

Abstract
Background: Understanding effects of acute smoke exposure (ASE) on airway epithelial gene expression and their relationship with the effects of chronic smoke exposure may provide biological insights into the development of smoking-related respiratory diseases. Methods: Bronchial airway epithelial cell brushings were collected from 63 individuals without recent cigarette smoke exposure and before and 24 h after smoking three cigarettes. RNA from these samples was profiled on Affymetrix Human Gene 1.0 ST microarrays. Results: We identified 91 genes differentially expressed 24 h after ASE (false discovery rate < 0.25). ASE induced genes involved in xenobiotic metabolism, oxidative stress, and inflammation and repressed genes related to cilium morphogenesis and cell cycle. While many genes altered by ASE are altered similarly in chronic smokers, metallothionein genes are induced by ASE and suppressed in chronic smokers. Metallothioneins are also suppressed in current and former smokers with lung cancer relative to those without lung cancer. Conclusions: Acute exposure to as little as three cigarettes and chronic smoking induce largely concordant changes in airway epithelial gene expression. Differences in short-term and long-term effects of smoking on metallothionein expression and their relationship to lung cancer requires further study given these enzymes’ role in the oxidative stress response.
Keywords: airway, cigarettes, epithelial cells, gene expression, smoking
BACKGROUND
Worldwide, there are ~1.25 billion adults who smoke cigarettes (26), and cigarette smoking remains the leading preventable risk factor for the development of chronic obstructive pulmonary disease (COPD) and lung cancer. Furthermore, smoking accounts for approximately six million deaths per year worldwide (6). The airway epithelium is the first tissue to encounter cigarette smoke. Numerous studies have demonstrated dramatic gene expression alterations in the bronchial airway epithelium of individuals with chronic exposure to cigarette smoke (1, 14, 34, 35). Importantly, we have found that the majority of the gene expression changes seen in current smokers revert to never-smoker levels among former smokers, although there are a number of genes whose expression is persistently altered by cigarette smoke (1). Strulovici-Barel et al. (37) further demonstrated that chronic exposure to secondhand smoke is associated with gene expression alterations in the small airway epithelium. Finally, we and others have identified airway gene expression differences associated with COPD and lung cancer (32, 35, 36).
While the intrathoracic airway response to smoke exposure has been well characterized among chronic smokers by gene expression profiling, it is unclear how acute exposure to cigarette smoke affects the airway epithelium. Previous literature has primarily identified responses to acute smoke exposure in blood, exhaled air, and bronchoalveolar fluid in human samples though notably not in bronchial epithelium (19, 27, 31, 43, 45). For instance, several studies have defined the effect of acute cigarette smoking on circulating and lung inflammatory cells from chronic smokers with normal lung function. Acute effects of cigarette smoke exposure (defined as effects measured within 24 h of cigarette use) include increased circulating neutrophils and a decreased number of blood lymphocytes, eosinophils, and basophils (43, 45). Within the lung, acute cigarette smoke increases the number of neutrophils and macrophages observed in bronchoalveolar lavage, suggesting an increased inflammatory response (19). Furthermore, previously published literature showed that smoking produces acute effects on bronchial epithelium from animal models (27, 31) and human bronchial epithelial cells (HBECs) in vitro (23, 29). With the exception of a previous in vivo study on acute smoke exposure effects using proteomic profiling of pooled samples (11), we are unaware of other ‘omic profiling studies of the effect of acute smoke exposure on human bronchial epithelium in vivo and no other studies that have measured the transcriptomic effect of acute smoke exposure.
In this study, we evaluated the effects of acute exposure to three cigarettes on gene expression in the bronchial airway epithelium. These data provide a unique opportunity to determine the relationship between ASE (acute cigarette smoke exposure) and CSE (chronic cigarette smoke exposure) potentially allowing us to identify some of the earliest biological abnormalities in response to cigarette smoke. Demonstration of the effects of ASE may provide biological insights into the development of smoking-related respiratory diseases as well as inform public perception regarding the dangers of even infrequent smoking.
METHODS
Study Population.
Current smokers were recruited (n = 63) from the Netherlands at the University Medical Center at Groningen (UMCG) between August 2011 and November 2014. These individuals were aged between 18 and 75 yr old. All subjects reported infrequent smoke exposure that was typically limited to social settings. Subjects were included based on their ability to abstain from smoking cigarettes for a period of 48 h. Individuals with a history of underlying lung disease such as alpha-1 antitrypsin deficiency or pulmonary fibrosis were excluded, as were individuals with active infections such as pneumonia or tuberculosis. While individuals with any type of carcinoma were excluded, a prior history of pulmonary nodules or COPD was not exclusion criterion. Institutional Review Board of the UMCG approved the study, and all participants provided written informed consent. The study is registered at clinicaltrials.gov under the National Clinical Trial (NCT) identifier: NCT00850863.
Airway epithelial cell collection.
Study participants were asked to refrain from cigarette smoking for at least 2 days (verified by exhaled CO < 6 ppm) and then smoke three cigarettes in the span of 1 h. Subjects then underwent fiber optic bronchoscopy after 24 h where samples were collected. In like manner, subjects abstained from smoking and underwent bronchoscopy at a separate time at least 6 wk from the postsmoking bronchoscopy to serve as an unexposed baseline sample (also verified by exhaled CO < 6 ppm). In both instances, bronchial brushings were collected from the 1st and 2nd subsegmental branches of the left lower lobe. The brushes were immediately placed in TRIzol reagent (Invitrogen) after removal from the bronchoscope and kept at −80°C until RNA isolation was performed. Total RNA was isolated with the miRNeasy Mini Kit (Qiagen, Valencia, CA). Integrity of the RNA samples was assessed by Agilent BioAnalyzer, and purity of the RNA was confirmed using a NanoDrop spectrophotometer.
Microarray data acquisition.
For each sample, 100–200 ng total RNA was processed as described in the GeneChip Whole Transcript (WT) Sense Target Labeling Assay Manual (Affymetrix, Santa Clara, CA) and Ambion WT Expression Kit Protocol (Life Technologies). The resulting material was analyzed with Affymetrix GeneChip Human Gene 1.0 ST arrays according to the manufacturer’s standard protocol.
Microarray analysis.
Probe-level hybridization intensities were normalized and summarized to produce gene-level expression values by the implementation of the robust multiarray average (16) in the affy package (version 1.36.1) (12), included in the Bioconductor software suite (version 2.11) (13), and an Entrez Gene-specific probe-set mapping (16.0.0) from the Molecular and Behavioral Neuroscience Institute (Brainarray) at the University of Michigan (9). Array quality was assessed by computing relative log expression and normalized unscaled standard error with the affyPLM package (version 1.34.0) (4). Batch correction was applied by the implementation of the ComBat algorithm in the sva package (version 3.6.0) (22). These analyses were performed by using the R environment for statistical computing (version 2.15.1). The gene expression data generated in this study are available from the National Center for Biotechnology Information’s Gene Expression Omnibus under accession GSE97010.
Determination of gene expression changes associated with ASE.
