

The effect of the incorporation of a methylhydroxyl moiety into a synthetic antisickling agent in facilitating αF-helix interactions to destabilize the sickle hemoglobin polymer and enhance antisickling properties is described.
Keywords: sickle cell disease, hemoglobin, relaxed state, aromatic aldehydes, antisickling, oxygen equilibrium, crystal structure, polymerization, F-helix
Abstract
Increasing the affinity of hemoglobin for oxygen represents a feasible and promising therapeutic approach for sickle cell disease by mitigating the primary pathophysiological event, i.e. the hypoxia-induced polymerization of sickle hemoglobin (Hb S) and the concomitant erythrocyte sickling. Investigations on a novel synthetic antisickling agent, SAJ-310, with improved and sustained antisickling activity have previously been reported. To further enhance the biological effects of SAJ-310, a structure-based approach was employed to modify this compound to specifically inhibit Hb S polymer formation through interactions which perturb the Hb S polymer-stabilizing αF-helix, in addition to primarily increasing the oxygen affinity of hemoglobin. Three compounds, TD-7, TD-8 and TD-9, were synthesized and studied for their interactions with hemoglobin at the atomic level, as well as their functional and antisickling activities in vitro. X-ray crystallographic studies with liganded hemoglobin in complex with TD-7 showed the predicted mode of binding, although the interaction with the αF-helix was not as strong as expected. These findings provide important insights and guidance towards the development of molecules that would be expected to bind and make stronger interactions with the αF-helix, resulting in more efficacious novel therapeutics for sickle cell disease.
1. Introduction
Sickle cell disease (SCD) occurs as a consequence of a single point mutation of βGlu6 in normal hemoglobin (Hb A) to βVal6 in sickle hemoglobin (Hb S). When deoxygenated, Hb S polymerizes, which leads to the characteristic sickling of red blood cells (RBCs), triggering multiple secondary pathological events including vaso-occlusion, painful crises, organ damage, oxidative stress, hemolysis of RBCs, decreased vascular nitric oxide (NO) bioavailability, inflammation, impaired microvascular blood flow, hemolytic anemia, morbidity and mortality (Thein et al., 2017 ▸; Ware et al., 2017 ▸; Rees et al., 2010 ▸; Kato et al., 2007 ▸; Chaturvedi & DeBaun, 2016 ▸; Pauling et al., 1949 ▸; Sedrak & Kondamudi, 2018 ▸). The hypoxia-induced polymerization process is exacerbated by the low affinity of Hb S for oxygen, presumably owing to high levels of 2,3-diphosphoglycerate (2,3-DPG) and/or sphingosine phosphate (S1P) in sickle RBCs (Rogers et al., 2013 ▸; Poillon et al., 1986 ▸; Poillon & Kim, 1990 ▸; Jensen, 2009 ▸; Zhang et al., 2014 ▸). The polymerization process is initiated by a primary interaction between the pathological β2Val6 from one Hb S molecule and a hydrophobic acceptor pocket in the region of β1Ala70, β1Phe85 and β1Leu88 of another proximate Hb S molecule, and is further stabilized by several secondary contacts between the Hb S molecules in the polymer (Ferrone, 2004 ▸; Ghatge et al., 2016 ▸; Cretegny & Edelstein, 1993 ▸; Eaton & Hofrichter, 1990 ▸; Harrington et al., 1997 ▸; Watowich et al., 1993 ▸). The importance of the secondary interactions is demonstrated by a number of naturally occurring mutations that reduce Hb S polymerization and RBC sickling by disrupting these polymer-stabilizing contacts (Bunn & Forget, 1986 ▸). For example, αAsn78→Lys (Hb Stanleyville) leads to an increase in the solubility of deoxy Hb S heterotetramers, mitigating the severity of SCD (Nagel et al., 1980 ▸; Rhoda et al., 1983 ▸; Benesch et al., 1979 ▸).
Hemoglobin functions in equilibrium between the tense state (T-state) possessing low oxygen (O2) affinity and an ensemble of R-states (classical R, R2, R3 etc.) possessing high O2 affinity (Perutz, 1972 ▸, 1978 ▸; Perutz et al., 1998 ▸; Silva et al., 1992 ▸; Jenkins et al., 2009 ▸; Safo & Bruno, 2011 ▸; Safo et al., 2011 ▸). Stabilization of the R-state by aromatic aldehydes in the form of the R2 conformation increases the oxygen affinity of Hb, which inhibits the hypoxia-induced Hb S polymerization (Safo & Kato, 2014 ▸; Abraham et al., 1991 ▸; Safo et al., 2004 ▸; Abdulmalik et al., 2005 ▸, 2011 ▸). Several candidates have been studied in the clinic for the treatment of SCD (Safo & Kato, 2014 ▸; Arya et al., 1996 ▸; Kato et al., 2013 ▸; Stern et al., 2012 ▸; Oder et al., 2016 ▸; Rolan et al., 1993 ▸, 1995 ▸; Oksenberg et al., 2016 ▸; Metcalf et al., 2017 ▸), with one, GBT440, currently in phase III clinical studies (https://clinicaltrials.gov; NCT03036813). However, the historical focus on exclusively shifting oxygen affinity to prevent sickling has posed important challenges, primarily related to toxicity and/or moderate efficacy, considering the large doses that are required to effectively modify a fraction of the abundant Hb S protein (Safo & Kato, 2014 ▸). Consequently, alternative strategies to boost the activities of the candidate molecules are currently being undertaken. For example, GBT440 was designed to bind to one α-subunit and occupy the entire cavity, reducing the drug:Hb stoichiometry by half (Oksenberg et al., 2016 ▸; Metcalf et al., 2017 ▸). Additionally, it has been reported to partition at a high rate into RBCs, further enhancing its efficacy (Oksenberg et al., 2016 ▸). Our group has been investigating approaches to modify candidate molecules to make additional interactions with Hb that would be expected to destabilize polymer contacts, in addition to the primary mechanism of increasing the oxygen affinity of Hb (Abdulmalik et al., 2011 ▸; Pagare et al., 2018 ▸). For example, our previously studied aromatic aldehyde INN-312 made a hydrophobic interaction with the αF-helix and moderately contributed to its antisickling activity by destabilizing the polymer (Abdulmalik et al., 2011 ▸).
