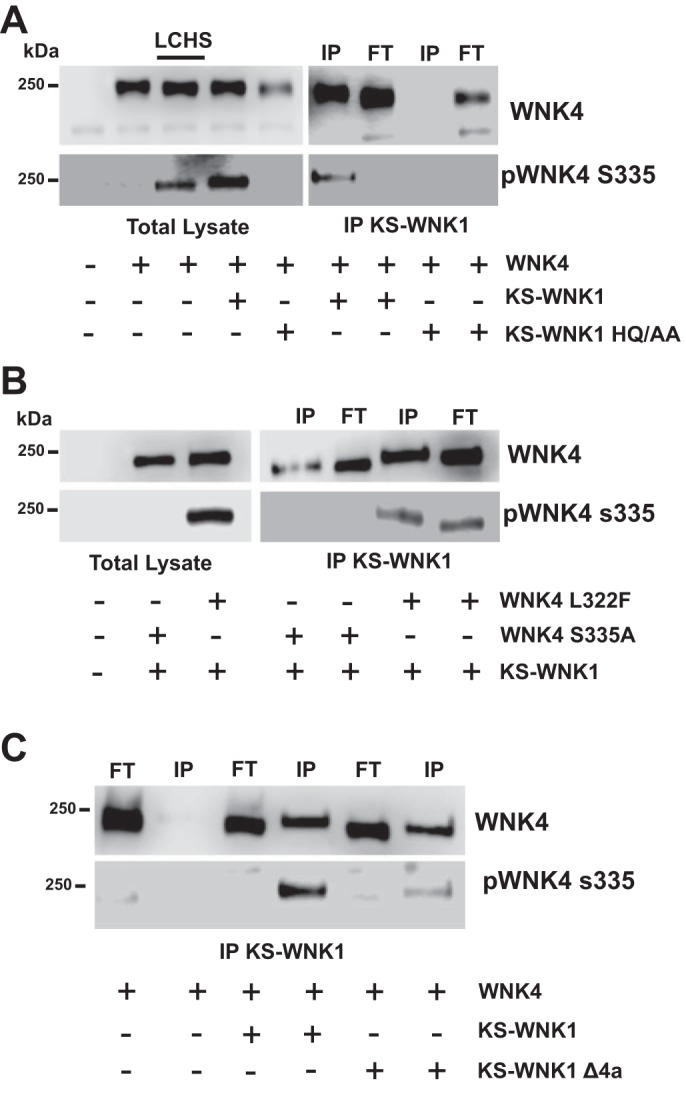
Kidney-specific with no lysine kinase 1 (KS-WNK1) promotes WNK4 S335 phosphorylation through heterodimer formation. A: representative Western blot of the total lysate and KS-WNK1 lacking exon 11 (KS-WNK1-Δ11) immunoprecipitation (IP) from oocytes injected with WNK4 cRNA alone or together with KS-WNK1-Δ11 cRNA or KS-WNK1-Δ11 HQ/AA, as stated. IP was performed using the c-Myc tag antibody, which exclusively recognizes KS-WNK1-Δ11. Left: blot from total lysate shows total WNK4 and pWNK4 expression, as stated. Lanes were loaded as follows: lane 1: H20-injected oocytes; lane 2: WNK4-injected oocytes; lane 3: WNK4-injected oocytes exposed to low-chloride hypotonic stress (LCHS) conditions; lane 4: WNK4 + KS-WNK1-Δ11-injected oocytes; lane 5: WNK4 + KS-WNK1-Δ11 HQ/AA-injected oocytes. Right: blot from the IP assay. IP was performed against the c-Myc tag, which recognizes KS-WNK1-Δ11 and revealed against WNK4 or pWNK4, as stated. Lanes were loaded as follows: lane 1: IP from WNK4 + KS-WNK1-Δ11-injected oocytes; lane 2: flowthrough of WNK4 + KS-WNK1-Δ11-injected oocytes; lane 3: IP from WNK4 + KS-WNK1-Δ11 HQ/AA-injected oocytes (the absence of WNK4 in the IP confirms the specificity of the c-Myc IP); lane 4: flowthrough from WNK4 + KS-WNK1-Δ11 HQ/AA-injected oocytes. Identical results were observed in 3 independent experiments. B: representative Western blot of the total lysate and KS-WNK1-Δ11 IP from oocytes injected with WNK4 L322F cRNA or WNK4 S335A alone or together with KS-WNK1-Δ11 cRNA, as stated. Left: blot from total lysate shows total WNK4 and pWNK4, as stated. Right: blot from the IP assay. Lanes 1 and 3 were loaded with proteins from the IP fraction, whereas lanes 2 and 4 were loaded with proteins from the flowthrough, as stated. The blot shows pWNK4 and WNK4 expression, as stated. Identical results were observed in 3 different