We used mixed-effects linear modeling and ANOVA to detect gene expression changes associated with smoking (baseline vs. postsmoking) after adjusting for RIN [RNA integrity number (RIN), a measure of RNA quality obtained from Bioanalyzer analysis] and intersubject variability. RIN was included to control for sources of variation in RNA quality. The P values from the ANOVA were converted to false discovery rate (FDR) q-values (2), and genes were considered differentially expressed between the baseline and postsmoking samples if they had an ANOVA q-value < 0.25. From this analysis, we also obtained a t-statistic for the difference between time points that was used to rank genes according to the effect of ASE in some of the gene set enrichment analyses described below. Gene expression was visualized by generating heat maps of data that was first mean normalized per study participant. These analyses were conducted with lme and anova.lme from the nlme package (version 3.1-118). These analyses were performed with R version 3.0.0.
Gene set enrichment analysis.
Gene set enrichment analysis (GSEA) (version 2.2.1) (39) was used to determine the relationship between this study and other data sets and to identify pathways that are altered by ASE. For this purpose, we created five ranked lists. The first list ranked genes from most repressed by ASE to most induced by the t-statistic from the comparison between ASE and baseline samples. The second list ranked genes by the t-statistic from the comparison of current chronic smokers (n = 52) to noncurrent chronic smokers (former chronic smokers and never smokers; n = 52) with data from a study of bronchial brushes previously published by our group (1). The third list ranked genes by the t-statistic computed from the comparison of HBECs exposed to cigarette smoke (n = 3) or air (n = 3) with data that we recently published (24). The fourth ranked genes by the t-statistic computed from comparison between current (n = 99) and former (n = 139) chronic smokers from another of our previously published bronchial brush data sets (36). The fifth list ranked genes by the t-statistic from the comparison of gene expression in bronchial brushes from subjects with lung cancer (n = 490) to subjects without lung cancer (n = 190) after accounting for smoking status (32). These ranked lists were used in GSEA together with either gene sets defined from data from the current study (genes significantly induced after ASE and those that significantly repressed after ASE), gene sets defined in these publications, the C2 and C5 collections of curated gene sets obtained from the Molecular Signatures Database (MSigDB), version 5.1 (38), and/or a set of metallothionein-encoding genes we curated from the literature (MT1A, MT1B, MT1CP, MT1DP, MT1E, MT1F, MT1G, MT1H, MT1HL1, MT1IP, MT1JP, MT1L, MT1M, MT1P1, MT1P3, MT1X, MT2A, MT3, and MT4). The permutation based q-value from GSEA was used to establish the significance of the observed enrichments. For each significant GSEA result, the set of genes identified by GSEA as the leading edge was used in further analyses.
Quantitative RT-PCR.
Gene expression of selected candidate genes was validated by quantitative (q)RT-PCR. The analysis was performed using SYBR Green-based RT2 qPCR Primer Assays (Qiagen). Primers of the assays for candidate genes (MT1X, ALDH3A1, AOX1, GPX2, and LMO3) and endogenous control gene [glyceraldehyde 3-phosphate dehydrogenase (GAPDH)] were designed and experimentally verified by Qiagen to ensure uniform and high PCR efficiencies under standardized amplification condition. Primers for candidate gene AKR1C2 were adapted from QuantiTect Primer Assay (Qiagen) and were designed but not experimentally verified by Qiagen. PCR efficiency of the primer pair for AKR1C2 was verified in this experiment with the test samples under the standardized experimental condition. All real-time PCR experiments were carried out in triplicate on each sample. Relative gene expression levels were calculated with the comparative threshold cycle (CT) method where the housekeeping gene GAPDH was used as a reference gene because of its relative stability of expression levels (30). Given the paired sample study design, the relative gene expressions levels of baseline and postsmoking samples were compared by a one-sided Wilcoxon signed rank test.
RESULTS
Study population.
Individuals recruited in this study were between 18 and 74 yr old with a mean age of 41 yr and predominantly men (78%). Table 1 provides demographic and clinical data of the cohort.
Table 1.
Demographics and clinical characteristics of the cohort
| Total (n = 63) | |
|---|---|
| Demographics | |
| Age | 40.78 (18–74) |
| Men, n (%) | 49 (78%) |
| BMI, kg/m2 | 24.53 ± 3.20 |
| Daily cigarette usage | |
| Cigarettes per day | 8.84 ± 7.63 |
| Cumulative smoking exposure (pack-years) | 17.59 ± 16.79 |
| Lung function | |
| FEV1 absolute | 3.76 ± 1.14 |
| FEV1, % predicted | 95.64 ± 20.47 |
| FEV1/FVC | 0.74 ± 0.14 |
| Presence of COPD | 18 (29%) |
Data are n (%), mean (range), or mean (±SD). BMI, body mass index. Pack-years is defined as the number of packs per day smoked multiplied by smoking duration (in yr). FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity; COPD, chronic obstructive pulmonary disease.
Gene expression changes associated with ASE.
We conducted a paired differential gene expression analysis of all study participants, comparing gene expression in the presence and absence of acute smoke exposure. This analysis identified 91 genes whose expression was altered by acute smoke exposure (FDR q < 0.25, Fig. 1). Of these, 50 (55%) were upregulated following acute smoke exposure including genes with functions in xenobiotic metabolism such as AOX1 and FMO1, antioxidants such as GPX2, TXNRD1, MT1X, and ALDH3A1, and genes involved in electron transport such as AKR1C2 and TALDO1 (17, 25). The 41 genes that were downregulated include AKT2, LMO3, and FOXP1, which are involved in the regulation of apoptosis and differentiation and may function as tumor suppressors (21, 46). Gene expression differences in response to ASE did not seem to vary based on smoking history. To explore further, we stratified the study participants by smoking history [<2 pack-years (PY), 2–20 PY, >20 PY] and ranked genes by the effect of ASE in each of these subgroups. We found that the genes found to be affected by ASE when considering all study participants are equivalently enriched in all three subgroups (Fig. 2). To better understand the processes that might be affected by ASE, we used GSEA to identify pathway-related gene sets from the KEGG, Reactome, and Gene Ontology databases that are enriched among the genes most induced or repressed by ASE. Gene sets with significant enrichment (GSEA q < 0.05) included pathways involved in oxidative stress, xenobiotic metabolism, zinc-ion binding, cilium development, and the cell cycle (representative GSEA results are shown in Table 2).
Fig. 1.
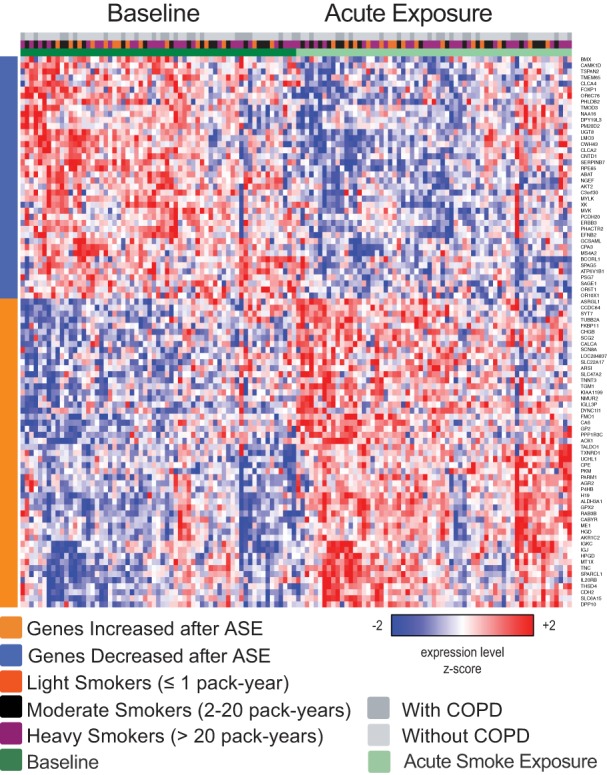
Genes with acute cigarette smoke exposure (ASE)-associated gene expression. Semisupervised heat map of the expression of 91 genes whose expression is significantly altered comparing the baseline samples with the post-ASE samples (ANOVA FDR q < 0.25). COPD, chronic obstructive pulmonary disease.
