

Abstract
Interleukin-2 (IL-2) has been used to treat diseases ranging from cancer to autoimmune disorders, but its concurrent immunostimulatory and immunosuppressive effects hinder efficacy. IL-2 orchestrates immune cell function through activation of a high-affinity heterotrimeric receptor (comprised of IL-2 receptor-α [IL-2Rα], IL-2Rβ, and common γ [γc]). IL-2Rα, which is highly expressed on regulatory T (TReg) cells regulates IL-2 sensitivity. Previous studies have shown that complexation of IL-2 with the JES6–1 antibody preferentially biases cytokine activity toward TReg cells through a unique mechanism whereby IL-2 is exchanged from the antibody to IL-2Rα. However, clinical adoption of a mixed antibody-cytokine complex regimen is limited by stoichiometry and stability concerns. Here, through structure-guided design, we engineered a single agent fusion of the IL-2 cytokine and JES6–1 antibody that, despite being covalently linked, preserves IL-2 exchange, selectively stimulating TReg expansion, and exhibiting superior disease control to the mixed IL-2/JES6–1 complex in a mouse colitis model. These studies provide an engineering blueprint for resolving a major barrier to the implementation of functionally similar IL-2/antibody complexes for treatment of human disease.
INTRODUCTION
Interleukin-2 (IL-2) is a pleiotropic cytokine that orchestrates the proliferation, survival, and function of both immune effector cells and regulatory T (TReg) cells to maintain immune homeostasis. IL-2 signals through activation of either a high-affinity (~100 pM) heterotrimeric receptor (composed of IL-2 receptor-α [IL-2Rα], IL-2Rβ, and the shared common gamma [γc]) or an intermediate-affinity (~1 nM) heterodimeric receptor (composed of only the IL-2Rβ and γc chains) (1–3). Consequently, IL-2 sensitivity is dictated by the non-signaling IL-2Rα chain, which is abundantly expressed on the surface of TReg cells, but virtually absent from naïve immune effector cells (i.e. natural killer [NK] cells and memory phenotype [MP] CD8+ T cells) (1, 2, 4). Formation of the IL-2 cytokine-receptor complex leads to activation of intracellular Janus kinase (JAK) proteins, which are constitutively associated with IL-2Rβ and γc. JAK proteins phosphorylate key tyrosine residues in the receptor intracellular domains, leading to recruitment and activation of signal transducer and activator of transcription (STAT)-5 to effect immune-related gene expression and regulate functional outcomes (1, 5, 6).
Due to its essential role in the differentiation and growth of TReg cells, the IL-2 cytokine has been extensively characterized in pre-clinical models to treat a range of autoimmune diseases, including diabetes and multiple sclerosis. These models have underlined the need to administer low doses of the cytokine to take advantage of the enhanced IL-2 sensitivity of TReg over effector cells (7, 8). More recently, proof-of-concept clinical trials backed by mechanistic studies have demonstrated that low-dose IL-2 therapy specifically activates and expands TReg cells to ameliorate autoimmune pathologies (9–11). However, careful dose titration is required for these studies and the off-target activation of effector cells (particularly activated cells with upregulated IL-2Rα expression) remains of concern.
Boyman and colleagues demonstrated that treating mice with complexes of IL-2 with the anti-IL-2 antibody JES6–1 biases cytokine activity toward TReg cells to orchestrate an immunosuppressive response (12), offering an exciting opportunity for targeted autoimmune disease therapy (13). Subsequent work has demonstrated that IL-2/JES6–1 complexes prevent development of autoimmune diseases (14–17) and promote graft tolerance (18, 19) in mice. We recently determined the molecular structure of the IL-2/JES6–1 complex to elucidate the mechanistic basis for its selective stimulation of TReg over effector cells. JES6–1 sterically obstructs IL-2 interaction with the IL-2Rβ and γc subunits to block signaling on IL-2RαLow effector cells, but also undergoes a unique allosteric exchange mechanism with the IL-2Rα subunit, wherein surface-expressed IL-2Rα displaces the JES6–1 antibody and liberates the cytokine to signal through the high-affinity heterotrimeric receptor on IL-2RαHigh TReg cells (Fig. 1a). This phenomenon occurs because key residues in the IL-2 AB interhelical loop engage the JES6–1 antibody and the IL-2Rα subunit in distinct orientations; thus, IL-2-antibody and IL-2-receptor binding are mutually exclusive, leading to bidirectional exchange. Activation of the IL-2 signaling pathway on IL-2RαHigh cells further upregulates IL-2Rα expression to create a positive feedback loop that exquisitely favors TReg expansion (17).
Figure 1. Unique antibody-receptor exchange mechanism underlies TReg bias of mixed IL-2/JES6–1 complex.
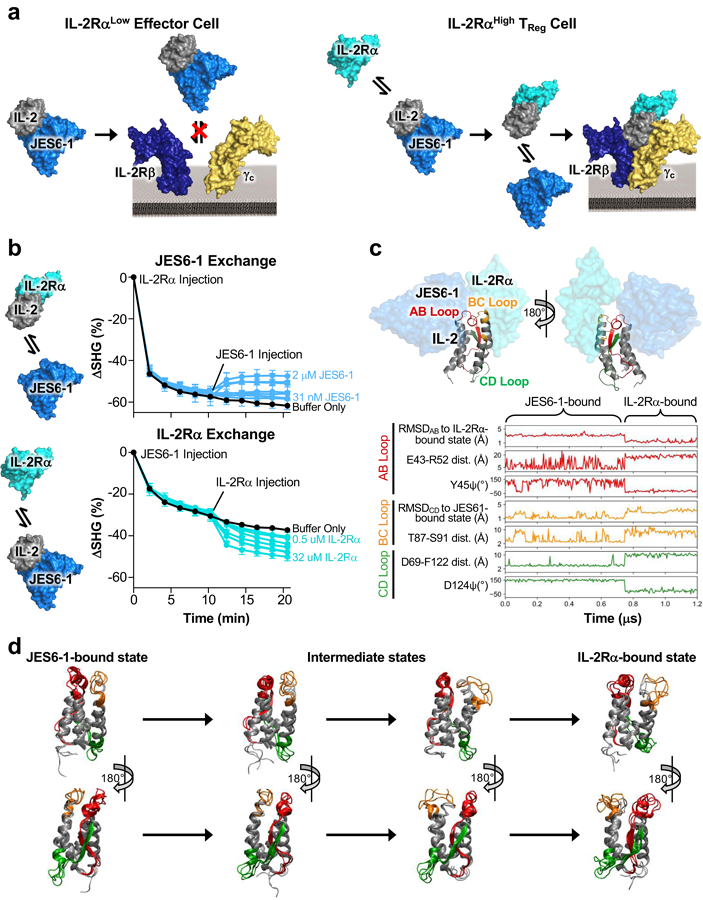
(a) Schematic of the mechanistic rationale for IL-2/JES6–1 complex-mediated selective potentiation of TReg cells. The JES6–1 antibody (shown in single-chain format) sterically obstructs IL-2 engagement of the IL-2Rβ and γc subunits, preventing activation of IL-2RαLow effector cells (left). However, allosteric exchange between JES6–1 and the IL-2Rα subunit allows for exclusive signaling on IL-2RαHigh TRegs, biasing toward an immunosuppressive response (right). (b) IL-2 was immobilized and 500 nM IL-2Rα (top) or 500 nM JES6–1 antibody (bottom) was injected at time 0 min. After 10 minutes, various concentrations of JES6–1 antibody ranging from 31 nM to 2 μM (top) or various concentrations of IL-2Rα ranging from 0.5 μM to 32 μM (bottom) were added and second-harmonic generation signal change (ΔSHG) was monitored. Exchange schemes are shown at left. (c) Molecular structure of the IL-2 cytokine bound to JES6–1 (PDB ID 4YQX) (17) overlaid with the IL-2Rα subunit from the IL-2 cytokine-receptor quaternary complex structure (PDB ID 2B5I) (3), highlighting the AB (red), BC (orange), and CD (green) interhelical loops of the cytokine (top). Molecular dynamics simulations of free IL-2 were conducted starting from the cytokine’s conformations in the crystallographic structures of IL-2 bound to the JES6–1 and S4B6 antibodies and the IL-2Rα subunit. Root mean square deviation (RMSD), dihedral angles, and inter-residue distances for the interhelical loops and flanking residues are plotted for a representative transition between the JES6–1-bound and IL-2Rα-bound states (bottom). (d) Overlay of three representative simulated conformations each from the JES6–1-bound state, the intermediate states, and the IL-2Rα-bound state of IL-2 that form the primary transition path, with interhelical regions colored as in (c).
