

Abstract
Acute myocardial ischemia/reperfusion (I/R) injury is a significant, unsolved clinical puzzle. In the disease context of acute myocardial infarction, reperfusion remains the only effective strategy to salvage ischemic myocardium, but it also causes additional damage. Myocardial I/R injury is composed of four types of damage, and these events attenuate the benefits of reperfusion therapy. Thus, inventing new strategies to conquer I/R injury is an unmet clinical need. A variety of pathological processes and mediators, including changes in the pH, generation of reactive oxygen radicals, and intracellular calcium overload, are proposed to be crucial in I/R-related cell injury. Among the intracellular events that occur during I/R, we stress the importance of protein phosphorylation signaling and elaborate its regulation. A variety of protein kinase pathways could be activated in I/R, including reperfusion injury salvage kinase and survivor-activating factor enhancement pathways, which are critical to cardiomyocyte survival. In addition to serine/threonine phosphorylation signaling, protein tyrosine phosphorylation is also critical in multiple cell functions and survival. However, the roles of protein kinases and phosphatases in I/R have not been extensively studied yet. By better understanding the mechanisms of I/R injury, we may have a better chance to develop new strategies for I/R injury and apply them in the clinical patient care.
KEYWORDS: Acute myocardial infarction, Ischemia/reperfusion injury, Protein phosphorylation
BACKGROUND OF MYOCARDIAL ISCHEMIA/REPERFUSION INJURY
Despite recent improvements in medical knowledge and management, acute myocardial infarction (AMI) and its consequences are still significant health problems worldwide. The myocardium is considered to have negligible regenerative capacity, and scar formation occurs when a significant amount of cardiac muscle is lost [1]. Timely restoration of blood flow to the ischemic myocardium is the standard treatment to prevent the death of threatened cardiomyocytes. Reperfusion is the only established strategy in the current clinical practice to limit the infarct size and decrease mortality and morbidity. In line with the increment of successful reperfusion, the in-hospital mortality rate for AMI declined from 20% in 1979 to <10% in 2008 in a study in Japan [2]. However, much evidence has shown that reperfusion itself causes additional injury to the ischemic myocardium [3]. These insults significantly diminish the benefits of reperfusion therapy. Over the past decades, great effort had been put into developing strategies to attenuate ischemia/reperfusion (I/R) injury. However, in spite of progress in the comprehension of I/R injury and promising results of some strategies in animal models, the outcomes of clinical studies for translating these findings into patient care have been largely disappointing.
Myocardial I/R injury, first reported by Jennings et al. in 1960 [4], is characterized by cell swelling, contracture of myofibrils, and disruption of the sarcolemma in myocardium, which has been subjected to ischemia followed by reperfusion. After the first report of I/R injury, evidence in the scientific literature accumulated over the past few decades. In this review article, we summarize the clinical manifestations and various basic mechanisms of myocardial I/R injury. After getting a better understanding of the mediators and mechanisms of I/R injury, we could develop more suitable strategies to minimize myocardial damage.
CLINICAL MANIFESTATIONS OF ISCHEMIA/REPERFUSION INJURY
I/R can result in four types of injury, including myocardial stunning, the no-reflow phenomenon, reperfusion arrhythmia, and lethal reperfusion injury [3].
Myocardial stunning
Myocardial stunning is a condition of persistent mechanical dysfunction after opening the occluded blood vessel, regardless of the absence of irreversible damage [5]. It is fully reversible. This functional abnormality in contractility can last for days, with eventual recovery. Several studies of AMI patients demonstrated that the systolic and diastolic function of the postischemic myocardium may not recover immediately after reperfusion. However, most improvement occurs within the first 1 week after the indicated infarction episode [6]. Factors that determine the severity of stunning are the degree and duration of blood flow deprivation, myocardial temperature, and loading conditions of the heart [5]. The mechanisms involve calcium overload, decreasing calcium responsiveness of the myofilaments, degradation of troponin I, and excitation-contraction uncoupling [7,8]. In the clinical setting, myocardial stunning can be diagnosed using various image modalities such as dobutamine echocardiography, myocardial contrast echocardiography [9], and gated Tc-99m single-photon emission computed tomography [10].
No-reflow phenomenon
The no-reflow phenomenon is incomplete and uneven reperfusion at the microvascular level even though the proximal artery has been re-opened after a period of ischemia [11]. This phenomenon occurs in 0.6%–3.2% of percutaneous coronary intervention (PCI) cases in the current practice [12]. Its development is associated with a significantly increased risk of death [13,14]. Hyperglycemia has been proposed and demonstrated as a factor that associates with the no-reflow phenomenon [15]. This phenomenon is considered to result from endothelial damage, leukocyte plugging, and mechanical compression [16]. The endothelial damage which occurs during ischemia results in an increasing infarct size and augmentation of microvascular hypoperfusion at the time of reperfusion. There are a variety of mechanisms which contribute to the endothelial injury, including acute inflammatory response, reactive oxygen species (ROS) generation, and intracellular calcium overload. It is confined to the necrotic zone, mainly located in the subendocardium, but also progresses toward the subepicardium after a long period of ischemia. In addition to endothelial damage, myocyte swelling as well as tissue edema can also occlude the microvasculature by external compression. Furthermore, the downstream embolization of thrombi and leukocytes also accounts for the no-reflow phenomenon. This finally leads to compromised tissue perfusion in spite of successful restoration of blood flow to the epicardial blood vessel.