experiments. C: representative Western blot of the total lysate and KS-WNK1-Δ11 IP from oocytes injected with WNK4 S335A cRNA together with KS-WNK1-Δ11 or KS-WNK1-Δ11-Δ4a cRNA, as stated. Blot from the IP assay is shown. Lanes 1, 3, and 5 were loaded with proteins from the flowthrough, whereas lanes 2, 4, and 6 were loaded with proteins from the IP fraction, as stated. The blot shows total WNK4 and pWNK4 expression, as stated. Identical results were obtained from 3 independent experiments.
The observations presented in this report appear to contradict previously published data obtained using genetically altered animals. Huang and coworkers (19) have shown that transgenic mice overexpressing the 1-257-amino-terminal residues of KS-WNK1 exhibited decreased NCC phosphorylation. The authors thus suggested an inhibitory role for KS-WNK1 in the regulation of NCC. However, in contrast to full-length KS-WNK1-Δ11, which activates NCC (Fig. 1A), the 1-257 fragment, when coexpressed with L-WNK1 or WNK4, exerts a dominant-negative effect as a WNK1 inhibitor (Fig. 8). Thus it is possible that the inhibition of NCC observed in 1-257-overexpressing mice is a reflection of the inhibitory property of this fragment of KS-WNK1 on L-WNK1 and/or WNK4.
Fig. 8.
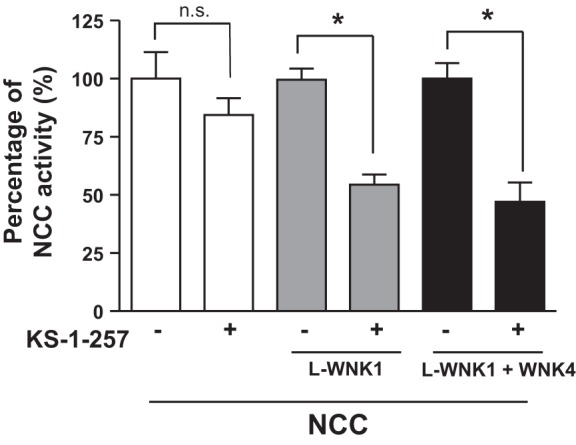
Kidney-specific with no lysine kinase 1 (KS-WNK1)-1-257 reduces the effect of long-variant (L)-WNK1 and WNK4 on Na+:Cl− cotransporter (NCC). Functional expression assay shows the NCC activity in groups of oocytes injected with NCC cRNA alone (open bars), NCC + L-WNK1 lacking exon 11 (L-WNK1-Δ11) cRNA (shaded bars), or NCC + WNK4 cRNA (shaded bars) in the absence or presence of KS-1-257 cRNA, as stated. The level of uptake in oocytes injected with NCC alone, NCC + L-WNK1-Δ11, or NCC + WNK4 was taken as 100%, and the groups coinjected with KS-1-257 were normalized accordingly. *P < 0.05; n = 3.