To further this line of investigation, we have systematically modified our lead antisickling agent, vanillin (Fig. 1 ▸ a), resulting in several generations of potent derivatives (Abdulmalik et al., 2011 ▸; Pagare et al., 2018 ▸; Nnamani et al., 2008 ▸). One such recently reported derivative is SAJ-310 (Fig. 1 ▸ a), which appeared to be a promising lead candidate in extensive in vitro pharmacokinetic (PK) and pharmacodynamic (PD) studies (Pagare et al., 2018 ▸). The co-crystal structure of SAJ-310 with liganded Hb led to stabilization of the R-state Hb, explaining its biological effect of increasing Hb oxygen affinity and significantly inhibiting hypoxia-induced Hb S polymerization and RBC sickling (Pagare et al., 2018 ▸). The crystal structure only showed a weak hydrophobic interaction with the αF-helix of Hb when compared with INN-312 (Fig. 1 ▸ a; Abdulmalik et al., 2011 ▸). Nonetheless, SAJ-310 showed significantly better in vitro pharmacokinetic activity than INN-312, making it a superior lead compound (Pagare et al., 2018 ▸). In this article, we further derivatized SAJ-310 into three compounds (TD-7, TD-8 and TD-9; Fig. 1 ▸ a) with the intent of making stronger interactions with the αF-helix than we observed for SAJ-310, and therefore enhancing potency. We investigated the compounds for their overall interactions with liganded Hb at the atomic level, as well as their functional and biological activities in vitro.
Figure 1.

(a) Structures of aromatic aldehydes, including the studied compounds. (b) Schematic representations of the anticipated binding modes of aromatic aldehydes to Hb.
2. Experimental procedures
2.1. Materials and general procedure
Normal whole blood was collected from adult donors at the Virginia Commonwealth University after informed consent, in accordance with the regulations of the IRB for Protection of Human Subjects. Hb was purified from discarded normal blood samples using standard procedures (Safo & Abraham, 2003 ▸). Leftover blood samples from patients with homozygous (SS) SCD were obtained and utilized, based on an approved IRB protocol at the Children’s Hospital of Philadelphia, with informed consent. All reagents used in the syntheses and functional assays were purchased from Sigma–Aldrich (St Louis, Missouri, USA) and ThermoFisher Scientific (Waltham, Massachusetts, USA) and were utilized without additional purification.
2.2. Synthesis
The TD compounds were synthesized according to standard procedures (Nnamani et al., 2008 ▸; Abdulmalik et al., 2011 ▸; Pagare et al., 2018 ▸) for similar compounds; the syntheses are reported in detail in the Supporting information.
2.3. Crystallization, data collection and structure determination of liganded Hb in complex with compounds
Co-crystals of liganded Hb with TD-7, TD-8 and TD-9 were obtained following procedures similar to that reported for SAJ-310 (Pagare et al., 2018 ▸). Briefly, freshly made solutions of TD-7, TD-8 or TD-9 in DMSO were separately incubated with carbonmonoxy Hb (30 mg ml−1 protein) at an Hb (tetramer):TD molar ratio of 1:10. The Schiff-base interaction formed between the aldehyde of the TD compounds and the αVal1 N-terminal N atom of Hb was reduced to the corresponding irreversible alkylamine covalent bond using sodium cyanoborohydride (NaCNBH3). The corresponding complex solution was crystallized with 10–20% PEG 6000, 100 mM HEPES pH 7.4 using the batch method, resulting in cherry-red needle crystals. X-ray diffraction data were collected at 100 K using a Rigaku MicroMax-007 HF X-ray generator, an EIGER R 4M detector and an Oxford Cobra cryosystem (Rigaku, The Woodlands, Texas, USA). The crystals were first cryoprotected with 80 µl mother liquor mixed with 50–60 µl 50% PEG 6000. The data sets were processed with CrysAlisPro (Rigaku) and the CCP4 suite of programs (Winn et al., 2011 ▸).
The isomorphous crystal structure of SAJ-310 in complex with liganded Hb (PDB entry 6bnr; P. P. Pagare, F. N. Musayev & M. K. Safo, unpublished work) was used as a starting model for the refinement of the TD-7, TD-8 and TD-9 data sets using both PHENIX and CNS (Adams et al., 2011 ▸; Echols et al., 2012 ▸; Brünger et al., 1998 ▸). Initial refinement of TD-7 in complex with liganded Hb identified two molecules of the compound at the α-cleft, which were included in the model along with several water molecules and CO bound to the hemes, resulting in a final R factor and R free of 19.5% and 24.9%, respectively, at 1.9 Å resolution. Unlike TD-7 in complex with liganded Hb, which showed very strong initial difference density for the bound pair of compounds, both TD-8 and TD-9 in complex with liganded Hb showed very weak and sparse density at the expected binding sites. Repeated refinement of these two complexes did not improve the density of the compound to allow fitting, and the refinements were terminated. The atomic coordinate and structure factors of TD-7 in complex with liganded Hb have been deposited in the RCSB Protein Data Bank as entry 6di4. Detailed crystallographic and refinement parameters are reported in Table 1 ▸. Model building and correction were carried out using Coot (Emsley et al., 2010 ▸).