Reperfusion arrhythmia
Reperfusion of the ischemic heart may also lead to arrhythmias, including the most life-threatening, ventricular tachycardia (VT) and ventricular fibrillation (VF) [17]. It is not unusual as up to 80% of AMI patients have at least one reperfusion arrhythmia in the first 48 hours [18]. The most common arrhythmias in AMI patients during and immediately after primary PCI are accelerated idioventricular rhythm, sinus bradycardia, nonsustained VT, and sinus tachycardia [19]. Some reperfusion arrhythmias are well tolerated, and no specific treatment is required. However, VT/VF is associated with a high risk of mortality [19]. The electrophysiological consequences of I/R depend on the duration of ischemia, and most reperfusion arrhythmias occur after a short ischemic duration. It is proposed that oxygen-derived-free radicals play a critical role in the generation of reperfusion arrhythmia [20,21]. Inhomogeneous changes in the intracellular and extracellular calcium, potassium, and sodium levels during I/R also lead to the dispersion of cardiomyocyte refractoriness, which potentiates the formation of re-entry. In addition to the re-entry mechanism, the triggered activity caused by early and delayed afterdepolarizations also accounts for reperfusion arrhythmias [22]. Accelerated idioventricular rhythm is common in the clinical practice and may result from increasing autonomic stimulation of Purkinje fibers near the ischemic region, thus enhancing automaticity or triggered activity [23]. Several factors increase the susceptibility of ischemic myocardium to reperfusion arrhythmia, including acidosis, α-adrenergic stimulation, and angiotensin II release [24].
Lethal reperfusion injury
Lethal reperfusion injury is defined as myocardial injury caused by the restoration of coronary blood flow after an ischemic episode [25]. It can cause immediate cardiomyocyte death at the beginning of reperfusion. It is the most serious consequence of I/R injury and is the major cause which prevents recovery of the ischemic myocardium after reperfusion therapy. However, it is difficult to identify in the clinical practice, and thus, the incidence in humans is not clear [26]. According to the previous studies, myocardial damage caused by lethal reperfusion injury may account for half of the final infarct size [3]. During the process of ischemia and reperfusion, various programs of cell death are activated, including apoptosis, necrosis, necroptosis, and autophagy-associated cell death [27]. Apoptosis, a process of programmed cell death characterized by typical deoxyribonucleic acid strand breaks, causes distinctive cell morphology changes including cell blebbing and shrinkage [28]. Necrosis is characterized by cell and organelle swelling followed by the rupture of surface membranes and the spilling of their intracellular contents [28]. Notably, necrotic cells are potent stimulators of the immune system and often lead to massive inflammatory cell infiltration and cytokine production in the injured area [29]. This process may further potentiate the severity of myocardial I/R injury. Necroptosis, sharing some features with necrosis and apoptosis, is regarded as one type of regulated cell death [30]. Autophagy, which is regarded as an adaptive response to sublethal stress, is a process of lysosomal degradation and characterized by the presence of double-membrane vesicles and increased expression of autophagy-related genes [28]. Although all these types of cell death account for lethal reperfusion injury, cardiomyocyte death during the myocardial I/R process is mainly mediated by necrosis [31].
MECHANISMS AND MEDIATORS IN MYOCARDIAL ISCHEMIA/REPERFUSION INJURY
After the presence and importance of myocardial I/R injury were identified, accumulating studies addressed the mechanisms and mediators that cause cardiac injury during I/R. Various pathological processes and mediators are proposed to be crucial in ischemia-related and reperfusion-related cell injury. These mediators contribute to particular pathological effects in each stage of I/R. These pathological processes relate to each other and work together to result in cell damage [Figure 1]. In the following section, we review some important pathological processes during cardiac I/R.
Figure 1.
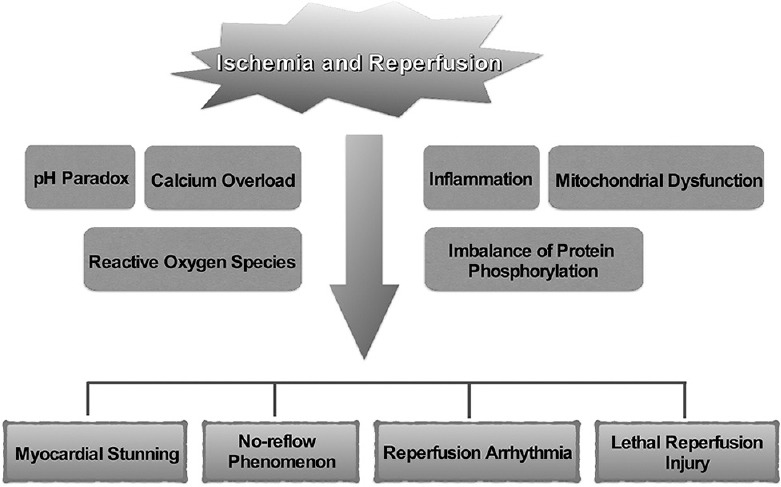
Schematic illustration of the mediators and consequences of ischemia/reperfusion injury
pH paradox
During ischemia, accumulation of intracellular sodium, hydrogen, and calcium ions results in increasing tissue acidosis. Once the blood flow is restored, acidosis is corrected rapidly by the activation of the Na+-H+ exchanger as well as the Na+-HCO− symporter [32]. Evidence has suggested that the rapid correction of pH during reperfusion can lead to enhanced cytotoxicity [33] because this rapid pH shift permits opening of mitochondrial permeability transition pores (MPTPs) [34], further resulting in cardiomyocyte death.
Calcium overload
Intracellular and mitochondrial calcium overload begins at the onset of ischemia [35] and is exacerbated during reperfusion [36]. During reperfusion, the Na+-H+ exchanger and the Na+-HCO3− transporter are activated, leading to intracellular sodium accumulation. A high sodium concentration, in turn, drives the increment of cytosolic calcium through reverse Na+-Ca2+ exchange [37]. Due to lack of adenosine triphosphate (ATP) during the I/R process, further cytosolic calcium accumulation occurs through enhancing calcium entry via sarcolemmal L-type Ca2+ channels and the deficient import of cytosolic calcium into the sarcoplasmic reticulum by sarco/endoplasmic reticulum Ca2+-ATPase [38]. Calcium overload could result in myofibrillar hypercontractility [39], mitochondrial damage [40], and myocardial stunning [41].
