

Abstract
Cardiac fibroblasts are critical mediators of fibrotic remodeling in the failing heart and transform into myofibroblasts in the presence of profibrotic factors such as transforming growth factor-β. Myocardial fibrosis worsens cardiac function, accelerating the progression to decompensated heart failure (HF). We investigated the effects of a novel inhibitor (NM922; NovoMedix, San Diego, CA) of the conversion of normal fibroblasts to the myofibroblast phenotype in the setting of pressure overload-induced HF. NM922 inhibited fibroblast-to-myofibroblast transformation in vitro via a reduction of activation of the focal adhesion kinase-Akt-p70S6 kinase and STAT3/4E-binding protein 1 pathways as well as via induction of cyclooxygenase-2. NM922 preserved left ventricular ejection fraction (P < 0.05 vs. vehicle) and significantly attenuated transverse aortic constriction-induced LV dilation and hypertrophy (P < 0.05 compared with vehicle). NM922 significantly (P < 0.05) inhibited fibroblast activation, as evidenced by reduced myofibroblast counts per square millimeter of tissue area. Picrosirius red staining demonstrated that NM922 reduced (P < 0.05) interstitial fibrosis compared with mice that received vehicle. Similarly, NM922 hearts had lower mRNA levels (P < 0.05) of collagen types I and III, lysyl oxidase, and TNF-α at 16 wk after transverse aortic constriction. Treatment with NM922 after the onset of cardiac hypertrophy and HF resulted in attenuated myocardial collagen formation and adverse remodeling with preservation of left ventricular ejection fraction. Future studies are aimed at further elucidation of the molecular and cellular mechanisms by which this novel antifibrotic agent protects the failing heart.
NEW & NOTEWORTHY Our data demonstrated that a novel antifibrotic agent, NM922, blocks the activation of fibroblasts, reduces the formation of cardiac fibrosis, and preserves cardiac function in a murine model of heart failure with reduced ejection fraction.
Keywords: heart failure, hypertrophy, pressure overload, myocardial fibrosis, transverse aortic constriction
INTRODUCTION
Remodeling of the myocardium during cardiovascular disease is a dynamic and complex process that alters myocardial structure and function of the heart at the cellular and subcellular levels (25). Cardiac fibroblasts are abundant in the heart, comprising 10–20% of total cardiac cells (38). Within the heart, cardiac fibroblasts play critical roles in development and remodeling and respond to different stimuli to coordinate signals throughout the heart. Their primary role in regulating extracellular matrix (ECM) remodeling has a definitive impact on cardiac structure and function (39). The ECM is a complex structure composed of various structural, adhesive, and matricellular proteins, with collagen being the predominant component (23, 39). During the manifestation and progression of heart failure, there are changes in the ECM at every stage to compensate for the increased stresses on the heart (39). Furthermore, there is an increase in growth factors and inflammatory cytokines activating the transformation of fibroblasts to myofibroblasts (41). The presence of myofibroblasts results in further exacerbation of cardiac remodeling, leading to excessive collagen turnover. Although these changes initially are beneficial, the chronic remodeling and stress on the heart become maladaptive, leading to exacerbation of heart failure symptoms (25).
Transforming growth factor (TGF)-β is a primary regulator in cardiac remodeling, having a central role in fibroblast activation (14, 15, 26). Because of its elevated expression in response to cardiac injury, TGF-β has been a candidate for antifibrotic therapies. Previous studies (17, 19, 27, 29) have demonstrated that while TGF-β-neutralizing antibodies, receptor antagonists, and deficient mice exhibit reduced fibrosis, direct inhibition of TGF-β results in profound left ventricular (LV) dilation and increased mortality in the setting of heart failure. Taken together, these preclinical studies suggest that targeting TGF-β directly may not be ideal for treating heart failure.
In the present study, we examined the effects of NM922 on several profibrotic pathways such as the focal adhesion kinase (FAK)-Akt-p70S6 kinase (p70S6K) and mammalian target of rapamycin (mTOR)/STAT3/ 4E-binding protein 1 (4E-BP1) pathways in vitro. We also tested the efficacy of NM922 in the development of LV fibrosis and cardiac function in an in vivo murine model of pressure overload-induced cardiac hypertrophy and heart failure. To simulate clinically relevant treatment conditions in heart failure, NM922 was administered starting at 6 wk after transverse aortic constriction (TAC).
MATERIALS AND METHODS
Experimental animals.
Male C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME) at 8–10 wk of age. All animals were housed in a temperature-controlled animal facility with a 12:12-h light-dark cycle and water and rodent chow provided ad libitum. All experimental protocols were approved by the Institutional Animal Care and Use Committee of Louisiana State University Health Sciences Center. All animals received humane care in compliance with the National Society of Medical Research’s Principles of Laboratory Animal Care and the National Institutes of Health Guide for the Care and Use of Laboratory Animals (8th ed., Revised 2011) and with federal and state regulations.
Experimental compound.
NM922 was provided by NovoMedix. The chemical structure of NM922 is shown in Fig. 1A.
Fig. 1.

NM922 prevents the conversion of normal human lung fibroblasts to the transforming growth factor-β (TGF-β)-induced myofibroblast phenotype in vitro. A: chemical structure of NM922. B and C: representative blots of phosphorylated (p-)focal adhesion kinase (FAK), cyclooxygenase-2 (COX-2), α-smooth muscle actin (α-SMA), cyclin D3, and p-4E-binding protein 1 (4E-BP1) (B) and p-STAT3, p-p70S6 kinase (p70S6K), and p-Akt (C). D: relative fold changes in the intensity of blots for α-SMA, p-FAK, p-Akt, p-P70S6K, p-STAT3, p-4E-BP1, cyclin D, and COX-2 in the TGF-β + vehicle (Veh)-treated group. Numbers inside columns denote sample sizes. NS, not significant.