Fig. 2.
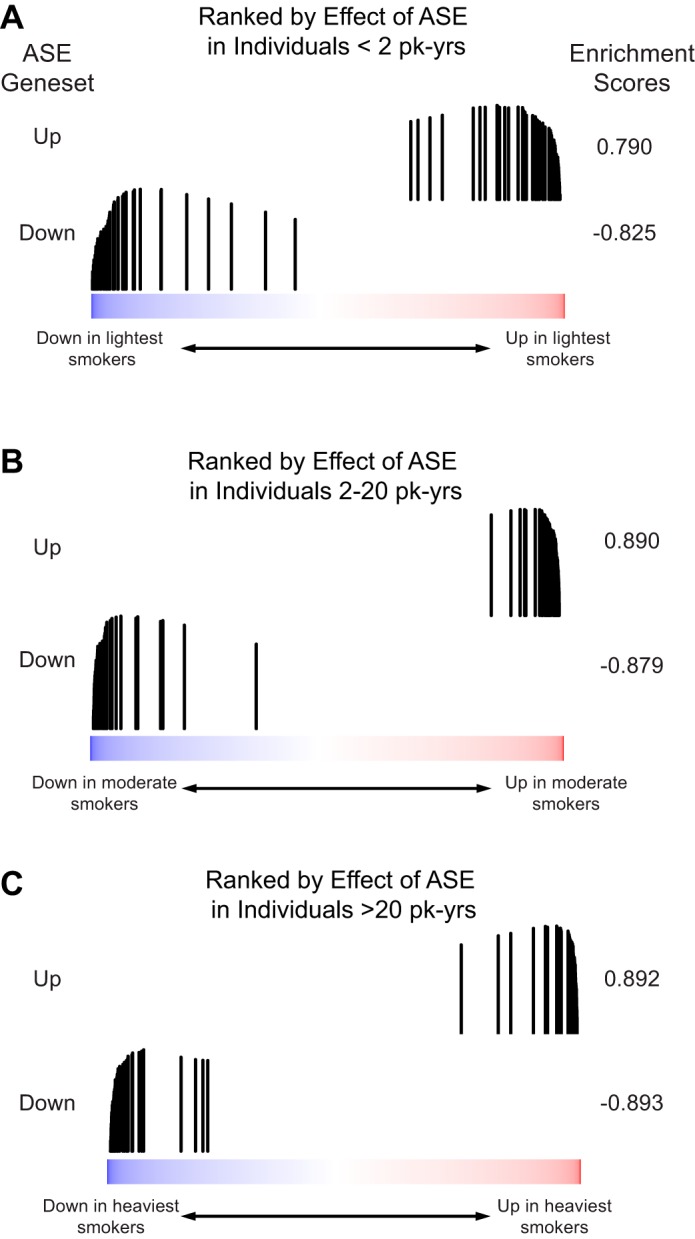
The effects of ASE are detectable independent of smoking history. The distribution of genes either significantly increased (ASE Geneset Up) or significantly decreased (ASE Geneset Down) from Fig. 1 was examined in a list of all genes ranked by the difference in expression between baseline and post-ASE samples in participants with A: <2 pack-year (PY) of lifetime smoking exposure (n = 15); B: 2–20 PY of lifetime smoking exposure (n = 21); C: >20 PY of lifetime smoking exposure (n = 27). Both gene sets were significantly enriched in all three ranked lists (each q < 0.001), and the enrichment scores are similar independent of lifetime smoking exposure.
Table 2.
Acute smoke exposure pathway-related gene sets identified by GSEA
| Pathway | NES | q-Value |
|---|---|---|
| KEGG Pathways | ||
| Upregulated genes | ||
| Pentose phosphate pathway | 2.179 | <0.001 |
| Glutathione metabolism | 1.712 | 0.029 |
| MAPK signaling pathway | 1.621 | 0.048 |
| Metabolism of xenobiotics – cytochrome P450 | 1.622 | 0.049 |
| Reactome Pathways | ||
| Upregulated genes | ||
| Antigen presentation folding and assembly | 1.885 | 0.012 |
| Cell cycle and G protein activation | 1.743 | 0.03 |
| Downregulated genes | ||
| Cell cycle | −1.913 | 0.016 |
| Mitosis | −1.793 | 0.027 |
| DNA repair | −1.767 | 0.028 |
| Apoptotic execution | −1.754 | 0.029 |
| DNA replication | −1.655 | 0.043 |
| Gene Ontology Pathways | ||
| Upregulated genes | ||
| Metal ion transmembrane transporter | 2.297 | <0.001 |
| Reactive oxygen species metabolism | 1.895 | 0.021 |
| Response to zinc ion | 1.963 | 0.012 |
| Acute inflammatory response | 1.873 | 0.026 |
| Downregulated genes | ||
| Cilium morphogenesis | −2.703 | <0.001 |
| Chromatin modification | −2.213 | 0.001 |
| Epigenetic regulation of gene expression | −2.158 | 0.002 |
GSEA, gene set enrichment analysis; KEGG, Kyoto Encyclopedia of Genes and Genomes; NES, normalized enrichment score; FDR, false discovery rate.
Relationship between ASE and CSE.
We next examined whether gene expression changes in the bronchial airways associated with ASE are among the genes altered by CSE. Using GSEA and a previously published (1), data set of chronic current smokers compared with noncurrent smokers (former smokers and never-smokers), we found that genes significantly increased or decreased by ASE were concordantly enriched among the genes most increased or decreased in chronic current smokers (GSEA q < 0.001 for both comparisons; Fig. 3). Furthermore, genes we previously identified as being induced in chronic smokers and rapidly repressed upon smoking cessation (1) were enriched among the genes most upregulated in response to ASE (GSEA q < 0.001; Fig. 4).
Fig. 3.