DISCUSSION
The results in the present study suggest that, when expressed alone, WNK4 is inhibited by the high [Cl−]i present in oocytes (1) and thus is unable to activate SPAK and NCC; however, in the presence of KS-WNK1, WNK4 exhibits reduced sensitivity to [Cl−]i, resulting in the activation of this kinase and thus SPAK/NCC, despite no change in [Cl−]i (illustrated in Fig. 9). Given that KS-WNK1 possesses no kinase domain and thus no chloride-binding sites (21), it is possible that, if one component of the dimer is insensitive to chloride, the entire dimer becomes active despite no change in chloride concentration. Thus, by interacting with other WNKs, KS-WNK1 is able to induce activation of NCC. The observation that KS-WNK1 effect on NCC is completely abrogated by the specific WNK inhibitor WNK463 strongly supports the hypothesis that activation of NCC by KS-WNK1 is due to KS-WNK1-induced activation of an endogenous WNK kinase.
Fig. 9.

A schematic illustrating the role of kidney-specific with no lysine kinase 1 (KS-WNK1) in the activation of WNK4. Left: in the absence of KS-WNK1, WNK4-WNK4 dimers are highly sensitive to chloride, which binds to WNK4 in the chloride motif of the kinase domain, thereby precluding the autophosphorylation and thus activation of WNK4. Right: in the presence of KS-WNK1, WNK4 is able to form dimers with KS-WNK1, which are likely unable to bind chloride ions because of the absence of the kinase domain (KD) and thus the absence of the chloride-binding site in KS-WNK1. The resulting KS-WNK1-WNK4 heterodimer is thus insensitive to chloride, allowing WNK4 to become phosphorylated and thus to gain activity toward STE20-proline-alanine rich kinase-Na+:Cl− cotransporter (SPAK-NCC).
The observations in the present study were all done using the Xenopus laevis expression system. This is not a mammalian cell, and results observed in this system are not necessarily translated to mammalian cells or to in vivo models. However, many observations in oocytes have been corroborated in mammalian systems over the years. In addition, observations in oocytes have been used in many occasions as a starting point of enlightening observations that are worth exploring with more detail using mammalian cells or in vivo models. This could be the case of KS-WNK1 whose role in DCT physiology remains elusive. In this regard, previous reports have proposed that KS-WNK1 might act as a dominant-negative regulator of L-WNK1. However, we observed that coexpression of L-WNK1-Δ11 with KS-WNK1-Δ11 did not result in the inhibition of L-WNK1-Δ11 (Fig. 2B). In this regard, a recent study at the cellular level strongly suggests that in DCT KS-WNK1 is required for WNK activation. In this work, Boyd-Shiwarski et al. (3) showed that stimuli known to activate the WNK-SPAK-NCC pathway, such as hypokalemia, promote the fact that WNKs concentrate in small punctate that are referred to as WNK bodies and that formation of these bodies is a hallmark of WNK activation. The authors show that the presence of KS-WNK1 is critical for WNK body formation, suggesting that it is involved in WNK activation.
The observations presented in the present study also appear counterintuitive in light of previous work showing that KS-WNK1 knockout mice exhibit increased expression/phosphorylation of NCC (15). Although we cannot yet offer an explanation for this finding, we believe that the increased abundance of NCC in KS-WNK1 knockout mice might not be directly related to the absence of KS-WNK1 and could represent a compensatory mechanism. It is well documented that an increase in NCC activity leads to the development of hypertension and hyperkalemia. It is believed that increased blood pressure and hyperkalemia in patients with FHHt are due to increased activity of NCC associated with decreased degradation of WNKs. Transgenic mice expressing WNK4 or WNK1 FHHt-type mutations exhibit increased NCC activity (phosphorylation), which is associated with hypertension and hyperkalemia (17, 33, 41). In support of this finding, Grimm et al. (13) recently reported that constitutive activation of SPAK exclusively in DCT1 results in increased activation/phosphorylation of NCC, with the consequent development of arterial hypertension and hyperkalemia. These phenotypes were resolved by inhibiting NCC with thiazide-type diuretics. Thus it is clear that a primary activation of NCC results in hypertension and hyperkalemia. In this regard, it is interesting to note that, in addition to increased NCC expression/phosphorylation exhibited by KS-WNK1 knockout mice, these mice also display a reduction in epithelial Na channel abundance and function, a physiological state that leads to decreased potassium loss in the urine (15). The fact that hypertension and hyperkalemia are not observed in these animals supports the possibility that the observed increase in NCC is not the primary phenotypic alteration because it is not accompanied by hypertension and hyperkalemia. Instead, it is possible that a potassium loss phenotype caused by the absence of KS-WNK1 is compensated by a secondary increase of NCC activity. Several similar conditions are consistently observed in physiology, including, for instance, primary vs. secondary aldosteronism or hyperparathyroidism. In light of our evidence that KS-WNK1 is a powerful activator of NCC, further studies are necessary to reevaluate the phenotype of KS-WNK1 knockout mice and fully characterize the physiological role of KS-WNK1.