Cell cultures.
Normal human lung fibroblasts were plated at 400,000 cells/well in 6-well plates and starved overnight. The wells were then randomized into the following three groups: unstimulated, TGF-β + vehicle, and TGF-β + NM922. At 24 h after the various treatments, cells were harvested for Western blot analysis.
TAC protocol.
Heart failure was created with a well-established pressure-overload hypertrophy model induced by TAC (34a). Briefly, mice were fully anesthetized (2 mg/kg xylazine and 20 mg/kg ketamine), orally intubated, and mechanically ventilated (MiniVent type 845, Hugo-Sachs Elektronik) with supplemental oxygen. Chest hair was removed, and mice were prepped with a series of Betadine and alcohol wipes. A small incision was made proximally to the left of the midline of the sternum, below the clavicle. With blunt dissection, the aorta was exposed and a constriction was made against a 27-gauge needle with a 7-0 silk suture between the brachiocephalic artery and left common carotid. Once the needle was removed, the chest and skin were closed and the mouse was placed in a recovery cage supplemented with 100% oxygen until normal behavior returned.
Heart failure protocol.
Thirty male C57BL/6J mice were included in the NM922 experiments. Baseline echocardiography was performed 3 days before the TAC surgery and every 2 wk for 16 wk. Six weeks after surgery, mice were randomized to treatment or vehicle-treated groups. The treatment groups received NM922 (50 mg·kg−1·day−1 ip) and the vehicle-treated group received only the drug vehicle (DMSO + HS-15). All animals were euthanized at 16 wk for tissue and blood collection for molecular analysis (Fig. 2A). Echocardiographic assessment of LV structure and function was performed in a fully blinded manner.
Fig. 2.

Pressure-overload heart failure experimental protocol and survival rate. A: mice were subjected to transverse aortic constriction (TAC) surgery. NM922 or vehicle treatment was administered daily via intraperitoneal injection starting 6 wk after TAC and continued for 10 wk until the 16-wk end point. B: percent survival over the course of the study revealed that administration of NM922 resulted in 87% survival compared with 64% survival in vehicle-treated mice. LV, left ventricular.
Echocardiographic assessment of LV structure and function.
In vivo transthoracic echocardiography was performed with a Visual Sonics VEVO 2100 echocardiography system. Isoflurane (0.5–1% supplemented with 100% oxygen) was used for anesthesia to minimize any cardiac effects of anesthesia. Heart rates were carefully maintained at 450–500 beats/min for all mice. M-mode images from long-axis views were used to measure LV posterior wall thickness at systole and diastole (LVPWs and LVPWd, respectively) and LV chamber diameters at end systole and end diastole (LVESD and LVEDD, respectively). LV ejection fraction (LVEF) was calculated from ECG-gated kilohertz visualization analysis.
Histological assessment of collagen and fibrosis.
Myocardial collagen content and fibrosis were assessed at the 16-wk end point in LV tissue (10). LV sections were fixed in 4% paraformaldehyde, paraffin embedded, sliced into 5-μm sections, and then attached to slides. Slides were stained with picrosirius red (PSR) to detect collagen and fibrosis. To quantify the collagen volume fraction (CVF) from PSR-stained slides, fluorescent images for PSR were obtained (×20) with a Nikon Eclipse (TE2000-U) and processed with NIS Elements software. The mean CVF for each heart was expressed as the percent total area and then group averaged.
Immunohistochemical assessment of cardiac fibroblast activation.
To elucidate the level of cardiac fibroblast activation, the number of α-smooth muscle actin-positive/von Willebrand factor-negative (α-SMA+/VWF−) cells was determined immunohistochemically at the 16-wk end point in LV tissue. Briefly, paraformaldehyde-fixed, paraffin-embedded heart tissues were sliced into 5-μm sections at the short axis and then attached to slides. After heat-mediated antigen retrieval in EDTA buffer (Abcam, pH 9), slides were probed with antibodies against VWF (ab11713, Abcam, 1:50) and α-SMA (ab5694, Abcam, 1:200) followed by rhodamine red-X- or fluorescein-conjugated secondary antibodies (nos. 132023 and 134533, Jackson ImmunoResearch Laboratories). Nuclei were stained with DAPI (ThermoFisher). Fluorescent images (×20) for α-SMA, VWF, and DAPI were taken with an Olympus BX53-DP80 system and merged with cellSens software. α-SMA+/VWF− cells were counted, and data are expressed as numbers of cells per square millimeter of tissue area.
Plasma brain natriuretic peptide.
Plasma levels of brain natriuretic peptide (BNP) were measured via ELISA (BNP EIA kit, Phoenix Pharmaceuticals) at 16 wk after TAC.
Quantitative PCR and mRNA isolation.
RNA was isolated from the heart tissue of vehicle- and NM922-treated mice at 16 wk after TAC. One milligram of RNA was transcribed with the I-script cDNA synthesis kit from Bio-Rad. TaqMan primers for TNF-α, lysyl oxidase (LOX), collagen type I, and collagen type III from Life Technology were used to amplify quantitative PCR. For real-time quantitative PCR experiments, 18S was used as the housekeeping gene and values were corrected to 18S. The method (where CT is threshold cycle) was used for the data analysis of all quantitative PCR data.
Western blot analysis.