The immunosuppressive effects of IL-2/JES6–1 complexes make them enticing candidates for autoimmune disease treatment in humans, but clinical administration of mixed IL-2/antibody complexes is complicated by logistical challenges in drug formulation including optimization of the dosing ratio and instability of the cytokine/antibody complex. Previously, IL-2 has been covalently linked to an anti-IL-2 antibody to enhance its in vivo half-life and stability (20). However, this approach is incompatible with the allosteric exchange mechanism enacted by the IL-2/JES6–1 complex as tethering IL-2 to the JES6–1 antibody greatly enhances the apparent antibody-cytokine affinity, obstructing the triggered release that is essential for TReg bias. To overcome this obstacle to therapeutic development, we utilized a structure-based engineering strategy to design a single-agent IL-2/JES6–1 fusion that preserves antibody-receptor exchange. Through modulation of the cytokine-antibody affinity, we successfully recapitulated the selective TReg potentiation elicited by mixed IL-2/JES6–1 complex treatment and we demonstrated that our engineered cytokine-antibody fusion controlled autoimmune disease better than the mixed complex in an induced mouse model of colitis. Collectively, our biophysical and functional studies present a mechanism-driven biomolecular engineering approach that enables the therapeutic translation of a cytokine-antibody complex, and that can readily be adapted to other systems for a range of immune disease applications.
MATERIALS AND METHODS
Protein expression and purification.
The sequence encoding hexahistidine-tagged mouse IL-2 (mIL-2, amino acids 1–149) was cloned into the pMal vector with an N-terminal maltose-binding protein (MBP) followed by a 3C protease site. mIL-2 was expressed in the periplasm of BL21(DE3) Escherichia coli cells by 20 h induction at 22°C with 1 mM isopropyl β-d-thiogalactopyranoside (IPTG). Protein in the periplasmic compartment was isolated by osmotic shock and purified by nickel-nitrilotriacetic acid (Ni-NTA) (Qiagen) affinity chromatography. Purity was improved via size-exclusion chromatography on a Superdex-75 column (GE Healthcare) in HEPES-buffered saline (HBS, 150 mM NaCl in 10 mM HEPES pH 7.3).
mIL-2Rα (amino acids 1–213) ectodomain, mIL-2Rβ ectodomain (amino acids 1–215), and mγc ectodomain (amino acids 34–233) were secreted and purified using a baculovirus expression system, as previously described (3). All proteins were purified to >98% homogeneity with a Superdex 200 sizing column (GE Healthcare) equilibrated in HBS. Purity was verified by SDS-PAGE analysis.
Recombinant JES6–1 antibody and immunocytokine and mutants thereof were co-expressed using the previously described BacMam technique (21) adapted for 293F human embryonic kidney cells (Thermo Life Technologies). For JES6–1 antibody and variants thereof, the JES6–1 variable heavy (VH) chain followed by the rat immunoglobulin (IgG) 2a constant domains was cloned into the pVLAD6 vector with a C-terminal hexahistidine tag. The JES6–1 variable light (VL) chain followed by the rat kappa light chain constant domain was separately cloned into the pVLAD6 vector with with a C-terminal hexahistidine tag. For immunocytokine constructs, mIL-2 was cloned N-terminal to the VL domain of the light chain construct, spaced by a (Gly4Ser)2 linker. Heavy and light chain constructs were separately transfected into Spodoptera frugiperda insect (SF9) cells as previously described (21) and the resulting viral supernatants were used to infect 293F cells (Thermo Life Technologies) in the presence of 10 mM sodium butyrate (Sigma). Heavy and light chain viruses were co-titrated to determine optimal infection ratios for equivalent expression of the two chains. Infected 293F cells were harvested after 72 hours and secreted protein was captured from the supernatant via Ni-NTA (Qiagen) affinity chromatography. Proteins were further purified to >98% homogeneity with a Superdex 200 sizing column (GE Healthcare) equilibrated in HBS, and purity was confirmed by SDS-PAGE analysis. Rat IgG2a isotype control antibody (Clone eBR2a) was purchased commercially (eBioscience).
For expression of biotinylated mouse IL-2 and mouse IL-2 receptor subunits, proteins containing a C-terminal biotin acceptor peptide (BAP)-LNDIFEAQKIEWHE were expressed and purified via Ni-NTA affinity chromatography and then biotinylated with the soluble BirA ligase enzyme in 0.5 mM Bicine pH 8.3, 100 mM ATP, 100 mM magnesium acetate, and 500 mM biotin (Sigma). Excess biotin was removed by size exclusion chromatography on a Superdex 200 column equilibrated in HBS. Antibodies and immunocytokines were biotinylated using the EZ-Link Sulfo-NHS-LC-Biotinylation kit (Pierce) according to the manufacturer’s protocol.
Cell Lines.
Unmodified YT-1 (22) and IL-2Rα+ YT-1 human natural killer cells (23) were cultured in RPMI complete medium (RPMI 1640 medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine, minimum non-essential amino acids, sodium pyruvate, 25 mM HEPES, and penicillin-streptomycin [Gibco]) and maintained at 37ºC in a humidified atmosphere with 5% CO2.
The subpopulation of YT-1 cells expressing IL-2Rα was purified via magnetic selection as described previously (24). Ten million unsorted IL-2Rα+ YT-1 cells were washed with FACS buffer (phosphate-buffered saline [PBS] pH 7.2 containing 0.1% bovine serum albumin) and incubated in FACS buffer with PE-conjugated anti-human IL-2Rα antibody (Biolegend, clone BC96) for 2 hr at 4°C. PE-labeled IL-2Rα+ cells were then incubated with paramagnetic microbeads coated with an anti-PE IgG for 20 min at 4º C, washed once with cold FACS buffer, and sorted on an LS MACS separation column (Miltenyi Biotec) according to the manufacturer’s protocol. Purified eluted cells were re-suspended and grown in RPMI complete medium. Enrichment of IL-2Rα+ cells was evaluated using an Accuri C6 flow cytometer (BD Biosciences) and persistence of IL-2Rα expression was monitored by PE-conjugated anti-human IL-2Rα antibody labeling and flow cytometric analysis of sorted IL-2Rα+ YT-1 cells.
Second harmonic generation antibody-receptor exchange studies.
For cytokine labeling, hexahistidine-tagged mIL-2 was exchanged into labeling buffer (50 mM HEPES pH 8.2, 150 mM NaCl) with a 0.5 mL, 7000 molecular weight cut-off Zeba™ Spin Desalting Column (Thermo). A five-fold molar excess of lysine-reactive (succinimidyl ester chemistry) second harmonic-active dye SHG1-SE (25) (Biodesy, Inc.) was added to the protein. The reaction proceeded on ice for 5 min and was then stopped by removal of unreacted SHG1-SE by buffer exchange into HBS with a 0.5 mL, 7000 molecular weight cut-off Zeba™ Spin Desalting Column (Thermo). The reacted protein was centrifuged at 16,000×g at 4 °C for 20 minutes to pellet any precipitate and the supernatant was analyzed by UV-Vis spectroscopy. The degree of labeling was determined to be 1 (dye:protein stoichiometry).
For target immobilization, Ni-NTA lipid-containing small unilamellar vesicles (SUVs) were generated in tris-buffered saline (TBS, pH 7.6) from the Ni-NTA Bilayer Surface reagent (Biodesy, Inc) according to the manufacturer’s protocol. Supported lipid bilayers were formed by fusion of the Ni-NTA SUVs on the glass surface of a 384-well Biodesy Delta plate. The formed bilayer was washed into HBS. SHG1-SE labeled hexahistidine-tagged mIL-2 was added to each well at 500 nM final concentration (20 uL well volume). The protein was allowed to attach to the surface through the Ni-NTA:Hexahistidine-tag interaction overnight at 4°C. The plate was then equilibrated to room temperature for 30 min and unbound protein was washed out. Following 20 min incubation at room temperature, the plate was transferred to the Biodesy Delta for data collection.