Our observations could help to explain the mechanism of FHHt disease caused by mutations in WNK4. The hypertension and hyperkalemia present in this disease result in reduced angiotensin II levels and increased [Cl−]i in DCT. These changes would in turn be expected to inactivate WNK4. KS-WNK1 is highly expressed in DCT cells. Thus any interference with WNK4 degradation that upregulates the expression of the kinase, as has been postulated to occur with WNK4, KLHL3, or CUL3 mutations (22, 27, 36, 39), increases the amount of WNK4 in the cells and thus the probability that KS-WNK1-WNK4 dimers formation that will remain active regardless of hypertensive and hyperkalemic state. This could also help to explain the exquisite phenotype observed in FHHt upon increased WNK4 in DCT1, even though the kinase is expressed in many other tissues without producing clinical consequences. Perhaps the absence of KS-WNK1 outside the DCT renders WNK4 highly sensitive to hyperkalemia, thereby inhibiting an effect in other tissues.
Perspectives
The results in the present study show that KS-WNK1 is an activator of the WNK-SPAK-NCC pathway. In addition to our observations, a recent study suggests that formation of WNK bodies in DCT is associated with WNK activation and that KS-WNK1 seems to be part of the WNK bodies. These studies together suggest a previously unsuspected role for KS-WNK1 in the modulation of other WNK kinases and thus the SPAK-NCC pathway. Given that KS-WNK1 is expressed almost exclusively in DCT, this protein could play an important role to explain the fact that most if not all the clinical consequences of FHHt are due to NCC activation. The existence of several genetically altered mice of the WNK-SPAK-NCC pathway could be used in the near future to elucidate the role of KS-WNK1 in DCT physiology.
GRANTS
This work was supported by the collaborative grant no. 188712 from CONACyT (Mexico) and ANR-12-ISVS1-0001-01 (France) to G. Gamba and J. Hadchouel, the grant IN207716 from DGAPA, UNAM to G. Gamba, and the NIDDK RO1 grant no. DK051496-15 to D. Ellison, G, Gamba, and J. Hadchouel.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
E.R.A., M.C.-C., J.G.-V., J.H., D.H.E., and G.G. conceived and designed research; E.R.A., M.C.-C., M.O.-F., A.R.-G., N.V., and X.G.-R. performed experiments; E.R.A., M.C.-C., M.O.-F., A.R.-G., N.V., X.G.-R., J.G.-V., J.H., and G.G. analyzed data; E.R.A., M.C.-C., M.O.-F., X.G.-R., J.G.-V., D.H.E., and G.G. interpreted results of experiments; E.R.A., M.O.-F., N.V., and G.G. prepared figures; E.R.A. and G.G. drafted manuscript; E.R.A., M.C.-C., D.H.E., and G.G. edited and revised manuscript; E.R.A., M.C.-C., M.O.-F., A.R.-G., N.V., X.G.-R., J.G.-V., J.H., D.H.E., and G.G. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dario Alessi for the kind gift of WNK468 inhibitor. E. Argaiz was supported by a scholarship from CONACyT-Mexico and is a graduate student in the Doctorado en Ciencias Biomédicas program of the Universidad Nacional Autónoma de México.