Reactive oxygen species
ROS are critical mediators in myocardial I/R injury as evidenced by (1) interventions that enhance ROS scavenging protect against reperfusion injury, (2) artificial generation of ROS reproduces the I/R injury response, and (3) enhanced ROS production and their characteristic products can be detected in postischemic tissues [42]. In the first few minutes of myocardial reperfusion, there is a burst of ROS [43] which could be from a variety of sources. Among all potential ROS sources, xanthine oxidase, nicotinamide adenine dinucleotide phosphate oxidase, mitochondria, and uncoupled nitric oxide synthase are the most likely contributors to reperfusion-induced oxidative stress [42]. These oxygen species are highly reactive and overwhelm the cell's endogenous-free radical scavenging system quickly. This harmful oxidative stress contributes to myocardial injury and cardiomyocyte death through a variety of mechanisms. They can not only cause direct damage to membranes and proteins but also induce indirect damage by opening of the MPTP and the following activation of pro-apoptotic pathways.
Mitochondria dysfunction
Cardiomyocytes consume lots of energy, and thus, the density of mitochondria inside the cells is relatively high. The function of mitochondria is regulated by several factors, including Ca2+ [44]. At a normal physiological status, mitochondrial Ca2+ is a positive effector that can trigger the activation of mitochondrial metabolic machinery to generate higher ATP output [45]. However, during the I/R process, the cytosolic calcium overload mentioned above results in subsequent mitochondrial calcium overload. Ca2+ cycling across the mitochondrial membrane can lead to amplification of mitochondrial Ca2+ overload [46] and cause mitochondrial dysfunction. Mitochondrial Ca2+ overload acts to dissipate the electrical membrane potential [38], facilitates the opening of MPTP, and further results in impairment of ATP synthesis. MPTP, a nonselective channel of the inner mitochondrial membrane, has been considered a critical participant in I/R injury [47]. The inner mitochondrial membrane, normally impermeable to ions and proteins, is responsible for maintaining the mitochondrial transmembrane potential [48]. Formation of the pores creates a nonselective channel and results in dissipation of the electrical potential across this membrane. In the setting of acute I/R, the MPTPs remain closed during ischemia and open at reperfusion in response to mitochondrial calcium and phosphate overload, oxidative stress, and rapid pH correction [48,49]. This opening results in loss of electrochemical gradient, generation of ROS, release of cytochrome c, and subsequent activation of downstream pro-apoptosis pathway, which eventually lead to cell death [50].
Inflammation
Inflammation is also proposed to be an another important player in I/R injury [51] although the I/R process typically occurs in a sterile environment. This sterile inflammation which is also characterized by the accumulation of inflammation cells involves in the recruitment and activation of innate and adaptive immune responses and contributes to myocardial injury [52]. The consequences of I/R share many phenotypic features with activation of the host immune response toward invading microorganisms [53]. The innate immune system is activated by attaching of ligands, termed “damage-associated molecular patterns,” to pattern-recognition molecules such as Toll-like receptors [54,55]. The I/R process also elicits a drastic adaptive immune response involving lymphocytes activated by antigen-specific and antigen-independent mechanisms [56]. Recruitment and activation of immune cells of the innate and adaptive immune system cause pernicious effects to the injured myocardium during the I/R process.
REGULATION OF PROTEIN PHOSPHORYLATION DURING ISCHEMIA/REPERFUSION
The status of protein phosphorylation can be very important in myocardial I/R injury since it involves in regulating a variety of cell functions including the onset and mode of cell death. There are several kinases and phosphatases which have been studied in the I/R process. In the following section, we examine the regulation of protein phosphorylation during myocardial I/R.
Current evidence on protein kinases
A variety of protein kinase pathways can be activated during myocardial ischemia and the reperfusion process. This activation potentially relates to the severity of myocardial injury, and thus, it is worthwhile to get a better understanding of the regulations of these signaling pathways. The protein kinase pathways activated during I/R process include serine/threonine kinases such as protein kinase C (PKC), pro-survival kinases (the mitogen-activated protein kinases [extracellular signal-regulated kinase (ERK) 1/2], PI-3 kinase/Akt), and tyrosine kinases [57]. Among them, the serine/threonine kinases have been most extensively studied.
Protein kinase C
The PKC family of enzymes is composed of isoenzymes that are classified into three groups based on their structure and cofactor regulation. The best characterized group is the conventional PKCs. The other two less understood groups are novel PKCs and atypical PKCs [58]. This was the first protein kinase which was examined in detail in I/R and ischemic preconditioning. Oxidative stress causes the translocation and activation of PKC [59]. This activation of PKC plays a role in myocardial I/R injury as evidence shows that inhibition of PKC activity could protect against I/R injury [60]. In ischemic preconditioning, however, blocking PKC activity by pharmacological agents abolishes the cardioprotective effect [61,62], indicating the contribution of PKC to preconditioning. The discrepancy in these results may come from the different effects of PKC isoenzymes. Which isoenzymes are the most critical mediators remains controversial. In one animal study using rabbit heart, selective εPKC and ηPKC translocation occurred in the ischemic preconditioned myocardium [63]. In another study using isolated rat heart, however, PKC-δ and PKC-ε translocation was noted after brief ischemic stimulation [64]. The studies show that activation of PKC-ε protects against myocardial I/R injury [65] and ablation of PKC-ε abolishes the protective effect of ischemic preconditioning [66]. Based on this evidence, PKC-ε is considered cardioprotective. In contrast, the studies of PKC-δ are more controversial. Some have demonstrated detrimental effects in activating PKC-δ in I/R, but others have suggested a protective role of PKC-δ in ischemic preconditioning [67].
The reperfusion injury salvage kinase pathway
During the I/R process, pro-survival kinase signaling cascades, including the PI-3K/Akt and ERK 1/2 pathways, are activated [68]. This has been termed the reperfusion injury salvage kinase (RISK) pathway, and it promotes cellular survival in part by phosphorylating and inactivating a variety of proteins which are involved in cell death. For example, the phosphorylation and inactivation of downstream effectors such as Bcl-2-associated death promoter, glycogen synthase kinase-3, Bax, Bim, and caspases have been reported [68]. Because the activation of the RISK pathway exerts a significant cardioprotective effect, great effort has been put into exploring strategies that can activate this pathway. Both ischemic preconditioning and postconditioning are documented mechanical strategies which activate the RISK pathway [69,70]. As for pharmacological intervention, an increasing number of agents show a cardioprotective potential which is linked to the recruitment of the RISK pathway. These agents include growth factors, natriuretic peptides, estrogen, volatile anesthetics, and statins [70].