Table 1. Data-collection and refinement statistics for carbonmonoxy Hb in complex with TD-7.
Values in parentheses are for the highest resolution shell.
| Data-collection statistics | |
| Space group | P212121 |
| Unit-cell parameters (Å) | a = 62.70, b = 83.40, c = 104.93 |
| Resolution (Å) | 29.34–1.90 (1.97–1.90) |
| Unique reflections | 43079 |
| Multiplicity | 4.13 (3.68) |
| Completeness (%) | 97.8 (95.0) |
| Average I/σ(I) | 15.7 (3.8) |
| R merge † (%) | 4.7 (33.6) |
| Refinement statistics | |
| Resolution (Å) | 29.34–1.90 (2.02–1.90) |
| No. of reflections | 42867 (6491) |
| R work (%) | 19.5 (32.2) |
| R free ‡ (%) | 24.9 (37.2) |
| R.m.s.d., bonds (Å) | 0.007 |
| R.m.s.d., angles (°) | 1.7 |
| Dihedral angles | |
| Most favored (%) | 96.29 |
| Allowed (%) | 3.53 |
| Average B factors (Å2) | |
| All atoms | 33.81 |
| Protein | 32.07 |
| Hemes | 29.61 |
| TD-7 | 45.25 |
| Water | 43.92 |
| PDB code | 6di4 |
R
merge = 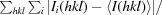
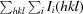 .
.
R free was calculated from a randomly selected 5% of reflections for cross-validation. All other measured reflections were used during refinement.
2.4. In vitro time-dependent Hb-adduct formation
We investigated the compounds in vitro for their time- and concentration-dependent effect on Hb-adduct formation (Hb modification). A 200 mM stock solution of each compound was made in DMSO. In a 96-well 1.2 ml polypropylene deep-well plate (Thermo Scientific), 600 µl of whole blood from normal healthy donors (adjusted to 30% final hematocrit) was supplemented with each compound to a final concentration of 2 mM and incubated at 37°C for 24 h with shaking (100 rev min−1). A control sample was supplemented only with an equivalent volume of DMSO. At defined (0.5, 1, 2, 4 and 12 h) time intervals, 75 µl aliquot samples of blood were simultaneously removed from individual wells using a multichannel pipette and immediately added to respective tubes containing 75 µl sodium cyanoborohydride and borohydride mixture [1:1(v:v) 50 mM stock] to terminate the Schiff-base reaction, fix the Schiff-base adducts and reduce the free reactive aldehyde. We had previously established this condition as optimal for our assays (Pagare et al., 2018 ▸). After mixing, the tubes were stored immediately at −80°C until ready for analysis to determine the degree of Hb modification.
2.5. Hemoglobin-modification, oxygen-equilibrium and antisickling studies using human sickle blood
We tested the antisickling properties, the Hb-modification properties and the shift in the oxygen-equilibrium curve (OEC) utilizing samples from consented individuals with homozygous SCD. Briefly, in 96-well plates, blood samples were suspended in Hemox buffer supplemented with glucose and bovine serum albumin to a final hematocrit of 20%. The samples were incubated under room air in the absence or presence of 0.5, 1, 1.5 or 2 mM concentrations of the test compounds at 37°C for 1 h to ensure that binding had attained equilibrium. Following this, the reaction plates were incubated in a Thermomixer R (Eppendorf) under hypoxia (4% oxygen/96% nitrogen) at 37°C for 2 h. Aliquot samples (5 µl) were obtained at the conclusion, fixed with 2% glutaraldehyde solution without exposure to air and then subjected to microscopic morphological analysis of bright-field images (at 40× magnification) of single-layer cells on an Olympus BX40 microscope fitted with an Infinity Lite B camera (Olympus) and the coupled Image Capture software. The residual samples were washed in phosphate-buffered saline and subjected to hypotonic lysis for OEC and Hb-modification analyses.
OEC studies were conducted using approximately 100 µl aliquot samples from the clarified lysates obtained from the antisickling study resuspended in 4 ml 0.1 M potassium phosphate buffer pH 7.0. Samples were transferred into a cuvette and subjected to hemoximetry analysis using a HEMOX Analyzer (TCS Scientific Corporation) to assess the shifts in the OEC or P 50 values, where P 50 is defined as the partial pressure at which half of the Hb is oxygenated (or deoxygenated). The degree of P 50 shift (ΔP 50) was expressed as the percentage fraction of control DMSO-treated samples.
Finally, for the Hb-modification (into the high-affinity adduct form) study, clarified lysates, also from the above antisickling study, were subjected to cation-exchange HPLC (Hitachi D-7000 Series, Hitachi Instruments Inc., San Jose, California, USA) using a weak cation-exchange column (Poly CAT A, 30 × 4.6 mm; Poly LC Inc., Columbia, Maryland, USA). A commercial standard consisting of approximately equal amounts of composite Hb F, Hb A, Hb S and Hb C (Helena Laboratories, Beaumont, Texas, USA) was utilized as reference isotypes. The areas of new peaks representing Hb S adducts were obtained, calculated as a percentage fraction of the total Hb area and reported as levels of modified Hb. All assays were conducted in five biological replicates on samples from different subjects on different days. Results are reported as mean values with standard deviations from five analyses.