REFERENCES
- 1.Bazúa-Valenti S, Chávez-Canales M, Rojas-Vega L, González-Rodríguez X, Vázquez N, Rodríguez-Gama A, Argaiz ER, Melo Z, Plata C, Ellison DH, García-Valdés J, Hadchouel J, Gamba G. The effect of WNK4 on the Na+-Cl- cotransporter is modulated by intracellular chloride. J Am Soc Nephrol 26: 1781–1786, 2015. doi: 10.1681/ASN.2014050470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bazúa-Valenti S, Gamba G. Revisiting the NaCl cotransporter regulation by with-no-lysine kinases. Am J Physiol Cell Physiol 308: C779–C791, 2015. doi: 10.1152/ajpcell.00065.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boyd-Shiwarski CR, Shiwarski DJ, Roy A, Nkashama LJ, Namboodiri HN, Xie J, McClain KL, Marciszyn A, Kleyman TR, Tan RJ, Stolz DB, Puthenveedu MA, Huang CL, Subramanya AR. Potassium-regulated distal tubule WNK bodies are kidney-specific WNK1 dependent. Mol Biol Cell 29: 499-509, 2017. doi: 10.1091/mbc.E17-08-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Castañeda-Bueno M, Cervantes-Perez LG, Rojas-Vega L, Arroyo-Garza I, Vázquez N, Moreno E, Gamba G. Modulation of NCC activity by low and high K+ intake: insights into the signaling pathways involved. Am J Physiol Renal Physiol 306: F1507–F1519, 2014. doi: 10.1152/ajprenal.00255.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Castañeda-Bueno M, Cervantes-Pérez LG, Vázquez N, Uribe N, Kantesaria S, Morla L, Bobadilla NA, Doucet A, Alessi DR, Gamba G. Activation of the renal Na+:Cl- cotransporter by angiotensin II is a WNK4-dependent process. Proc Natl Acad Sci USA 109: 7929–7934, 2012. doi: 10.1073/pnas.1200947109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang MH, Plata C, Zandi-Nejad K, Sindić A, Sussman CR, Mercado A, Broumand V, Raghuram V, Mount DB, Romero MF. Slc26a9–anion exchanger, channel and Na+ transporter. J Membr Biol 228: 125–140, 2009. doi: 10.1007/s00232-009-9165-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chávez-Canales M, Zhang C, Soukaseum C, Moreno E, Pacheco-Alvarez D, Vidal-Petiot E, Castañeda-Bueno M, Vázquez N, Rojas-Vega L, Meermeier NP, Rogers S, Jeunemaitre X, Yang CL, Ellison DH, Gamba G, Hadchouel J. WNK-SPAK-NCC cascade revisited: WNK1 stimulates the activity of the Na-Cl cotransporter via SPAK, an effect antagonized by WNK4. Hypertension 64: 1047–1053, 2014. doi: 10.1161/HYPERTENSIONAHA.114.04036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiga M, Rai T, Yang SS, Ohta A, Takizawa T, Sasaki S, Uchida S. Dietary salt regulates the phosphorylation of OSR1/SPAK kinases and the sodium chloride cotransporter through aldosterone. Kidney Int 74: 1403–1409, 2008. doi: 10.1038/ki.2008.451. [DOI] [PubMed] [Google Scholar]
- 9.Delaloy C, Lu J, Houot AM, Disse-Nicodeme S, Gasc JM, Corvol P, Jeunemaitre X. Multiple promoters in the WNK1 gene: one controls expression of a kidney-specific kinase-defective isoform. Mol Cell Biol 23: 9208–9221, 2003. doi: 10.1128/MCB.23.24.9208-9221.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ellison DH, Terker AS, Gamba G. Potassium and its discontents: new insight, new treatments. J Am Soc Nephrol 27: 981–989, 2016. doi: 10.1681/ASN.2015070751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gamba G. Regulation of the renal Na+-Cl− cotransporter by phosphorylation and ubiquitylation. Am J Physiol Renal Physiol 303: F1573–F1583, 2012. doi: 10.1152/ajprenal.00508.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gamba G, Miyanoshita A, Lombardi M, Lytton J, Lee WS, Hediger MA, Hebert SC. Molecular cloning, primary structure, and characterization of two members of the mammalian electroneutral sodium-(potassium)-chloride cotransporter family expressed in kidney. J Biol Chem 269: 17713–17722, 1994. [PubMed] [Google Scholar]
