

Abstract
Several lines of evidence suggest that gut bacterial microbiota is altered in patients with chronic kidney disease (CKD), though the mechanism of which this dysbiosis takes place is not well understood. Recent studies delineated changes in gut microbiota in both CKD patients and experimental animal models using microarray chips. We present 16S ribosomal RNA gene sequencing of both stool pellets and small bowel contents of C57BL/6J mice that underwent a remnant kidney model and establish that changes in microbiota take place in the early gastrointestinal tract. Increased intestinal urea concentration has been hypothesized as a leading contributor to dysbiotic changes in CKD. We show that urea transporters (UT)-A and UT-B mRNA are both expressed throughout the whole gastrointestinal tract. The noted increase in intestinal urea concentration appears to be independent of UTs’ expression. Urea supplementation in drinking water resulted in alteration in bacterial gut microbiota that is quite different than that seen in CKD. This indicates that increased intestinal urea concentration might not fully explain the CKD- associated dysbiosis.
Keywords: chronic kidney disease, gut bacterial microbiota, microbiology, urea transporters, uremic milieu
INTRODUCTION
The human gut microbiome is a diverse, microscopic ecosystem located in the confines of the gastrointestinal (GI) tract. Within these borders, the residing organismal fauna serve intricate roles in a number of physiological processes that can impact the overall health of their host (6). However, disruptions in the homeostasis between host and microbiome have been connected to the onset and progression of several disease states that include diabetes mellitus type 2, dyslipidemia, and obesity (47, 56). Research has shown that a principal way to initiate this “dysbiosis” is through alterations in the local luminal environment of the intestine, such as those exhibited through changes in diet or antibiotic use.
Emerging evidence suggests the gut microbiota is also modified in chronic kidney disease (CKD) (9, 23, 31, 58, 67, 76). Of note, a landmark study identified the effects of CKD on dysbiosis in both human subjects and a remnant kidney model in rats (67). Our current work builds upon these studies, aiming to quantitatively evaluate CKD-mediated changes in the microbiome. Although qualitative/semiquantitative differences in the gut microbiota have been shown in the context of CKD, previous studies relied upon either in vitro cultivation (23, 58) or microarray-based detection of gut bacteria (67). Both of these approaches have limitations that prevent accurate, quantitative interpretation. Here, we applied recent advances in high-throughput sequencing and attendant computational tools to quantitatively and phylogenetically evaluate the bacterial biodiversity in C57Bl/6J mice with CKD by analyzing the 16S ribosomal subunit (54). Unlike rats subjected to 5/6 nephrectomy, which can result in severe uremic comorbidities and rapid overall decline postoperatively (40, 41, 78), C57Bl/6J mice seem resistant to advanced uremia when subjected to a remnant kidney model wherein they continue to consume the same amount of food, exhibit similar weight gain as in controls, and their estimated glomerular filtration rate (eGFR) drops to half, making this strain a perfect model to study microbiota changes in moderate CKD (33).
Of the numerous events that occur following the initial decline in renal function, the buildup of intestinal urea has been hypothesized as a leading contributor for dysbiotic changes in CKD (22, 28, 42, 65, 67). Typically, the kidneys are responsible for the majority of urea excretion from the body. As renal function worsens in CKD patients, blood urea nitrogen (BUN) can accumulate because of the growing inability of the kidneys to remove the metabolic waste product. The increase in circulating urea can lead to its elevation in the intestines, where urease-expressing bacteria have the ability to subsequently metabolize the urea into ammonia and CO2 (22, 28, 42, 65, 67). Ammonia is later metabolized to ammonium hydroxide, which is believed to further damage the enterocytes, erodes the epithelial barrier, and triggers local inflammation and cytokine production (35, 66, 69). It is believed that these byproducts may directly impact the composition and overall function of the microbiome. Therefore, we additionally sought to evaluate if the oral administration of urea, on its own, could induce CKD-like dysbiotic changes in healthy mice.
METHODS
Animals and housing conditions.
Eight-week-old C57BL/6J male mice purchased from Jackson Laboratory (Bar Harbor, ME) were housed under specific pathogen-free conditions. Animal studies were performed in accordance with the guidelines approved by the University at Buffalo Institutional Animal Care and Use Committee. To account for cage effects on the gut bacterial microbiota and ensure similar gut microbiota between CKD and Sham cohort at baseline (bl), we used a standardized protocol (Fig. 1A). Briefly, upon arrival, mice were separated into two mice per cage and housed for 2 wk. Mice were then separated into individual cages with one mouse used for the CKD cohort and the other for Sham. Mice received standard laboratory chow ad libitum. Distilled autoclaved water was provided in bottles. Stool pellets were collected at bl and before euthanasia in a sterile fashion. A medial small bowel (SB) section was obtained at the time of euthanasia. Samples were snap frozen in liquid nitrogen and stored at −80°C.
Fig. 1.