3. Results and discussion
3.1. Rational design and synthesis of compounds
We have previously studied and reported a promising aromatic aldehyde, SAJ-310 (Fig. 1 ▸ a), with significantly enhanced in vitro antisickling and pharmacokinetic properties (Pagare et al., 2018 ▸). Based on crystallographic findings for liganded Hb in complex with SAJ-310, which showed that the compound bound and directed its ortho-positioned pyridinylmethoxy group (relative to the aldehyde moiety) to make weak hydrophobic interactions with the αF-helix of Hb (Fig. 1 ▸ b; Pagare et al., 2018 ▸), we designed and synthesized a derivative, TD-7, by incorporating a methylhydroxyl moiety on the pyridine ring (Fig. 1 ▸ a). We also derivatized TD-7 by moving the methoxy group on the benzaldehyde ring from the meta position (relative to the aldehyde group) to the para position to give TD-9 (Fig. 1 ▸ a). Like SAJ-310, we expected the ortho-positioned pyridinylmethoxy group in both TD-7 and TD-9 to direct towards the surface of the protein, but with the methylhydroxyl moiety making closer interaction with the αF-helix than observed in SAJ-310 (Fig. 1 ▸ b). Our primary intent was to perturb the αF-helix to weaken the secondary polymer-stabilization interaction mediated by αAsn78 of the αF-helix, and thus improve on the antisickling activities of these compounds. As noted above, the αF-helix is very important in stabilizing the polymer, and the natural mutant αAsn78→Lys (Hb Stanleyville) is known to significantly increase the solubility of Hb S by abrogating the αAsn78 hydrogen-bond interaction (Bunn & Forget, 1986 ▸; Nagel et al., 1980 ▸; Benesch et al., 1979 ▸; Rhoda et al., 1983 ▸). We also derivatized TD-9 by moving the ortho-positioned pyridinylmethoxy group (relative to the aldehyde) to the meta-position of the benzaldehyde to give TD-8 (Fig. 1 ▸ a). Unlike TD-7 and TD-9, we expected TD-8 to bind and direct its meta-positioned pyridinylmethoxy group further down the central water cavity and away from the αF-helix (Fig. 1 ▸ b). In summary, all three molecules are expected to make additional interactions with the protein that should lead to improvement in their allosteric activities, while TD-7 and TD-9 are also expected to increase the polymer-destabilization effect. Synthetic schemes for the compounds are shown in Figs. 2 ▸(a) and 2 ▸(b), respectively.
Figure 2.

Synthetic schemes for (a) TD-7 and (b) TD-9.
Briefly, substituted hydroxymethoxybenzaldehydes and bromomethylpyridinemethanol were reacted together under basic conditions at room temperature for 12 h to give TD-7, TD-8 and TD-9 in 97.8%, 96.9% and 99.5% yields, respectively. The detailed syntheses of these compounds are reported in the Supporting information. The synthesis of SAJ-310, which was used as a positive control, has been reported previously (Nnamani et al., 2008 ▸; Pagare et al., 2018 ▸). The compounds were subsequently investigated for their binding interactions with Hb and their in vitro pharmacokinetic and pharmacodynamic properties using both normal and sickle blood.
3.2. Compounds sustained the modification of hemoglobin
Aromatic aldehydes are highly susceptible to oxidative metabolism in the liver and blood, resulting in suboptimal pharmacokinetic properties, for example poor bioavailability and fast clearance, ultimately reducing their pharmacological effects (Parikh & Venitz, 2014 ▸; Godfrey et al., 1999 ▸; Stern et al., 2012 ▸; Abdulmalik et al., 2005 ▸). The pharmacokinetic properties of these compounds are in a large part dependent on the strength of the Schiff-base interaction between the aldehyde moiety and the N-terminal αVal1 N atom of Hb, and importantly the ability of the compound to resist oxidative metabolism of the active aldehyde into the corresponding inactive acid. To determine the duration of the effect of the compounds in whole blood, where significant metabolism of the aldehyde occurs, we conducted time-dependent (0–12 h) Hb-modification studies with the compound at 2 mM using normal adult whole blood (hematocrit 30%) at 37°C, followed by analyses by cation-exchange HPLC. The three compounds TD-7, TD-9 and SAJ-310 (positive control), all with an ortho-located pyridinylmethoxy group, showed the highest levels of Hb modification (∼50%), while TD-8 with the meta-located pyridinylmethoxy group showed the lowest level of modification (35%; Fig. 3 ▸ a). All compounds showed significantly sustained activity during the 12 h experiment; however, the activity of TD-9 continued to increase even after 12 h. Interestingly, TD-7 and SAJ-310 showed a quicker onset and attained peak levels of Hb modification at ∼2 h. In comparison, TD-8 and TD-9 showed a very slow onset, with TD-8 reaching a maximum level of Hb modification at 4 h, while the activity of TD-9 continued to increase. It is apparent that the Hb-modification property that was previously observed in SAJ-310 was conserved in TD-7 but not in TD-8 or TD-9. Nonetheless, it is clear that an ortho-substituted pyridinylmethoxy analog would be pharmacologically superior to a meta-substituted analog. For potentially efficacious SCD therapy, molecules with sustained activity and faster onset, e.g. TD-7 and SAJ-310, would be preferred over those with slower onsets, such as TD-9.
Figure 3.

(a) Time-dependent modification of Hb A in normal blood incubated with 2 mM test compounds. (b) Concentration-dependent inhibition of SS cell sickling under hypoxia for 2 h. (c) Concentration-dependent modification of Hb S incubated with test compounds. Hemolysates from the above antisickling assay were subjected to cation-exchange HPLC analyses and levels of modified Hb were assessed. (d) Concentration-dependent P 50 shift of Hb S incubated with test compounds. Hemolysates from the antisickling assay were used for the study.