To assess JES6–1 exchange, 500 nM IL-2Rα was injected at time t=0 and second harmonic generation (SHG) signal was monitored for 10 minutes. Two-fold serial dilutions of JES6–1 antibody ranging from 2 μM to 31 nM in assay buffer containing 500 nM IL-2Rα (to keep the receptor concentration constant) were injected and percent SHG signal change (ΔSHG) was tracked for 10 min. To assess IL-2Rα exchange, 500 nM JES6–1 was injected at time t=0 and SHG signal was monitored for 10 minutes. Two-fold serial dilutions of IL-2Rα ranging from 32 μM to 0.5 μM in assay buffer containing 500 nM JES6–1 (to keep the antibody concentration constant) were injected and ΔSHG was tracked for 10 min. The percent change in SHG intensity was calculated as (SHGF-SHGB)/SHGB × 100, where SHGB is the SHG signal at t=0 and SHGF is the SHG signal at each time point post-injection.
Molecular dynamics simulations.
Atomistic molecular dynamics simulations of mIL-2 were performed using the Gromacs 5.0.4 package (26) with the Amber99SB-ILDN force field (27) and the TIP3P water model (28). Initial conformations were obtained from the crystal structures of the mIL-2:JES6–1 scFv (PDB ID 4YQX), mIL-2:S4B6 Fab (PDB ID 4YUE), and hIL-2:hIL-2Rα (PDB ID: 1Z92) complexes, with the binding partner omitted. The Modeller 9.14 package (29) was used to mutate the hIL-2 conformation to the mouse sequence and to build crystallographically-unresolved loops, with five predicted configurations for each crystal structure. Starting conformations of mIL-2 were positioned in a dodecahedron box with a minimum of 10 Å between the protein’s surface and the edge of the box. The protein was solvated with approximately 8400 water molecules; five sodium ions were added to neutralize the charge. Energy minimization was performed using a steepest descent algorithm, and solvent was equilibrated for 500 ps at 300 K and 1 bar with the positions of the protein atoms held fixed. Production simulations were performed using a 2 fs timestep, with trajectory lengths of 150 ns and an aggregate simulation time of 19.4 μs. Constant temperature and pressure were maintained by applying a velocity-rescaling thermostat (30) and a Parrinello-Rahman barostat (31). Bonds were constrained using the LINCS algorithm (32), and electrostatic interactions were treated with the particle mesh Ewald method (33). Adaptive sampling was performed by reseeding simulations from poorly sampled regions of the mIL-2 conformational landscape; conformations in these regions were identified by time-structure independent component analysis (34), an unbiased method to determine the slowest degrees of conformational freedom.
A Markov State model (MSM) with a lag-time of 5 ns was constructed from the simulation data using the MSMBuilder 2.8.2 package (35). Similar to prior analysis of IL-2 simulations (24), conformations were clustered into 50 states using a hybrid k-centers k-medoids algorithm, with distances between all pairs of conformations determined from the root mean square deviation (RMSD) of the backbone atom positions. A representative trajectory of the predicted equilibrium dynamics was constructed using kinetic Monte Carlo sampling of the MSM transition probability matrix. Inter-residue distances, dihedral angles, and RMSD were computed using MDTraj 1.7.2 (36).
Surface plasmon resonance studies.
For IL-2 affinity titration studies, biotinylated mouse IL-2Rα and IL-2Rβ receptors, biotinylated JES6–1 antibody mutants, or biotinylated JES6–1 IC mutants were immobilized to streptavidin-coated chips for analysis on a Biacore T100 instrument (GE Healthcare). An irrelevant biotinylated protein was immobilized in the reference channel to subtract non-specific binding. Less than 100 response units (RU) of each ligand was immobilized to minimize mass transfer effects. Three-fold serial dilutions of mIL-2 were flowed over the immobilized ligands for 60 s and dissociation was measured for 240 s. Surface regeneration for all interactions was conducted using 15 s exposure to 1 M MgCl2 in 10 mM sodium acetate pH 5.5.
For immunocytokine receptor exchange studies, biotinylated mIL-2Rα or mIL-2Rβ receptors were immobilized to streptavidin-coated chips for analysis on a Biacore T100 instrument (GE Healthcare). An irrelevant biotinylated protein was immobilized in the reference channel to subtract non-specific binding. Less than 100 response units (RU) of each ligand was immobilized to minimize mass transfer effects. Three-fold serial dilutions of mIL-2 or JES6–1 IC mutants were flowed over the immobilized ligands for 60 s and dissociation was measured for 240 s. Surface regeneration for all interactions was conducted using 15 s exposure to 1 M MgCl2 in 10 mM sodium acetate pH 5.5.
SPR experiments were carried out in HBS-P+ buffer (GE Healthcare) supplemented with 0.2% bovine serum albumin (BSA) at 25ºC and all binding studies were performed at a flow rate of 50 μL/min to prevent analyte rebinding. Data was visualized and processed using the Biacore T100 evaluation software version 2.0 (GE Healthcare). Equilibrium titration curve fitting and equilibrium binding dissociation (KD) value determination was implemented using GraphPad Prism assuming all binding interactions to be first order.
YT-1 cell STAT5 phosphorylation studies.
Approximately 2×105 YT-1 or IL-2Rα+ YT-1 cells were plated in each well of a 96-well plate and re-suspended in RPMI complete medium containing serial dilutions of mIL-2, mIL-2/antibody complexes, JES6–1 IC, or JES6–1 IC mutants. Complexes were formed by incubating a 1:1 molar ratio of antibody or antibody fragment to mIL-2 for 30 min at room temperature. Cells were stimulated for 15 min at 37ºC and immediately fixed by addition of formaldehyde to 1.5% and 10 min incubation at room temperature. Permeabilization of cells was achieved by resuspension in ice-cold 100% methanol for 30 min at 4ºC. Fixed and permeabilized cells were washed twice with FACS buffer (phosphate-buffered saline [PBS] pH 7.2 containing 0.1% bovine serum albumin) and incubated with Alexa Fluor® 647-conjugated anti-STAT5 pY694 (BD Biosciences) diluted in FACS buffer for 2 hr at room temperature. Cells were then washed twice in FACS buffer and MFI was determined on a CytoFLEX flow cytometer (Beckman-Coulter). Dose-response curves were fitted to a logistic model and half-maximal effective concentrations (EC50s) were calculated using GraphPad Prism data analysis software after subtraction of the mean fluorescence intensity (MFI) of unstimulated cells and normalization to the maximum signal intensity. Experiments were conducted in triplicate and performed three times with similar results.
Immune cell subset proliferation studies.
For relative TReg:CD8+/CD4+ T cell proliferation studies, 12 weeks old C57BL/6 mice (3 per cohort) or NOD mice (4 per cohort) were injected i.p. with PBS, mixed IL-2/JES6–1 complex (prepared by pre-incubating 1.5 μg mIL-2 [eBioscience] with 6 μg JES6–1 [2:1 cytokine:antibody molar ratio] in PBS for 30 mins), or 6 μg of the indicated JES6–1 IC mutants on days 1, 2, 3 and 4. Mice were sacrificed on day 5 by cervical dislocation and spleens were harvested. Single-cell suspensions were prepared by mechanical homogenization and absolute count of splenocytes was assessed for each spleen by automated cell counter (Vicell, Beckman Coulter). Cells were resuspended in PBS and subsequently stained for 30 min on ice with fluorophore-conjugated anti-mouse monoclonal antibodies (mAbs) for phenotyping of TReg (CD4+IL-2Rα+Foxp3+) or CD8+ effector T cells (CD8+) using BV605-conjugated anti-CD4 (Biolegend, clone RM4–5), PeCy7-conjugated anti-IL-2Rα (eBioscience, clone PC61.5), PerCP-Cy5.5-conjugated anti-CD8 (eBioscience, clone 53–6.72), and mAbs. Fixable Blue Dead Cell Stain Kit (Life Techonologies) was used to assess live cells. Cells were then washed twice with FACS buffer (1% BSA, 1% Sodium Azide) and fixed in Foxp3 Transcription Factor Fixation/Permeabilization Buffer (eBioscience) for 30 mins on ice. After two washes in Permeabilization Buffer (eBioscience), TReg cells were stained with FITC-conjugated anti-mouse/rat Foxp3 mAb (eBioscience, clone FJK-16s) for 1 hour on ice. Two final washes were conducted in Permeabilization Buffer and cells were resuspended in FACS buffer for flow cytometric analysis on an LSRII (BD Biosciences). Data were analyzed using FlowJo X software (Tree Star). Ratios of the absolute numbers of TReg cells to either CD8+ effector T cells or total CD4+ T cells are presented. Statistical significance was determined by two-tailed unpaired Student’s t-test. Experiments were performed three times with similar results.