Characteristics of experimental chronic kidney disease animal model. A: housing strategy and experimental timeline. B: no significant postoperative weight gain was noted between the two groups; change in weight (∆weight) calculated based on postoperative day 1 weight. C: nine weeks postoperatively, 5/6 Nephrectomy (NPX) mice showed doubling serum creatinine (Scr). D: nine weeks postoperatively, NPX mice showed doubling blood urea nitrogen (BUN). *P < 0.05.
Experimental CKD model.
A midline dorsal skin incision was performed. The left kidney was exposed via a left flank incision and decapsulated to avoid ureter and adrenal damage, and the upper pole was partially resected with a Bovie medical high-temperature cautery system. The contralateral (right) kidney was nephrectomized. The Sham cohort underwent a similar surgical preparation procedure, anesthesia, skin, and flank incisions and kidney decapsulation without any tissue removal. Both groups received the standard postoperative treatment that included immediate subcutaneous buprenorphine (0.1 mg/kg) and carprofen (5 mg/kg) daily for three days. No antibiotics were given perioperatively. Mice were monitored for 9 wk, euthanized under isoflurane anesthesia, and tissues collected for further analysis.
Urea supplementation in drinking water.
We first sought to determine the proper dose of urea in drinking water by performing a kinetic study after a single gavage urea dose. 10–12-wk-old C57BL/6J mice (n = 5 for each group and time point) received 300 µl of either 4 g/l urea in water or water as the vehicle control. Mice were euthanized at 30 min, 1, 3, and 5 h postgavage. An additional group of mice that did not receive any gavage were also euthanized the same day to serve as the bl for BUN. Based on the previous experiment results, we selected 4 g/l urea in drinking water. Eight-week-old C57BL/6J male mice housed under the same conditions as above were divided into two mice per cage and housed for 2 wk. Mice from the same cage were then single housed to either continue consuming the same drinking water or 4 g/l urea (Sigma-Aldrich, St. Louis, MO, Cat. no. 33247) in drinking water for 7 wk.
Urease functional analysis.
To assess the capacity of microbial urea hydrolysis in various treatment regiments, we predicted the community metagenome using PICRUSt v1.1.13 (34). PICRUSt assumes that closely related microbes have similar gene content, which allows investigators to make functional predictions involving bacteria yet to be cultured in isolation. By incorporating the predicted gene content of these bacteria, many known only by their 16S RNA sequence, we can establish a more complete picture of the community's functional capacity. In this instance, we focused on assessing the capacity of communities under urea supplementation to hydrolyze urea.
After adjusting for 16S RNA and gene copy numbers, we applied a centered log-ratio transformation to account for the compositional nature of the data (1) and used a two-sided t-test to identify differential abundances in the α, β, and γ urease subunits, as well as five urease accessory proteins. P values <0.05 were considered significant. Transformations and statistical tests were performed using scikit-bio v0.5.1 [Scikit-Bio (2015), http://scikit-bio.org] and scipy v0.19.1 (25) in Python v3.5 (63). KEGG Orthology IDs tested were: urease α subunit (K01428), urease β subunit (K01429), urease accessory proteins (K03187, K03188, K03189, K03190, K03192), and urease γ subunit (K01430).
Urea measurements.
BUN was measured in serum samples using a commercially available kit (BioAssay Systems, Hayward, CA), according to the manufacturer’s protocol. SB harvested from each mouse was immediately placed on dry ice and stored at −80°C until all mice were euthanized. Intestinal urea measurement experiment was all completed on the same day. The SB was thawed on ice with 2 ml of ice-cold DNAse-/RNAse-free sterile phosphate-buffered saline (PBS) solution and 1.0 mm glass beads (BioSpec Products, Bartlesville, OK, Cat. no. 11079110). The tube was vortexed for 3 min in 4°C, centrifuged (16,000 g for 5 min twice, 4°C), and the supernatant urea concentration measured using QuantiChrom Urea Assay Kit (Cat. no. DIUR-100) following the manufacturer’s instructions.
Quantitative PCR.
Mice were divided into CKD and Sham groups (n = 5) using the standardized protocol (Fig. 1A). Nine weeks postnephrectomy, mice were euthanized under isoflurane anesthesia. The gastrointestinal tract (SB and colon) was harvested, dissected, and flushed with ice-cold DNAse-/RNAse-free sterile PBS solution. Small sections from duodenum, proximal and distal halves of the SB (postduodenum), and the colon were placed in RNAlater and stored at −80°C. Primers for quantitative PCR were as follows: Solute carrier family 14, member 2 urea transporter (UT)-A, (5′-AAGGAGATGTCTGACAGCAACA-3′ 5′-and GGGCTGGGTGTGTATCCTG-3′), solute carrier family 14, member 1 (UT-B) (5′-TTAAAGTAGACCGGGGTGAAAAC-3′ and 5′-ACCCGTGACGTAGCCAAGTA-3′), and GAPDH (5′-GCCATCAACGACCCCTTCAT-3′ and 5′-ATGATGACCCGTTTGGCTCC-3′). PCR was performed using SYBR Green Master Mix (Applied Biosystems) and the Applied Biosystems 7500 Real-time PCR system. Gene expression was normalized to GAPDH, and fold-change in expression relative to the control group was calculated using the 2-ΔΔCt method.
Bacterial DNA isolation, 16S sequencing, annotation, and operational taxonomic units statistical analysis.
The v3-v4 hypervariable region of 16S rRNA was extracted from samples. After PCR amplification, the targeted RNA was sequenced on an Illumina MiSeq with v3 chemistry and 2 × 300 paired-end reads. Reads were joined using the default parameters in Paired-End reAd mergeR (PEAR v0.9.6) (79) and filtered to retain sequences in which at least 90% of the bases have a quality score greater than Q30 using the FASTX Toolkit v0.0.13 (21). Closed reference operational taxonomic unit (OTU) picking was performed against the Green Genes v13.8 database (13).
Statistical analyses.
OTU tables were collapsed to the phyla, genera, and species levels, and frequency was normalized for downstream analysis. Ordination analyses and diversity measures were calculated using Python v3.5 (63) and the SciKit-Bio library [Scikit-Bio (2015), http://scikit-bio.org]. Data manipulation and statistical analysis was performed with Pandas v0.20.2 (44), and numerical measures were calculated using NumPy v1.12.1 (62a), and SciPy v0.19.0 (25).
To assess data separation, we performed principal component analysis (PCA) between cases and controls. OTUs of interest were identified through PCA component weights and Student’s t-tests. Dimension reduction through PCA was performed using Scikit-Learn v0.18.1 (50). To evaluate the significance and quantification of cluster separation in PCA plots, we used the Mahalanobis metric to quantify the distance between group centroids and calculated a two-sample Hotelling's T2 statistic from the data. Then, an F-test was applied to determine the significance of the centroid separation (19).
Rarefaction data was subsampled to a collection of fixed depths without replacement using a hypergeometric model trained on the data. Smooth curves were generated by fitting the data to a power function of the form:
Data are expressed as mean ± standard deviation of the mean (x ± SD). The two-sided unpaired t-test and Mann-Whitney U nonparametric tests were used to analyze data between two groups after determination of data distributions and variance. The ANOVA with Bonferroni correction was used when more than two groups were present. GraphPad Prism software was used for statistical analysis. P values <0.05 were considered to be statistically significant, whereas P values between 0.05 and 0.1 are denoted as trends.
RESULTS
Characteristics of experimental CKD animal model.
C57BL/6J mice underwent either sham operation (Sham) or unilateral nephrectomy with simultaneous ipsilateral upper pole cautery resection (NPX) at 8 wk of age (n = 10 per group, Fig. 1A). Consistent with previous reports, we did not observe a change in body weight gain between groups during the 9-wk postoperative period (Fig. 1B) (33). Furthermore, serum creatinine and BUN both increased ~2.5-fold in the NPX group compared with Sham at euthanasia (Fig. 1, C and D). These results demonstrate the applicability of the remnant kidney model for the induction of CKD and its neutrality on weight gain.
Characterization of the CKD-associated gut bacterial microbiota dysbiosis.
We evaluated the microbiome from each mouse to characterize dysbiotic shifts in NPX animals. Stool samples collected at bl and 9 wk postintervention displayed no significant difference in the α diversity or the richness of the bacterial communities within a single sample between NPX and Sham groups (Fig. 2, A and B). However, the analysis of the mid-SB sections revealed a higher α diversity compared with the corresponding stool samples at bl and postintervention in both the NPX SB (P < 0.005, Fig. 2, A and B) and Sham (P < 0.01, Fig. 2B) groups.
Fig. 2.
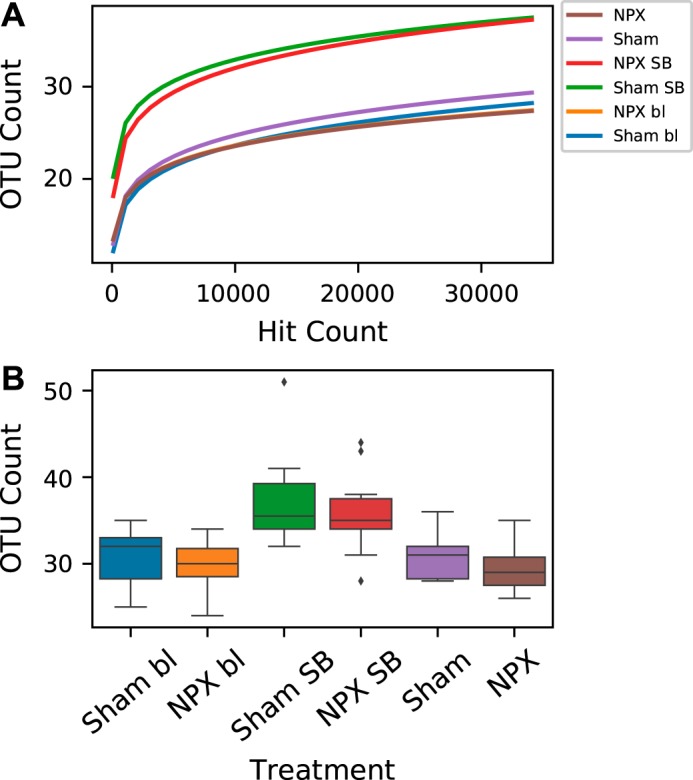
α diversity. α diversity based on operational taxonomic unit (OTU) hit count (A), and mean α diversity between all groups (B) indicates a significantly high richness in the small bowel (SB) of both groups when compared with the corresponding stool pellet richness. 5/6 Nephrectomy (NPX) mice at nine weeks old had insignificantly lower α diversity when compared with Sham group. No significant difference was measured between the two groups at baseline (bl).
Next, we examined the diversity in the microbial communities between biological groups, or β diversity. To evaluate the bacterial β diversity, we performed dimension reduction with PCA on the phyla, genera, and species levels between NPX mice and controls and evaluated the differences in the relative abundance of each annotated OTU on the same levels. Phyla, genera, and species level analysis revealed a significantly different projection onto principal components between NPX and Sham groups (P < 0.02, P < 0.001, P < 0.001; Fig. 3, A, B, and C, respectively). Bl bacterial diversity was similar in both groups (NPX bl vs. Sham bl) on the genera and species levels, as demonstrated by the nonsignificant separation (P > 0.1, Fig. 4, A, B, and C).
Fig. 3.

5/6 Nephrectomy (NPX)-induced changes in β diversity. Experimental chronic kidney disease model resulted in a significant alteration in the gut bacterial microbiota composition on the phyla (A), genera (B), and species (C) levels. **P < 0.01, ***P < 0.001. PC, Principal component.
Fig. 4.

