

Abstract
Lorenzo-Redondo et al. recently analyzed HIV RNA sequences in plasma virus and proviral DNA sequences in lymph nodes (LN) and peripheral blood mononuclear cells (PBMC) from samples collected over a 6-month period from 3 individuals following initiation of antiretroviral therapy (ART) and concluded that ongoing HIV replication occurred in LN despite ART and that this replication maintained the HIV reservoir. We analyzed the same sequences and found that the dataset was very limited (median of 5 unique RNA or DNA sequences per sample) after accounting for polymerase chain reaction resampling and hypermutation and that the few remaining DNA sequences after 3 and 6 months on ART were not more diverse or divergent from those in pre-ART in any of the individuals studied. These findings, and others, lead us to conclude that the claims of ongoing replication on ART made by Lorenzo-Redondo et al. are not justified from the dataset analyzed in their publication.
Keywords: clonal expansion, HIV evolution, HIV reservoir, lymph nodes, ongoing replication on ART, single-genome sequencing
ART suppresses HIV viremia and prolongs survival but does not eliminate the HIV reservoir, which leads to rebound viremia after antiretroviral therapy (ART) is stopped. There has been continued debate as to whether the reservoir is maintained through the proliferation and persistence of cells infected before initiation of ART or from ongoing viral replication in potential sanctuary sites, such as lymph nodes (LN). Previously, we sought evidence of viral replication during ART by comparing single-genome sequences (SGSs) in plasma from before ART with those that persisted on long-term ART, but we did not find evidence for viral evolution in the large majority of donors [1]. More recently, we showed that HIV-infected cells can expand clonally [2] and persist during ART, resulting in populations of identical viral variants in plasma and proviruses in peripheral blood mononuclear cells (PBMC) [1, 3]. We also found that a large infected cell clone capable of producing infectious virus was widely distributed anatomically, including in LN, and exhibited no evolution over 3 years on ART [3]. These observations, along with evidence that a large fraction (>95%) of the proviruses in patients on ART are defective [4], have led to our current model of HIV persistence on ART, in which a substantial fraction of the proviruses, derived from the rapidly replicating and evolving virus populations throughout the course of untreated infection, are either defective or latent and integrated in long-lived, often proliferating, cells, forming a fossil record of viral evolution dating back to the time of initial infection. Consequently, sequence variation of proviral DNA populations with time on ART (i.e., “evolution”) can arise from at least 5 different processes, only 1 of which is ongoing viral replication, with the other 4 being: (2) clearance of cells producing infectious virus, thereby exposing the archival or “fossil” HIV DNA; (3) selection of infected cells containing proviruses with mutations or deletions that facilitate escape from immune responses; (4) clonal amplification (or loss) of proviruses from cell proliferation (or death); and (5) inadequate sampling of diverse virus populations resulting in biased sequence detection.
In their report, Lorenzo-Redondo et al. performed deep sequencing on 454 bulk polymerase chain reaction (PCR)–amplified samples from blood and LN collected 3 and 6 months after initiating ART in 3 individuals and compared them with PBMCs and plasma sequences obtained from before ART [5]. They concluded that ongoing HIV replication occurs in the LN during ART and that this replication maintains the HIV reservoir. This conclusion contradicts what would be expected from the recent findings cited above. It has been demonstrated that HIV DNA decays for up to 1 year after initiating ART [6]; thus, looking at DNA populations after 3 to 6 months on ART may give the false appearance of viral replication when, in fact, proviral populations are shifting due to the loss of infected cells, revealing those proviruses that are in long-lived cells or proliferating T-cell clones. To accurately address the question of ongoing HIV replication on ART, samples must be collected over several years on ART and compared with proviral populations prior to ART initiation or, preferably, after a shorter time on ART.
Despite the inappropriate time points used to address the question of ongoing replication, we investigated the contradiction further by analyzing the published sequences from Lorenzo-Redondo et al. For brevity, we restricted our findings to the gag sequences in their Figure 1 and Extended Data Figure 2 [5]. Table S1 shows the total number of sequences they obtained (median 28 sequences/sample). Unlike SGS, which was developed to eliminate errors inherent in bulk PCR amplification, the method used by Lorenzo-Redondo et al. restores these errors by its inability to discern independent amplification of identical sequences from repeated amplification of a single original template (i.e., resampling) or by its inability to identify errors arising during PCR amplification. To account for this problem, we collapsed the identical sequences in the published data set into single variants for analysis, which is the only appropriate way to analyze sequences from bulk PCR amplifications. The viral DNA copy number in the samples was not reported in the paper, except that it was “low” and may have been inefficiently captured by PCR; thus, the default assumption is that there is likely to have been significant resampling of the few starting templates in the bulk PCR. It is up to the authors to prove they are not using resampled sequences, but they showed no data in support of their assumption that groups of identical sequences represent biologically meaningful “haplotypes.” In addition, HIV sequences are known to become diverse shortly after infection [7], suggesting that identical sequences are likely an artifact of the amplification procedure [8, 9]. Therefore, only unique sequences should be analyzed. This correction reduced the median number of sequences analyzed to only 6.5 per sample. As shown in the table, we also removed sequences that contained stop codons (mostly G-to-A hypermutants) as they cannot be a part of a replicating virus population, leaving a median of only 5 (range, 0–37) unique sequences per sample and no sequences at all in 4 of the 21 samples analyzed, many fewer than would be considered adequate for studies of diverse virus populations and far fewer than are necessary to assess the possibility of HIV replication during ART. The extremely limited sampling of the virus populations in the 3 individuals studied is a major flaw of the study and likely contributed to erroneous conclusions.