- 13.Grimm PR, Coleman R, Delpire E, Welling PA. Constitutively active SPAK causes hyperkalemia by activating NCC and remodeling distal tubules. J Am Soc Nephrol 28: 2597–2606, 2017. doi: 10.1681/ASN.2016090948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hadchouel J, Ellison DH, Gamba G. Regulation of renal electrolyte transport by WNK and SPAK-OSR1 kinases. Annu Rev Physiol 78: 367–389, 2016. doi: 10.1146/annurev-physiol-021115-105431. [DOI] [PubMed] [Google Scholar]
- 15.Hadchouel J, Soukaseum C, Büsst C, Zhou XO, Baudrie V, Zürrer T, Cambillau M, Elghozi JL, Lifton RP, Loffing J, Jeunemaitre X. Decreased ENaC expression compensates the increased NCC activity following inactivation of the kidney-specific isoform of WNK1 and prevents hypertension. Proc Natl Acad Sci USA 107: 18109–18114, 2010. doi: 10.1073/pnas.1006128107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang Y, Ferguson WB, Peng JB. WNK4 enhances TRPV5-mediated calcium transport: potential role in hypercalciuria of familial hyperkalemic hypertension caused by gene mutation of WNK4. Am J Physiol Renal Physiol 292: F545–F554, 2007. doi: 10.1152/ajprenal.00187.2006. [DOI] [PubMed] [Google Scholar]
- 17.Lalioti MD, Zhang J, Volkman HM, Kahle KT, Hoffmann KE, Toka HR, Nelson-Williams C, Ellison DH, Flavell R, Booth CJ, Lu Y, Geller DS, Lifton RP. Wnk4 controls blood pressure and potassium homeostasis via regulation of mass and activity of the distal convoluted tubule. Nat Genet 38: 1124–1132, 2006. doi: 10.1038/ng1877. [DOI] [PubMed] [Google Scholar]
- 18.Lifton RP, Gharavi AG, Geller DS. Molecular mechanisms of human hypertension. Cell 104: 545–556, 2001. doi: 10.1016/S0092-8674(01)00241-0. [DOI] [PubMed] [Google Scholar]
- 19.Liu Z, Xie J, Wu T, Truong T, Auchus RJ, Huang CL. Downregulation of NCC and NKCC2 cotransporters by kidney-specific WNK1 revealed by gene disruption and transgenic mouse models. Hum Mol Genet 20: 855–866, 2011. doi: 10.1093/hmg/ddq525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nessler S, Friedrich O, Bakouh N, Fink RH, Sanchez CP, Planelles G, Lanzer M. Evidence for activation of endogenous transporters in Xenopus laevis oocytes expressing the Plasmodium falciparum chloroquine resistance transporter, PfCRT. J Biol Chem 279: 39438–39446, 2004. doi: 10.1074/jbc.M404671200. [DOI] [PubMed] [Google Scholar]
- 21.O’Reilly M, Marshall E, Speirs HJ, Brown RW. WNK1, a gene within a novel blood pressure control pathway, tissue-specifically generates radically different isoforms with and without a kinase domain. J Am Soc Nephrol 14: 2447–2456, 2003. doi: 10.1097/01.ASN.0000089830.97681.3B. [DOI] [PubMed] [Google Scholar]