Baseline (bl) β diversity at the phyla, genera, and species levels. Principal component (PC) analysis of β diversity between 5/6 Nephrectomy (NPX) and sham groups at base (preoperatively) indicates no significant difference on the phyla (A), genera (B), and species (C) levels. P > 0.1.
To identify the OTUs that best separate the NPX from Sham mice, we first compared the relative abundance of these taxa on phyla, genera, and species levels. At 9 wk postintervention, the two major phyla (Firmicutes and Bacteroidetes) were almost identical between the groups, and insignificant differences were seen among other phyla (Fig. 5, A and B). In contrast, we detected observable changes on both the genera and species levels (Fig. 6, A and B and Fig. 7, A and B). At the genera level, NPX mice exhibited a significantly lower relative abundance of Anaerostipes Spp. (P < 0.05). A trend of decreased relative abundances in NPX mice was also observed in two uncultured genera in the Clostridiales order (Firmicutes phylum), as well as an increased relative abundance of Akkermansia Spp. and Lactobacillus Spp. (P < 0.1). On the species level, NPX mice showed a lower relative abundance of an unidentified species in the Anaerostipes genus (P < 0.05). NPX mice display trends toward a decreased relative abundance in Blautia producta, two uncultured species in the Clostridiales order, and one uncultured species from the Mogibacteriaceae family (all from the Firmicutes phylum), and an uncultured species in the Bacteroides genus (Bacteroidetes phylum) was observed. Finally, NPX mice showed trends toward a higher relative abundance of Akkermansia muciniphila and an uncultured species in the genus Lactobacillus (P < 0.1; Fig. 6, B and C, Fig. 7B).
Fig. 5.

Relative abundance differences at the phyla level. No significant differences were noted between 5/6 Nephrectomy (NPX) and Sham groups on the phyla level. A: individual data. B: mean and standard deviation of groups.
Fig. 6.

Genera and species alteration in stool of 5/6 Nephrectomy (NPX) mice. Anaerostipes Spp. (A) and an uncultured species (B) in this genus significantly decreased in NPX group. Multiple genera and species showed a trend toward increased or decreased relative abundance in the NPX groups as shown in A and B. *P < 0.05. OTU, operational taxonomic unit.
Fig. 7:

Individual data of relative abundance at the genera and species levels. Individual data of 5/6 Nephrectomy (NPX) and Sham groups’ relative abundance of a selected genera (A) and species (B).
Because notable shifts were noted in various bacterial categories, we wanted to further evaluate if the identified OTUs (genera and species levels) were responsible for the significant differences between the two groups. This was accomplished by performing a sensitivity analysis utilizing heat map, hierarchical clustering, and PCA first on selected OTUs, followed by the identical analyses on the remaining groups, minus the selected OTUs. Disappearance of statistically significant differences between Sham and NPX after removing the selected OTUs would thus confirm the positive findings. We first evaluated the genera that had either a trend or a significant separation between the two groups. PCA revealed a significant divergence when the above genera were isolated (P < 0.02), which disappeared when the projection of the remaining genera were included (P = 0.36) (Fig. 8A, Fig. 9A). Heat map and hierarchical clustering of the samples using the selected genera resulted in a good separation between NPX and Sham groups. Again, this separation disappeared when the nonselected genera were analyzed (Fig. 8B, Fig. 9B). Similar findings were noted on the species level (Fig. 8, C and D, Fig. 9, B and C).
Fig. 8.

Selected operational taxonomic unit (OTU) analysis of stool pellet samples. Principal component (PC) analysis, heat map, and hierarchical clustering of selected OTUs that were either statistically significant or showed a trend between 5/6 Nephrectomy (NPX) and Sham groups on genera and species levels reveals a significant difference between the two groups on the genera level (A), with excellent auto-clustering (B). Similar findings noted on the species level (C and D). **P < 0.01.
Fig. 9.

β diversity comparison of the nonselected genera and species. Principal component (PC) analysis of the nonselected operational taxonomic units (OTUs) (total minus selected OTUs from Fig. 5) shows no significant differences between the 5/6 Nephrectomy (NPX) and Sham groups on the genera (A) and species (C) levels. Heat map and hierarchical clustering resulted in random clustering of OTUs (B and D), confirming the loss of significance between the two groups after eliminating the selected OTUs and the importance of these selected OTUs in clustering the NPX and Sham groups.
CKD affects upper gastrointestinal gut bacterial composition.
The upper GI microbiome in humans is heavily molded and maintained by the rapid uptake and fermentation of simple carbohydrates ingested in the diet (81). Microbiome alterations in this location have recently been observed in different clinical conditions that include obesity, antibiotics use, and smoking (18, 46, 71, 77). We hypothesized that similar microbial shifts occur in mice with surgically-induced CKD, which could explain a portion of the dysbiosis that we measured in stool samples.
To characterize the β diversity, similar analyses as described above were performed on mid-SB samples resected from the distal jejunum/proximal ileum. Similar to the stool pellets, mid-SB sections also consisted predominately of Firmicutes (~40%) and Bacteroidetes (39%) phyla. However, the relative abundance of Firmicutes decreased with a notable increase in Verrucomicrobia phylum (18%) as compared with stool pellets (Fig. 10A, Fig. 5A). Collectively, no difference was found on the phyla level between the NPX SB and Sham SB groups (P > 0.4, Fig. 11A) in both multivariate PCA analysis and when analyzed individually (Fig. 10, B and C).
Fig. 10.

Postoperative small bowel (SB) diversity on phyla level. No significant difference between 5/6 Nephrectomy (NPX) and Sham SB bacterial diversity was noted on the phyla level when analyzed individually (A) or collectively (B).
Fig. 11.

Effects of 5/6 Nephrectomy (NPX) on upper gastrointestinal tract bacterial microbiota composition. Mid small bowel (SB) section of NPX and Sham mice showed no significant difference on the phyla level (A). Significant difference was seen between the two groups on the genera (B), and species (C) levels. ***P < 0.001. PC, principal component.
On the genera level, distinct bacterial compositions were observed (Fig. 11B). A significant increase in the relative abundance of Lactobacillus Spp. (P < 0.04) was seen in NPX SB versus Sham SB, whereas decreases were observed in the relative abundance of Dehalobacterium Spp. (P < 0.03) and an uncluttered genus in the order Clostridiales (P < 0.03). Trends toward the decreased relative abundance of Oscillospira Spp., Sutterella Spp., and the family Ruminococcaceae were noted (Fig. 12, A and B). Each of these belong to the Firmicutes phylum, except for Proteobacteria Sutterella Spp. The shift in the relative abundance of these genera was mostly compatible with the trend in the stool pellets. However, the relative abundance of both Akkermansia Spp. and Anaerostipes Spp. were similar between the two groups in the SB. SB analysis on the species levels revealed similar findings (Fig. 11C). A significant increase in an uncultured species from the Lactobacillus genus, in addition to a decrease in the relative abundance of two uncultured species from the Dehalobacterium genus and the Clostridiales order, was seen (P < 0.05). A trend toward decreased relative abundance in Prevotella copri, uncultured species from the Oscillospira and Coprococcus genera, and the Ruminococcaceae family was observed (P < 0.1). The shift in the relative abundance of these species was mostly similar to the trend in the stool pellets (Fig. 6, A and B).
Fig. 12.

Genera and species alteration in small bowel (SB) of 5/6 Nephrectomy (NPX) mice. Genera (A) and species (B) analysis of SB section of NPX mice reveals a significant increase in Lactobacillus Spp. and an uncultured species in that genus, a significant decrease in an uncultured species of the Dehalobacterium genus, and an uncultured species from the Clostridiales family. *P < 0.05.
Similar to the previous experiments, we conducted a sensitivity analysis using heat map, hierarchical clustering, and PCAs. We first evaluated the seven genera that largely connected with this divergence. PCA revealed a significant separation when the above genera were used alone (P < 0.001), which disappeared when the projection of the remaining genera was performed (P = 0.69) (Fig. 13A, Fig. 14A). Heat map and hierarchical clustering of the samples using the selected genera resulted in a muddled separation between NPX and Sham; individual clusters can be identified as mostly NPX or Sham, though we see an alternating pattern where the nearest neighbor of a Sham cluster is an NPX cluster. This separation did not change when the nonselected genera were analyzed (Fig. 14, B and C). Similar findings were noted on the species level (Fig. 13B, Fig. 14, D, E, and F). In summary, our results demonstrate that CKD-associated gut bacterial dysbiosis takes place proximal to the colon.
Fig. 13.
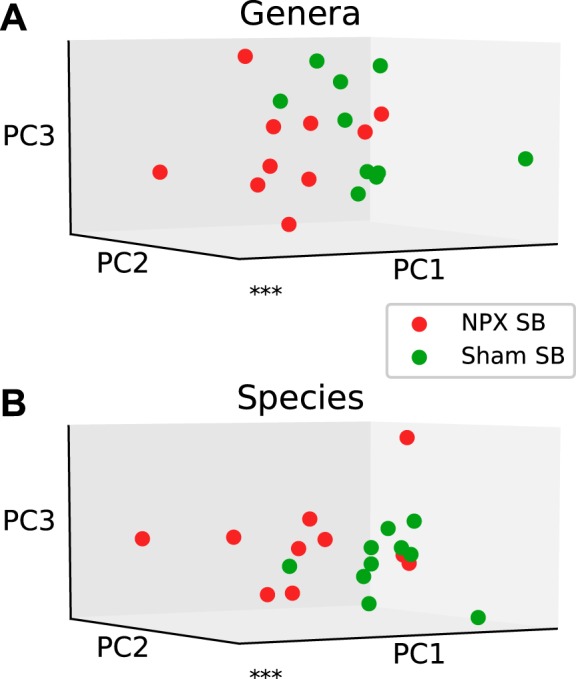
Small bowel (SB) selected operational taxonomic unit (OTU) analysis. Principal component (PC) analysis of selected OTUs that were either statistically significant or showed a trend between 5/6 Nephrectomy (NPX) and Sham SB samples on genera (A) and species (B) levels reveals a significant difference between the two groups. ***P < 0.001.
Fig. 14.