Figure 1.
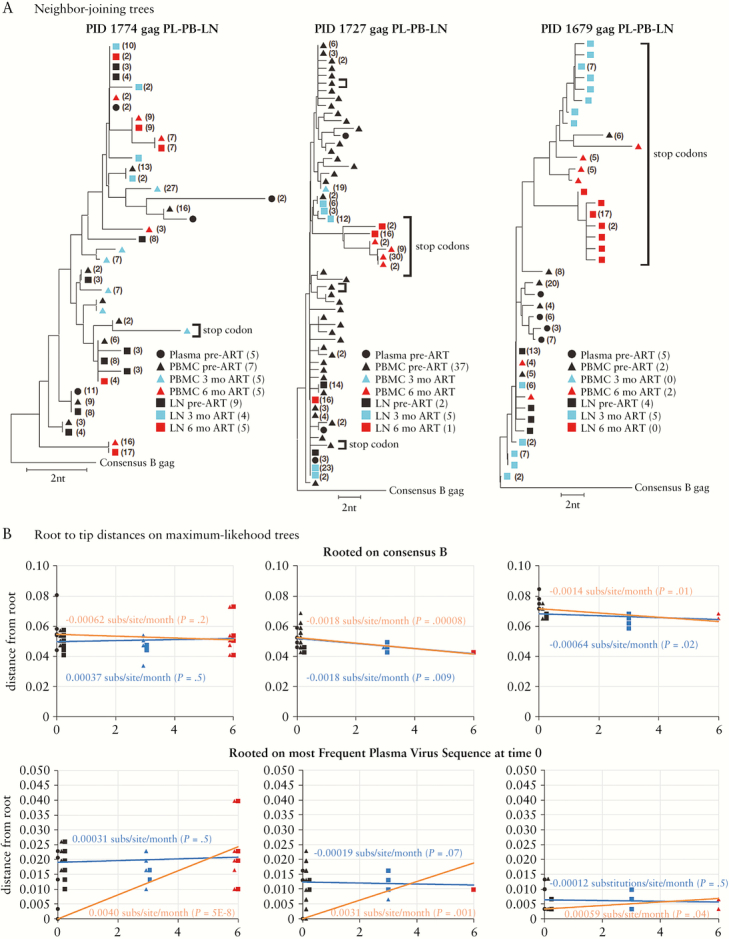
(A) Neighbor-joining trees of all gag sequences from Lorenzo-Redondo et al. (their Table S1) rooted on the consensus subgroup B HIV sequence. The numbers in parentheses indicate the frequencies of each variant (referred to by the authors [5] as “haplotype”) in the data set. Those without numbers were a single sequence in the dataset. Sequences with stop codons (mostly hypermutant) are labeled. The arrows indicate the most frequent plasma virus RNA sequences used by Lorenzo-Redondo [5], as described in the legend of their Extended Data Table 1, as the baseline for calculation of evolutionary rates. We note that 4 of the 5 the sequence sets comprising sequences from 6 months on antiretroviral therapy in peripheral blood mononuclear cell (red triangles) in PID 1774, and lymph node (red squares) are identical, which is unlikely to have occurred in vivo. (B) Plots of the distances of each sequence from the root of maximum likelihood trees versus time. Two different rootings were used as indicated. Distances of the DNA sequences from this root are plotted by linear regression using either the DNA sequences only (blue lines) or excluding the time 0 DNA sequences (orange lines), starting with the most frequent time 0 plasma virus RNA sequence (bottom panels), as was used by Lorenzo-Redondo et al. to calculate substitution rate. The slopes of the regression lines (in substitutions/site/month) are shown for each method, along with the P value relative to a line of 0 slope (in parentheses). Some of the 0-, 3-, and 6-month points are slightly shifted for clarity. ART, antiretroviral therapy; LN, lymph node, PBMC, peripheral blood mononuclear cell.