Adoptive T cell transfer studies.
OT-I Ly 5.1+ mice (3 per cohort) were sacrificed by cervical dislocation and lymph nodes (head, axillary, inquinal and mesenteric) were harvested. Single-cell suspensions were prepared by homogenization (GentleMACS Dissociator, Miltenyi Biotec) and CD8+ effector T cells were negatively sorted on AutoMACS (Miltenyi Biotec). Isolated CD8+ T cells were labeled with carboxyfluorescein succinimidyl ester (CFSE) and injected i.v. into C57BL/6 mice (Ly 5.2) (1×106 cells/mouse). Mice were then treated i.p. on day 1 with PBS or with 2 nmol SIINFEKL peptide alone or in combination with 75 μg polyI:C7, 7.5 μg mIL-2 plus 30 μg Rat IgG2a isotype control antibody (BioXcell, Clone 2A3), mixed mIL-2/JES6–1 complex (formed by pre-incubating 7.5 μg mIL-2 [Peprotech] with 30 μg JES6–1 [2:1 cytokine:antibody molar ratio] in PBS for 15 min), or 30 μg JY3 immunocytokine. On day 2, 3, and 4, mice were treated with the same doses of mIL-2 plus Rat IgG2a isotype control antibody, mixed mIL-2/JES6–1 complex, or JY3 immunocytokine. Mice were sacrificed on day 5 by cervical dislocation and spleens were harvested, homogenized, and analyzed by flow cytometry as described for Immune cell subset proliferation studies. Four immune populations were distinguished and profiled for IL-2Rα expression using the following fluorophore-conjugated anti-mouse mAbs: adoptively transferred CD8+ T cells - V500 Horizon®-conjugated anti-CD3 (BD Biosciences, clone 500A2), PerCP-Cy5.5-conjugated anti-CD8 (eBioscience, clone 53–6.72), APC-conjugated anti-CD45.1 (eBioscience, clone A20), and eFluor 450®-conjugated anti-IL-2Rα, TRegs (CD3+CD4+Foxp3+) - V500 Horizon®-conjugated anti-CD3 (BD Biosciences, clone 500A2), PerCP-conjugated anti-CD4 (BD Biosciences, clone RM4–5), PE-conjugated anti-mouse/rat Foxp3 (eBioscience, clone FJK-16s), and APC-conjugated anti-IL-2Rα mAbs (eBioscience, clone PC61.5); MP CD8+ T cells (CD3+CD8+CD44+IL-2Rβ+) - V500 Horizon®-conjugated anti-CD3, PerCP-Cy5.5-conjugated anti-CD8 (eBioscience, clone 53–6.72), APC-conjugated anti-CD44 (eBioscience, clone IM7), PE-conjugated anti-IL-2Rβ (eBioscience, clone 5H4), and eFluor 450®-conjugated anti-IL-2Rα (eBioscience, clone PC61.5) mAbs; and NK cells (CD3−CD49b+CD161) - V500 Horizon®-conjugated anti-CD3, PE-conjugated anti-CD49b (eBioscience, clone DX5), APC-conjugated anti-CD161 (eBioscience, clone PK136), and eFluor 450®-conjugated anti-IL-2Rα mAbs. Fixable Viability Dye eFluor™ 780 (eBioscience) was used to assess live cells. Data were analyzed using FlowJo X software (Tree Star). Relative number of cells and mean fluorescence intensity (MFI) of IL-2Rα are presented for each cohort in all four immune cell subsets. Statistical significance was determined by two-tailed unpaired Student’s t-test. The experiment was performed three times with similar results.
Mouse dextran sodium sulfate (DSS)-induced colitis model.
BALB/c mice (6 per cohort) were injected i.p. daily for seven days with PBS, 7.5 μg mIL-2 plus 30 μg isotype control antibody (BioXcell, clone 2A3), mixed mIL-2/JES6–1 complex (formed by pre-incubating 7.5 μg mIL-2 [Peprotech] with 30 μg JES6–1 [2:1 cytokine:antibody molar ratio] in PBS for 15 min), or 30 μg JY3 immunocytokine. Beginning on day 8, mice were administered 3% DSS (molecular weight=40000, MP Biomedicals Inc.) in their drinking water to induce colitis. On day 15, body weight was recorded and disease severity was assessed using clinical disease activity index, as described previously (4, 37). On day 16, mice were sacrificed and entire colons were removed (from cecum to anus). Colon length was measured and shortening was used as an indirect marker of pathological inflammation. Statistical significance was determined by one-way ANOVA + Dunnett’s multiple comparison post-test. The experiment was performed two times with similar results.
RESULTS
IL-2 undergoes bidirectional exchange between the JES6–1 antibody and the IL-2Rα receptor subunit.
The aforementioned allosteric exchange mechanism allows for displacement of JES6–1 in the cytokine/antibody complex by the surface-bound IL-2Rα receptor subunit (Fig. 1a). This mechanism was supported by structural and surface plasmon resonance (SPR)-based studies (17). To demonstrate the bidirectionality of the antibody-receptor exchange mechanism, we interrogated the capacity of both antibody and receptor to engage bound mouse IL-2 complexes. To this end, we used a second-harmonic generation (SHG) detection platform, which was previously used to detect conformational changes in proteins in time and space (25, 38, 39). IL-2 was labeled with a second-harmonic-active dye and immobilized to a surface. The tethered cytokine was then saturated with either mouse IL-2Rα (Fig. 1b, top) or JES6–1 (Fig. 1b, bottom). Subsequently, various concentrations of soluble JES6–1 (Fig. 1b, top) or IL-2Rα (Fig. 1b, bottom) were added and changes in SHG signal, indicative of modulations in average tilt angles of the dye particles conjugated to IL-2, were quantified. In both topologies, dose-dependent conformational changes were observed in IL-2 upon adding soluble protein to the immobilized complex, demonstrating the bidirectional exchange between antibody and receptor engagement of the cytokine.
To further corroborate the allosteric exchange mechanism, we performed molecular dynamics simulations to study the distinct conformational states of IL-2 when bound to the JES6–1 antibody versus the IL-2Rα receptor, as well as the transition between these states. IL-2, and in particular the IL-2Rα-binding epitope of the cytokine, is known to exhibit extensive conformational flexibility (40–42). We constructed an atomically-detailed Markov State model (MSM) of the conformational landscape of free IL-2. The equilibrium dynamics captured by the MSM predicted that IL-2 stably adopts a JES6–1-bound conformation even in the absence of antibody but occasionally relaxes to a distinct metastable state that resembles the IL-2Rα-bound conformation (Fig. 1c, Supplemental Video 1). The antibody-bound and receptor-bound states of the cytokine diverge significantly with respect to root mean square deviation (RMSD), inter-residue distances, and residue-specific dihedral angles in all three interhelical loops (Fig. 1c). The transition from the JES6–1-bound to the IL-2Rα-bound states involves significant conformational rearrangements and, in particular, destabilization of a salt bridge and a hydrogen bond in the AB and BC loops, respectively, that appear to rigidify these regions (Fig. 1c, red and orange). These changes coincide with the loss of a cation-pi interaction between the B helix and the beta strand of the CD region, accompanied by increased flexibility of the latter (Fig. 1c, green). Inspection of the primary transition path with higher temporal resolution suggests that loss of loop rigidity occurs in sequential fashion. Deformation of the BC loop, which interacts with IL-2Rα, is predicted to precede destabilization of the AB loop, which engages IL-2Rα at its C-terminal end and JES6–1 at its N-terminal end (Fig. 1d). Such a stepwise transition may facilitate allosteric exchange between the JES6–1 antibody and IL-2Rα subunit (17). Taken together, our biophysical and computational studies offer mechanistic insight into the antibody-receptor exchange that drives the TReg cell bias induced by stimulation with the mixed IL-2/JES6–1 complex.