Principal component (PC) and heat map of small bowel (SB) operational taxonomic units (OTUs). SB bacterial comparison of the nonselected OTUs between the two groups indicates no significant difference on the genera (A) and species (D) levels. Heat map and hierarchical clustering using the selected OTUs resulted in a muddled separation between NPX: 5/6 Nephrectomy and Sham on both genera (B) and species levels (E). Random auto-clustering was noted when the nonselected OTUs were analyzed on both genera (C) and species (F) levels.
Intestinal urea amount is passively increased in CKD.
As mentioned above, decreased urinary clearance and accumulation of nitrogenous wastes in advanced CKD have been reported to increase GI elimination of urea and other toxins (30, 37, 52, 53, 69, 75). We addressed this by measuring urea content of SBs collected from Sham and NPX groups. NPX SB urea amount was doubled in comparison with Sham (Fig. 15A). This data supports the hypothesis of increased intestinal urea in CKD. To establish if this increased accumulation was dependent on UTs, we performed Sham and NPX operations on a separate group of mice (n = 5) and harvested tissues for gene expression analysis 9 wk later. No significant differences in UT-A expression were seen between the two groups at any anatomical location. Higher relative expression of UT-B was noted in the distal SB of Sham mice compared with the proximal SB of the same group, and UT-A expression was higher in the distal SB compared with colon in both groups (Fig. 15, B and C). This indicates that the increased intestinal urea observed in our CKD model was independent of changes in intestinal and colonic UT mRNA expression.
Fig. 15.

Intestinal urea and its transporters in experimental chronic kidney disease model. NPX mice small bowel (SB) urea amount doubled in comparison with Sham group (A). Urea transporters A and B (UT-A, UT-B) mRNA distribution across that whole gastrointestinal tract showed no difference between NPX: 5/6 Nephrectomy and Sham. However, distal SB appear to have a higher mRNA expression of both UT-A (B) and UT-B (C) when compared with colonic proximal transporter expression in each of the respective groups. SB1 and SB2 are post duodenum small bowel halves.
Increased urease-processing bacteria in experimental CKD group.
To test the hypothesis that intestinal urea buildup contributes to the dysbiotic changes in CKD (22, 28, 42, 65, 67), we tested the abundance of bacteria with the capacity of urea hydrolysis in both groups. No significant bl differences were observed when SB bacterial species were studied for their ability to hydrolyze urea. In contrast, Sham stool pellet analysis showed a significantly lower abundance of bacteria with the capacity of urea hydrolysis compared with stool pellets from NPX mice (t-statistic, −2.375, P = 0.029).
Increased intestinal urea exposure affects gut bacterial composition.
We next examined if increased intestinal urea exposure contributed to CKD-associated dysbiosis through the administration of urea in the drinking water of mice. First, to ensure that oral urea load would result in increased BUN, we conducted a kinetic experiment aimed at evaluating BUN following a single oral dose. Compared with control, BUN increased 62% by 30 min, 90% by 60 min, then declined to bl 5 h post gavage (Fig. 16A). Based on this, an experiment was designed to evaluate if continuous urea supplementation in drinking water impacted the gut microbiota. Ten-week-old C57BL/6J mice (n = 5 per group, single housed, Fig. 16B) received autoclaved drinking water or autoclaved water supplemented with 4 g/l urea for seven weeks. Over this timeframe, there was a trend of increased weight gain between the two groups (Fig. 16C). Mice were euthanized after an overnight fast of 12 h.
Fig. 16.

Urea experiment outline and measured results. A: changes of blood urea nitrogen (BUN) after single 300 µL gavage of 4g/l urea. BUN rises to 62% and 90% over 30–60 min, respectively, then trends back to baseline over 5 h. B: housing and urea supplementation strategy. C: change in weight (∆weight) calculated based on day 1 (10 wk of age) weight shows an increase in urea supplementation mice weight over 7 wk. *P < 0.05.
Urea supplementation resulted in a significant change in bacterial gut microbiota on the genera (P < 0.01, Fig. 17A) and species levels (P < 0.01, Fig. 17B). A trend toward decreased Verrucomicrobia phylum in the 4 g/l urea group was noted (Fig. 18A). Genera analysis showed a significant decrease in Anaerostipes Spp., Clostridium Spp., uncultured genus of the Mogibacteriaceae family, and increases in Oscillospira Spp., Rumminococuss Spp., and an uncultured genus from the Lachnosiraceae family (P < 0.05) (Fig. 18B). Species analysis showed a significant decrease in the relative abundance of Clostridium perfringens, uncultured species from the Anaerostipes and Clostridium genera, and an uncultured species from the Mogibacteriaceae family. An increased relative abundance of Ruminococcus gravus, uncultured species from the Oscillospira and Rumminococuss genera, plus an uncultured species from the Lachnosiraceae family was noted in the urea supplementation group compared with control (P < 0.05) (Fig. 18C). Heat map and hierarchical clustering of the samples using the selected genera/species (Fig. 19A, Fig. 19B) resulted in a complete separation between control and urea supplementation groups.
Fig. 17.

Urea supplementation effects of gut microbiota. Urea supplementation (4 g/l) in drinking water for 7 wk resulted in a significant shift in the gut bacterial microbiota on the genera (A) and species (B) levels. **P < 0.01. PC, principal component.
Fig. 18.

Operational taxonomic unit (OTU) changes after urea supplementation in drinking water. Though urea supplementation resulted in a collective significant change on the phyla level, individual phylum changes were not as robust (A). A trend in Verrucomicrobia phylum was noted. However, both genus (B) and species (C) level analysis revealed multiple OTU changes associated with urea supplementation. *P < 0.05.
Fig. 19.