Even with the limitations of the data set, we re-analyzed the sequences and found no evidence for viral evolution in the 3 individuals reported. Because Figure 1 in the paper was plotted in a way that makes it difficult to discern the underlying relationships among the sequences analyzed, we generated neighbor-joining trees to show distance relationships among the gag sequences (Figure 1A). PBMC and LN sequences obtained from 3 and 6 months on ART (total = 33) were far more limited than pre-ART sequences (total = 75). In fact, in 2 of the 3 participants, no (PID 1679) or only a single variant (PID 1727) remained from LN at the 6-month time point after excluding those with stop codons (mostly hypermutants due to APOBEC3F or G and known to be incapable of viral replication). The few LN sequences that remained were not on longer branches than those from PBMC obtained at the same time points and were actually on branches shorter than those from the pre-ART samples, likely reflecting a loss of viral diversity coinciding with the decline in the number of infected cells on ART, rather than an increase in diversity that would be expected with ongoing cycles of viral replication as observed in untreated individuals [7].
Despite their use of complex evolutionary arguments to support their claim, Lorenzo-Redondo et al. actually calculated the “evolutionary rate” by the simplest possible method: using the p-distance of sequences at 3 and 6 months on ART from a pre-ART baseline sequence, chosen arbitrarily, to be the most frequent plasma virus RNA sequence at that time. It is evident that this choice of the baseline sequence strongly biases the analysis of viral DNA sequences at later time points in favor of showing the appearance of evolution, especially relative to the use of a more appropriate baseline, such as the consensus of pre-ART DNA sequences. To illustrate the error inherent in their approach, we repeated their analysis by: (1) aligning the sequences; (2) determining the best fit evolutionary model using jModelTest213; (3) generating maximum likelihood trees with the best fit model and with GTR+l+Gamma, as used by Lorenzo-Redondo et al.; (4) calculating the distance of each sequence to a common root; and (5) plotting the distances over time to determine if the branch lengths increased, resulting in a positive slope consistent with viral replication. The analyses were rooted either using the consensus subtype B HIV sequence (Figure 1B, top), which does not bias the analysis by assuming an evolutionary model other than a common ancestor for all sequences at the time of infection, or using the most prevalent plasma variant present at time 0, as did Lorenzo-Redondo et al. (their Extended Data Figure 2A and Table 2). The data were plotted 2 ways, either comparing the 3- and 6-month DNA sequences to the most frequent time 0 RNA sequences, as reported by Lorenzo-Redondo et al. (Figure 1B, orange lines), or basing the plots only on the DNA sequences at all time points (blue lines). Only in the DNA sequences from 1 participant (1774), who had been infected for 17 years and whose slow decline in plasma virus after ART initiation implied incomplete suppression of HIV replication in the blood during the period of observation, was there visible, but not significant, accumulation of genetic distance (ca. 3*10–4 substitutions/sites/month, P = .5) relative to the time 0 DNA sample (Figure 1B, blue lines in top and bottom graphs). By contrast, the 2 well-suppressed patients on ART (1727 and 1679) exhibited no detectable increase in root-to-tip distance of the proviral DNA with either rooting method (blue lines). Instead, they showed statistically significant evidence of a negative slope, becoming closer to the root with time. When we used the approach of Lorenzo-Redondo et al., however, we recreated the illusion of evolution with substitution rates similar to the ones reported in their Extended Data Table 2. This finding shows that the appearance of evolution clearly depends on the choice of routing method. Comparing the most prevalent viral RNA sequence at time 0 with the DNA sequences at later times, as reported by Lorenzo-Redondo et al., skews the results in favor of more distant branches at later sampling time points, makes the incorrect assumption that viral RNA and DNA sequences represent the same replicating virus population, and yields the erroneous perception of evolution due to virus replication (Figure 1B, orange lines in bottom graphs).
We also performed our own investigation of HIV sequence variation during suppressive ART by analyzing viral DNA sequences in PBMCs from 4 individuals whose viremia was suppressed on ART for >7 years and who showed clonal expansion of infected cells (Figure 2) [2]. The conclusion by Lorenzo-Redondo et al. of HIV evolution over a 6-month observation period in LN of patients on ART, which in their model is then transferred to PBMCs through cell migration, predicts that large proviral sequence changes would be evident during the approximately 10-fold longer periods of observation in the 4 individuals we studied. We plotted the distances of the sequences after long-term ART from those present before ART (Figure 2) and did not find significant divergence over time. The dashed lines on the plots in Figure 2B show the mean slope of the line, and the shaded areas represent the range of slopes expected after long-term suppression from the evolutionary rate reported by Lorenzo-Redondo et al. [5]. The solid lines show the evolutionary rates that we observed, which were not different from 0. We found only very slight evidence of sequence divergence, and that in only 1 individual (PID 3), which could easily have resulted from sampling error. These results show that, rather than an increase of viral diversity on ART from the pretherapy population, a loss of genetic diversity, including narrowing of the proviral populations toward identical sequences, is observed. These findings are consistent with our prior work showing no change in plasma viral diversity after long-term ART [1], and they are also consistent with HIV persistence on ART resulting from long-term survival and clonal proliferation of cells infected prior to ART initiation [2].