Design of a single-agent cytokine-antibody fusion.
To stabilize the IL-2/JES6–1 complex with an eye toward translation, we fused the IL-2 cytokine to the full-length JES6–1 antibody, tethering IL-2 to the N-terminal end of the light chain via a flexible (Gly4Ser)2 linker (Fig. 2a). Based on the IL-2/JES6–1 complex structure (17), the C-terminus of IL-2 is predicted to be 19.9 Å from the N-terminus of the JES6–1 light chain (Supplemental Fig. 1a). Our cytokine/antibody construction (hereafter denoted the JES6–1 immunocytokine [IC]) was designed to allow for intramolecular cytokine engagement. Interaction between IL-2 and JES6–1 within the immunocytokine was confirmed by SPR-based titrations of the IL-2Rα subunit. Whereas untethered IL-2 binds the IL-2Rα subunit with an equilibrium dissociation constant (KD) of 9.8 nM, JES6–1 IC has a 30-fold weaker IL-2Rα affinity (KD=290 nM) (Fig. 2b), reflective of cytokine sequestering by the tethered antibody.
Figure 2. IL-2/JES6–1 immunocytokine fusion fails to recapitulate TReg-promoting activity of the mixed antibody-cytokine complex.
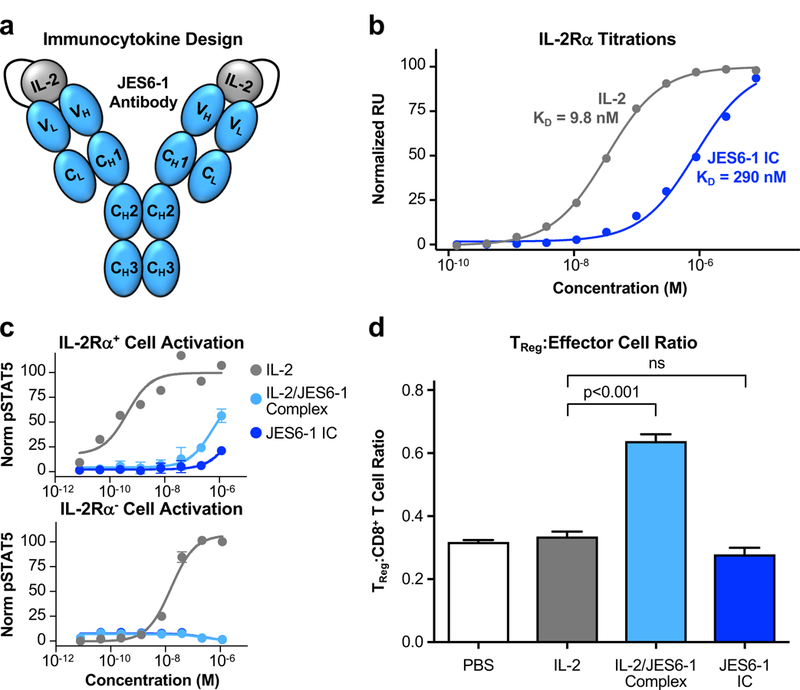
(a) Schematic of the IL-2/JES6–1 single-chain immunocytokine (IC) fusion with the C-terminus of the cytokine tethered to the N-terminus of the antibody light chain via a (Gly4Ser)2 flexible linker. (b) Equilibrium surface plasmon resonance titrations of soluble IL-2 (gray) or JES6–1 IC (blue) binding to immobilized IL-2Rα. Fitted equilibrium dissociation constants (KD) are indicated. (c) STAT5 phosphorylation response (mean ± S.D.) of IL-2Rα+ (top) or IL-2Rα− (bottom) YT-1 human NK cells stimulated with IL-2, IL-2/JES6–1 complex, or JES6–1 IC. (d) Ratio of TReg to CD8+ effector T cell abundance in spleens harvested from non-obese diabetic (NOD) mice (n=4 per cohort) treated with PBS, IL-2, IL-2/JES6–1 complex, or JES6–1 IC for four consecutive days. Data represents mean ± s.d. Statistical significance was determined by two-tailed unpaired Student’s t-test. The experiment was performed three times with similar results.
The IL-2-JES6–1 affinity (KD=5.6 nM) is similar to the IL-2-IL-2Rα affinity (KD=9.8 nM) and significantly stronger than the IL-2-IL-2Rβ affinity (KD=7.4 μM) (Supplemental Fig. 1b). Thus, effective exchange of the IL-2 cytokine between the JES6–1 antibody and the IL-2Rα subunit is observed when the affinities are closely matched. We hypothesized that tethering IL-2 to the JES6–1 antibody would enhance the apparent cytokine-antibody affinity due to avidity effects, and this increased cytokine-antibody affinity would in turn weaken IL-2-IL-2Rα interaction in the context of the IC. We speculated that changes in the antibody-cytokine affinity and, by consequence, the IC-IL-2Rα affinity, would impact on the exchange mechanism in a biphasic manner. If the affinity of the IL-2-JES6–1 complex was greatly reduced, the antibody would fall off constitutively, leading to the cytokine to behave the same as the naked IL-2 and activate both IL-2RαHigh TReg and IL-2RαLow effector cells, thus erasing the robust TReg cell IL-2 signaling bias conferred by JES6–1 (Supplemental Fig. 1c). Conversely, if the affinity of the IL-2-JES6–1 complex was significantly increased, to the limit of an irreversible interaction, the antibody would never be displaced by IL-2Rα, ablating the exchange mechanism and precluding cytokine activity on both TReg and effector cells (Supplemental Fig. 1c). Consequently, there exists an optimal IL-2-antibody affinity to maximize TReg over effector cell expansion, and substantial enhancement of the IL-2-antibody affinity through immunocytokine construction could push this affinity outside of the optimal range.
Parent IL-2-JES6–1 immunocytokine exhibits reduced activation of IL-2RαHigh cells and does not promote TReg expansion in vivo.
To examine the functional consequences of antibody-cytokine tethering on cell subset-selective activity, we tested activation of two genotypically matched cell lines based on the YT-1 human NK lineage that differ only in their expression of the IL-2Rα subunit (23), as a surrogate for stimulation of IL-2RαHigh TReg cells compared to IL-2RαLow effector cells. IL-2 exhibited over 30-fold more potent activation (as measured by STAT5 phosphorylation) on IL-2Rα+ cells compared to IL-2Rα− cells, as was expected due to the higher affinity of the heterotrimeric versus the heterodimeric IL-2 receptor complex. The mixed IL-2/JES6–1 complex induced weaker activation of both cell lines but, importantly, showed more pronounced obstruction of signaling on IL-2Rα− compared to IL-2Rα+ cells, rationalizing the complex’s IL-2RαHigh TReg bias. JES6–1 IC did not activate IL-2Rα− cells and induced much weaker activation of IL-2Rα+ cells compared to the mixed complex, consistent with its impaired interaction with the IL-2Rα subunit (Fig. 2c). We explored how this differential in vitro signaling would translate into in vivo immune cell subset bias. Administration of IL-2 alone to non-obese diabetic (NOD) mice did not induce an increase in TReg relative to CD8+ effector T cell abundance, but treatment with IL-2/JES6–1 complex doubled the TReg:CD8+ T cell ratio. However, this increase was completely absent for JES6–1 IC, indicating that stabilization of the IL-2-antibody affinity had disrupted the exchange mechanism, ablating cytokine activity on both TReg and effector cells (Fig. 2d, Supplemental Fig. 1c). Accordingly, enrichment of TReg in the total CD4+ T cell population was observed following treatment with IL-2/JES6–1 complex treatment but not JES6–1 IC (Supplemental Fig. 1d).