Heat map and hierarchical clustering of the urea supplementation samples using the selected genera/species. A complete separation and auto-clustering on both genera (A) and species (B) levels is noted between the urea supplementation and control groups.
Our results show that daily urea intake in drinking water affected the GI microbiome. To explore whether increased urea exposure is mostly responsible for the CKD-associated dysbiosis noted earlier, we compared the changes in relative abundance in OTUs (Table 1). Both urea supplementation and the experimental CKD model resulted in decreased relative abundance of the Anaerostipes genus. Except for the changes noted in Anaerostipes Spp., urea supplementation resulted in completely different changes in the gut microbiota. For example, urea supplementation was associated with decreased Clostridium and increased Oscillospira and Rumminococuss genera-relative abundances. In the CKD group, these genera were not changed. Additionally, other OTUs that displayed differences showed an opposite trend or no change when CKD and urea groups were compared. This can be seen for the Akkermansia Spp., which increased in the CKD model but decreased when urea was administered alone. Mice receiving urea in drinking water exhibited no significant difference in the abundance of bacteria with the capacity to hydrolyze urea (compared with same group bl). This reveals that increased intestinal urea content alone may not be the primary contributor to CKD-associated dysbiosis.
Table 1.
NPX vs. urea supplementation effects on bacterial gut microbiota
| OUT | NPX | 4 g/l Urea |
|---|---|---|
| Anaerostipes Spp.** | Decreased* | Decreased* |
| Clostridium Spp.** | No change | Decreased* |
| Oscillospira Spp.** | No change | Increased* |
| Rumminococuss Spp.** | No change | Increased* |
| Lactobacillus Spp.** | Increased* | No change |
| f_Lachnosiraceae;g_** | No change | Increased |
| Blautia producta | Decreased | No change |
| o_Clostridiales;;f_;g_ (1)** | Decreased | No change |
| o_Clostridiales;f_;g_ (2)** | Decreased | No change |
| g_Bacteroides;s_ | Decreased | No change |
| Akkermansia Spp. | Increased | Decreased |
| Rumminococuss gnavus | No change | Increased |
| Clostridium perfringens | No change | Decreased |
| Akkermansia muciniphila | Increased | Decreased |
| Verrucomicrobia phylum | No change | Decreased |
Comparisons between the changes seen in fecal samples of the unilateral nephrectomy (NPX) mice and urea supplementation groups indicates that only changes in Anaerostipes Spp. are similar between the two groups.
P < 0.05,
Similar changes in uncultured species in the corresponding genus.
DISCUSSION
Our work confirms previous findings that CKD is associated with significant dysbiotic changes in the GI microbiome. We also demonstrate that microbiome changes occurred in the SBs of mice with surgically-induced CKD. To our knowledge, this is the first report indicating CKD-associated alterations originating in the SB microbiome. The proximal SB is generally regarded as relatively sterile when compared with the colon, with sparse bacterial populations present (3, 60). Using 16S sequencing, these long lasting assumptions have been recently challenged. Limited data from patients with renal disease indicate bacterial overgrowth and shifts in the bacterial composition of the small intestines. Although the exact mechanisms are unknown, and more studies are required, both the uremic milieu and shifts in intestinal motility and permeability have been implicated (38, 58, 59, 68).
In this study, mice with surgically induced CKD exhibited increased intestinal urea amounts. UTs encoded by the solute carrier family 14, member 1 (UT-B) and solute carrier family 14, member 2 (UT-A) genes facilitate the movement of urea across plasma membranes. UT-A is mainly expressed in the kidneys, while UT-B has a wider distribution that includes the apical membrane of superficial epithelial cells and crypts of colon and small intestine (8, 39). We hypothesized that alterations in the intestinal expression of UT-A and UT-B could account for the increased urea amount in mice with CKD. We report here the presence of both UTs in the SB with significantly higher relative expression in the distal SB when compared with its colonic expression. However, mRNA expression of either transporter was unable to explain the increased urea intestinal load in CKD. This is consistent with a previous report that failed to show increased colonic UT-B expression under experimental CKD conditions (24).
We chronically administered urea to healthy mice to determine if elevated intestinal urea exposure alone could mirror the dysbiosis we observed in mice with CKD. This was founded on previous reports implicating elevated intestinal urea as a driver for bacterial dysbiosis in CKD (22, 28, 42, 65, 67). We selected 4 g/l because higher urea concentrations (20 g/l to result in increased BUN) were found to significantly inhibit insulin secretion, resulting in subsequent hyperglycemia (24, 32). We have also shown that 300 µl of 4 g/dl urea (1.2 mg) resulted in a rise in BUN for ~5 h. A 30 g C57BL/6J male mouse consumes on average 5–6 ml of drinking water daily (2, 62). This will result in a 20– to 24-mg daily urea load with an average hourly dose of 0.83–1 mg. Similar to findings in Fig. 16A, we expect that our experimental mice experienced a doubling of BUN most of the time during urea consumption in drinking water. This mimics our findings of doubling BUN in experimental CKD animal models (NPX group).
Our results show that urea did indeed cause shifts in the GI bacterial species, but these changes were almost entirely different when compared with those in experimental CKD mice. It is important to note that in addition to raising the gut urea level, advanced CKD causes numerous other biochemical and biophysical changes, which most likely act together with the rise in the intestinal urea concentration to impact the structure and function of the microbiome (30, 37, 52, 53, 69, 75). Importantly though, our findings of the difference in the changes of the composition of microbiome between the CKD versus urea supplementation alone does not diminish the possible impact of urea as one of the main causes of uremia-induced dysbiosis.
The oral administration of urea to mice without CKD was performed in an attempt to isolate and identify a single contributor of CKD-associated dysbiosis. However, it is important to note that mice used in the study had normal renal function. This was done to try to eliminate other factors associated with renal impairment that could affect the microbiome. This is not true in patients with advanced CKD and end-stage renal disease. Diet restriction and other therapeutic interventions play an important role in CKD-associated dysbiosis (74). It is possible that in CKD patients, the uremic milieu, alongside other pathophysiological factors, contributes simultaneously to impact the gut bacterial microbiota.
In CKD, the increased intestinal urea would be due to the influx from the increased circulating urea. In contrast, oral urea loading directly leads to the increase in the intestinal urea with subsequent influence on the circulating urea level. Such difference in the cause of increased intestinal urea would affect to the microbiome itself and limit our abilities to conclude with high certainty that increased intestinal urea in CKD is not the main contributing factor to dysbiosis. This is supported by the increased bacteria with urea hydrolysis capacity noted in our experimental CKD mice compared with control.
NPX mice showed a decreased relative abundance of Anaerostipes and Oscillospira (in SB only) and a higher relative abundance of Akkermansia. Additionally, we found an opposite trend between Anaerostipes and Lactobacillus. Both considered probiotics, we measured a decreased relative abundance of Anaerostipes and a trend toward increased relative abundance of Lactobacillus (5, 14, 17, 27, 48). Anaerostipes are gram-variable, obligate anaerobes which produce acetate, butyrate, and lactate as the end products of glucose metabolism (57). However, they have not yet been fully studied in clinical conditions. Increased relative abundance of both Anaerostipes and Lactobacillus genera results in increased short-chain fatty acid production as well as higher NaOH consumption, whereas ammonia and branched-chain fatty acids are decreased (4). Furthermore, clinical trials evaluating the effect of prebiotic use on quality of life improvements have shown that inulin-type fructans increased the relative abundance of Anaerostipes (29, 45, 64). This indicates a potential positive association between health and this genus. In our study, decreased Anaerostipes was the only similar change observed in both NPX mice and those with urea supplementation. This suggests that intestinal urea amounts may negatively impact Anaerostipes within the GI microbiome.