In the in vitro cell culture-based experiments, 24 h after TGF-β exposure, cells were harvested and lysed with M-PER. Protein expressions of α-SMA (catalog no. A5228, Sigma), phosphorylated (p-)FAK (catalog no. BD-611722, BD Biosciences), p-Akt (catalog no. 4060, Cell Signaling), p-p70S6K (catalog no. 9234, Cell Signaling), p-STAT3 (catalog no. 9145, Cell Signaling), p-4E-BP1 (catalog no. 13443, Cell Signaling), cyclin D3 (catalog no. 2936, Cell Signaling), and COX-2 (catalog no. 12282, Cell Signaling) were measured with Western blot techniques. Blots were probed with LI-COR-labeled secondary antibodies, and bands were analyzed with LI-COR software. All bands were normalized to β-tubulin, and data are expressed as fold changes relative to the TGF-β + vehicle-treated group.
In the in vivo heart failure experiments, myocardial tissue samples from vehicle- and NM922-treated mice were homogenized and lysates were used for Western blot analysis. The following primary antibodies were used: α-SMA (catalog no. A5228, Sigma), COX-2 (ab15191, Abcam), TGF-β (catalog no. 3711, Cell Signaling), and VEGF (ab46154, Abcam).
Statistical analysis.
All data in this study are expressed as means ± SE. Differences in data between the groups were compared by unpaired Student’s t-test or two-way ANOVA analysis with Prism 6 (GraphPad Software). When statistical significance was found with ANOVA, a Tukey multiple-comparison test was performed. P values of <0.05 were considered statistically significant.
RESULTS
NM922 reduces activation of profibrotic pathways and prevents activation of human lung fibroblasts in vitro.
Twenty-four hours after TGF-β exposure, we measured the level of profibrotic pathway activation. Primary human lung fibroblasts that received NM922 (20 µM) displayed reduced levels of p-FAK, p-Akt, p-p70S6K, p-STAT3, and p-4E-BP1 (Fig. 1, B–D). The marker of fibroblast activation, α-SMA expression, was reduced in cells treated with NM922 (Fig. 1, B and D). In addition, NM922 treatment induced the expression of COX-2 and downregulated cyclin D3 (Fig. 1, B and D).
Experimental design and survival.
Next, to investigate the cardioprotective effects of NM922 therapy in pressure overload-induced heart failure mice, we followed the protocol described in materials and methods (Fig. 2A). Data for survival during the 16-wk experimental protocol are shown in Fig. 2B. Survival was 64% in the vehicle-treated group and 87% in the NM922-treated group (P = 0.17 between groups).
NM922 preserves LV function, prevents cardiac dilation, and reduces cardiac hypertrophy after TAC.
Echocardiography was performed 3 days before TAC surgeries for baseline and then subsequently every 2 wk after TAC to assess cardiac structure and function. Administration of NM922 resulted in a significant attenuation of LV dilation with a 20–30% reduction in LVESD (P < 0.05; Fig. 3A) and a 16–20% reduction in LVEDD (P < 0.01; Fig. 3B) at 12, 14, and 16 wk after TAC. NM922-treated mice exhibited reduced wall thickening (LVPWd: 1.1 ± 0.03 vs. 1.2 ± 0.03 mm, P < 0.05) at 8 wk after TAC compared with vehicle-treated mice (Fig. 3C). LVEF was preserved in NM922-treated mice, with significant improvements observed in LVEF at 12 wk after TAC (P < 0.01) and continuing to the 16-wk end point (P < 0.01; Fig. 3D).
Fig. 3.

NM922 attenuates left ventricular (LV) dilation and dysfunction after transverse aortic constriction (TAC). Echocardiography was performed at baseline and every 2 wk after TAC to assess LV end-systolic diameter (LVESD; A), LV end-diastolic diameter (LVEDD; B), LV posterior wall thickness at diastole (LVPWd; C), and LV ejection fraction (LVEF; D). n = 15 mice/study group. *P < 0.05 and **P < 0.01 vs. vehicle.
Postmortem morphometric data (ventricle weight/tibia length and atria weight/tibia length (Fig. 4, A and B) of the effects of NM922 revealed a reduction in cardiac hypertrophy. NM922-treated mice had significantly reduced heart weight-to-tibia length (8.7 ± 0.5 vs. 12.0 ± 0.9 mg/mm, P < 0.01) and atria weight-to-tibia length (0.5 ± 0.04 vs. 0.8 ± 0.08 mg/mm, P < 0.01) ratios compared with vehicle-treated mice.
Fig. 4.
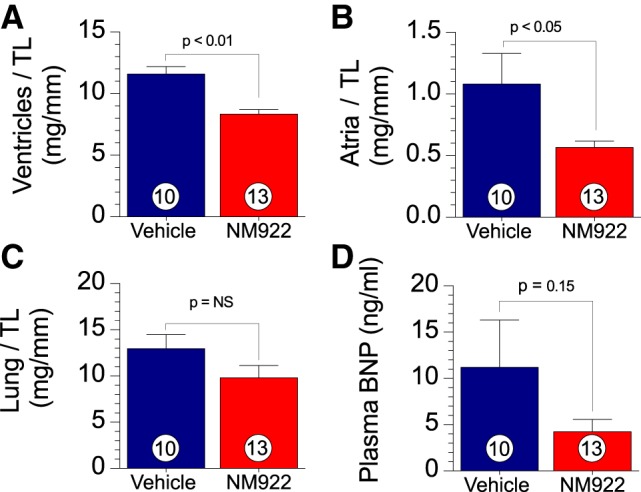
Delayed treatment with NM922 reduces transverse aortic constriction (TAC)-induced hypertrophy. A–C: postmortem morphological assessment of hypertrophy at 16 wk after TAC of ventricular weight-to-tibia length (TL) ratio (A), atrial weight-to-TL ratio (B), and lung weight-to-TL ratio (C) from NM922- and vehicle-treated mice at 16 wk after TAC. D: circulating levels of brain natriuretic peptide (BNP). Numbers inside bars denote numbers of animals/group. NS, not significant.
Effects of NM922 on pulmonary edema and circulating BNP levels.