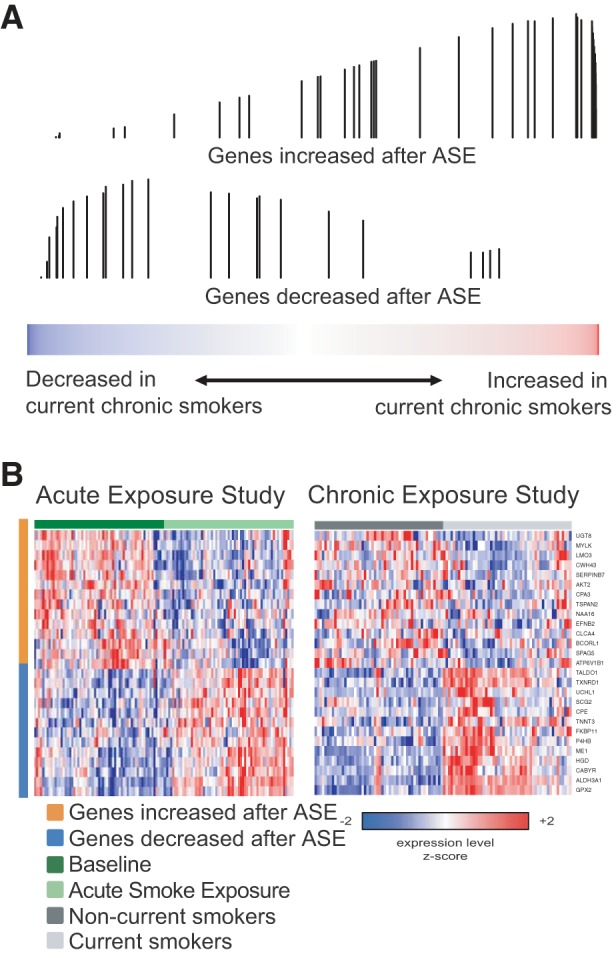
The effects of ASE are significantly concordant with the effects of chronic cigarette smoke exposure (CSE). A: the distribution of genes either significantly increased (top) or significantly decreased (bottom) by ASE was examined in a list of all genes ranked by their expression difference between current chronic and noncurrent chronic smokers (former chronic smokers and never-smokers) from the data from Beane et al. (1) using gene set enrichment analysis (GSEA). The genes increased by ASE are enriched among the genes most induced in current chronic smokers (q < 0.001), and the genes decreased by ASE are enriched among the genes most decreased in current chronic smokers (q < 0.001). Each vertical bar indicates the position of a gene from an ASE gene set within the gene list ranked by the effect of CSE. The height of the bar represents the running enrichment score. B: expression of the most enriched genes from the GSEA analysis in the ASE (left) and CSE data sets (right).
Fig. 4.
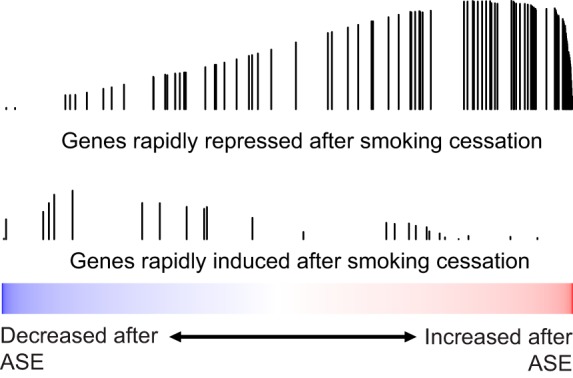
Genes that change rapidly after smoking cessation are enriched among the genes changed by ASE. The distribution of genes identified by Beane et al. (1) as being either rapidly repressed (top) or rapidly induced (bottom) after smoking cessation were examined in a list of all genes ranked by their expression difference between baseline and post-ASE samples was examined by GSEA. Genes that are rapidly repressed toward the levels seen in nonsmokers after smoking cessation are enriched among the genes most increased by ASE (q < 0.001). We did not observe significant enrichment of genes that are rapidly induced after smoking cessation among the genes that are most decreased by ASE (q = 0.23).
Relationship between ASE and in vitro exposure to cigarette smoke.
We next sought to determine whether the gene expression changes in airway epithelium we found associated with ASE are likely to be the direct consequence of smoke exposure on epithelial cells. We compared the genes changed with ASE in our volunteers with the genes altered in HBECs following in vitro exposure to cigarette smoke. To do this, we made use of a recently published study from our laboratory in which HBECs cultured from nonsmoking donors were grown at an air-liquid interface and exposed to 1 puff of cigarette smoke per minute (n = 3) or 1 puff of air per minute (n = 3) for 48 min and profiled for gene expression 24 h later (24). Using GSEA, we found that genes significantly increased or decreased by ASE were significantly concordantly enriched among the genes most increased or decreased (GSEA q < 0.05 for both comparisons) by short-term exposure of HBECs to cigarette smoke in vitro (Fig. 5).
Fig. 5.
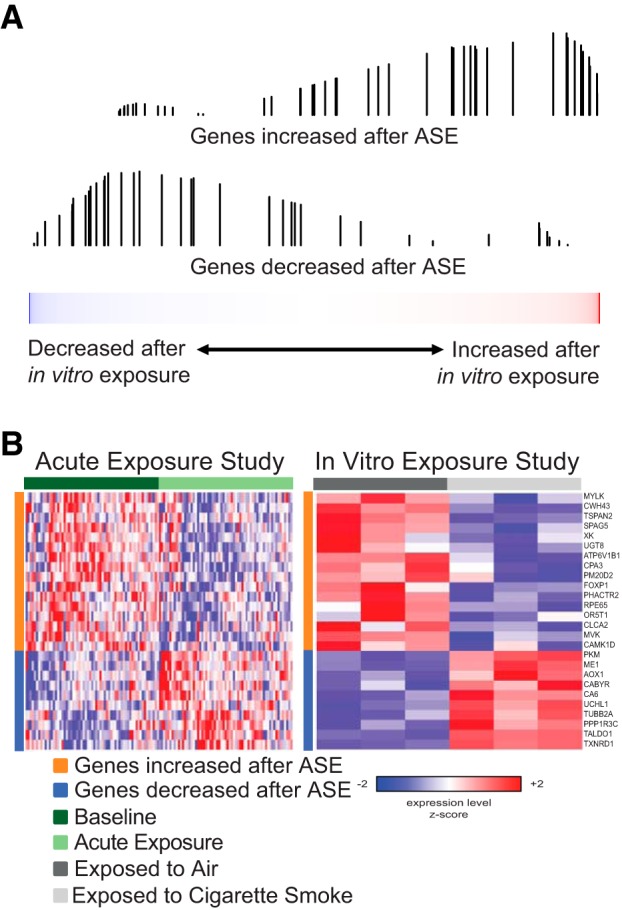
The effects of ASE are significantly concordant with the effects of in vitro exposure to cigarette smoke. A: the distribution of genes we identified as either significantly increased (top) or significantly decreased (bottom) by ASE was examined in a list of all genes ranked by their expression difference between in vitro cigarette-smoke exposed and air-exposed human bronchial epithelial cells (HBECs) using data we have previously published (24). The genes increased by ASE are enriched among the genes most induced by in vitro exposure to cigarette smoke (q = 0.02), and the genes decreased by ASE are enriched among the genes most repressed by in vitro exposure to cigarette smoke (q = 0.01). B: expression of the most enriched genes from the GSEA in the ASE (left) and in vitro exposure (right) data sets.
Discordant effect of ASE and CSE on metallothionein expression.
We found significant enrichment of zinc-binding genes and genes involved in the cellular response to other metallic ions among the genes that are induced by ASE (Table 2). Many of these genes encode for metallothioneins. This finding was surprising as we have previously identified several metallothionein genes as being repressed in chronic smokers relative to never smokers (1). The induction of metallothionein genes occurs both in response to ASE and in vitro exposure to cigarette smoke (GSEA q < 0.05 for both analyses; Fig. 6, A and B), which is in stark contrast to the downregulation of these same genes in chronic smokers (GSEA q < 0.05; Fig. 6, C and D). Consistent with a potential relevance for repressed metallothionein expression in the pathogenesis of smoking-related lung diseases, using data from the AEGIS trials (32), we also observed that the metallothionein genes are significantly enriched among the genes most repressed in the airways of current and former chronic smokers with lung cancer relative to current and former chronic smokers with benign diseases (GSEA q < 0.05; Fig. 6E).