Affinity mutant immunocytokines demonstrate improved exchange and elicit biased IL-2Rα+ cell activation.
To rescue the TReg-biased activity of the immunocytokine, we used crystallographic insights to rationally design a panel of eight single-point alanine mutants of the JES6–1 antibody. We selected four variable heavy (VH) and four variable light (VL) chain residues at the cytokine/antibody interface, intentionally avoiding residues proximal to the IL-2Rα chain to circumvent disruption of the allosteric exchange mechanism (Fig. 3a). We formatted each alanine mutant as a full-length antibody and characterized binding to the IL-2 cytokine via SPR titrations. All mutants with the exception of R62A decreased the antibody-cytokine affinity, with a maximum affinity impairment of 89-fold relative to the parent JES6–1 antibody (Fig. 3b and Table I).
Figure 3. Disruption of antibody-cytokine affinity enhances immunocytokine activity on IL-2Rα+ cells.
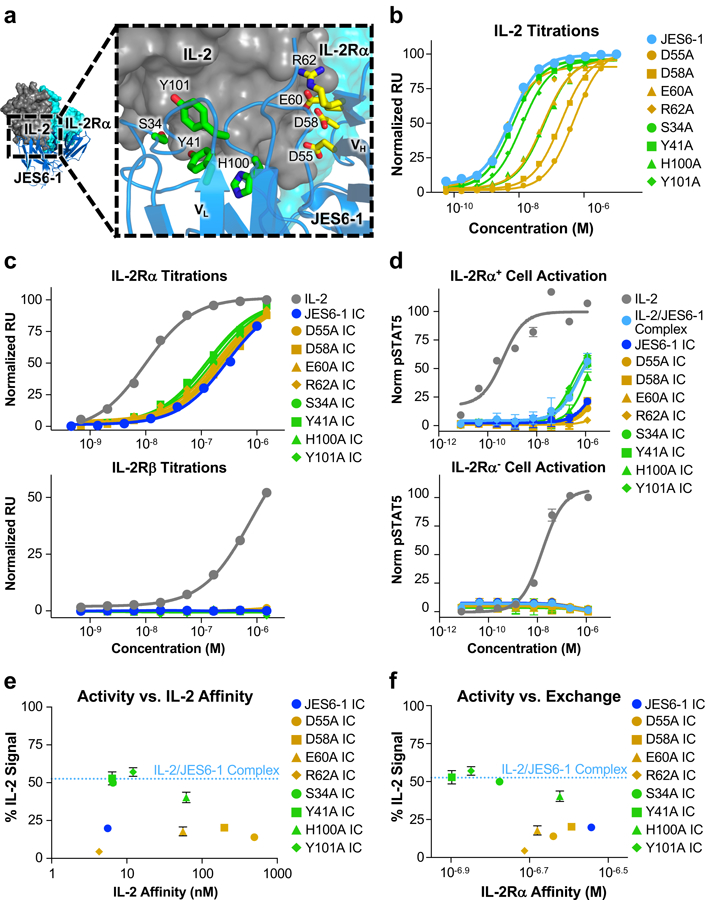
(a) Crystallographic structure of the IL-2/JES6–1 interface (PDB ID 4YQX) (17) with interfacial antibody residues that were mutated to alanine highlighted in yellow (heavy chain) or green (light chain). Human IL-2Rα is overlaid from the IL-2 cytokine-receptor quaternary complex structure for reference (PDB ID 2B5I) (3). (b) Equilibrium surface plasmon resonance titrations of soluble IL-2 binding to immobilized JES6–1 or the indicated antibody variants. (c) Equilibrium surface plasmon resonance titrations of soluble IL-2, JES6–1 IC, or JES6–1 IC mutants binding to immobilized IL-2Rα (top) or IL-2Rβ (bottom). (d) STAT5 phosphorylation response of IL-2Rα+ (top) or IL-2Rα− (bottom) YT-1 human NK cells treated with IL-2, JES6–1 IC, or JES6–1 IC mutants. Data represent mean ± s.d. (e) Comparison of the STAT5 phosphorylation activity of the indicated JES6–1 IC variants (% IL-2-induced signal at 1.2 μM concentration) versus IL-2 affinity of their corresponding antibodies. Activity of the IL-2/JES6–1 complex is indicated by the dashed blue line. Data represent mean ± s.d. (f) Comparison of the STAT5 phosphorylation activity of the indicated JES6–1 IC variants (% IL-2-induced signal at 1.2 μM concentration) to their IL-2Rα affinities (representative of their exchanging propensities). Activity of the IL-2/JES6–1 complex is indicated by the dashed blue line. Data represent mean ± s.d. Heavy chain mutations are colored yellow and light chain mutations are colored green throughout the figure.
Table I. Cytokine affinity and receptor exchange properties of engineered JES6–1 antibody and immunocytokine point mutants.
The JES6–1 antibody chains on which the mutations are located are indicated in parentheses.
| Construct | KD of IL-2/Antibody Complex (nM) | KD of IL-2Rα/IC Complex (nM) |
|---|---|---|
| JES6–1 | 5.6 | 290 |
| D55A (VH) | 500 | 230 |
| D58A (VH) | 200 | 260 |
| E60A (VH) | 56 | 210 |
| R62A (VH) | 4.3 | 190 |
| S34A (VL) | 6.5 | 170 |
| Y41A (VL) | 6.4 | 130 |
| H100A (VL) | 62 | 240 |
| Y101A (VL) | 12 | 140 |
To probe the effects of reduced cytokine/antibody affinity on IL-2Rα exchange, we reformatted each of the alanine mutants as IC fusions and measured the binding of these soluble IC variants to immobilized IL-2Rα using SPR. Each of the eight IC mutants tested exhibited increased receptor affinity compared to the parent JES6–1 IC, indicative of improved exchange due to enhanced antibody displacement. The most pronounced affinity improvement was observed for the Y41A IC mutant, which had a 2.2-fold tighter affinity for IL-2Rα than JES6–1 IC (Fig. 3c, top and Table I). Notably, neither the parent JES6–1 IC nor any of the mutant IC constructs bound the immobilized IL-2Rβ subunit, indicating that JES6–1-mediated blockade of the IL-2Rβ remained intact for our engineered IC mutants (Fig. 3c, bottom).
We predicted that improved antibody displacement by the IL-2Rα subunit would potentiate mutant JES6–1 IC activity on IL-2Rα+ cells and, indeed, we observed that many of our immunocytokine mutants enhanced STAT5 signaling on IL-2Rα+ YT-1 cells relative to the parent JES6–1 IC. Three IC mutants (S34A, Y41A, and Y101A) recovered the extent of STAT5 signaling induced by the mixed IL-2/JES6–1 complex (Fig. 3d, top). None of the engineered IC mutants activated IL-2Rα− YT-1 cells, consistent with the behavior of the mixed complex (Fig. 3d, bottom). Our JES6–1 IC mutant cellular activation assays also offered insight into the relationships between affinity, exchange, and functional response. We hypothesized that activity would correlate with affinity in a biphasic manner (Supplemental Fig. 1c), and our data support this postulate (Fig. 3e). However, additional structural factors appear to contribute to signaling output, as is evidenced by the much greater potency of activation induced by JES6–1 IC mutants with alanine substitutions in the VL versus the VH domain. Since the cytokine was linked to the JES6–1 light chain, heavy chain mutants disrupt the cytokine/antibody interface further from the tether and we would thus expect function to be more dramatically affected for VH versus VL mutants. Accordingly, the VH mutants generally impair affinity to a greater extent than do VL mutants (Fig. 3b, Table I). The discrepancy between VH and VL JES6–1 IC mutant constructs is even more apparent when comparing IL-2Rα+ cell activity to exchange (as determined by IL-2Rα affinity). Although activity correlates with exchange within the JES6–1 IC VL mutants, the JES6–1 IC VH mutants all elicit weak stimulation of IL-2Rα+ cells, independent of their IL-2Rα exchange propensities (Fig. 3f).
Multi-site immunocytokine mutants exhibit enhanced IL-2Rα exchange, cellular activation, and in vivo TReg-biased expansion.