We measured lung weight/tibia length and circulating BNP levels as additional indexes of heart failure severity. Lung weights and plasma BNP levels were not significantly attenuated after treatment with NM922 compared with animals that received vehicle (Fig. 4, C and D).
NM922 treatment decreases myocardial fibrosis and collagen deposition in heart failure.
Histological assessment of total cardiac collagen was performed at 16 wk after TAC to examine the level of myocardial fibrosis. Representative photomicrographs are shown in Fig. 5A. Treatment with NM922 resulted in a 51% reduction (P < 0.05; Fig. 5B) in interstitial collagen as quantified via the percentage of CVF (Fig. 5B) of PSR-stained slides. We next examined the relative mRNA levels of the two predominant collagens in the ECM, collagen types I and III, and other ECM regulators at 16 wk after TAC. Treatment with NM922 reduced mRNA levels (P < 0.05) of collagen type I (Fig. 5C), collagen type III (Fig. 5D), TNF-α (Fig. 5E), and LOX (Fig. 5F) compared with the vehicle-treated group of mice. LOX is a copper-dependent amine oxidase that covalently cross-links fibrillar collagens (36).
Fig. 5.
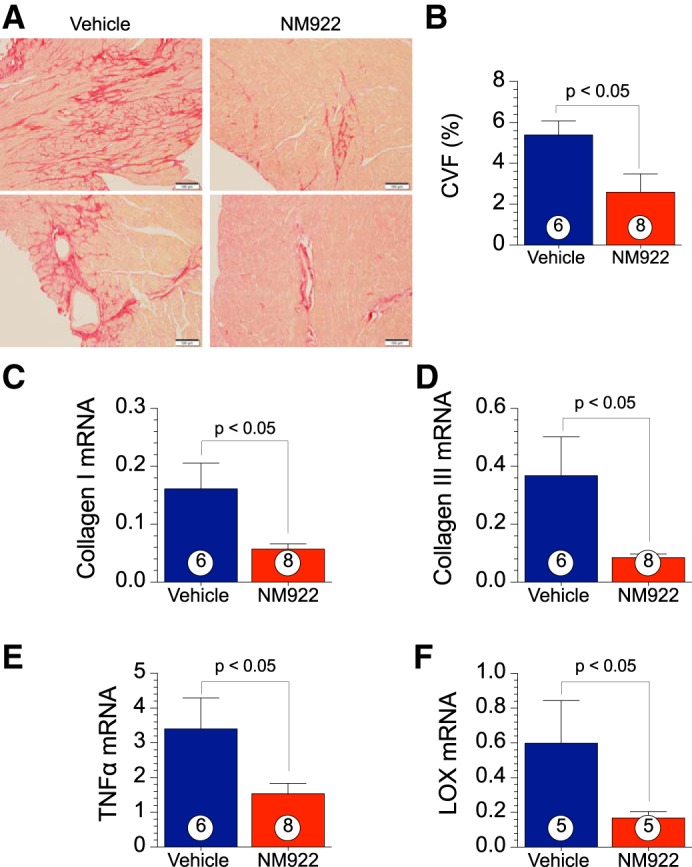
NM922 attenuates transverse aortic constriction (TAC)-induced myocardial collagen deposition and fibrosis. A: representative images of cardiac tissue from vehicle- and NM922-treated animals at 16 wk after TAC stained for picrosirius red for fibrosis. B: quantification of the percent collagen volume fraction (CVF). C–F: mRNA levels of collagen type I (C), collagen type III (D), TNF-α (E), and lysyl oxidase (LOX; F) were measured in ventricular homogenates from mice treated with NM922 or vehicle at 16 wk after TAC. Numbers inside bars denote numbers of animals/group.
NM922 inhibits cardiac fibroblast activation in vivo.
A key phenotypic feature of myofibroblast differentiation is α-SMA (24). Examination of myocardial α-SMA (Fig. 6D) revealed significant reductions (P < 0.05) in cardiac protein expression at 16 wk after TAC. In addition, through an immunohistochemical approach, we observed that hearts from NM922-treated mice displayed a significantly reduced number of α-SMA+/VWF− cells (Fig. 6, A–C). There are a number of growth factors that induce myofibroblast differentiation, including TGF-β. In the present study, we did not observe any reduction in protein expression of TGF-β (Fig. 6F) in myocardial tissue after treatment with NM922 (P = not significant between groups). In contrast, NM922 treatment significantly increased (P < 0.05) the protein expression of COX-2 (Fig. 6E) in the heart at 16 wk after TAC. COX-2-mediated prostaglandin production inhibits myocardial fibrosis and exerts cardioprotective effects under pathological conditions such as heart failure (7, 28, 30).
Fig. 6.

Delayed treatment with NM922 reduces myofibroblast abundance in vivo. A and B: representative ×20 fluorescent images of slides costained for α-smooth muscle actin (α-SMA; red), von Willebrand factor (VWF; green), and DAPI (blue). Arrows denote αSMA+/VWF− cells. C: αSMA+/VWF− cell counts at 16 wk after TAC for mice treated with NM922 or vehicle. D–F: protein expression of α-SMA (D), cyclooxygenase-2 (COX-2; E), and transforming growth factor-β (TGF-β; F). GAPDH was used as a loading control. Numbers inside bars denote numbers of animals/group. RU, relative units; NS, not significant.