The survivor-activating factor enhancement pathway
In addition to the RISK pathway, an alternative pathway called the survivor-activating factor enhancement (SAFE) pathway involving the activation of Janus kinase (JAK) and signal transducer and activator of transcription-3 (STAT-3) is also activated during I/R [71]. The activation of STAT-3 following AMI was observed after ligation of the left coronary artery of rats [72]. Administration of the JAK/STAT-3 pathway inhibitor suppresses the phosphorylation of STAT-3 and results in an increased number of apoptotic cells [73], therefore suggesting a protective role of STAT-3 in I/R. Through inhibition of MPTP opening, the SAFE pathway promotes cardiomyocyte survival [74]. Cross-talk between the RISK and SAFE pathways has already been addressed based on the finding that inhibiting critical mediators in either pathway abolishes the cardioprotective effect raised from the other [74]. It has been demonstrated that tumor necrosis factor alpha can conquer I/R injury through activation of the SAFE pathway [75]. Some agents, including apolipoprotein A-I [76] and ethanolamine [77], are also considered activators of this pathway.
Protein tyrosine phosphorylation
Protein tyrosine phosphorylation is dynamically regulated by protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs). The dynamic regulation of PTKs and PTPs is crucial for maintaining the homeostasis of tyrosine phosphorylation, and it is involved in critical cellular functions such as cell survival [78]. Imbalance between PTKs and PTPs can lead to pathological consequences. It has been shown that treatment with orthovanadate protects the heart from I/R injury by inhibiting PTPs with subsequent increasing intracellular tyrosine phosphorylation levels [79]. Ischemic preconditioning is proposed as a powerful strategy to diminish myocardial I/R injury, and one of its protective mechanisms is increasing tyrosine phosphorylation by activating tyrosine kinases [80]. All this evidence suggests that the status of protein tyrosine phosphorylation plays a critical role in myocardial I/R injury. Although evidence suggests that the status of protein tyrosine phosphorylation is crucial, the role of each specific tyrosine kinase and phosphatase in myocardial I/R injury has not yet been extensively elucidated.
Tyrosine kinases
Among the family of tyrosine kinases, the role of Src is particularly mentioned. The activity of Src is decreased by the I/R process [81], and a significant increment of Src activity is observed in the heart subjected to ischemic preconditioning [80,82]. After exposing to oxidative stress, Src family tyrosine kinases can activate ERKs, which further promote cardiomyocyte survival [83]. Src activation, as well as the beneficial effects of ischemic preconditioning, is abrogated by tyrosine kinase inhibitors [80]. Other strategies in addition to ischemic preconditioning may activate Src. Urocortin is one of the various compounds that are proposed to be beneficial in I/R conditions. It achieves its protective effect through recruitment of the Src-ERK pathway [84]. Ischemic preconditioning also activates Lck which suggests that Lck may also participate in the I/R process [80].
Tyrosine phosphatases
PTPs, encoded by the largest family of phosphatase genes, are the predominant enzymes that mediate the removal of the phosphate moiety from tyrosine residues [85]. There is limited evidence suggesting the participating of PTPs in myocardial I/R injury. According to a series of studies, treating I/R mice with pan-PTP inhibitors exerts a significant cardioprotective effect [79,86,87]. Genetic deletion of PTP1B, a classic nonreceptor type PTP, showed benefit in protecting the heart against MI-induced injury [88]. Similarly, ablation of PTP1B with RNA interference resulted in enhanced cardiac survival when cultured cardiomyocytes were exposed to hypoxia and reoxygenation [89]. Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) has also been studied under the setting of myocardial I/R. PTEN, which was first identified as a tumor suppressor gene, is an essential regulator of cell proliferation, differentiation, growth, and apoptosis [90]. PTEN is the main phosphatase which negatively regulates the PI3K/Akt pathway [91]; thus, its activity is inversely related to cell survival. In hearts undergoing I/R, PTEN activity is downregulated during the ischemia phase and restored gradually after reperfusion [92]. Ischemic preconditioning also reduces PTEN activity, thus promoting cardiomyocyte survival [92].
CONCLUSION AND FUTURE DIRECTIONS
Early and successful restoration of blood flow to the infarct area in AMI is the most effective strategy to save the ischemic myocardium and decrease mortality and morbidity. However, reperfusion can paradoxically cause tissue injury, thereby reducing the benefits of therapy. There has been great progress in understanding the mechanisms of cardiac I/R injury over the past few decades. According to the existing evidence, cellular signaling transduction is critical to cell survival and is tightly regulated. The setting of ischemia followed by reperfusion alters this carefully orchestrated homeostasis. The events that result from the I/R processes, including rapid pH shifts, calcium overload, mitochondrial dysfunction, inflammation, and imbalance of protein phosphorylation, work together to cause the detrimental effects in injured cardiomyocytes. Translation of cardioprotective strategies from successful experiments to clinical practice has been largely disappointing. There is a need for additional insight into the molecular events triggered by the I/R process. Through investigating the mechanisms of I/R and better understanding the underlying signaling pathway, we would have a better chance to develop treatments to protect against I/R injury. This is an active area of research. We look forward to developing new therapies to diminish myocardial I/R injury and integrate them into clinical practice.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Pfeffer MA. Left ventricular remodeling after acute myocardial infarction. Annu Rev Med. 1995;46:455–66. doi: 10.1146/annurev.med.46.1.455. [DOI] [PubMed] [Google Scholar]