3.3. TD compounds prevent hypoxia-induced RBC sickling by modifying Hb into high O2-affinity Hb
The antisickling properties of aromatic aldehydes are primarily dependent on their ability to increase the affinity of Hb for oxygen, which is directly dependent on the levels of Schiff-base adduct formation (Hb modification) in the high-affinity Hb form. We therefore tested the antisickling activities of the compounds in vitro as described and assessed sickling by microscopy (Abdulmalik et al., 2005 ▸). Successively, we conducted cation-exchange HPLC analyses to measure the degree of Hb modification and OEC to assess P 50 shifts using aliquots from the sickling studies. Consistent with the observations from the time-dependent Hb-modification studies, TD-7 and SAJ-310 demonstrated strong antisickling activities when compared with TD-8 and TD-9, with TD-8 showing the lowest potency (Fig. 3 ▸ b, Table 2 ▸, Supplementary Fig. S1). At 2 mM concentrations, TD-7 and SAJ-310 inhibited 85% and 97% of sickling, compared with 27% and 10% for TD-9 and TD-8, respectively. Remarkably, the antisickling effect of SAJ-310 was already near-maximal at 1.5 mM, while TD-7 only showed 60% inhibition, suggesting SAJ-310 to be the most superior. Similarly, the observed levels of Hb modification and degree of P 50 shifts correlated with the antisickling activities, as shown in Figs. 3 ▸(c) and 3 ▸(d) (see also Supplementary Fig. S2) and summarized in Table 2 ▸. Clearly, our modification of placing a methylhydroxyl moiety on the pyridine ring of SAJ-310 at best only led to similar functional and/or biological activities as in TD-7.
Table 2. Hemoglobin-modification, change in oxygen equilibrium and antisickling studies on test compounds using human sickle blood.
All studies were conducted with SS cell suspensions (20% hematocrit) incubated with 0.5, 1, 1.5 and 2 mM of each test compound. The results are the mean values ± SD for five individual replicate experiments. The final concentration of DMSO was <2% in all samples, including in control samples.
| Modified Hb† (%) | ΔP 50 ‡ (%) | Sickling inhibition§ (%) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.5 mM | 1 mM | 1.5 mM | 2 mM | 0.5 mM | 1 mM | 1.5 mM | 2 mM | 0.5 mM | 1 mM | 1.5 mM | 2 mM | |
| TD8 | 13.1 ± 2.9 | 19.7 ± 7.2 | 18.0 ± 2.6 | 29.3 ± 12.7 | 7.1 ± 4.4 | 16.4 ± 11.5 | 13.2 ± 3.5 | 23.3 ± 14.5 | 4.7 ± 4.4 | 6.5 ± 7.6 | 9.2 ± 6.8 | 9.9 ± 6.2 |
| TD9 | 17.0 ± 3.5 | 27.8 ± 3.8 | 35.2 ± 4.7 | 43.9 ± 5.5 | 7.9 ± 2.7 | 18.4 ± 4.9 | 29.1 ± 9.3 | 35.2 ± 2.0 | 8.0 ± 7.8 | 10.6 ± 7.3 | 19.6 ± 5.5 | 26.6 ± 9.5 |
| TD7 | 25.9 ± 7.6 | 45.3 ± 11.6 | 59.5 ± 10.7 | 74.0 ± 8.0 | 10.0 ± 2.9 | 27.6 ± 11.7 | 29.3 ± 7.2 | 48.2 ± 9.5 | 16.4 ± 6.3 | 29.1 ± 6.1 | 57.5 ± 10.9 | 84.7 ± 13.6 |
| SAJ310 | 30.9 ± 9.5 | 54.0 ± 13.4 | 75.2 ± 7.2 | 83.3 ± 6.7 | 20.8 ± 10.9 | 35.7 ± 12.1 | 37.5 ± 6.7 | 58.1 ± 12.6 | 19.2 ± 4.8 | 47.7 ± 10.8 | 92.4 ± 5.9 | 97.3 ± 0.6 |
Hb S adduct values obtained from HPLC elution patterns of individual hemolysates after incubation of compounds with SS cells.
P 50 is the oxygen pressure at which the hemolysates are 50% saturated with oxygen. ΔP 50 was determined as ΔP 50 (%) = (P 50 of lysates from untreated cells − P 50 of lysates from treated cells × 100)/(P 50 of lysates from untreated cells).
Antisickling studies with SS cells were conducted under hypoxia (4% oxygen/96% nitrogen).
TD-7 only differs from TD-9 in the position of the methoxy group on the benzaldehyde ring, suggesting that the ortho location of the methoxy moiety confers superior antisickling properties on the compound, likely owing to improved binding and a faster onset of action. Moving the pyridinylmethoxy group from the ortho position in TD-9 to the meta position in TD-8 decreased the antisickling potency of the compound. In summary, while our structural modifications largely conserved the functional properties of SAJ-310 in TD-7, they did not result in our intended consequence, i.e. lead to an improvement in the antisickling properties of SAJ-310. This observation could be explained by the crystallographic result (see below), which in conjunction with the functional and biological results suggests future structural modifications to improve the antisickling activities of these compounds.
3.4. Compounds bind to the α-cleft of hemoglobin and stabilize the R-state
To ascertain whether the TD molecules bind as predicted, as well as to gain atomic insight into their functional and biological activities, we co-crystallized all three molecules, TD-7, TD-8 and TD-9, with carbonmonoxy Hb for structural studies. Detailed crystallographic parameters are reported in Table 1 ▸. The crystal structure of SAJ-310 (PDB entry 6bnr), the precursor of these molecules, has previously been reported by our group (Pagare et al., 2018 ▸). The use of all three compounds, TD-7, TD-8 and TD-9, resulted in R2 crystals that were isomorphous to those of SAJ-310, with typical unit-cell parameters a = 63, b = 83, c = 105 Å and space group P212121. As expected, the TD-7 complex structure showed two molecules bound as Schiff bases to the N-terminal αVal1 N atoms in a symmetry-related fashion to the two α-chains (Figs. 4 ▸, 5 ▸ and 6 ▸). Nonetheless, the electron density of the bound TD-8 and TD-9 were very weak and sparse, and further refinement of the model with or without water molecules did not improve the respective densities to allow model fitting; therefore, the refinements were prematurely terminated. The weaker density observed for TD-8 and TD-9 could explain the less potent effect of these two compounds compared with TD-7, perhaps owing to weaker interactions with Hb.