Based on our three most active single-point mutant JES6–1 IC constructs (the VL domain mutants S34A, Y41A, and Y101A) (Fig. 3d), we designed three double-alanine mutant IC constructs and one triple-alanine JES6–1 IC mutant construct to enhance IL-2Rα exchanging capacity and IL-2Rα+ cell-selective signaling. All multi-residue mutant IC constructs potentiated IL-2Rα exchange compared to the parent JES6–1 IC, with the most actively-exchanging mutants (Y41A+Y101A and S34A+Y101A) exhibiting a 2.6-fold IL-2Rα affinity enhancement relative to the parent JES6–1 IC (Fig. 4a, top, Table II). Several of the multi-residue mutants (including Y41A+Y101A) also bound to immobilized IL-2Rβ due to the weakened cytokine-antibody interaction, but all IC mutants bound weaker to IL-2Rβ than to IL-2Rα and had lower IL-2Rβ affinities than the free IL-2 cytokine (Fig. 4a, bottom). Signaling activity of the multi-residue IC mutants on IL-2Rα+ YT-1 cells correlated directly with IL-2Rα exchange; all mutants activated IL-2Rα+ cells more potently than the parent JES6–1 IC and three of the four constructs had greater activity than the mixed IL-2/JES6–1 complex (Fig. 4b, top). The Y41A+Y101A and S34+Y101A mutants were again the most active and none of the mutants triggered appreciable activation of IL-2Rα− YT-1 cells (Fig. 4b, bottom).
Figure 4. Engineered double mutant immunocytokine recovers TReg-biased activity of the IL-2/JES6–1 complex.
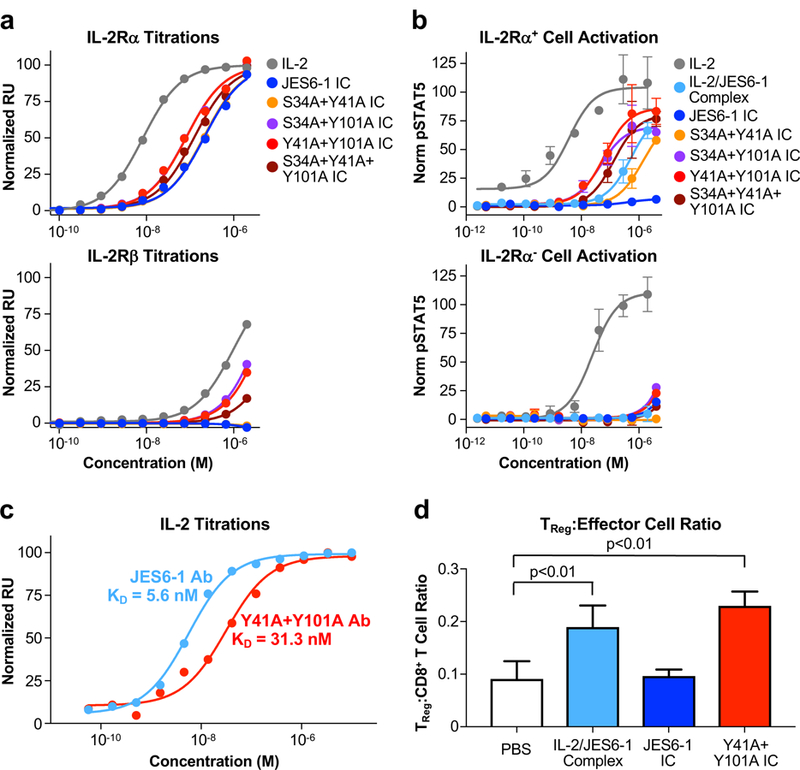
(a) Equilibrium surface plasmon resonance titrations of soluble IL-2, JES6–1 IC, or double/triple mutant JES6–1 IC mutants binding to immobilized IL-2Rα (top) or IL-2Rβ (bottom). (b) STAT5 phosphorylation response of IL-2Rα+ (top) or IL-2Rα− (bottom) YT-1 human NK cells treated with IL-2, JES6–1 IC, or double/triple mutant JES6–1 IC mutants. Data represent mean ± s.d. (c) Equilibrium surface plasmon resonance titrations of soluble IL-2 binding immobilized JES6–1 antibody (light blue) or the JY3 antibody variant (red). (d) Ratio of TReg to CD8+ effector T cell abundance in spleens harvested from C57BL/6 mice (n=3 per cohort) treated with PBS, IL-2/JES6–1 complex, JES6–1 IC, or the Y41A+Y101A IC mutant for four consecutive days, as determined by flow cytometry analysis. Data represent mean ± s.d. Statistical significance was determined by two-tailed unpaired Student’s t-test. The experiment was performed three times with similar results.
Table II. Receptor exchange properties of engineered JES6–1 antibody and immunocytokine multi-site mutants.
All mutations are located on the JES6–1 variable light chain.
| Construct | KD of IL-2Rα/IC Complex (nM) |
|---|---|
| JES6–1 | 220 |
| S34A+Y41A | 210 |
| S34A+Y101A | 82 |
| Y41A+Y101A | 82 |
| S34A+Y41A+Y101A | 130 |
Guided by our cellular activation results, we chose to further characterize the Y41A+Y101A mutant, which has a 5.6-fold weaker cytokine-antibody affinity than the unmodified JES6–1 (Fig. 4c), and the S34A+Y101A mutant, which had a 1.9-fold weaker cytokine-antibody affinity than the parent JES6–1 (Supplemental Fig. 2a). We assessed the ability of the Y41A+Y101A IC mutant to expand immune cell subset populations in C57BL/6 mice and found that, in contrast with JES6–1 IC, Y41A+Y101A IC induced preferential TReg versus CD8+ effector T cell expansion to the same extent as the mixed IL-2/JES6–1 complex (Fig. 4d and Supplemental Fig. 3a), indicating that reducing cytokine-antibody affinity had restored biased activity on IL-2RαHigh cells. Consistent results were observed for the TReg versus CD4+ T cell ratio (Supplemental Fig. 3b), confirming specific expansion of TReg by the Y41A+Y101A IC variant. IC behavior in vivo was found to be highly sensitive to affinity and structural modifications, as the S34A+Y101A IC mutant did not elicit biased TReg expansion (Supplemental Figs. 2b and 3), despite its similar exchange (Fig. 4a) and signaling (Fig 4b) properties to the Y41A+Y101A IC mutant in vitro. Further studies demonstrated that the Y41A+Y101A IC mutant (hereafter denoted JY3 IC) effected preferential TReg over CD8+ effector T cell growth in a dose-dependent manner in C57BL/6 (Supplemental Fig. 2c) and NOD mice (Supplemental Fig. 2e). Moreover, JY3 treatment upregulated IL-2Rα expression on TReg cells to a much greater extent than the mixed IL-2/JES6–1 complex (Supplemental Figs. 2d and 2f). IC formulation also had benefits in enhancing maximum tolerated dose of the IL-2 cytokine compared to the mixed complex, as administration of a 7.4 μg dose of IL-2 in mixed complex format was lethal to NOD mice (3/4 mice died), but an equivalent dose of the JY3 IC (30 μg) was well tolerated (0/4 mice died). This suggests that tethering the cytokine to the antibody may mitigate toxicity relative to the mixed complex, presumably through increased complex stability, which reduces off-target effects elicited by free IL-2.
Engineered immunocytokine induces cell subset-specific expansion in an adoptive T cell transfer model.
Given that JY3 IC selectively expanded IL-2RαHigh cells in a mixed immune population, we wondered whether our construct would be able to precisely control immune cell subset populations in an adoptive T cell transfer model. We purified and CFSE-labeled CD8+ T cells from OT-I mice, transferred the cells into congenic B6 mice, and treated recipient mice with a low dose of the SIINFEKL peptide plus various IL-2-antibody regimens for analysis of recipient immune cell expansion (Fig. 5a). Peptide stimulation with or without poly-L-lysine and IL-2 co-administration with an isotype control antibody failed to expand adoptively transferred activated CD8+ T cells, whereas the mixed IL-2/JES6–1 complex induced robust proliferation of transferred cells, and JY3 IC further enhanced this expansion at equivalent doses (Fig. 5b). An identical trend was observed for IL-2Rα expression on transferred CD8+ T cells, with higher surface levels of IL-2Rα elicited by JY3 IC versus IL-2/JES6–1 complex treatment (Supplemental Fig. 4a).