DISCUSSION
We investigated the effects of the novel antifibrotic drug NM922 in the setting of pressure-overload hypertrophy and heart failure induced by TAC. Our in vitro data demonstrate that NM922 reduced the conversion of normal fibroblasts to myofibroblasts in the presence of TGF-β by inhibiting the activation of FAK-Akt-p70S6K (18, 37) and mTOR/STAT3/4E-BP1 (4, 5, 21) profibrotic pathways. Meanwhile, NM922 treatment reduced the levels of the cell cycle regulator protein cyclin D3, which is a downstream effector of mTOR, corroborating the inhibitory effects of NM922 on the STAT3/4E-BP1 pathway (2). We also demonstrated that daily administration of NM922 initiated at 6 wk after TAC attenuated maladaptive LV remodeling and preserved LV function compared with vehicle. Moreover, these changes in cardiac structure and function were correlated with a reduction in α-SMA+/VWF− cell counts and in collagen deposition as shown through both histological and biochemical approaches.
Fibroblasts play a critical role in regulating myocardial remodeling through the mediation of ECM components including collagen, elastin, and fibronectin (11). During cardiovascular disease, cytokines, hormones, and growth factors stimulate the activation of fibroblasts to myofibroblasts (6). This change in phenotype to myofibroblasts exacerbates ECM remodeling in response to the additional stress on the heart. Despite this initial beneficial effect, myofibroblast stimulation ultimately leads to excessive production of collagen and alterations in regulators of the ECM, including matrix metalloproteinases to their tissue inhibitors (6, 39).
It is well documented that TGF-β is a primary regulator of fibroblast activation. Previous studies have shown that inhibition of TGF-β signaling by anti-TGF-β antibodies (19, 27), genetic knockout mice (26, 29), or small-molecule antagonism of the TGF-β1 receptor (ALK5) (17) attenuates fibroblast activation, thereby decreasing collagen deposition and fibrosis; however, these are not without serious consequences. Significant reduction of myocardial fibrosis as a result of TGF-β inhibition also resulted in LV dilation and increased mortality in response to myocardial ischemia-reperfusion injury (19) and pressure overload-induced heart failure (29). In the present study, it was observed that NM922 inhibited fibroblast activation as measured by a reduction in α-SMA expression, the primary marker for myofibroblasts, after induction with TGF-β in vitro. Inhibition of cardiac fibroblast activation was also observed in vivo at 16 wk after TAC, with a reduction in α-SMA protein levels and lower α-SMA+/VWF− cell counts in the heart after daily administration of NM922 starting 6 wk after TAC. Myocardial levels of TGF-β were not altered by NM922 treatment at 16 wk after TAC, indicating that NM922 works in a TGF-β-independent manner. Histological assessment of PSR-stained heart sections confirmed that NM922 treatment significantly reduced collagen deposition. To further access the antifibrotic properties of NM922, we examined the relative expressions of collagen types I and III, the primary components of the cardiac ECM, and observed that mice treated with NM922 displayed significantly reduced expressions of both. Increases in TNF-α during the progression of cardiovascular disease have been linked to the activation of various regulators of ECM remodeling (8, 9). Voloshenyuk et al. (42) demonstrated that stimulation with TNF-α in cardiac fibroblasts resulted in increased expression of collagen types I and III and LOX, a collagen cross-linking enzyme, which were attenuated after TNF-α antagonism. Before deposition into the ECM collagen must be cross-linked, and inhibition of LOX during compensatory remodeling in the setting of volume-overload stress can preserve cardiac function (16). Inhibition of TNF-α in vivo demonstrated cardioprotective properties in both volume-overload (22) and pressure-overload (40) models of heart failure with inhibited pathological myocardial remodeling. In the present study, we observed that administration of NM922 resulted in a significant reduction of cardiac TNF-α mRNA levels along with a decrease in cardiac LOX mRNA levels. These reductions in TNF-α and LOX mRNA levels as a result of NM922 treatment may be critical in understanding the mechanisms behind its cardioprotection.
Echocardiographic studies have revealed that NM922 treatment attenuated TAC-induced LV dilation and hypertrophy in addition to preserving LV function, contrary to some studies that used TGF-β inhibitors. Similar to our findings with NM922, treatment with pirfenidone, an antifibrotic agent, in animal studies showed reduced scar formation after myocardial infarction injury (34) and attenuated myocardial fibrosis in both DOCA-salt hypertensive rats (31) and TAC (43). Pirfenidone is a Federal Drug Administration-approved antifibrotic agent primarily used for idiopathic pulmonary fibrosis (35). Moreover, administration of pirfenidone had significant effects on preserving cardiac function (34, 43). Besides inhibition of TGF-β signaling, pirfenidone has significant anti-inflammatory properties such as reducing TNF-α, which has a role in regulating ECM remodeling (33).
In various disease states, it has been documented that PGE2 exerts powerful antifibrotic properties through inhibition of fibroblast function (3, 28, 44, 45). Under these pathological conditions, COX-2 is induced and mediates the synthesis of prostaglandins, including PGE2, from arachidonic acid (20, 28, 44). Investigators have shown that mice deficient in COX-2 have inadequate levels of endothelial nitric oxide synthase to maintain normal vascular function (1, 46). COX-2-deficient mice also demonstrate impaired recovery of contractile function after myocardial ischemia injury (12). Also, in the clinical setting, COX-2 inhibitors have been linked to increased risk of cardiovascular events including myocardial infarction and stroke (32). On the other hand, it has been shown that activation of COX-2 induces atheroprotection in female mice by reducing oxidative stress and platelet activation (15). Moreover, COX-2 activation during ischemic preconditioning has been linked to the protective effects during late-phase preconditioning during both myocardial infarction and myocardial stunning (7). In the present study, NM922 treatment induced COX-2 expression in TGF-β-treated human lung fibroblasts, and COX-2 expression was also enhanced in the hearts 16 wk after TAC.