Figure 2.

(A) Neighbor-joining trees of sequences from PIDs 2–5 of Figure 1 of Maldarelli et al. (GenBank accession numbers KX018120 to KX018257) [2]. Single-genome sequences was performed as described on peripheral blood mononuclear cell (PBMC) DNA samples [2]. Sequence alignments were performed by Muscle or manually. NJ trees were generated in MEGA 6.06. Open circles denote baseline PBMC DNA sequences; filled circles, PBMC sequences after ≥7 years on suppressive antiretroviral therapy (ART). (B) The best fit evolutionary model was determined using jModelTest2 and ML trees were generated with PAUP for root-to-tip distances, which were calculated with TreeStat (v. 1.6.2). Plots of the distances of each sequence from the root (consensus B) of ML trees versus time (solid lines) is shown, as compared with the rate of evolution during ART reported by Lorenzo-Redondo et al. (mean dashed line, range in shading (6.24 to 10.3 × 10–4 substitutions/site/month)). Note that the mean root-to-tip distance of the only participant showing any positive difference between the first and last points (0.0008 substitutions/site/year) is about 12-fold less than the rate reported by Lorenzo-Redondo et al.
In summary, from our re-analysis of the sequences published by Lorenzo-Redondo et al. [5], combined with evidence from prior studies [1–3] and current analyses of samples whose dominant proviruses are known to arise from cellular proliferation rather than viral replication [2], we find that the conclusions in the publication by Lorenzo-Redondo et al. are not valid and that the concept of ongoing HIV replication during ART remains unproven.
Supplementary Data
Supplementary materials are available at Open Forum Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
Supplementary Material
Acknowledgments
Author contributions. M.F.K. performed data analyses and wrote the paper. A.W., W.R.M., and M.B. performed data analyses. W.S. wrote programs used for data analysis and performed data analyses. B.L. performed statistical analyses. F.M. provided clinical samples and interpreted results. J.W.M. interpreted data and wrote the paper. J.M.C. interpreted data and wrote the paper.
Competing financial interests. J.W.M. is a consultant to Gilead Sciences and a shareholder of Co-Crystal, Inc. The remaining authors have no potential conflicts.
Financial support. Funding was provided by intramural NIH funding to the Center for Cancer Research, NCI and Leidos contract # HHSN261200800001E to the University of Pittsburgh.
Potential conflicts of interest. All authors: no reported conflicts of interest.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1. Kearney MF, Spindler J, Shao W, et al. Lack of detectable HIV-1 molecular evolution during suppressive antiretroviral therapy. PLoS Pathog. 2014;10:e1004010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Maldarelli F, Wu X, Su L, et al. HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science. 2014;345:179–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Simonetti FR, Sobolewski MD, Fyne E, et al. Clonally expanded CD4+ T cells can produce infectious HIV-1 in vivo. Proc Natl Acad Sci U S A. 2016;113:1883–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ho YC, Shan L, Hosmane NN, et al. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell. 2013;155:540–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lorenzo-Redondo R, Fryer HR, Bedford T, et al. Persistent HIV-1 replication maintains the tissue reservoir during therapy. Nature. 2016;530:51–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Besson GJ, Lalama CM, Bosch RJ, et al. HIV-1 DNA decay dynamics in blood during more than a decade of suppressive antiretroviral therapy. Clin Infect Dis. 2014;59:1312–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kearney M, Maldarelli F, Shao W, et al. Human immunodeficiency virus type 1 population genetics and adaptation in newly infected individuals. J Virol. 2009;83:2715–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boltz VF, Hattori J, Shao W, et al. Linkage of rare drug resistance mutations detected by new ultrasensitive SGS. Paper presented at: Conference on Retroviruses and Opportunistic Infections (CROI); February 13–16, 2017; 2016; Boston, MA Available at: http://www.croiwebcasts.org/ [Google Scholar]
- 9. Jabara CB, Jones CD, Roach J, et al. Accurate sampling and deep sequencing of the HIV-1 protease gene using a Primer ID. Proc Natl Acad Sci U S A. 2011;108:20166–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.