Figure 4.

Crystal structure of Hb in the R2 conformation in complex with two molecules of TD-7 bound at the α-cleft. Hb subunits are shown as ribbons (α1 subunit in pink, α2 in purple, β1 in yellow and β2 in gray) and hemes are shown as sticks. For clarity, not all binding-site residues are shown but are described in the text. (a) Overall structure of the complex showing a pair of bound TD-7 molecules (green sticks) in the central water cavity. (b) Two-dimensional contacts between one TD-7 molecule, the protein and the second TD-7 molecule as described in the text.
Figure 5.

Close-up view of TD-7 (green sticks) bound at the α-cleft of R2 Hb with contoured electron density. Hb subunits are shown as ribbons (α1 subunit in pink, α2 subunit in purple). Note how the pyridine rings are engaged in a π–π stacking interaction to stabilize the binding and the R-state Hb (see text). For clarity, not all binding-site residues are shown but are described in the text. (a) Final 2F o − F c refined electron-density map contoured at 1.0σ. (b) Initial F o − F c electron-density map (prior to addition of TD-7 to the model) contoured at 3.0σ.
Figure 6.

Structural comparison of molecules bound at the α-cleft of Hb. The α1 subunit is shown in pink and the α2 subunit in purple. (a) Superposition of TD-7 (green) and SAJ-310 (orange) molecules showing interactions with residues from the A-helix and/or F-helix. (b) 90° rotated view of (a).
The SAJ-310 and TD-7 tetramer structures superposed on each other with a root-mean-square deviation (r.m.s.d.) of 0.15 Å, suggesting similar quaternary R2 conformations. The geometry of the heme pockets is also very similar, with each heme iron bound to a CO molecule. As for SAJ-310, and as predicted, formation of the Schiff-base interaction between the aldehyde of TD-7 and the N-terminal amines of αVal1 directed the ortho-located pyridinylmethoxy group of the pair of compounds out of the central water cavity towards the αF-helix (Fig. 6 ▸ b). Based on the orientation of the pyridine moiety in SAJ-310, we expected the methylhydroxyl moiety on the pyridine in TD-7 to point towards and make closer contacts with the αF-helix through hydrophobic and/or hydrogen-bond interactions. Unexpectedly, the pyridine group of TD-7 rotated 180° from that of SAJ-310, precluding such interactions (Fig. 5 ▸ b). In fact, like SAJ-310, only the pyridine ring of TD-7 made hydrophobic contacts of 3.5–4.5 Å with the αF-helix. This compares with INN-312, which is also a close analog of SAJ-310, which made a 3.3–4.0 Å hydrophobic interaction with the αF-helix, explaining the ability of INN-312 to exhibit a moderate polymer-destabilization effect (Abdulmalik et al., 2011 ▸). Detailed interactions between TD-7 and the protein will only be described for the α2 subunit interactions, noting that the second molecule (symmetry-related) is involved in similar interactions with the protein (Fig. 4 ▸ b). The methylhydroxyl moiety of TD-7 made a very weak water-mediated hydrogen-bond interaction with the αLeu2 amide N atom. In SAJ-310, the ortho-located pyridine N atom made a direct intra-subunit hydrogen-bond interaction with the hydroxyl group of α2Ser131, while that of TD-7 made no such interaction owing to the ring rotation. The other interactions between TD-7 and the protein are very similar to those observed for SAJ-310. These include intra- and inter-subunit hydrophobic interactions between the benzaldehyde and α2Ser131 and α1Thr134, respectively, while the pyridinylmethoxy O atom appears to make a weak intrasubunit hydrogen-bond interaction with the hydroxyl group of α2Ser131. The pyridine rings from the two symmetry-related bound compounds make extensive 3.4–4.0 Å face-to-face π–π stacking interactions with each other (Figs. 4 ▸, 5 ▸ and 6 ▸). In summary, the interactions involving the compound and protein provide additional interactions across the two α-subunit interfaces of liganded Hb in the R2 conformation that lead to further stabilization of the R-state and increase the affinity of Hb for O2. Because of the weak hydrophobic interactions with the αF-helix, TD-7, like SAJ-310, did not show any significant antisickling effect at 100% nitrogen, which is a measure of the direct polymer-destabilization effect (data not shown). We also note that TD-7 and SAJ-310 make similar contacts with the protein in the most part; nonetheless, the direct hydrogen-bond interaction between the pyridine N atom and the hydroxyl group of α2Ser131 in SAJ-310 may explain its better functional/biological activities.
Detailed interactions of TD-8 and TD-9 are not possible because of the weak and sparse electron densities, which are consistent with the significantly low functional and biological activities of these two compounds. Nonetheless, from the weak electron densities for TD-8 and TD-9 in their Hb adducts, we could reasonably ascertain the binding orientations of these two molecules. Like TD-7 and SAJ-310, the ortho-positioned pyridinylmethoxy group of TD-9 is also directed towards the αF-helix. In contrast, and also as expected from a similar compound with meta-positioned pyridinylmethoxy, INN-298 (Abdulmalik et al., 2011 ▸), binding of TD-8 directed the group further down the central water cavity.