Conclusions
Our results demonstrate that NM922 inhibited the activation of two well-established profibrotic pathways in TGF-β-treated human lung fibroblasts in vitro, namely, the FAK-Akt-P70S6K and mTOR/STAT3/4E-BP1 pathways. In addition, NM922 treatment induced the expression of a known antifibrotic factor, COX-2, both in vitro and in vivo. Importantly, inhibition of fibroblast activation by NM922 treatment resulted in attenuation of adverse ECM remodeling, reduction in cardiac fibrosis, and preservation of cardiac function in an in vivo murine model of heart failure.
GRANTS
This study was funded by National Heart, Lung, and Blood Institute Grant 1R43 HL-131356-01A1 (to NovoMedix and D. J. Lefer).
DISCLOSURES
L. G. Corral, R. W. Sullivan, L. Fung, K. W. H. Chan, C. A. Sindlehurst, and D. J. Lefer are employed by or served on the advisory board of NovoMedix LLC.
AUTHOR CONTRIBUTIONS
J.M.B., Z.L., T.E.S., T.T.G., L.G.C., L.F., K.W.C., R.W.S., C.A.S., and D.J.L. conceived and designed research; J.M.B., P.S., Z.L., and L.G.C. performed experiments; J.M.B., P.S., Z.L., T.E.S., and L.G.C. analyzed data; J.M.B., P.S., Z.L., C.A.S., and D.J.L. interpreted results of experiments; J.M.B., P.S., Z.L., and C.A.S. prepared figures; J.M.B. and Z.L. drafted manuscript; J.M.B., Z.L., T.E.S., T.T.G., and D.J.L. edited and revised manuscript; T.T.G., R.W.S., C.A.S., and D.J.L. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Jean Carnal and Craig Zibilich for assistance during the course of the study.
REFERENCES
- 1.Ahmetaj-Shala B, Kirkby NS, Knowles R, Al’Yamani M, Mazi S, Wang Z, Tucker AT, Mackenzie L, Armstrong PC, Nüsing RM, Tomlinson JA, Warner TD, Leiper J, Mitchell JA. Evidence that links loss of cyclooxygenase-2 with increased asymmetric dimethylarginine: novel explanation of cardiovascular side effects associated with anti-inflammatory drugs. Circulation 131: 633–642, 2015. doi: 10.1161/CIRCULATIONAHA.114.011591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson AA, Child ES, Prasad A, Elphick LM, Mann DJ. Cyclin D1 and cyclin D3 show divergent responses to distinct mitogenic stimulation. J Cell Physiol 225: 638–645, 2010. doi: 10.1002/jcp.22207. [DOI] [PubMed] [Google Scholar]
- 3.Baird AC, Lloyd F, Lawrance IC. Prostaglandin E2 and polyenylphosphatidylcholine protect against intestinal fibrosis and regulate myofibroblast function. Dig Dis Sci 60: 1603–1616, 2015. doi: 10.1007/s10620-015-3552-9. [DOI] [PubMed] [Google Scholar]
- 4.Banerjee S, Gianino SM, Gao F, Christians U, Gutmann DH. Interpreting mammalian target of rapamycin and cell growth inhibition in a genetically engineered mouse model of Nf1-deficient astrocytes. Mol Cancer Ther 10: 279–291, 2011. doi: 10.1158/1535-7163.MCT-10-0654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baybis M, Yu J, Lee A, Golden JA, Weiner H, McKhann G 2nd, Aronica E, Crino PB. mTOR cascade activation distinguishes tubers from focal cortical dysplasia. Ann Neurol 56: 478–487, 2004. doi: 10.1002/ana.20211. [DOI] [PubMed] [Google Scholar]
- 6.Berk BC, Fujiwara K, Lehoux S. ECM remodeling in hypertensive heart disease. J Clin Invest 117: 568–575, 2007. doi: 10.1172/JCI31044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bolli R, Shinmura K, Tang XL, Kodani E, Xuan YT, Guo Y, Dawn B. Discovery of a new function of cyclooxygenase (COX)-2: COX-2 is a cardioprotective protein that alleviates ischemia/reperfusion injury and mediates the late phase of preconditioning. Cardiovasc Res 55: 506–519, 2002. doi: 10.1016/S0008-6363(02)00414-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bradham WS, Bozkurt B, Gunasinghe H, Mann D, Spinale FG. Tumor necrosis factor-alpha and myocardial remodeling in progression of heart failure: a current perspective. Cardiovasc Res 53: 822–830, 2002. doi: 10.1016/S0008-6363(01)00503-X. [DOI] [PubMed] [Google Scholar]
- 9.Bradham WS, Moe G, Wendt KA, Scott AA, Konig A, Romanova M, Naik G, Spinale FG. TNF-alpha and myocardial matrix metalloproteinases in heart failure: relationship to LV remodeling. Am J Physiol Heart Circ Physiol 282: H1288–H1295, 2002. doi: 10.1152/ajpheart.00526.2001. [DOI] [PubMed] [Google Scholar]
- 10.Bradley JM, Li Z, Organ CL, Polhemus DJ, Otsuka H, Islam KN, Bhushan S, Gorodnya OM, Ruchko MV, Gillespie MN, Wilson GL, Lefer DJ. A novel mtDNA repair fusion protein attenuates maladaptive remodeling and preserves cardiac function in heart failure. Am J Physiol Heart Circ Physiol 314: H311–H321, 2018. doi: 10.1152/ajpheart.00515.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Camelliti P, Green CR, LeGrice I, Kohl P. Fibroblast network in rabbit sinoatrial node: structural and functional identification of homogeneous and heterogeneous cell coupling. Circ Res 94: 828–835, 2004. doi: 10.1161/01.RES.0000122382.19400.14. [DOI] [PubMed] [Google Scholar]