- 2.Takii T, Yasuda S, Takahashi J, Ito K, Shiba N, Shirato K, et al. Trends in acute myocardial infarction incidence and mortality over 30 years in Japan: Report from the MIYAGI-AMI registry study. Circ J. 2010;74:93–100. doi: 10.1253/circj.cj-09-0619. [DOI] [PubMed] [Google Scholar]
- 3.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–35. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 4.Jennings RB, Sommers HM, Smyth GA, Flack HA, Linn H. Myocardial necrosis induced by temporary occlusion of a coronary artery in the dog. Arch Pathol. 1960;70:68–78. [PubMed] [Google Scholar]
- 5.Bolli R, Marbán E. Molecular and cellular mechanisms of myocardial stunning. Physiol Rev. 1999;79:609–34. doi: 10.1152/physrev.1999.79.2.609. [DOI] [PubMed] [Google Scholar]
- 6.Bolli R. Myocardial ‘stunning’ in man. Circulation. 1992;86:1671–91. doi: 10.1161/01.cir.86.6.1671. [DOI] [PubMed] [Google Scholar]
- 7.Bolli R. Mechanism of myocardial “stunning”. Circulation. 1990;82:723–38. doi: 10.1161/01.cir.82.3.723. [DOI] [PubMed] [Google Scholar]
- 8.Kim SJ, Kudej RK, Yatani A, Kim YK, Takagi G, Honda R, et al. A novel mechanism for myocardial stunning involving impaired Ca(2+) handling. Circ Res. 2001;89:831–7. doi: 10.1161/hh2101.098547. [DOI] [PubMed] [Google Scholar]
- 9.Cheirif J. Diagnosis of myocardial stunning: A new use for myocardial contrast and dobutamine echocardiography. Am J Card Imaging. 1995;9:261–8. [PubMed] [Google Scholar]
- 10.AlJaroudi WA, Hage FG. Myocardial stunning by gated SPECT: An old tool reinvented in a stunning turn. J Nucl Cardiol. 2017 doi: 10.1007/s12350-017-1127-1. doi: 10.1007/s12350-017-1127-1. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 11.Ito H. No-reflow phenomenon and prognosis in patients with acute myocardial infarction. Nat Clin Pract Cardiovasc Med. 2006;3:499–506. doi: 10.1038/ncpcardio0632. [DOI] [PubMed] [Google Scholar]
- 12.Ramjane K, Han L, Jin C. The diagnosis and treatment of the no-reflow phenomenon in patients with myocardial infarction undergoing percutaneous coronary intervention. Exp Clin Cardiol. 2008;13:121–8. [PMC free article] [PubMed] [Google Scholar]
- 13.Abbo KM, Dooris M, Glazier S, O’Neill WW, Byrd D, Grines CL, et al. Features and outcome of no-reflow after percutaneous coronary intervention. Am J Cardiol. 1995;75:778–82. doi: 10.1016/s0002-9149(99)80410-x. [DOI] [PubMed] [Google Scholar]
- 14.Resnic FS, Wainstein M, Lee MK, Behrendt D, Wainstein RV, Ohno-Machado L, et al. No-reflow is an independent predictor of death and myocardial infarction after percutaneous coronary intervention. Am Heart J. 2003;145:42–6. doi: 10.1067/mhj.2003.36. [DOI] [PubMed] [Google Scholar]
- 15.Iwakura K, Ito H, Ikushima M, Kawano S, Okamura A, Asano K, et al. Association between hyperglycemia and the no-reflow phenomenon in patients with acute myocardial infarction. J Am Coll Cardiol. 2003;41:1–7. doi: 10.1016/s0735-1097(02)02626-8. [DOI] [PubMed] [Google Scholar]
- 16.Reffelmann T, Kloner RA. The no-reflow phenomenon: A basic mechanism of myocardial ischemia and reperfusion. Basic Res Cardiol. 2006;101:359–72. doi: 10.1007/s00395-006-0615-2. [DOI] [PubMed] [Google Scholar]
- 17.Wit AL, Janse MJ. Reperfusion arrhythmias and sudden cardiac death: A century of progress toward an understanding of the mechanisms. Circ Res. 2001;89:741–3. [PubMed] [Google Scholar]
- 18.Tatli E, Alicik G, Buturak A, Yilmaztepe M, Aktoz M. Arrhythmias following revascularization procedures in the course of acute myocardial infarction: Are they indicators of reperfusion or ongoing ischemia? ScientificWorldJournal. 2013;2013:160380. doi: 10.1155/2013/160380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gorenek B, Blomström Lundqvist C, Brugada Terradellas J, Camm AJ, Hindricks G, Huber K, et al. Cardiac arrhythmias in acute coronary syndromes: Position paper from the joint EHRA, ACCA, and EAPCI task force. EuroIntervention. 2015;10:1095–108. doi: 10.4244/EIJY14M08_19. [DOI] [PubMed] [Google Scholar]
- 20.Meerson FZ, Belkina LM, Sazontova TG, Saltykova VA, Arkhipenko YuV. The role of lipid peroxidation in pathogenesis of arrhythmias and prevention of cardiac fibrillation with antioxidants. Basic Res Cardiol. 1987;82:123–37. doi: 10.1007/BF01907060. [DOI] [PubMed] [Google Scholar]
- 21.Bernier M, Manning AS, Hearse DJ. Reperfusion arrhythmias: Dose-related protection by anti-free radical interventions. Am J Physiol. 1989;256:H1344–52. doi: 10.1152/ajpheart.1989.256.5.H1344. [DOI] [PubMed] [Google Scholar]
- 22.Priori SG, Mantica M, Napolitano C, Schwartz PJ. Early afterdepolarizations induced in vivo by reperfusion of ischemic myocardium. A possible mechanism for reperfusion arrhythmias. Circulation. 1990;81:1911–20. doi: 10.1161/01.cir.81.6.1911. [DOI] [PubMed] [Google Scholar]