- 22.Ohta A, Schumacher FR, Mehellou Y, Johnson C, Knebel A, Macartney TJ, Wood NT, Alessi DR, Kurz T. The CUL3-KLHL3 E3 ligase complex mutated in Gordon’s hypertension syndrome interacts with and ubiquitylates WNK isoforms: disease-causing mutations in KLHL3 and WNK4 disrupt interaction. Biochem J 451: 111–122, 2013. doi: 10.1042/BJ20121903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pacheco-Alvarez D, Cristóbal PS, Meade P, Moreno E, Vazquez N, Muñoz E, Díaz A, Juárez ME, Giménez I, Gamba G. The Na+:Cl- cotransporter is activated and phosphorylated at the amino-terminal domain upon intracellular chloride depletion. J Biol Chem 281: 28755–28763, 2006. doi: 10.1074/jbc.M603773200. [DOI] [PubMed] [Google Scholar]
- 24.Piala AT, Moon TM, Akella R, He H, Cobb MH, Goldsmith EJ. Chloride sensing by WNK1 involves inhibition of autophosphorylation. Sci Signal 7: ra41, 2014. doi: 10.1126/scisignal.2005050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richardson C, Rafiqi FH, Karlsson HK, Moleleki N, Vandewalle A, Campbell DG, Morrice NA, Alessi DR. Activation of the thiazide-sensitive Na+-Cl- cotransporter by the WNK-regulated kinases SPAK and OSR1. J Cell Sci 121: 675–684, 2008. doi: 10.1242/jcs.025312. [DOI] [PubMed] [Google Scholar]
- 26.San-Cristobal P, Ponce-Coria J, Vázquez N, Bobadilla NA, Gamba G. WNK3 and WNK4 amino-terminal domain defines their effect on the renal Na+-Cl− cotransporter. Am J Physiol Renal Physiol 295: F1199–F1206, 2008. doi: 10.1152/ajprenal.90396.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shibata S, Zhang J, Puthumana J, Stone KL, Lifton RP. Kelch-like 3 and Cullin 3 regulate electrolyte homeostasis via ubiquitination and degradation of WNK4. Proc Natl Acad Sci USA 110: 7838–7843, 2013. doi: 10.1073/pnas.1304592110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Subramanya AR, Yang CL, Zhu X, Ellison DH. Dominant-negative regulation of WNK1 by its kidney-specific kinase-defective isoform. Am J Physiol Renal Physiol 290: F619–F624, 2006. doi: 10.1152/ajprenal.00280.2005. [DOI] [PubMed] [Google Scholar]
- 29.Terker AS, Zhang C, Erspamer KJ, Gamba G, Yang CL, Ellison DH. Unique chloride-sensing properties of WNK4 permit the distal nephron to modulate potassium homeostasis. Kidney Int 89: 127–134, 2016. doi: 10.1038/ki.2015.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Terker AS, Zhang C, McCormick JA, Lazelle RA, Zhang C, Meermeier NP, Siler DA, Park HJ, Fu Y, Cohen DM, Weinstein AM, Wang WH, Yang CL, Ellison DH. Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab 21: 39–50, 2015. doi: 10.1016/j.cmet.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thastrup JO, Rafiqi FH, Vitari AC, Pozo-Guisado E, Deak M, Mehellou Y, Alessi DR. SPAK/OSR1 regulate NKCC1 and WNK activity: analysis of WNK isoform interactions and activation by T-loop trans-autophosphorylation. Biochem J 441: 325–337, 2012. doi: 10.1042/BJ20111879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vidal-Petiot E, Cheval L, Faugeroux J, Malard T, Doucet A, Jeunemaitre X, Hadchouel J. A new methodology for quantification of alternatively spliced exons reveals a highly tissue-specific expression pattern of WNK1 isoforms. PLoS One 7: e37751, 2012. doi: 10.1371/journal.pone.0037751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vidal-Petiot E, Elvira-Matelot E, Mutig K, Soukaseum C, Baudrie V, Wu S, Cheval L, Huc E, Cambillau M, Bachmann