- 12.Camitta MG, Gabel SA, Chulada P, Bradbury JA, Langenbach R, Zeldin DC, Murphy E. Cyclooxygenase-1 and -2 knockout mice demonstrate increased cardiac ischemia/reperfusion injury but are protected by acute preconditioning. Circulation 104: 2453–2458, 2001. doi: 10.1161/hc4401.098429. [DOI] [PubMed] [Google Scholar]
- 14.Dobaczewski M, Chen W, Frangogiannis NG. Transforming growth factor (TGF)-β signaling in cardiac remodeling. J Mol Cell Cardiol 51: 600–606, 2011. doi: 10.1016/j.yjmcc.2010.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duan J, Gherghe C, Liu D, Hamlett E, Srikantha L, Rodgers L, Regan JN, Rojas M, Willis M, Leask A, Majesky M, Deb A. Wnt1/βcatenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair. EMBO J 31: 429–442, 2012. doi: 10.1038/emboj.2011.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.El Hajj EC, El Hajj MC, Ninh VK, Gardner JD. Cardioprotective effects of lysyl oxidase inhibition against volume overload-induced extracellular matrix remodeling. Exp Biol Med 241: 539–549, 2016. doi: 10.1177/1535370215616511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Engebretsen KV, Skårdal K, Bjørnstad S, Marstein HS, Skrbic B, Sjaastad I, Christensen G, Bjørnstad JL, Tønnessen T. Attenuated development of cardiac fibrosis in left ventricular pressure overload by SM16, an orally active inhibitor of ALK5. J Mol Cell Cardiol 76: 148–157, 2014. doi: 10.1016/j.yjmcc.2014.08.008. [DOI] [PubMed] [Google Scholar]
- 18.Fan GP, Wang W, Zhao H, Cai L, Zhang PD, Yang ZH, Zhang J, Wang X. Pharmacological inhibition of focal adhesion kinase attenuates cardiac fibrosis in mice cardiac fibroblast and post-myocardial-infarction models. Cell Physiol Biochem 37: 515–526, 2015. doi: 10.1159/000430373. [DOI] [PubMed] [Google Scholar]
- 19.Frantz S, Hu K, Adamek A, Wolf J, Sallam A, Maier SK, Lonning S, Ling H, Ertl G, Bauersachs J. Transforming growth factor beta inhibition increases mortality and left ventricular dilatation after myocardial infarction. Basic Res Cardiol 103: 485–492, 2008. doi: 10.1007/s00395-008-0739-7. [DOI] [PubMed] [Google Scholar]
- 20.Grosser T, Fries S, FitzGerald GA. Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J Clin Invest 116: 4–15, 2006. doi: 10.1172/JCI27291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harries LW, Fellows AD, Pilling LC, Hernandez D, Singleton A, Bandinelli S, Guralnik J, Powell J, Ferrucci L, Melzer D. Advancing age is associated with gene expression changes resembling mTOR inhibition: evidence from two human populations. Mech Ageing Dev 133: 556–562, 2012. doi: 10.1016/j.mad.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jobe LJ, Meléndez GC, Levick SP, Du Y, Brower GL, Janicki JS. TNF-alpha inhibition attenuates adverse myocardial remodeling in a rat model of volume overload. Am J Physiol Heart Circ Physiol 297: H1462–H1468, 2009. doi: 10.1152/ajpheart.00442.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jugdutt BI. Remodeling of the myocardium and potential targets in the collagen degradation and synthesis pathways. Curr Drug Targets Cardiovasc Haematol Disord 3: 1–30, 2003. doi: 10.2174/1568006033337276. [DOI] [PubMed] [Google Scholar]
- 24.Kapoun AM, Liang F, O’Young G, Damm DL, Quon D, White RT, Munson K, Lam A, Schreiner GF, Protter AA. B-type natriuretic peptide exerts broad functional opposition to transforming growth factor-beta in primary human cardiac fibroblasts: fibrosis, myofibroblast conversion, proliferation, and inflammation. Circ Res 94: 453–461, 2004. doi: 10.1161/01.RES.0000117070.86556.9F. [DOI] [PubMed] [Google Scholar]
- 25.Kehat I, Molkentin JD. Extracellular signal-regulated kinase 1/2 (ERK1/2) signaling in cardiac hypertrophy. Ann NY Acad Sci 1188: 96–102, 2010. doi: 10.1111/j.1749-6632.2009.05088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, Vagnozzi RJ, Liu R, Huynh T, Lee SJ, Karch J, Molkentin JD. Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest 127: 3770–3783, 2017. doi: 10.1172/JCI94753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuwahara F, Kai H, Tokuda K, Kai M, Takeshita A, Egashira K, Imaizumi T. Transforming growth factor-beta function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation 106: 130–135, 2002. doi: 10.1161/01.CIR.0000020689.12472.E0. [DOI] [PubMed] [Google Scholar]
- 28.Lama V, Moore BB, Christensen P, Toews GB, Peters-Golden M. Prostaglandin E2 synthesis and suppression of fibroblast proliferation by alveolar epithelial cells is cyclooxygenase-2-dependent. Am J Respir Cell Mol Biol 27: 752–758, 2002. doi: 10.1165/rcmb.4857. [DOI] [PubMed] [Google Scholar]
- 29.Lucas JA, Zhang Y, Li P, Gong K, Miller AP, Hassan E, Hage F, Xing D, Wells B, Oparil S, Chen YF. Inhibition of transforming growth factor-beta signaling induces left ventricular dilation and dysfunction in the pressure-overloaded heart. Am J Physiol Heart Circ Physiol 298: H424–H432, 2010. doi: 10.1152/ajpheart.00529.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mendez M, LaPointe MC. PPARgamma inhibition of cyclooxygenase-2, PGE2 synthase, and inducible nitric oxide synthase in cardiac myocytes. Hypertension 42: 844–850, 2003. doi: 10.1161/01.HYP.0000085332.69777.D1. [DOI] [PubMed] [Google Scholar]