- 23.Kaplinsky E, Ogawa S, Michelson EL, Dreifus LS. Instantaneous and delayed ventricular arrhythmias after reperfusion of acutely ischemic myocardium: Evidence for multiple mechanisms. Circulation. 1981;63:333–40. doi: 10.1161/01.cir.63.2.333. [DOI] [PubMed] [Google Scholar]
- 24.Thomas DE, Jex N, Thornley AR. Ventricular arrhythmias in acute coronary syndromes – Mechanisms and management. Contin Cardiol Educ. 2017;3:22–9. [Google Scholar]
- 25.Piper HM, García-Dorado D, Ovize M. A fresh look at reperfusion injury. Cardiovasc Res. 1998;38:291–300. doi: 10.1016/s0008-6363(98)00033-9. [DOI] [PubMed] [Google Scholar]
- 26.Kloner RA. Does reperfusion injury exist in humans? J Am Coll Cardiol. 1993;21:537–45. doi: 10.1016/0735-1097(93)90700-b. [DOI] [PubMed] [Google Scholar]
- 27.Konstantinidis K, Whelan RS, Kitsis RN. Mechanisms of cell death in heart disease. Arterioscler Thromb Vasc Biol. 2012;32:1552–62. doi: 10.1161/ATVBAHA.111.224915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361:1570–83. doi: 10.1056/NEJMra0901217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eltzschig HK, Eckle T. Ischemia and reperfusion – from mechanism to translation. Nat Med. 2011;17:1391–401. doi: 10.1038/nm.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–14. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 31.Zhang T, Zhang Y, Cui M, Jin L, Wang Y, Lv F, et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat Med. 2016;22:175–82. doi: 10.1038/nm.4017. [DOI] [PubMed] [Google Scholar]
- 32.Tani M, Neely JR. Role of intracellular Na+in Ca2+ overload and depressed recovery of ventricular function of reperfused ischemic rat hearts.Possible involvement of H+-Na+and Na+-Ca2+exchange. Circ Res. 1989;65:1045–56. doi: 10.1161/01.res.65.4.1045. [DOI] [PubMed] [Google Scholar]
- 33.Lemasters JJ, Bond JM, Chacon E, Harper IS, Kaplan SH, Ohata H, et al. The pH paradox in ischemia-reperfusion injury to cardiac myocytes. EXS. 1996;76:99–114. doi: 10.1007/978-3-0348-8988-9_7. [DOI] [PubMed] [Google Scholar]
- 34.Kim JS, Jin Y, Lemasters JJ. Reactive oxygen species, but not Ca2+ overloading, trigger pH- and mitochondrial permeability transition-dependent death of adult rat myocytes after ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2006;290:H2024–34. doi: 10.1152/ajpheart.00683.2005. [DOI] [PubMed] [Google Scholar]
- 35.Steenbergen C, Murphy E, Levy L, London RE. Elevation in cytosolic free calcium concentration early in myocardial ischemia in perfused rat heart. Circ Res. 1987;60:700–7. doi: 10.1161/01.res.60.5.700. [DOI] [PubMed] [Google Scholar]
- 36.Jennings RB, Schaper J, Hill ML, Steenbergen C, Jr, Reimer KA. Effect of reperfusion late in the phase of reversible ischemic injury. Changes in cell volume, electrolytes, metabolites, and ultrastructure. Circ Res. 1985;56:262–78. doi: 10.1161/01.res.56.2.262. [DOI] [PubMed] [Google Scholar]
- 37.Garcia-Dorado D, Ruiz-Meana M, Inserte J, Rodriguez-Sinovas A, Piper HM. Calcium-mediated cell death during myocardial reperfusion. Cardiovasc Res. 2012;94:168–80. doi: 10.1093/cvr/cvs116. [DOI] [PubMed] [Google Scholar]
- 38.Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Piper HM. Energy deficiency, calcium overload or oxidative stress: Possible causes of irreversible ischemic myocardial injury. Klin Wochenschr. 1989;67:465–76. doi: 10.1007/BF01721672. [DOI] [PubMed] [Google Scholar]
- 40.Halestrap AP. Calcium, mitochondria and reperfusion injury: A pore way to die. Biochem Soc Trans. 2006;34:232–7. doi: 10.1042/BST20060232. [DOI] [PubMed] [Google Scholar]
- 41.Kusuoka H, Porterfield JK, Weisman HF, Weisfeldt ML, Marban E. Pathophysiology and pathogenesis of stunned myocardium. Depressed Ca2+ activation of contraction as a consequence of reperfusion-induced cellular calcium overload in ferret hearts. J Clin Invest. 1987;79:950–61. doi: 10.1172/JCI112906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Granger DN, Kvietys PR. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015;6:524–51. doi: 10.1016/j.redox.2015.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Becker LB. New concepts in reactive oxygen species and cardiovascular reperfusion physiology. Cardiovasc Res. 2004;61:461–70. doi: 10.1016/j.cardiores.2003.10.025. [DOI] [PubMed] [Google Scholar]
- 44.Tarasov AI, Griffiths EJ, Rutter GA. Regulation of ATP production by mitochondrial Ca(2+) Cell Calcium. 2012;52:28–35. doi: 10.1016/j.ceca.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jouaville LS, Pinton P, Bastianutto C, Rutter GA, Rizzuto R. Regulation of mitochondrial ATP synthesis by calcium: Evidence for a long-term metabolic priming. Proc Natl Acad Sci U S A. 1999;96:13807–12. doi: 10.1073/pnas.96.24.13807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, AT, and ROS: A mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 2004;287:C817–33. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- 47.Halestrap AP. Mitochondria and reperfusion injury of the heart – A holey death but not beyond salvation. J Bioenerg Biomembr. 2009;41:113–21. doi: 10.1007/s10863-009-9206-x. [DOI] [PubMed] [Google Scholar]
- 48.Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion – A target for cardioprotection. Cardiovasc Res. 2004;61:372–85. doi: 10.1016/S0008-6363(03)00533-9. [DOI] [PubMed] [Google Scholar]