Oscillospira is known to digest resistant starches and fibers. Its presence has an effect on the metabolism of nutritional fibers (43, 61). The decreased relative abundance of this genus in our study is in accordance with a shift from carbohydrate to proteolytic metabolism and increased intestinal nitrogenous products seen in CKD. Though the biological importance and health effects of this genus, beyond its metabolism function, have not yet been studied in renal diseases, a significant body of evidence highlights its importance. It is associated with decreased body mass index (leanness) in children and adults (15, 20, 61, 70), and its relative abundance was increased in disease states such as Crohn disease (26).
Akkermansia muciniphila is one of the most abundant members of the human gut microbiota, representing up to 5% of all species (7, 11). A high-protein diet in experimental animal models expanded the Akkermansia genus and resulted in decreased fat content (73). Studies have shown that Akkermansia muciniphila prevents high-fat diet-induced obesity and diabetes in experimental animal models and protected the gut barrier (10, 16, 36, 49, 51, 80). It has been shown to adhere to intestinal cells, strengthening its integrity, decreasing leaky gut, and activating the immune system (12, 55). High urea supplementation in drinking water almost eradicated Akkermansia muciniphila in contrast to its increased abundance in CKD, indicating a possible alternative mechanism other than increased nitrogenous products.
In conclusion, our study depicts measurable changes in both the SB and stool pellet microbiome in experimental CKD. Urea supplementation to mice was unable to explain these alterations. Further mechanistic studies are warranted to evaluate if these variations can explain CKD-associated comorbidities and explore the impact of additional factors outside of elevated intestinal urea to explain shifts in the GI microbiome.
GRANTS
This work was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health (NIH) under award number UL1-TR-001412 to the Univ. at Buffalo and by the NIH. This work was also supported by the University at Buffalo Genome, Environment and Microbiome, and department fund (to R. Yacoub and L. Chaves), Innovative Micro-Programs Accelerating Collaboration in Themes (to R. Yacoub), and NIH Grants RO1-AI-125982 and RO1-DE-024523 (to Y. Sun).
DISCLAIMERS
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
L.D.C., D.I.M., M.B., Y.S., R.J.G., R.J.Q., and R.Y. conceived and designed research; L.D.C., A.M.H., S.A., S.A.T., S.W., and R.Y. performed experiments; L.D.C., D.I.M., M.A.B., A.M.H., and R.Y. analyzed data; L.D.C., D.I.M., M.B., Y.S., R.J.G., R.J.Q., and R.Y. interpreted results of experiments; L.D.C., D.I.M., and R.Y. prepared figures; L.D.C., D.I.M., A.M.H., and R.Y. drafted manuscript; L.D.C., D.I.M., M.A.B., A.M.H., S.A., S.A.T., S.W., M.B., Y.S., R.J.G., R.J.Q., and R.Y. edited and revised manuscript; L.D.C., D.I.M., M.A.B., A.M.H., S.A., S.A.T., S.W., M.B., Y.S., R.J.G., R.J.Q., and R.Y. approved final version of manuscript.
REFERENCES
- 1.Aitchison J. The Statistical Analysis of Compositional Data. London, UK: Chapman & Hall, Ltd, 1986. doi: 10.1007/978-94-009-4109-0. [DOI] [Google Scholar]
- 2.Bachmanov AA, Reed DR, Beauchamp GK, Tordoff MG. Food intake, water intake, and drinking spout side preference of 28 mouse strains. Behav Genet 32: 435–443, 2002. doi: 10.1023/A:1020884312053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bornside GH, Welsh JS, Cohn I Jr. Bacterial flora of the human small intestine. JAMA 196: 1125–1127, 1966. doi: 10.1001/jama.1966.03100260063018. [DOI] [PubMed] [Google Scholar]
- 4.Bothe MK, Maathuis AJH, Bellmann S, van der Vossen JMBM, Berressem D, Koehler A, Schwejda-Guettes S, Gaigg B, Kuchinka-Koch A, Stover JF. Dose-dependent prebiotic effect of lactulose in a computer-controlled in vitro model of the human large intestine. Nutrients 9: 767, 2017. doi: 10.3390/nu9070767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ciorba MA, Riehl TE, Rao MS, Moon C, Ee X, Nava GM, Walker MR, Marinshaw JM, Stappenbeck TS, Stenson WF. Lactobacillus probiotic protects intestinal epithelium from radiation injury in a TLR-2/cyclo-oxygenase-2-dependent manner. Gut 61: 829–838, 2012. doi: 10.1136/gutjnl-2011-300367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: an integrative view. Cell 148: 1258–1270, 2012. doi: 10.1016/j.cell.2012.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collado MC, Derrien M, Isolauri E, de Vos WM, Salminen S. Intestinal integrity and Akkermansia muciniphila, a mucin-degrading member of the intestinal microbiota present in infants, adults, and the elderly. Appl Environ Microbiol 73: 7767–7770, 2007. doi: 10.1128/AEM.01477-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coyle J, McDaid S, Walpole C, Stewart GS. UT-B urea transporter localization in the bovine gastrointestinal tract. J Membr Biol 249: 77–85, 2016. doi: 10.1007/s00232-015-9850-5. [DOI] [PubMed] [Google Scholar]
- 9.Crespo-Salgado J, Vehaskari VM, Stewart T, Ferris M, Zhang Q, Wang G, Blanchard EE, Taylor CM, Kallash M, Greenbaum LA, Aviles DH. Intestinal microbiota in pediatric patients with end stage renal disease: a Midwest Pediatric Nephrology Consortium study. Microbiome 4: 50, 2016. doi: 10.1186/s40168-016-0195-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dao MC, Everard A, Aron-Wisnewsky J, Sokolovska N, Prifti E, Verger EO, Kayser BD, Levenez F, Chilloux J, Hoyles L, Dumas ME, Rizkalla SW, Doré J, Cani PD, Clément K; MICRO-Obes Consortium . Akkermansia muciniphila and improved metabolic health during a dietary intervention in obesity: relationship with gut microbiome richness and ecology. Gut 65: 426–436, 2016. doi: 10.1136/gutjnl-2014-308778. [DOI] [PubMed] [Google Scholar]
- 11.Derrien M, Collado MC, Ben-Amor K, Salminen S, de Vos WM. The Mucin degrader Akkermansia muciniphila is an abundant resident of the human intestinal tract. Appl Environ Microbiol 74: 1646–1648, 2008. doi: 10.1128/AEM.01226-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Derrien M, Van Baarlen P, Hooiveld G, Norin E, Müller M, de Vos WM. Modulation of mucosal immune response, tolerance, and proliferation in mice colonized by the mucin-degrader Akkermansia muciniphila. Front Microbiol 2: 166, 2011. doi: 10.3389/fmicb.2011.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72: 5069–5072, 2006. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duncan SH, Flint HJ. Probiotics and prebiotics and health in ageing populations. Maturitas 75: 44–50, 2013. doi: 10.1016/j.maturitas.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 15.Escobar JS, Klotz B, Valdes BE, Agudelo GM. The gut microbiota of Colombians differs from that of Americans, Europeans and Asians. BMC Microbiol 14: 311, 2014. doi: 10.1186/s12866-014-0311-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, Guiot Y, Derrien M, Muccioli GG, Delzenne NM, de Vos WM, Cani PD. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci USA 110: 9066–9071, 2013. doi: 10.1073/pnas.1219451110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferrario C, Taverniti V, Milani C, Fiore W, Laureati M, De Noni I, Stuknyte M, Chouaia B, Riso P, Guglielmetti S. Modulation of fecal Clostridiales bacteria and butyrate by probiotic intervention with Lactobacillus paracasei DG varies among healthy adults. J Nutr 144: 1787–1796, 2014. doi: 10.3945/jn.114.197723. [DOI] [PubMed] [Google Scholar]