4. Conclusion
Aromatic aldehydes are currently one of the classes of compounds being studied for the treatment of SCD, with one such compound, GBT440, in phase III clinical studies. These compounds increase the affinity of Hb for oxygen, and as a result prevent hypoxia-induced Hb S polymerization and the consequent secondary adverse pathophysiology. We have systematically and rationally developed several such aromatic aldehydes to increase their potency and bioavailability, and importantly have obtained derivatives that in addition to their ability to increase Hb affinity for oxygen also directly destabilize polymer formation by interacting with the surface-located αF-helix (Abdulmalik et al., 2011 ▸; Pagare et al., 2018 ▸). In this study, we structurally modified our most promising compound, SAJ-310, by placing a methylhydroxyl moiety on the pyridine ring not only to increase its interaction with the protein but more importantly to make closer interactions with the αF-helix and increase its polymer-destabilization effect. Although one of the derivatives, TD-7, showed significant in vitro pharmacological activity, it unexpectedly did not improve its interaction with the αF-helix, nor its consequent antisickling activity through direct polymer destabilization. Nonetheless, our findings with TD-9 and TD-8 suggest possible modifications that would help to guide the design of slower, but longer-acting candidate molecules as needed. Overall, the results of the structural study suggest possible modifications to allow closer interaction with the αF-helix by placing the methylhydroxyl group at the para position instead of the meta position of the pyridine ring and/or incorporating disubstituents on the ring.
Supplementary Material
PDB reference: carbonmonoxy hemoglobin in complex with TD-7, 6di4
Synthesis of compounds and Supplementary Figures.. DOI: 10.1107/S2059798318009919/ag5018sup1.pdf
Acknowledgments
Conflict-of-interest disclosure: Virginia Commonwealth University owns a patent related to TD-7, TD-8 and TD-9. These compounds have been licensed to Baxalta/Shire.
Funding Statement
This work was funded by National Institutes of Health, National Institute on Minority Health and Health Disparities grant MD009124. National Institutes of Health, National Center for Research Resources grant S10-OD021756. Virginia General Assembly Higher Education Equipment Trust Fund grant .
References
- Abdulmalik, O., Ghatge, M. S., Musayev, F. N., Parikh, A., Chen, Q., Yang, J., Nnamani, I., Danso-Danquah, R., Eseonu, D. N., Asakura, T., Abraham, D. J., Venitz, J. & Safo, M. K. (2011). Acta Cryst. D67, 920–928. [DOI] [PMC free article] [PubMed]
- Abdulmalik, O., Safo, M. K., Chen, Q., Yang, J., Brugnara, C., Ohene-Frempong, K., Abraham, D. J. & Asakura, T. (2005). Br. J. Haematol. 128, 552–561. [DOI] [PubMed]
- Abraham, D. J., Mehanna, A. S., Wireko, F. C., Whitney, J., Thomas, R. P. & Orringer, E. P. (1991). Blood, 77, 1334–1341. [PubMed]
- Adams, P. D. et al. (2011). Methods, 55, 94–106.
- Arya, R., Rolan, P. E., Ootton, R., Posner, J. & Bellingham, A. J. (1996). Br. J. Haematol. 93, 817–821. [DOI] [PubMed]
- Benesch, R. E., Kwong, S., Edalji, R. & Benesch, R. (1979). J. Biol. Chem. 254, 8169–8172. [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Bunn, H. F. & Forget, B. G. (1986). Hemoglobin: Molecular, Genetic and Clinical Aspects. Philadelphia: W. B. Saunders & Co.
- Chaturvedi, S. & DeBaun, M. R. (2016). Am. J. Hematol. 91, 5–14. [DOI] [PubMed]
- Cretegny, I. & Edelstein, S. J. (1993). J. Mol. Biol. 230, 733–738. [DOI] [PubMed]
- Eaton, W. A. & Hofrichter, J. (1990). Adv. Protein Chem. 40, 63–279. [DOI] [PubMed]
- Echols, N., Grosse-Kunstleve, R. W., Afonine, P. V., Bunkóczi, G., Chen, V. B., Headd, J. J., McCoy, A. J., Moriarty, N. W., Read, R. J., Richardson, D. C., Richardson, J. S., Terwilliger, T. C. & Adams, P. D. (2012). J. Appl. Cryst. 45, 581–586. [DOI] [PMC free article] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Ferrone, F. A. (2004). Microcirculation, 11, 115–128. [DOI] [PubMed]
- Ghatge, M. S., Ahmed, M. H., Omar, A. S. M., Pagare, P. P., Rosef, S., Kellogg, G. E., Abdulmalik, O. & Safo, M. K. (2016). J. Struct. Biol. 194, 446–450. [DOI] [PMC free article] [PubMed]
- Godfrey, V. B., Chen, L. J., Griffin, R. J., Lebetkin, E. H. & Burka, L. T. (1999). J. Toxicol. Environ. Health A, 57, 199–210. [DOI] [PubMed]
- Harrington, D. J., Adachi, K. & Royer, W. E. (1997). J. Mol. Biol. 272, 398–407. [DOI] [PubMed]
- Jenkins, J. D., Musayev, F. N., Danso-Danquah, R., Abraham, D. J. & Safo, M. K. (2009). Acta Cryst. D65, 41–48. [DOI] [PubMed]
- Jensen, F. B. (2009). J. Exp. Biol. 212, 3387–3393. [DOI] [PubMed]
- Kato, G. J., Gladwin, M. T. & Steinberg, M. H. (2007). Blood Rev. 21, 37–47. [DOI] [PMC free article] [PubMed]
- Kato, G. J., Lawrence, M. P., Mendelsohn, L. G., Saiyed, R., Wang, X., Conrey, A. K., Starling, J. M., Grimes, G., Taylor, J. G., McKew, J., Minniti, C. P. & Stern, W. (2013). Blood, 122, 1009.