Fig. 6.
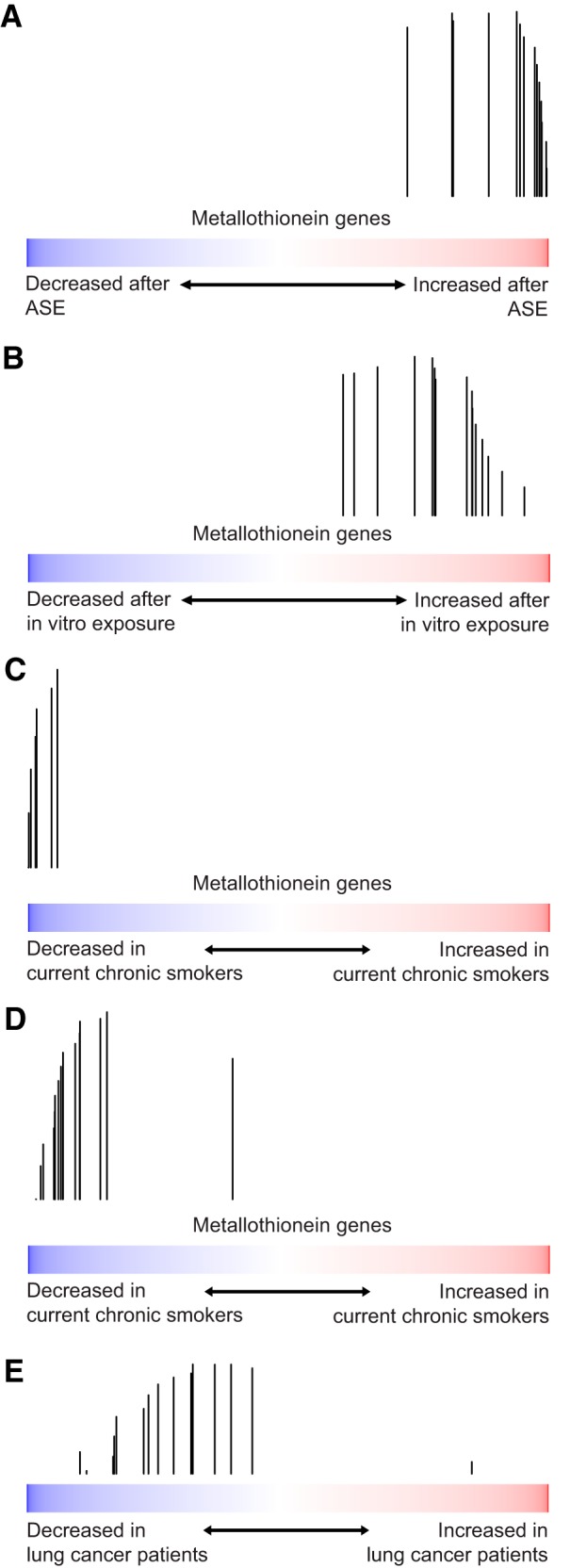
Metallothioneins are downregulated in CSE and upregulated in ASE (both in vivo and in vitro). The distribution of metallothionein genes was examined in lists of all genes ranked by their smoking-related gene expression in four different data sets. A: genes ranked by effect of ASE. B: genes ranked by effect of in vitro cigarette smoke exposure (24). C: genes ranked by comparison of current chronic smokers to noncurrent chronic smokers (1). D: genes ranked by comparison of current chronic smokers to former chronic smokers (36). E: genes ranked by comparison of subjects with lung cancer to those without lung cancer (32). While genes from the metallothionein family are enriched among the genes induced by ASE (q < 0.001) and after in vitro cigarette smoke exposure (q = 0.02), they are among the genes most repressed in chronic smokers in two independent data sets (q < 0.001 and q < <0.001). Similarly, they are also significantly repressed in smokers with lung cancer (q < 0.05).
Validation of differential expression of selected genes by qRT-PCR.
Gene expression of selected genes was measured by qRT-PCR including MT1X, ALDH3A1, AKR1C2, GPX2, AOX1, and LMO3. These genes were found to be differentially expressed between baseline and ASE samples, thus forming the basis of their selection for validation by qRT-PCR. Samples from 25 patients (50 samples total) were used for the qRT-PCR. We found significant concordant differential expression for five of the six genes (Table 3), including the metallothionein encoded by MT1X. The one gene that was not concordant was AKR1C2, though this may in part be due to technical issues. As mentioned previously, the other candidate genes were analyzed with primers designed and experimentally verified by Qiagen, while that of AKR1C2 was only designed but never experimentally verified.
Table 3.
Mean LOG2 fold changes in gene expression and qRT-PCR for selected genes
| Gene | Mean LOG2 Fold Change Gene Expression | Mean LOG2 Fold Change qRT-PCR | P Value PCR |
|---|---|---|---|
| MT1X | 0.262 | 0.213 | 0.031 |
| ALDH3A1 | 0.693 | 0.959 | 3.575e-4 |
| AKR1C2 | 0.251 | 0.205 | 0.132 |
| GPX2 | 0.459 | 0.485 | 3.183e-5 |
| AOX1 | 0.131 | 0.370 | 2.98e-8 |
| LMO3 | −0.973 | −1.586 | 2.98e-8 |
qRT-PCR, quantitative reverse transcription polymerase chain reaction.
DISCUSSION
Our data indicate that smoking as little as three cigarettes induces many of the same gene expression changes as seen in CSE. By performing whole-genome gene expression profiling of bronchial brushings of individuals after smoking three cigarettes, we defined a gene expression signature of ASE. There are several important characteristics of this response. First, these gene expression alterations are characterized by increased expression of genes involved in xenobiotic metabolism and oxidative stress. Second, acute smoking-associated changes in gene expression in the bronchial airway epithelium are similar to those previously reported to be associated with CSE, suggesting that some of the gene expression differences observed in chronic smokers are the consequence of repeated acute exposure. Finally, acute smoking-associated gene expression in vivo mirrors gene expression changes seen following ASE exposure to epithelial cells in vitro, suggesting that the gene expression changes observed with ASE are particularly due to the direct effects of smoking on airway epithelial cells.
Many of the gene expression alterations we identified are consistent with the known effects of cigarette smoke. For example, cigarette smoking has previously been shown to cause the formation of reactive aldehydes (42). These reactive aldehydes have further been shown to induce the production of neutrophil and eosinophil chemoattractants as part of an acute inflammatory response (41). Consistent with these observations, we found that, as measured by microarrays and qRT-PCR, ASE increases expression of ALDH3A1, which encodes an aldehyde dehydrogenase involved in the oxidation of toxic aldehydes produced from oxidative stress and exposure to cigarette smoke. Proteomic profiling on a subset of these same patients similarly found an upregulation of ALDH3A1 after ASE when measured by ELISA (11). Several studies have previously demonstrated that lung tissue obtained from smokers (25) and HBECs exposed to cigarette smoke extract exhibit a marked increase in ALDH3A1 gene and protein expression (17). Furthermore, this same study showed that HBECs overexpressing ALDH3A1 were protected from cigarette smoke extract-induced cytotoxicity and exhibited reduced DNA damage, suggesting that the upregulation of ALDH3A1 protects against the reactive aldehydes produced by cigarette combustion.