S, Doucet A, Jeunemaitre X, Hadchouel J. WNK1-related familial hyperkalemic hypertension results from an increased expression of L-WNK1 specifically in the distal nephron. Proc Natl Acad Sci USA 110: 14366–14371, 2013. doi: 10.1073/pnas.1304230110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vitari AC, Deak M, Morrice NA, Alessi DR. The WNK1 and WNK4 protein kinases that are mutated in Gordon’s hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem J 391: 17–24, 2005. doi: 10.1042/BJ20051180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vitari AC, Thastrup J, Rafiqi FH, Deak M, Morrice NA, Karlsson HK, Alessi DR. Functional interactions of the SPAK/OSR1 kinases with their upstream activator WNK1 and downstream substrate NKCC1. Biochem J 397: 223–231, 2006. doi: 10.1042/BJ20060220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wakabayashi M, Mori T, Isobe K, Sohara E, Susa K, Araki Y, Chiga M, Kikuchi E, Nomura N, Mori Y, Matsuo H, Murata T, Nomura S, Asano T, Kawaguchi H, Nonoyama S, Rai T, Sasaki S, Uchida S. Impaired KLHL3-mediated ubiquitination of WNK4 causes human hypertension. Cell Reports 3: 858–868, 2013. doi: 10.1016/j.celrep.2013.02.024. [DOI] [PubMed] [Google Scholar]
- 37.Wilson FH, Disse-Nicodème S, Choate KA, Ishikawa K, Nelson-Williams C, Desitter I, Gunel M, Milford DV, Lipkin GW, Achard JM, Feely MP, Dussol B, Berland Y, Unwin RJ, Mayan H, Simon DB, Farfel Z, Jeunemaitre X, Lifton RP. Human hypertension caused by mutations in WNK kinases. Science 293: 1107–1112, 2001. doi: 10.1126/science.1062844. [DOI] [PubMed] [Google Scholar]
- 38.Wilson FH, Kahle KT, Sabath E, Lalioti MD, Rapson AK, Hoover RS, Hebert SC, Gamba G, Lifton RP. Molecular pathogenesis of inherited hypertension with hyperkalemia: the Na-Cl cotransporter is inhibited by wild-type but not mutant WNK4. Proc Natl Acad Sci USA 100: 680–684, 2003. doi: 10.1073/pnas.242735399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu G, Peng JB. Disease-causing mutations in KLHL3 impair its effect on WNK4 degradation. FEBS Lett 587: 1717–1722, 2013. doi: 10.1016/j.febslet.2013.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamada K, Park HM, Rigel DF, DiPetrillo K, Whalen EJ, Anisowicz A, Beil M, Berstler J, Brocklehurst CE, Burdick DA, Caplan SL, Capparelli MP, Chen G, Chen W, Dale B, Deng L, Fu F, Hamamatsu N, Harasaki K, Herr T, Hoffmann P, Hu QY, Huang WJ, Idamakanti N, Imase H, Iwaki Y, Jain M, Jeyaseelan J, Kato M, Kaushik VK, Kohls D, Kunjathoor V, LaSala D, Lee J, Liu J, Luo Y, Ma F, Mo R, Mowbray S, Mogi M, Ossola F, Pandey P, Patel SJ, Raghavan S, Salem B, Shanado YH, Trakshel GM, Turner G, Wakai H, Wang C, Weldon S, Wielicki JB, Xie X, Xu L, Yagi YI, Yasoshima K, Yin J, Yowe D, Zhang JH, Zheng G, Monovich L. Small-molecule WNK inhibition regulates cardiovascular and renal function. Nat Chem Biol 12: 896–898, 2016. doi: 10.1038/nchembio.2168. [DOI] [PubMed] [Google Scholar]
- 41.Yang SS, Morimoto T, Rai T, Chiga M, Sohara E, Ohno M, Uchida K, Lin SH, Moriguchi T, Shibuya H, Kondo Y, Sasaki S, Uchida S. Molecular pathogenesis of pseudohypoaldosteronism type II: generation and analysis of a Wnk4(D561A/+) knockin mouse model. Cell Metab 5: 331–344, 2007. doi: 10.1016/j.cmet.2007.03.009. [DOI] [PubMed] [Google Scholar]