- 18.Gall A, Fero J, McCoy C, Claywell BC, Sanchez CA, Blount PL, Li X, Vaughan TL, Matsen FA, Reid BJ, Salama NR. Bacterial composition of the human upper gastrointestinal tract microbiome is dynamic and associated with genomic instability in a Barrett’s esophagus cohort. PLoS One 10: e0129055, 2015. doi: 10.1371/journal.pone.0129055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goodpaster AM, Kennedy MA. Quantification and statistical significance analysis of group separation in NMR-based metabonomics studies. Chemom Intell Lab Syst 109: 162–170, 2011. doi: 10.1016/j.chemolab.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, Beaumont M, Van Treuren W, Knight R, Bell JT, Spector TD, Clark AG, Ley RE. Human genetics shape the gut microbiome. Cell 159: 789–799, 2014. doi: 10.1016/j.cell.2014.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gordon A, Hannon G. Fastx-toolkit. FASTQ/A short-reads preprocessing tools. http://hannonlab.cshl.edu/fastx_toolkit/ [3 January 2018].
- 22.Hatch M, Vaziri ND. Enhanced enteric excretion of urate in rats with chronic renal failure. Clin Sci (Lond) 86: 511–516, 1994. doi: 10.1042/cs0860511. [DOI] [PubMed] [Google Scholar]
- 23.Hida M, Aiba Y, Sawamura S, Suzuki N, Satoh T, Koga Y. Inhibition of the accumulation of uremic toxins in the blood and their precursors in the feces after oral administration of Lebenin, a lactic acid bacteria preparation, to uremic patients undergoing hemodialysis. Nephron 74: 349–355, 1996. doi: 10.1159/000189334. [DOI] [PubMed] [Google Scholar]
- 24.Inoue H, Kozlowski SD, Klein JD, Bailey JL, Sands JM, Bagnasco SM. Regulated expression of renal and intestinal UT-B urea transporter in response to varying urea load. Am J Physiol Renal Physiol 289: F451–F458, 2005. doi: 10.1152/ajprenal.00376.2004. [DOI] [PubMed] [Google Scholar]
- 25.Jones E, Oliphant T, Peterson P. SciPy: Open Source Scientific Tools for Python, 2001-. https://www.scipy.org/ [3 January 2018].
- 26.Kaakoush NO, Day AS, Huinao KD, Leach ST, Lemberg DA, Dowd SE, Mitchell HM. Microbial dysbiosis in pediatric patients with Crohn’s disease. J Clin Microbiol 50: 3258–3266, 2012. doi: 10.1128/JCM.01396-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kale-Pradhan PB, Jassal HK, Wilhelm SM. Role of Lactobacillus in the prevention of antibiotic-associated diarrhea: a meta-analysis. Pharmacotherapy 30: 119–126, 2010. doi: 10.1592/phco.30.2.119. [DOI] [PubMed] [Google Scholar]
- 28.Kang JY. The gastrointestinal tract in uremia. Dig Dis Sci 38: 257–268, 1993. doi: 10.1007/BF01307542. [DOI] [PubMed] [Google Scholar]
- 29.Kant R, Rasinkangas P, Satokari R, Pietilä TE, Palva A. Genome sequence of the butyrate-producing anaerobic bacterium Anaerostipes hadrus PEL 85. Genome Announc 3: e00224-15, 2015. doi: 10.1128/genomeA.00224-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kes P. Serum gastrin concentration in chronic renal failure. Acta Med Croatica 46: 47–58, 1992. [PubMed] [Google Scholar]
- 31.Kieffer DA, Piccolo BD, Vaziri ND, Liu S, Lau WL, Khazaeli M, Nazertehrani S, Moore ME, Marco ML, Martin RJ, Adams SH. Resistant starch alters gut microbiome and metabolomic profiles concurrent with amelioration of chronic kidney disease in rats. Am J Physiol Renal Physiol 310: F857–F871, 2016. doi: 10.1152/ajprenal.00513.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koppe L, Nyam E, Vivot K, Manning Fox JE, Dai XQ, Nguyen BN, Trudel D, Attané C, Moullé VS, MacDonald PE, Ghislain J, Poitout V. Urea impairs β cell glycolysis and insulin secretion in chronic kidney disease. J Clin Invest 126: 3598–3612, 2016. doi: 10.1172/JCI86181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kren S, Hostetter TH. The course of the remnant kidney model in mice. Kidney Int 56: 333–337, 1999. doi: 10.1046/j.1523-1755.1999.00527.x. [DOI] [PubMed] [Google Scholar]
- 34.Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, Beiko RG, Huttenhower C. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31: 814–821, 2013. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lau WL, Savoj J, Nakata MB, Vaziri ND. Altered microbiome in chronic kidney disease: systemic effects of gut-derived uremic toxins. Clin Sci (Lond) 132: 509–522, 2018. doi: 10.1042/CS20171107. [DOI] [PubMed] [Google Scholar]
- 36.Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, Almeida M, Arumugam M, Batto JM, Kennedy S, Leonard P, Li J, Burgdorf K, Grarup N, Jørgensen T, Brandslund I, Nielsen HB, Juncker AS, Bertalan M, Levenez F, Pons N, Rasmussen S, Sunagawa S, Tap J, Tims S, Zoetendal EG, Brunak S, Clément K, Doré J, Kleerebezem M, Kristiansen K, Renault P, Sicheritz-Ponten T, de Vos WM, Zucker JD, Raes J, Hansen T, Bork P, Wang J, Ehrlich SD, Pedersen O; MetaHIT Consortium . Richness of human gut microbiome correlates with metabolic markers. Nature 500: 541–546, 2013. doi: 10.1038/nature12506. [DOI] [PubMed] [Google Scholar]
- 37.Lee YT. Urea concentration in intestinal fluids in normal and uremic dogs. J Surg Oncol 3: 163–168, 1971. doi: 10.1002/jso.2930030210. [DOI] [PubMed] [Google Scholar]
- 38.Lefebvre HP, Ferré JP, Watson AD, Brown CA, Serthelon JP, Laroute V, Concordet D, Toutain PL. Small bowel motility and colonic transit are altered in dogs with moderate renal failure. Am J Physiol Regul Integr Comp Physiol 281: R230–R238, 2001. doi: 10.1152/ajpregu.2001.281.1.R230. [DOI] [PubMed] [Google Scholar]
- 39.Li X, Chen G, Yang B. Urea transporter physiology studied in knockout mice. Front Physiol 3: 217, 2012. doi: 10.3389/fphys.2012.00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma LJ, Fogo AB. Model of robust induction of glomerulosclerosis in mice: importance of genetic background. Kidney Int 64: 350–355, 2003. doi: 10.1046/j.1523-1755.2003.00058.x. [DOI] [PubMed] [Google Scholar]
- 41.Ma LJ, Nakamura S, Aldigier JC, Rossini M, Yang H, Liang X, Nakamura I, Marcantoni C, Fogo AB. Regression of glomerulosclerosis with high-dose angiotensin inhibition is linked to decreased plasminogen activator inhibitor-1. J Am Soc Nephrol 16: 966–976, 2005. doi: 10.1681/ASN.2004060492. [DOI] [PubMed] [Google Scholar]
- 42.Macfarlane GT, Macfarlane S. Fermentation in the human large intestine: its physiologic consequences and the potential contribution of prebiotics. J Clin Gastroenterol 45, Suppl: S120–S127, 2011. doi: 10.1097/MCG.0b013e31822fecfe. [DOI] [PubMed] [Google Scholar]
- 43.Mackie RI, Aminov RI, Hu W, Klieve AV, Ouwerkerk D, Sundset MA, Kamagata Y. Ecology of uncultivated Oscillospira species in the rumen of cattle, sheep, and reindeer as assessed by microscopy and molecular approaches. Appl Environ Microbiol 69: 6808–6815, 2003. doi: 10.1128/AEM.69.11.6808-6815.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McKinney W. Data structures for statistical computing in python. In: Proceedings of the 9th Python in Science Conference (SciPy 2010). SciPy: Austin, TX, June 28–July 3 2010, 51–56. [Google Scholar]
- 45.Moens F, Verce M, De Vuyst L. Lactate- and acetate-based cross-feeding interactions between selected strains of lactobacilli, bifidobacteria and colon bacteria in the presence of inulin-type fructans. Int J Food Microbiol 241: 225–236, 2017. doi: 10.1016/j.ijfoodmicro.2016.10.019. [DOI] [PubMed] [Google Scholar]
- 46.Mu C, Yang Y, Su Y, Zoetendal EG, Zhu W. Differences in microbiota membership along the gastrointestinal tract of piglets and their differential alterations following an early-life antibiotic intervention. Front Microbiol 8: 797, 2017. doi: 10.3389/fmicb.2017.00797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Musso G, Gambino R, Cassader M. Obesity, diabetes, and gut microbiota: the hygiene hypothesis expanded? Diabetes Care 33: 2277–2284, 2010. doi: 10.2337/dc10-0556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ohashi Y, Ushida K. Health-beneficial effects of probiotics: its mode of action. Anim Sci J 80: 361–371, 2009. doi: 10.1111/j.1740-0929.2009.00645.x. [DOI] [PubMed] [Google Scholar]
- 49.Org E, Parks BW, Joo JW, Emert B, Schwartzman W, Kang EY, Mehrabian M, Pan C, Knight R, Gunsalus R, Drake TA, Eskin E, Lusis AJ. Genetic and environmental control of host-gut microbiota interactions. Genome Res 25: 1558–1569, 2015. doi: 10.1101/gr.194118.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V. Scikit-learn: Machine learning in Python. J Mach Learn Res 12: 2825–2830, 2011. [Google Scholar]