Cigarette smoking has also been shown to cause significant lung oxidative stress, with up to 1014 free radicals generated from each puff of cigarette smoke (8). Key players in the oxidative stress response are glutathione peroxidases (GPXs), which reduce hydrogen peroxide to protect against DNA injury, protein damage, and lipid peroxidation (40). Among the genes we measured by gene expression analysis and qRT-PCR that are induced by ASE, we identified GPX2, which encodes one of four GPX isoenzymes responsible for the majority of glutathione-dependent hydrogen peroxide-reducing activity. Of the four different glutathione peroxidases, GPX2 is the most strongly induced in the airway epithelium of smokers compared with nonsmokers (33). Many of the other genes that are upregulated by ASE are involved in the detoxification and metabolism of additional cigarette smoke components, including aldehyde oxidase 1 (AOX1) and flavin-containing monooxygenase 1 (FMO1): FMO1 catalyzes the oxidation of nicotine to nicotine N-oxide (15), while AOX1 catalyzes the conversion of the nicotine-∆5′(1’)-iminium ion to cotinine (3).
The aldo-keto reductase AKR1C1 is also upregulated in response to ASE. Van Dyck et al. (44) showed that the expression of AKR1C1 was induced in histologically normal bronchial biopsy specimens obtained from smokers relative to those obtained from nonsmokers. The NADPH oxidoreductases encoded by the four genes in the AK1RC family serve several functions, including the detoxification of drugs and metabolism of xenobiotics (10). Within the lung, AKR1C1 is the most highly expressed member of this family and is responsible for the activation of procarcinogenic polycyclic aromatic hydrocarbons (5) and the detoxification of carcinogenic nitrosamino-ketone compounds (7).
We identified striking similarity between the gene expression changes associated with ASE and the gene expression differences between chronic smokers and nonsmokers. This suggests that some of the gene expression changes associated with chronic exposure to cigarette smoke are the consequence of ongoing acute exposure. We have previously shown that the majority of genes differentially expressed between current and never smokers are rapidly reversible upon smoking cessation, while the remainder are either slowly reversible or persistently altered (1). The genes rapidly repressed following smoking cessation (which is the majority of rapidly reversible genes) are enriched among the genes that are upregulated in response to acute smoke exposure. This suggests that many of the genes that are rapidly induced by ASE are similarly rapidly reversed with smoking cessation. We also found significant enrichment for genes altered by ASE among the genes whose expression is altered in HBECs by cigarette smoke exposure in vitro, suggesting that much of the impact of ASE on gene expression is likely due to the direct and transient effect of cigarette smoke components on the epithelial cells.
Metallothioneins are a particularly interesting class of genes that are discordant between acute and chronic smoke exposure. While these genes are downregulated in chronic smokers, they are surprisingly induced by ASE in gene expression analysis, qRT-PCR, and after in vitro cigarette smoke exposure. Metallothioneins bind essential metals (such as copper, iron, and zinc) as well as toxic metals (such as cadmium and mercury) and are involved in the response to oxidative stress (18). Increased levels of cadmium, zinc, and MT1 and MT2 metallothionein isoforms have been observed in placental tissue obtained from postpartum female smokers (28), and increased levels of metallothionein have been observed in the periodontal tissue of smokers, where they may play a role in defense against free radicals (20). The reason for the discordance between the expression of metallothioneins in acute and chronic exposure is presently unclear. One possible explanation is that at baseline, metallothionein gene expression is induced in response to smoke exposure but that the repeated cellular injury that occurs with CSE interferes with the ability of these genes to be induced in response to environmental or smoking-related stressors. Alternatively, CSE might prevent metallothionein gene expression either through an epigenetic mechanism or decreases in the number of metallothionein expressing cells. Interestingly, we previously found that in former smokers, metallothionein gene expression returns to baseline more slowly than the majority of genes that are rapidly reversible (1). An inability to induce metallothioneins in the context of CSE might impair the epithelial cell’s ability to repair injury from oxidative stress, which in turn could contribute to the pathogenesis of diseases such as COPD and lung cancer. Our observation that metallothionein gene expression is decreased in the airways of patients with lung cancer independently of smoking status is consistent with this hypothesis.
Our study has several notable strengths. One particularly powerful aspect is that it captures the effects of ASE in individuals without recent exposure. This allows for an assessment of gene expression changes that occur with relatively rapid kinetics following ASE. Additionally, the collection of paired baseline and postsmoking samples allows for a paired analysis that accounts for heterogeneity due to interindividual differences at baseline, including heterogeneity due to differences in historical tobacco exposure and heterogeneity due to differences in lung health.
While our study provides a unique opportunity to understand the acute effects of cigarette exposure, there are a number of important limitations to our findings. The participants in the study are current smokers who report cigarette usage primarily in social settings and who were able to abstain from smoking for >48 h before each study visit; but smoking history varies dramatically between study participants. Another limitation lies within the clinical heterogeneity of the cohort both with respect to age and lung function. However, despite the diversity of the study participants, we were able to use a paired-sample study design and identify genes whose expression differs between baseline and after ASE. But the small sample size prevented us from being able to detect differences in the response to ASE between clinical subgroups (e.g., participants with and without COPD).
In summary, our findings suggest that as few as three cigarettes impacts gene expression in the bronchial airway epithelium within 24 h of exposure and that there is significant similarity between the effects of ASE and the changes seen among chronic smokers. These findings may affect public perception of the lung health risks arising from occasional smoking and influence public policy related to tobacco health education. The observation that ASE induces metallothionein expression but that chronic smokers exhibit reduced metallothionein expression suggests a potential effect of CSE that might contribute to the pathogenesis of diseases associated with chronic smoking. Further study of this phenomenon and other differences between the effects of ASE and CSE may provide an avenue for understanding the potentially disease-relevant biological consequences of persistent cigarette smoke exposure.
GRANTS
This study was funded by the Top Institute Pharma consortium, project T1-108: Foundation TI Pharma, Nycomed BV, GlaxoSmithKline, UMCG, University of Groningen (RUG). A. Faiz was funded by Longfonds Junior Investigators Grant 4.2.16.132JO.
DISCLOSURES
D. S. Postma: The University of Groningen has received money for Professor Postma regarding a grant for research from Astra Zeneca, Chiesi, Genentec, GSK, and Roche. Fees for consultancies were given to the University of Groningen by Astra Zeneca, Boehringer Ingelheim, Chiesi, GSK, Takeda, and TEVA.
W. Timens reports grants from Merck during the conduct of the study; personal fees from Pfizer, GSK, Chiesi, and Roche Diagnostics/Ventana, grants from Dutch Asthma Fund, personal fees from Biotest, Merck Sharp Dohme, Novartis, Lilly Oncology, and Boehringer Ingelheim, outside the submitted work.
M. van den Berge reports research grants paid to the university from GSK, Astra Zeneca, TEVA, and Chiesi outside the submitted work.
A. Spira reports personal fees from Veracyte, Inc. and Janssen Pharma and Allegro Diagnostics, Inc., outside the submitted work.