- 31.Mirkovic S, Seymour AM, Fenning A, Strachan A, Margolin SB, Taylor SM, Brown L. Attenuation of cardiac fibrosis by pirfenidone and amiloride in DOCA-salt hypertensive rats. Br J Pharmacol 135: 961–968, 2002. doi: 10.1038/sj.bjp.0704539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mukherjee D, Nissen SE, Topol EJ. Risk of cardiovascular events associated with selective COX-2 inhibitors. JAMA 286: 954–959, 2001. doi: 10.1001/jama.286.8.954. [DOI] [PubMed] [Google Scholar]
- 33.Nakazato H, Oku H, Yamane S, Tsuruta Y, Suzuki R. A novel anti-fibrotic agent pirfenidone suppresses tumor necrosis factor-alpha at the translational level. Eur J Pharmacol 446: 177–185, 2002. doi: 10.1016/S0014-2999(02)01758-2. [DOI] [PubMed] [Google Scholar]
- 34.Nguyen DT, Ding C, Wilson E, Marcus GM, Olgin JE. Pirfenidone mitigates left ventricular fibrosis and dysfunction after myocardial infarction and reduces arrhythmias. Heart Rhythm 7: 1438–1445, 2010. doi: 10.1016/j.hrthm.2010.04.030. [DOI] [PubMed] [Google Scholar]
- 34a.Polhemus DJ, Kondo K, Bhushan S, Bir SC, Kevil CG, Murohara T, Lefer DJ, Calvert JW. Hydrogen sulfide attenuates cardiac dysfunction after heart failure via induction of angiogenesis. Circ Heart Fail 6: 1077–1086, 2013. doi: 10.1161/CIRCHEARTFAILURE.113.000299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raghu G, Johnson WC, Lockhart D, Mageto Y. Treatment of idiopathic pulmonary fibrosis with a new antifibrotic agent, pirfenidone: results of a prospective, open-label Phase II study. Am J Respir Crit Care Med 159: 1061–1069, 1999. doi: 10.1164/ajrccm.159.4.9805017. [DOI] [PubMed] [Google Scholar]
- 36.Rucker RB, Kosonen T, Clegg MS, Mitchell AE, Rucker BR, Uriu-Hare JY, Keen CL. Copper, lysyl oxidase, and extracellular matrix protein cross-linking. Am J Clin Nutr 67, Suppl: 996S–1002S, 1998. doi: 10.1093/ajcn/67.5.996S. [DOI] [PubMed] [Google Scholar]
- 37.Sandilands E, Schoenherr C, Frame MC. p70S6K is regulated by focal adhesion kinase and is required for Src-selective autophagy. Cell Signal 27: 1816–1823, 2015. doi: 10.1016/j.cellsig.2015.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith CL, Baek ST, Sung CY, Tallquist MD. Epicardial-derived cell epithelial-to-mesenchymal transition and fate specification require PDGF receptor signaling. Circ Res 108: e15–e26, 2011. doi: 10.1161/CIRCRESAHA.110.235531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Souders CA, Bowers SL, Baudino TA. Cardiac fibroblast: the renaissance cell. Circ Res 105: 1164–1176, 2009. doi: 10.1161/CIRCRESAHA.109.209809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun M, Chen M, Dawood F, Zurawska U, Li JY, Parker T, Kassiri Z, Kirshenbaum LA, Arnold M, Khokha R, Liu PP. Tumor necrosis factor-alpha mediates cardiac remodeling and ventricular dysfunction after pressure overload state. Circulation 115: 1398–1407, 2007. doi: 10.1161/CIRCULATIONAHA.106.643585. [DOI] [PubMed] [Google Scholar]
- 41.van den Borne SW, Diez J, Blankesteijn WM, Verjans J, Hofstra L, Narula J. Myocardial remodeling after infarction: the role of myofibroblasts. Nat Rev Cardiol 7: 30–37, 2010. doi: 10.1038/nrcardio.2009.199. [DOI] [PubMed] [Google Scholar]
- 42.Voloshenyuk TG, Hart AD, Khoutorova E, Gardner JD. TNF-α increases cardiac fibroblast lysyl oxidase expression through TGF-β and PI3Kinase signaling pathways. Biochem Biophys Res Commun 413: 370–375, 2011. doi: 10.1016/j.bbrc.2011.08.109. [DOI] [PubMed] [Google Scholar]
- 43.Wang Y, Wu Y, Chen J, Zhao S, Li H. Pirfenidone attenuates cardiac fibrosis in a mouse model of TAC-induced left ventricular remodeling by suppressing NLRP3 inflammasome formation. Cardiology 126: 1–11, 2013. doi: 10.1159/000351179. [DOI] [PubMed] [Google Scholar]
- 44.Wilborn J, Crofford LJ, Burdick MD, Kunkel SL, Strieter RM, Peters-Golden M. Cultured lung fibroblasts isolated from patients with idiopathic pulmonary fibrosis have a diminished capacity to synthesize prostaglandin E2 and to express cyclooxygenase-2. J Clin Invest 95: 1861–1868, 1995. doi: 10.1172/JCI117866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoon YS, Lee YJ, Choi JY, Cho MS, Kang JL. Coordinated induction of cyclooxygenase-2/prostaglandin E2 and hepatocyte growth factor by apoptotic cells prevents lung fibrosis. J Leukoc Biol 94: 1037–1049, 2013. doi: 10.1189/jlb.0513255. [DOI] [PubMed] [Google Scholar]
- 46.Yu SY, Liu L, Li P, Li J. Rapamycin inhibits the mTOR/p70S6K pathway and attenuates cardiac fibrosis in adriamycin-induced dilated cardiomyopathy. Thorac Cardiovasc Surg 61: 223–228, 2013. [DOI] [PubMed] [Google Scholar]