- 49.Griffiths EJ, Halestrap AP. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J. 1995;307(Pt 1):93–8. doi: 10.1042/bj3070093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 51.Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol. 2014;11:255–65. doi: 10.1038/nrcardio.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Boros P, Bromberg JS. New cellular and molecular immune pathways in ischemia/reperfusion injury. Am J Transplant. 2006;6:652–8. doi: 10.1111/j.1600-6143.2005.01228.x. [DOI] [PubMed] [Google Scholar]
- 53.Chen GY, Nuñez G. Sterile inflammation: Sensing and reacting to damage. Nat Rev Immunol. 2010;10:826–37. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oyama J, Blais C, Jr, Liu X, Pu M, Kobzik L, Kelly RA, et al. Reduced myocardial ischemia-reperfusion injury in toll-like receptor 4-deficient mice. Circulation. 2004;109:784–9. doi: 10.1161/01.CIR.0000112575.66565.84. [DOI] [PubMed] [Google Scholar]
- 55.Timmers L, Pasterkamp G, de Hoog VC, Arslan F, Appelman Y, de Kleijn DP, et al. The innate immune response in reperfused myocardium. Cardiovasc Res. 2012;94:276–83. doi: 10.1093/cvr/cvs018. [DOI] [PubMed] [Google Scholar]
- 56.Linfert D, Chowdhry T, Rabb H. Lymphocytes and ischemia-reperfusion injury. Transplant Rev (Orlando) 2009;23:1–0. doi: 10.1016/j.trre.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Armstrong SC. Protein kinase activation and myocardial ischemia/reperfusion injury. Cardiovasc Res. 2004;61:427–36. doi: 10.1016/j.cardiores.2003.09.031. [DOI] [PubMed] [Google Scholar]
- 58.Newton AC. Protein kinase C: Structure, function, and regulation. J Biol Chem. 1995;270:28495–8. doi: 10.1074/jbc.270.48.28495. [DOI] [PubMed] [Google Scholar]
- 59.Gopalakrishna R, Jaken S. Protein kinase C signaling and oxidative stress. Free Radic Biol Med. 2000;28:1349–61. doi: 10.1016/s0891-5849(00)00221-5. [DOI] [PubMed] [Google Scholar]
- 60.Inagaki K, Chen L, Ikeno F, Lee FH, Imahashi K, Bouley DM, et al. Inhibition of delta-protein kinase C protects against reperfusion injury of the ischemic heart in vivo. Circulation. 2003;108:2304–7. doi: 10.1161/01.CIR.0000101682.24138.36. [DOI] [PubMed] [Google Scholar]
- 61.Liu Y, Cohen MV, Downey JM. Chelerythrine, a highly selective protein kinase C inhibitor, blocks the anti-infarct effect of ischemic preconditioning in rabbit hearts. Cardiovasc Drugs Ther. 1994;8:881–2. doi: 10.1007/BF00877409. [DOI] [PubMed] [Google Scholar]
- 62.Ytrehus K, Liu Y, Downey JM. Preconditioning protects ischemic rabbit heart by protein kinase C activation. Am J Physiol. 1994;266:H1145–52. doi: 10.1152/ajpheart.1994.266.3.H1145. [DOI] [PubMed] [Google Scholar]
- 63.Ping P, Zhang J, Qiu Y, Tang XL, Manchikalapudi S, Cao X, et al. Ischemic preconditioning induces selective translocation of protein kinase C isoforms epsilon and eta in the heart of conscious rabbits without subcellular redistribution of total protein kinase C activity. Circ Res. 1997;81:404–14. doi: 10.1161/01.res.81.3.404. [DOI] [PubMed] [Google Scholar]
- 64.Mitchell MB, Meng X, Ao L, Brown JM, Harken AH, Banerjee A, et al. Preconditioning of isolated rat heart is mediated by protein kinase C. Circ Res. 1995;76:73–81. doi: 10.1161/01.res.76.1.73. [DOI] [PubMed] [Google Scholar]
- 65.Ping P, Song C, Zhang J, Guo Y, Cao X, Li RC, et al. Formation of protein kinase C (epsilon)-lck signaling modules confers cardioprotection. J Clin Invest. 2002;109:499–507. doi: 10.1172/JCI13200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Saurin AT, Pennington DJ, Raat NJ, Latchman DS, Owen MJ, Marber MS, et al. Targeted disruption of the protein kinase C epsilon gene abolishes the infarct size reduction that follows ischaemic preconditioning of isolated buffer-perfused mouse hearts. Cardiovasc Res. 2002;55:672–80. doi: 10.1016/s0008-6363(02)00325-5. [DOI] [PubMed] [Google Scholar]
- 67.Inagaki K, Churchill E, Mochly-Rosen D. Epsilon protein kinase C as a potential therapeutic target for the ischemic heart. Cardiovasc Res. 2006;70:222–30. doi: 10.1016/j.cardiores.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 68.Hausenloy DJ, Yellon DM. New directions for protecting the heart against ischaemia-reperfusion injury: Targeting the reperfusion injury salvage kinase (RISK)-pathway. Cardiovasc Res. 2004;61:448–60. doi: 10.1016/j.cardiores.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 69.Hausenloy DJ, Tsang A, Yellon DM. The reperfusion injury salvage kinase pathway: A common target for both ischemic preconditioning and postconditioning. Trends Cardiovasc Med. 2005;15:69–75. doi: 10.1016/j.tcm.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 70.Hausenloy DJ, Yellon DM. Reperfusion injury salvage kinase signalling: Taking a RISK for cardioprotection. Heart Fail Rev. 2007;12:217–34. doi: 10.1007/s10741-007-9026-1. [DOI] [PubMed] [Google Scholar]
- 71.Lecour S. Activation of the protective survivor activating factor enhancement (SAFE) pathway against reperfusion injury: Does it go beyond the RISK pathway? J Mol Cell Cardiol. 2009;47:32–40. doi: 10.1016/j.yjmcc.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 72.Omura T, Yoshiyama M, Ishikura F, Kobayashi H, Takeuchi K, Beppu S, et al. Myocardial ischemia activates the JAK-STAT pathway through angiotensin II signaling in in vivo myocardium of rats. J Mol Cell Cardiol. 2001;33:307–16. doi: 10.1006/jmcc.2000.1303. [DOI] [PubMed] [Google Scholar]