The remaining authors have nothing to disclose.
AUTHOR CONTRIBUTIONS
E.B. and Y.O.G. analyzed data; E.B., Y.O.G., and M.E.L. interpreted results of experiments; E.B. prepared figures; E.B. drafted manuscript; E.B., A.F., Y.O.G., A.L., Y.A., X.X., G.L., N.H.t.H., I.H., W.T., C.-A.B., D.S.P., M.v.d.B., A.S., and M.E.L. edited and revised manuscript; E.B., A.F., Y.O.G., A.L., Y.A., X.X., G.L., N.H.t.H., I.H., W.T., C.-A.B., D.S.P., M.v.d.B., A.S., and M.E.L. approved final version of manuscript; A.F., N.H.t.H., I.H., W.T., C.-A.B., D.S.P., and M.v.d.B. conceived and designed research; A.F., A.L., Y.A., X.X., G.L., N.H.t.H., I.H., W.T., C.-A.B., D.S.P., and M.v.d.B. performed experiments.
REFERENCES
- 1.Beane J, Sebastiani P, Liu G, Brody JS, Lenburg ME, Spira A. Reversible and permanent effects of tobacco smoke exposure on airway epithelial gene expression. Genome Biol 8: 1–17, 2007. doi: 10.1186/gb-2007-8-9-r201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J R Stat Soc B 57: 289–300, 1995. [Google Scholar]
- 3.Brandänge S, Lindblom L. The enzyme “aldehyde oxidase” is an iminium oxidase. Reaction with nicotine delta 1‘(5’) iminium ion. Biochem Biophys Res Commun 91: 991–996, 1979. doi: 10.1016/0006-291X(79)91977-6. [DOI] [PubMed] [Google Scholar]
- 4.Brettschneider J, Collin F, Bolstad BM, Speed TP. Quality Assessment for Short Oligonucleotide Microarray Data. Technometrics 50: 241–264, 2008. doi: 10.1198/004017008000000334. [DOI] [Google Scholar]
- 5.Cantin AM. Cellular response to cigarette smoke and oxidants: adapting to survive. Proc Am Thorac Soc 7: 368–375, 2010. doi: 10.1513/pats.201001-014AW. [DOI] [PubMed] [Google Scholar]
- 6.Jamal A, Homa DM, O’Connor E, Babb SD, Caraballo RS, Singh T, Hu SS, King BA; Centers for Disease Control and Prevention . Current cigarette smoking among adults - United States, 2005-2014. MMWR Morb Mortal Wkly Rep 64: 1233–1240, 2015. doi: 10.15585/mmwr.mm6444a2. [DOI] [PubMed] [Google Scholar]
- 7.Chen W-D, Zhang Y. Regulation of aldo-keto reductases in human diseases. Front Pharmacol 3: 35–40, 2012. doi: 10.3389/fphar.2012.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Church DF, Pryor WA. Free-radical chemistry of cigarette smoke and its toxicological implications. Environ Health Perspect 64: 111–126, 1985. doi: 10.1289/ehp.8564111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dai M, Wang P, Boyd AD, Kostov G, Athey B, Jones EG, Bunney WE, Myers RM, Speed TP, Akil H, Watson SJ, Meng F. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res 33: e175, 2005. doi: 10.1093/nar/gni179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ebert B, Kisiela M, Wsól V, Maser E. Proteasome inhibitors MG-132 and bortezomib induce AKR1C1, AKR1C3, AKR1B1, and AKR1B10 in human colon cancer cell lines SW-480 and HT-29. Chem Biol Interact 191: 239–249, 2011. doi: 10.1016/j.cbi.2010.12.026. [DOI] [PubMed] [Google Scholar]
- 11.Franciosi L, Postma DS, van den Berge M, Govorukhina N, Horvatovich PL, Fusetti F, Poolman B, Lodewijk ME, Timens W, Bischoff R, ten Hacken NHT. Susceptibility to COPD: differential proteomic profiling after acute smoking. PLoS One 9: e102037, 2014. doi: 10.1371/journal.pone.0102037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gautier L, Cope L, Bolstad BM, Irizarry RA. affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20: 307–315, 2004. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- 13.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JYH, Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5: 1–16, 2004. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harvey B-G, Heguy A, Leopold PL, Carolan BJ, Ferris B, Crystal RG. Modification of gene expression of the small airway epithelium in response to cigarette smoking. J Mol Med (Berl) 85: 39–53, 2007. doi: 10.1007/s00109-006-0103-z. [DOI] [PubMed] [Google Scholar]
- 15.Hinrichs AL, Murphy SE, Wang JC, Saccone S, Saccone N, Steinbach JH, Goate A, Stevens VL, Bierut LJ. Common polymorphisms in FMO1 are associated with nicotine dependence. Pharmacogenet Genomics 21: 397–402, 2011. doi: 10.1097/FPC.0b013e328346886f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4: 249–264, 2003. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 17.Jang J-H, Bruse S, Liu Y, Duffy V, Zhang C, Oyamada N, Randell S, Matsumoto A, Thompson DC, Lin Y, Vasiliou V, Tesfaigzi Y, Nyunoya T. Aldehyde dehydrogenase 3A1 protects airway epithelial cells from cigarette smoke-induced DNA damage and cytotoxicity. Free Radic Biol Med 68: 80–86, 2014. doi: 10.1016/j.freeradbiomed.2013.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kägi JHR. Overview of metallothionein, in Methods in Enzymology, edited by Riordan JF, Vallee BL. Elsevier, 1991, p. 613–626. [69], https://www.sciencedirect.com/science/article/pii/007668799105145L. [DOI] [PubMed] [Google Scholar]
- 19.Karimi R, Tornling G, Grunewald J, Eklund A, Sköld CM. Cell recovery in bronchoalveolar lavage fluid in smokers is dependent on cumulative smoking history. PLoS One 7: e34232, 2012. doi: 10.1371/journal.pone.0034232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Katsuragi H, Hasegawa A, Saito K. Distribution of metallothionein in cigarette smokers and non-smokers in advanced periodontitis patients. J Periodontol 68: 1005–1009, 1997. doi: 10.1902/jop.1997.68.10.1005. [DOI] [PubMed] [Google Scholar]
- 21.Kim S-Y, Lee J-H, Huh JW, Ro JY, Oh Y-M, Lee S-D, An S, Lee Y-S. Cigarette smoke induces Akt protein degradation by the ubiquitin-proteasome system. J Biol Chem 286: 31932–31943, 2011. doi: 10.1074/jbc.M111.267633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28: 882–883, 2012. doi: 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mio T, Romberger DJ, Thompson AB, Robbins RA, Heires A, Rennard SI. Cigarette smoke induces interleukin-8 release from human bronchial epithelial cells. Am J Respir Crit Care Med 155: 1770–1776, 1997. doi: 10.1164/ajrccm.155.5.9154890. [DOI] [PubMed] [Google Scholar]