- Metcalf, B. et al. (2017). ACS Med. Chem. Lett. 8, 321–326. [DOI] [PMC free article] [PubMed]
- Nagel, R. L., Johnson, J., Bookchin, R. M., Garel, M. C., Rosa, J., Schiliro, G., Wajcman, H., Labie, D., Moo-Penn, W. & Castro, O. (1980). Nature (London), 283, 832–834. [DOI] [PubMed]
- Nnamani, I. N., Joshi, G. S., Danso-Danquah, R., Abdulmalik, O., Asakura, T., Abraham, D. J. & Safo, M. K. (2008). Chem. Biodivers. 5, 1762–1769. [DOI] [PubMed]
- Oder, E., Safo, M. K., Abdulmalik, O. & Kato, G. J. (2016). Br. J. Haematol. 175, 24–30. [DOI] [PMC free article] [PubMed]
- Oksenberg, D., Dufu, K., Patel, M. P., Chuang, C., Li, Z., Xu, Q., Silva-Garcia, A., Zhou, C., Hutchaleelaha, A., Patskovska, L., Patskovsky, Y., Almo, S. C., Sinha, U., Metcalf, B. W. & Archer, D. R. (2016). Br. J. Haematol. 175, 141–153. [DOI] [PubMed]
- Pagare, P. P., Ghatge, M. S., Musayev, F. N., Deshpande, T. M., Chen, Q., Braxton, C., Kim, S., Venitz, J., Zhang, Y., Abdulmalik, O. & Safo, M. K. (2018). Bioorg. Med. Chem. 26, 2530–2538. [DOI] [PMC free article] [PubMed]
- Parikh, A. & Venitz, J. (2014). Clin. Pharmacol. Ther. 95, S83–S84.
- Pauling, L., Itano, H. A., Singer, S. J. & Wells, I. C. (1949). Science, 110, 543–548. [DOI] [PubMed]
- Perutz, M. F. (1972). Nature (London), 237, 495–499. [DOI] [PubMed]
- Perutz, M. F. (1978). Sci. Am. 239, 92–125. [DOI] [PubMed]
- Perutz, M. F., Wilkinson, A. J., Paoli, M. & Dodson, G. G. (1998). Annu. Rev. Biophys. Biomol. Struct. 27, 1–34. [DOI] [PubMed]
- Poillon, W. N. & Kim, B. C. (1990). Blood, 76, 1028–1036. [PubMed]
- Poillon, W. N., Kim, B. C., Welty, E. V. & Walder, J. A. (1986). Arch. Biochem. Biophys. 249, 301–305. [DOI] [PubMed]
- Rees, D. C., Williams, T. N. & Gladwin, M. T. (2010). Lancet, 376, 2018–2031. [DOI] [PubMed]
- Rhoda, M.-D., Martin, J., Blouquit, Y., Garel, M.-C., Edelstein, S. J. & Rosa, J. (1983). Biochem. Biophys. Res. Commun. 111, 8–13. [DOI] [PubMed]
- Rogers, S. C., Ross, J. G. C., d’Avignon, A., Gibbons, L. B., Gazit, V., Hassan, M. N., McLaughlin, D., Griffin, S., Neumayr, T., DeBaun, M., DeBaun, M. R. & Doctor, A. (2013). Blood, 121, 1651–1662. [DOI] [PMC free article] [PubMed]
- Rolan, P. E., Mercer, A. J., Wootton, R. & Posner, J. (1995). Br. J. Clin. Pharmacol. 39, 375–380. [DOI] [PMC free article] [PubMed]
- Rolan, P. E., Parker, J. E., Gray, S. J., Weatherley, B. C., Ingram, J., Leavens, W., Wootton, R. & Posner, J. (1993). Br. J. Clin. Pharmacol. 35, 419–425. [DOI] [PMC free article] [PubMed]
- Safo, M. K., Abdulmalik, O., Danso-Danquah, R., Burnett, J. C., Nokuri, S., Joshi, G. S., Musayev, F. N., Asakura, T. & Abraham, D. J. (2004). J. Med. Chem. 47, 4665–4676. [DOI] [PubMed]
- Safo, M. K. & Abraham, D. J. (2003). Methods Mol. Med. 82, 1–19. [DOI] [PubMed]
- Safo, M. K., Ahmed, M. H., Ghatge, M. S. & Boyiri, T. (2011). Biochim. Biophys. Acta, 1814, 797–809. [DOI] [PubMed]
- Safo, M. K. & Bruno, S. (2011). Chemistry and Biochemistry of Oxygen Therapeutics: From Transfusion to Artificial Blood, edited by A. Mozzarelli & S. Bettati, pp. 285–300. Chichester: John Wiley & Sons.
- Safo, M. K. & Kato, G. J. (2014). Hematol. Oncol. Clin. North Am. 28, 217–231. [DOI] [PMC free article] [PubMed]
- Sedrak, A. & Kondamudi, N. P. (2018). StatPearls. Treasure Island: StatPearls Publishing.
- Silva, M. M., Rogers, P. H. & Arnone, A. (1992). J. Biol. Chem. 267, 17248–17256. [PubMed]
- Stern, W., Mathews, D., McKew, J., Shen, X. & Kato, G. J. (2012). Blood, 120, 3210.
- Thein, M. S., Igbineweka, N. E. & Thein, S. L. (2017). Pathology, 49, 1–9. [DOI] [PMC free article] [PubMed]
- Ware, R. E., de Montalembert, M., Tshilolo, L. & Abboud, M. R. (2017). Lancet, 390, 311–323. [DOI] [PubMed]
- Watowich, S. J., Gross, L. J. & Josephs, R. (1993). J. Struct. Biol. 111, 161–179. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Zhang, Y. et al. (2014). J. Clin. Invest. 124, 2750–2761. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: carbonmonoxy hemoglobin in complex with TD-7, 6di4
Synthesis of compounds and Supplementary Figures.. DOI: 10.1107/S2059798318009919/ag5018sup1.pdf