- 51.Plovier H, Everard A, Druart C, Depommier C, Van Hul M, Geurts L, Chilloux J, Ottman N, Duparc T, Lichtenstein L, Myridakis A, Delzenne NM, Klievink J, Bhattacharjee A, van der Ark KC, Aalvink S, Martinez LO, Dumas ME, Maiter D, Loumaye A, Hermans MP, Thissen JP, Belzer C, de Vos WM, Cani PD. A purified membrane protein from Akkermansia muciniphila or the pasteurized bacterium improves metabolism in obese and diabetic mice. Nat Med 23: 107–113, 2017. doi: 10.1038/nm.4236. [DOI] [PubMed] [Google Scholar]
- 52.Quintero E, Kaunitz J, Nishizaki Y, De Giorgio R, Sternini C, Guth PH. Uremia increases gastric mucosal permeability and acid back-diffusion injury in the rat. Gastroenterology 103: 1762–1768, 1992. doi: 10.1016/0016-5085(92)91432-4. [DOI] [PubMed] [Google Scholar]
- 53.Quintero E, Ohning GV, Del Rivero M, Wong HC, Walsh JH, Guth PH. Gastrin mediates the increase in gastric cell growth in uremic rats. Am J Physiol 268: G586–G591, 1995. doi: 10.1152/ajpgi.1995.268.4.G586. [DOI] [PubMed] [Google Scholar]
- 54.Rescigno M. The microbiota revolution: excitement and caution. Eur J Immunol 47: 1406–1413, 2017. doi: 10.1002/eji.201646576. [DOI] [PubMed] [Google Scholar]
- 55.Reunanen J, Kainulainen V, Huuskonen L, Ottman N, Belzer C, Huhtinen H, de Vos WM, Satokari R. Akkermansia muciniphila adheres to enterocytes and strengthens the integrity of the epithelial cell layer. Appl Environ Microbiol 81: 3655–3662, 2015. doi: 10.1128/AEM.04050-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, Muehlbauer MJ, Ilkayeva O, Semenkovich CF, Funai K, Hayashi DK, Lyle BJ, Martini MC, Ursell LK, Clemente JC, Van Treuren W, Walters WA, Knight R, Newgard CB, Heath AC, Gordon JI. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341: 1241214, 2013. doi: 10.1126/science.1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schwiertz A, Hold GL, Duncan SH, Gruhl B, Collins MD, Lawson PA, Flint HJ, Blaut M. Anaerostipes caccae gen. nov., sp. nov., a new saccharolytic, acetate-utilising, butyrate-producing bacterium from human faeces. Syst Appl Microbiol 25: 46–51, 2002. doi: 10.1078/0723-2020-00096. [DOI] [PubMed] [Google Scholar]
- 58.Simenhoff ML, Dunn SR, Zollner GP, Fitzpatrick ME, Emery SM, Sandine WE, Ayres JW. Biomodulation of the toxic and nutritional effects of small bowel bacterial overgrowth in end-stage kidney disease using freeze-dried Lactobacillus acidophilus. Miner Electrolyte Metab 22: 92–96, 1996. [PubMed] [Google Scholar]
- 59.Strid H, Simrén M, Stotzer PO, Ringström G, Abrahamsson H, Björnsson ES. Patients with chronic renal failure have abnormal small intestinal motility and a high prevalence of small intestinal bacterial overgrowth. Digestion 67: 129–137, 2003. doi: 10.1159/000071292. [DOI] [PubMed] [Google Scholar]
- 60.Thadepalli H, Lou MA, Bach VT, Matsui TK, Mandal AK. Microflora of the human small intestine. Am J Surg 138: 845–850, 1979. doi: 10.1016/0002-9610(79)90309-X. [DOI] [PubMed] [Google Scholar]
- 61.Tims S, Derom C, Jonkers DM, Vlietinck R, Saris WH, Kleerebezem M, de Vos WM, Zoetendal EG. Microbiota conservation and BMI signatures in adult monozygotic twins. ISME J 7: 707–717, 2013. doi: 10.1038/ismej.2012.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tordoff MG, Bachmanov AA, Reed DR. Forty mouse strain survey of water and sodium intake. Physiol Behav 91: 620–631, 2007. doi: 10.1016/j.physbeh.2007.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62a.van der Walt S, Colbert SC, Varoquaux G. The NumPy array: a structure for efficient numerical computation. Comput Sci Eng 13: 22–30, 2011. doi: 10.1109/MCSE.2011.37. [DOI] [Google Scholar]
- 63.Van Rossum G, Drake FL. Python Language Reference Manual. Bristol, UK: Network Theory, 2003. [Google Scholar]
- 64.Vandeputte D, Falony G, Vieira-Silva S, Wang J, Sailer M, Theis S, Verbeke K, Raes J. Prebiotic inulin-type fructans induce specific changes in the human gut microbiota. Gut 66: 1968–1974, 2017. doi: 10.1136/gutjnl-2016-313271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vaziri ND, Dure-Smith B, Miller R, Mirahmadi MK. Pathology of gastrointestinal tract in chronic hemodialysis patients: an autopsy study of 78 cases. Am J Gastroenterol 80: 608–611, 1985. [PubMed] [Google Scholar]
- 66.Vaziri ND, Goshtasbi N, Yuan J, Jellbauer S, Moradi H, Raffatellu M, Kalantar-Zadeh K. Uremic plasma impairs barrier function and depletes the tight junction protein constituents of intestinal epithelium. Am J Nephrol 36: 438–443, 2012. doi: 10.1159/000343886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vaziri ND, Wong J, Pahl M, Piceno YM, Yuan J, DeSantis TZ, Ni Z, Nguyen TH, Andersen GL. Chronic kidney disease alters intestinal microbial flora. Kidney Int 83: 308–315, 2013. doi: 10.1038/ki.2012.345. [DOI] [PubMed] [Google Scholar]
- 68.Vaziri ND, Yuan J, Nazertehrani S, Ni Z, Liu S. Chronic kidney disease causes disruption of gastric and small intestinal epithelial tight junction. Am J Nephrol 38: 99–103, 2013. doi: 10.1159/000353764. [DOI] [PubMed] [Google Scholar]
- 69.Vaziri ND, Yuan J, Norris K. Role of urea in intestinal barrier dysfunction and disruption of epithelial tight junction in chronic kidney disease. Am J Nephrol 37: 1–6, 2013. doi: 10.1159/000345969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Verdam FJ, Fuentes S, de Jonge C, Zoetendal EG, Erbil R, Greve JW, Buurman WA, de Vos WM, Rensen SS. Human intestinal microbiota composition is associated with local and systemic inflammation in obesity. Obesity (Silver Spring) 21: E607–E615, 2013. doi: 10.1002/oby.20466. [DOI] [PubMed] [Google Scholar]
- 71.Vogtmann E, Flores R, Yu G, Freedman ND, Shi J, Gail MH, Dye BA, Wang GQ, Klepac-Ceraj V, Paster BJ, Wei WQ, Guo HQ, Dawsey SM, Qiao YL, Abnet CC. Association between tobacco use and the upper gastrointestinal microbiome among Chinese men. Cancer Causes Control 26: 581–588, 2015. doi: 10.1007/s10552-015-0535-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang L, Jacobs JP, Lagishetty V, Yuan PQ, Wu SV, Million M, Reeve JR Jr, Pisegna JR, Taché Y. High-protein diet improves sensitivity to cholecystokinin and shifts the cecal microbiome without altering brain inflammation in diet-induced obesity in rats. Am J Physiol Regul Integr Comp Physiol 313: R473–R486, 2017. doi: 10.1152/ajpregu.00105.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wong J, Piceno YM, DeSantis TZ, Pahl M, Andersen GL, Vaziri ND. Expansion of urease- and uricase-containing, indole- and p-cresol-forming and contraction of short-chain fatty acid-producing intestinal microbiota in ESRD. Am J Nephrol 39: 230–237, 2014. doi: 10.1159/000360010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wu MJ, Chang CS, Cheng CH, Chen CH, Lee WC, Hsu YH, Shu KH, Tang MJ. Colonic transit time in long-term dialysis patients. Am J Kidney Dis 44: 322–327, 2004. doi: 10.1053/j.ajkd.2004.04.048. [DOI] [PubMed] [Google Scholar]
- 76.Xu KY, Xia GH, Lu JQ, Chen MX, Zhen X, Wang S, You C, Nie J, Zhou HW, Yin J. Impaired renal function and dysbiosis of gut microbiota contribute to increased trimethylamine-N-oxide in chronic kidney disease patients. Sci Rep 7: 1445, 2017. doi: 10.1038/s41598-017-01387-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yang H, Huang X, Fang S, Xin W, Huang L, Chen C. Uncovering the composition of microbial community structure and metagenomics among three gut locations in pigs with distinct fatness. Sci Rep 6: 27427, 2016. doi: 10.1038/srep27427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang HC, Zuo Y, Fogo AB. Models of chronic kidney disease. Drug Discov Today Dis Models 7: 13–19, 2010. doi: 10.1016/j.ddmod.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang J, Kobert K, Flouri T, Stamatakis A. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 30: 614–620, 2014. doi: 10.1093/bioinformatics/btt593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang Z, Wu X, Cao S, Cromie M, Shen Y, Feng Y, Yang H, Li L. Chlorogenic acid ameliorates experimental colitis by promoting growth of Akkermansia in mice. Nutrients 9: 677, 2017. doi: 10.3390/nu9070677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zoetendal EG, Raes J, van den Bogert B, Arumugam M, Booijink CC, Troost FJ, Bork P, Wels M, de Vos WM, Kleerebezem M. The human small intestinal microbiota is driven by rapid uptake and conversion of simple carbohydrates. ISME J 6: 1415–1426, 2012. doi: 10.1038/ismej.2011.212. [DOI] [PMC free article] [PubMed] [Google Scholar]