- 24.Moses E, Wang T, Corbett S, Jackson GR, Drizik E, Perdomo C, Perdomo C, Kleerup E, Brooks D, O’Connor G, Dubinett S, Hayden P, Lenburg ME, Spira A. Molecular Impact of Electronic Cigarette Aerosol Exposure in Human Bronchial Epithelium. Toxicol Sci 155: 248–257, 2017. doi: 10.1093/toxsci/kfw198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patel M, Lu L, Zander DS, Sreerama L, Coco D, Moreb JS. ALDH1A1 and ALDH3A1 expression in lung cancers: correlation with histologic type and potential precursors. Lung Cancer 59: 340–349, 2008. doi: 10.1016/j.lungcan.2007.08.033. [DOI] [PubMed] [Google Scholar]
- 26.Proctor RN. Tobacco and the global lung cancer epidemic. Nat Rev Cancer 1: 82–86, 2001. doi: 10.1038/35094091. [DOI] [PubMed] [Google Scholar]
- 27.Robbins RA, Koyama S, Spurzem JR, Rickard KA, Nelson KJ, Gossman GL, Thiele GM, Rennard SI. Modulation of neutrophil and mononuclear cell adherence to bronchial epithelial cells. Am J Respir Cell Mol Biol 7: 19–29, 1992. doi: 10.1165/ajrcmb/7.1.19. [DOI] [PubMed] [Google Scholar]
- 28.Ronco AM, Arguello G, Muñoz L, Gras N, Llanos M. Metals content in placentas from moderate cigarette consumers: correlation with newborn birth weight. Biometals 18: 233–241, 2005. doi: 10.1007/s10534-005-0583-2. [DOI] [PubMed] [Google Scholar]
- 29.Rusznak C, Mills PR, Devalia JL, Sapsford RJ, Davies RJ, Lozewicz S. Effect of cigarette smoke on the permeability and IL-1beta and sICAM-1 release from cultured human bronchial epithelial cells of never-smokers, smokers, and patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 23: 530–536, 2000. doi: 10.1165/ajrcmb.23.4.3959. [DOI] [PubMed] [Google Scholar]
- 30.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3: 1101–1108, 2008. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 31.Shoji S, Ertl RF, Koyama S, Robbins R, Leikauf G, Von Essen S, Rennard SI. Cigarette smoke stimulates release of neutrophil chemotactic activity from cultured bovine bronchial epithelial cells. Clin Sci (Lond) 88: 337–344, 1995. doi: 10.1042/cs0880337. [DOI] [PubMed] [Google Scholar]
- 32.Silvestri GA, Vachani A, Whitney D, Elashoff M, Porta Smith K, Ferguson JS, Parsons E, Mitra N, Brody J, Lenburg ME, Spira A, AEGIS Study Team . A Bronchial Genomic Classifier for the Diagnostic Evaluation of Lung Cancer. N Engl J Med 373: 243–251, 2015. doi: 10.1056/NEJMoa1504601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singh A, Rangasamy T, Thimmulappa RK, Lee H, Osburn WO, Brigelius-Flohé R, Kensler TW, Yamamoto M, Biswal S. Glutathione peroxidase 2, the major cigarette smoke-inducible isoform of GPX in lungs, is regulated by Nrf2. Am J Respir Cell Mol Biol 35: 639–650, 2006. doi: 10.1165/rcmb.2005-0325OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spira A, Beane J, Shah V, Liu G, Schembri F, Yang X, Palma J, Brody JS. Effects of cigarette smoke on the human airway epithelial cell transcriptome. Proc Natl Acad Sci USA 101: 10143–10148, 2004. doi: 10.1073/pnas.0401422101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spira A, Beane JE, Shah V, Steiling K, Liu G, Schembri F, Gilman S, Dumas Y-M, Calner P, Sebastiani P, Sridhar S, Beamis J, Lamb C, Anderson T, Gerry N, Keane J, Lenburg ME, Brody JS. Airway epithelial gene expression in the diagnostic evaluation of smokers with suspect lung cancer. Nat Med 13: 361–366, 2007. doi: 10.1038/nm1556. [DOI] [PubMed] [Google Scholar]
- 36.Steiling K, van den Berge M, Hijazi K, Florido R, Campbell J, Liu G, Xiao J, Zhang X, Duclos G, Drizik E, Si H, Perdomo C, Dumont C, Coxson HO, Alekseyev YO, Sin D, Pare P, Hogg JC, McWilliams A, Hiemstra PS, Sterk PJ, Timens W, Chang JT, Sebastiani P, O’Connor GT, Bild AH, Postma DS, Lam S, Spira A, Lenburg ME. A dynamic bronchial airway gene expression signature of chronic obstructive pulmonary disease and lung function impairment. Am J Respir Crit Care Med 187: 933–942, 2013. doi: 10.1164/rccm.201208-1449OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Strulovici-Barel Y, Omberg L, O’Mahony M, Gordon C, Hollmann C, Tilley AE, Salit J, Mezey J, Harvey B-G, Crystal RG. Threshold of biologic responses of the small airway epithelium to low levels of tobacco smoke. Am J Respir Crit Care Med 182: 1524–1532, 2010. doi: 10.1164/rccm.201002-0294OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Subramanian A, Kuehn H, Gould J, Tamayo P, Mesirov JP. GSEA-P: a desktop application for Gene Set Enrichment Analysis. Bioinformatics 23: 3251–3253, 2007. doi: 10.1093/bioinformatics/btm369. [DOI] [PubMed] [Google Scholar]
- 39.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550, 2005. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun Y. Free radicals, antioxidant enzymes, and carcinogenesis. Free Radic Biol Med 8: 583–599, 1990. doi: 10.1016/0891-5849(90)90156-D. [DOI] [PubMed] [Google Scholar]
- 41.van der Toorn M, Slebos DJ, de Bruin HG, Gras R, Rezayat D, Jorge L, Sandra K, van Oosterhout AJ. Critical role of aldehydes in cigarette smoke-induced acute airway inflammation. Respir Res 14: 45–56, 2013. doi: 10.1186/1465-9921-14-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Troszok A, van der Toorn M, Rezayat D, Gras R, de Bruin H, Slebos D-J, van Oosterhout A. Role of highly reactive aldehydes in cigarette smoke induced airway inflammation. Eur Respir J 38: 3831, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van der Vaart H, Postma DS, Timens W, ten Hacken NH. Acute effects of cigarette smoke on inflammation and oxidative stress: a review. Thorax 59: 713–721, 2004. doi: 10.1136/thx.2003.012468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van Dyck E, Nazarov PV, Muller A, Nicot N, Bosseler M, Pierson S, Van Moer K, Palissot V, Mascaux C, Knolle U, Ninane V, Nati R, Bremnes RM, Vallar L, Berchem G, Schlesser M. Bronchial airway gene expression in smokers with lung or head and neck cancer. Cancer Med 3: 322–336, 2014. doi: 10.1002/cam4.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Winkel P, Statland BE. The acute effect of cigarette smoking on the concentrations of blood leukocyte types in healthy young women. Am J Clin Pathol 75: 781–785, 1981. doi: 10.1093/ajcp/75.6.781. [DOI] [PubMed] [Google Scholar]
- 46.Woenckhaus M, Klein-Hitpass L, Grepmeier U, Merk J, Pfeifer M, Wild P, Bettstetter M, Wuensch P, Blaszyk H, Hartmann A, Hofstaedter F, Dietmaier W. Smoking and cancer-related gene expression in bronchial epithelium and non-small-cell lung cancers. J Pathol 210: 192–204, 2006. doi: 10.1002/path.2039. [DOI] [PubMed] [Google Scholar]