- 73.Xuan YT, Guo Y, Han H, Zhu Y, Bolli R. An essential role of the JAK-STAT pathway in ischemic preconditioning. Proc Natl Acad Sci U S A. 2001;98:9050–5. doi: 10.1073/pnas.161283798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hadebe N, Cour M, Lecour S. The SAFE pathway for cardioprotection: Is this a promising target? Basic Res Cardiol. 2018;113:9. doi: 10.1007/s00395-018-0670-5. [DOI] [PubMed] [Google Scholar]
- 75.Lecour S, Suleman N, Deuchar GA, Somers S, Lacerda L, Huisamen B, et al. Pharmacological preconditioning with tumor necrosis factor-alpha activates signal transducer and activator of transcription-3 at reperfusion without involving classic prosurvival kinases (Akt and extracellular signal-regulated kinase) Circulation. 2005;112:3911–8. doi: 10.1161/CIRCULATIONAHA.105.581058. [DOI] [PubMed] [Google Scholar]
- 76.Kalakech H, Hibert P, Prunier-Mirebeau D, Tamareille S, Letournel F, Macchi L, et al. RISK and SAFE signaling pathway involvement in apolipoprotein A-I-induced cardioprotection. PLoS One. 2014;9:e107950. doi: 10.1371/journal.pone.0107950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kelly RF, Lamont KT, Somers S, Hacking D, Lacerda L, Thomas P, et al. Ethanolamine is a novel STAT-3 dependent cardioprotective agent. Basic Res Cardiol. 2010;105:763–70. doi: 10.1007/s00395-010-0125-0. [DOI] [PubMed] [Google Scholar]
- 78.Hunter T. Protein kinases and phosphatases: The yin and yang of protein phosphorylation and signaling. Cell. 1995;80:225–36. doi: 10.1016/0092-8674(95)90405-0. [DOI] [PubMed] [Google Scholar]
- 79.Takada Y, Hashimoto M, Kasahara J, Aihara K, Fukunaga K. Cytoprotective effect of sodium orthovanadate on ischemia/reperfusion-induced injury in the rat heart involves akt activation and inhibition of fodrin breakdown and apoptosis. J Pharmacol Exp Ther. 2004;311:1249–55. doi: 10.1124/jpet.104.070839. [DOI] [PubMed] [Google Scholar]
- 80.Ping P, Zhang J, Zheng YT, Li RC, Dawn B, Tang XL, et al. Demonstration of selective protein kinase C-dependent activation of Src and lck tyrosine kinases during ischemic preconditioning in conscious rabbits. Circ Res. 1999;85:542–50. doi: 10.1161/01.res.85.6.542. [DOI] [PubMed] [Google Scholar]
- 81.Takeishi Y, Abe Ji, Lee JD, Kawakatsu H, Walsh RA, Berk BC, et al. Differential regulation of p90 ribosomal S6 kinase and big mitogen-activated protein kinase 1 by ischemia/reperfusion and oxidative stress in perfused guinea pig hearts. Circ Res. 1999;85:1164–72. doi: 10.1161/01.res.85.12.1164. [DOI] [PubMed] [Google Scholar]
- 82.Takeishi Y, Huang Q, Wang T, Glassman M, Yoshizumi M, Baines CP, et al. Src family kinase and adenosine differentially regulate multiple MAP kinases in ischemic myocardium: Modulation of MAP kinases activation by ischemic preconditioning. J Mol Cell Cardiol. 2001;33:1989–2005. doi: 10.1006/jmcc.2001.1463. [DOI] [PubMed] [Google Scholar]
- 83.Aikawa R, Komuro I, Yamazaki T, Zou Y, Kudoh S, Tanaka M, et al. Oxidative stress activates extracellular signal-regulated kinases through Src and Ras in cultured cardiac myocytes of neonatal rats. J Clin Invest. 1997;100:1813–21. doi: 10.1172/JCI119709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yuan Z, McCauley R, Chen-Scarabelli C, Abounit K, Stephanou A, Barry SP, et al. Activation of Src protein tyrosine kinase plays an essential role in urocortin-mediated cardioprotection. Mol Cell Endocrinol. 2010;325:1–7. doi: 10.1016/j.mce.2010.04.013. [DOI] [PubMed] [Google Scholar]
- 85.Tonks NK. Protein tyrosine phosphatases: From genes, to function, to disease. Nat Rev Mol Cell Biol. 2006;7:833–46. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 86.Liem DA, Gho CC, Gho BC, Kazim S, Manintveld OC, Verdouw PD, et al. The tyrosine phosphatase inhibitor bis (maltolato) oxovanadium attenuates myocardial reperfusion injury by opening ATP-sensitive potassium channels. J Pharmacol Exp Ther. 2004;309:1256–62. doi: 10.1124/jpet.103.062547. [DOI] [PubMed] [Google Scholar]
- 87.Bhuiyan MS, Takada Y, Shioda N, Moriguchi S, Kasahara J, Fukunaga K, et al. Cardioprotective effect of vanadyl sulfate on ischemia/reperfusion-induced injury in rat heart in vivo is mediated by activation of protein kinase B and induction of FLICE-inhibitory protein. Cardiovasc Ther. 2008;26:10–23. doi: 10.1111/j.1527-3466.2008.00039.x. [DOI] [PubMed] [Google Scholar]
- 88.Besnier M, Galaup A, Nicol L, Henry JP, Coquerel D, Gueret A, et al. Enhanced angiogenesis and increased cardiac perfusion after myocardial infarction in protein tyrosine phosphatase 1B-deficient mice. FASEB J. 2014;28:3351–61. doi: 10.1096/fj.13-245753. [DOI] [PubMed] [Google Scholar]
- 89.Song H, Zhang Z, Wang L. Small interference RNA against PTP-1B reduces hypoxia/reoxygenation induced apoptosis of rat cardiomyocytes. Apoptosis. 2008;13:383–93. doi: 10.1007/s10495-008-0181-1. [DOI] [PubMed] [Google Scholar]
- 90.Hopkins BD, Hodakoski C, Barrows D, Mense SM, Parsons RE. PTEN function: The long and the short of it. Trends Biochem Sci. 2014;39:183–90. doi: 10.1016/j.tibs.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hlobilková A, Knillová J, Bártek J, Lukás J, Kolár Z. The mechanism of action of the tumour suppressor gene PTEN. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2003;147:19–25. [PubMed] [Google Scholar]
- 92.Cai Z, Semenza GL. PTEN activity is modulated during ischemia and reperfusion: Involvement in the induction and decay of preconditioning. Circ Res. 2005;97:1351–9. doi: 10.1161/01.RES.0000195656.52760.30. [DOI] [PubMed] [Google Scholar]