Figure 5. Engineered immunocytokine selectively potentiates the growth of activated adoptively transferred CD8+ T cells while boosting immunosuppression in the recipient.
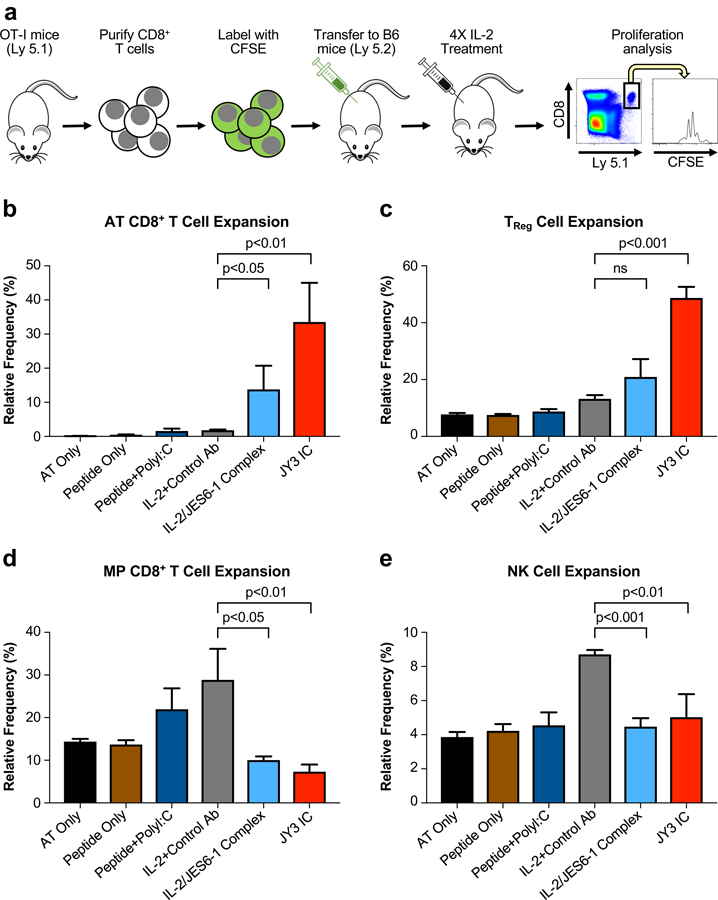
(a) Schematic of the adoptive transfer procedure. CD8+ T cells were purified from OT-I/Ly 5.1 mice and adoptively transferred into C57BL/6 mice (Ly 5.2) (n=4 per cohort), which were then stimulated by SIINFEKL peptide and subjected to the indicated treatments for four consecutive days. Mice were sacrificed 48 hours after the final injection and relative expansion was quantified via flow cytometry for the adoptively transferred (AT) CD8+ T cells (b) and the recipient TReg cells (c), MP CD8+ T cells (d), and NK cells (e). Data represent mean ± s.d. Statistical significance was determined by two-tailed unpaired Student’s t-test. The experiment was performed three times with similar results.
Characterization of recipient mouse T cell subsets supported our hypothesis that JY3 IC biases IL-2 activity to IL-2RαHigh TReg cells over IL-2RαLow naïve effector cells. No TReg cell expansion was induced by IL-2/isotype control antibody treatment, but we observed an increase in TReg cell number following IL-2/JES6–1 complex treatment and an even more profound increase following JY3 IC treatment (Fig. 5c). In contrast, IL-2/isotype control antibody treatment expanded both MP CD8+ T cells and NK cells, whereas neither IL-2/JES6–1 complex nor JY3 IC promoted proliferation of these effector subsets (Fig. 5d, e). IL-2/JES6–1 complex also upregulated IL-2Rα expression on TReg and MP CD8+ T cells, although not on NK cells, and JY3 IC increased surface IL-2Rα levels on TReg and MP CD8+ T cells to a greater extent than the complex and also robustly upregulated IL-2Rα on NK cells (Supplemental Fig. 4b-d). Overall, our adoptive transfer studies establish that JY3 IC specifically targets IL-2 activity to IL-2RαHigh immune cell subsets, and that it promotes more robust expansion and receptor upregulation on these subsets compared to the mixed IL-2/JES6–1 complex.
Engineered immunocytokine prevents the development of autoimmune disease in mice.
To explore the therapeutic potential of our engineered IC mutant, we compared the efficacy of JY3 IC to the mixed IL-2/JES6–1 complex in a dextran sodium sulfate (DSS)-induced colitis model. Mice were pre-treated with PBS, IL-2 with an isotype control antibody, IL-2/JES6–1 complex, or JY3 IC for seven consecutive days and disease was induced beginning on day 8 (Fig. 6a). One week post-colitis induction, as compared to IL-2/isotype control antibody-treated mice, IL-2/JES6–1 complex-treated mice exhibited significant reductions in disease severity, including attenuated weight loss, increased colon length, and lower disease activity index (Fig. 6b–d), consistent with previous findings (17). JY3 IC further enhanced autoimmune disease prevention, with more pronounced improvements in weight loss, colon length, and disease activity score compared to the IL-2/JES6–1 complex (Fig. 6b–d). These results suggest that the JY3 IC could have therapeutic advantages to the mixed cytokine-antibody complex beyond the logistical considerations of stability and ease of formulation.
Figure 6. Engineered immunocytokine reduces disease severity in a mouse model of colitis.
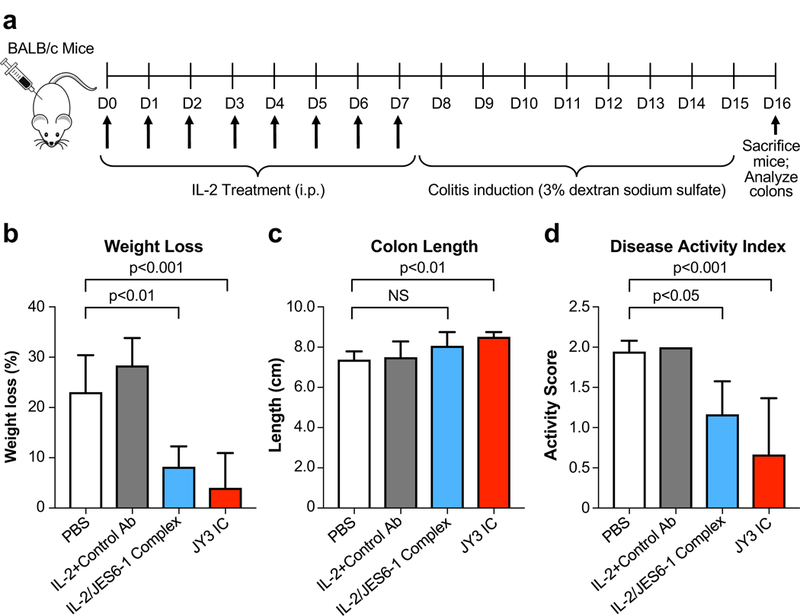
(a) Schematic of the mouse colitis study. BALB/c mice (n=6 per cohort) were treated once daily for 7 days with PBS, IL-2 plus a control antibody, IL-2/JES6–1 complex, or JY3 IC. Beginning on day 8, mice were subjected to 3% DSS in their drinking water to induce colitis. Weight loss (b) and disease activity index (c) were assessed on day 15. Mice were sacrificed on day 16 and colon length (d) was measured. Data represent mean ± s.d. Statistical significance by one-way ANOVA + Dunnett’s multiple comparison post-test is indicated. The experiment was performed twice with similar results.
DISCUSSION
There is a growing interest in the development of antibody-cytokine fusions (immunocytokines) to empower cytokines as drugs (43). Whereas cytokines have short in vivo half-lives (often less than five minutes) and thus require frequent dosing (44–46), antibodies benefit from prolonged serum persistence due to neonatal Fc receptor-mediated recycling (47, 48). In addition, fusion to surface antigen-binding antibodies can allow for targeted cytokine delivery tailored to particular disease indications (43, 49–54). However, clinical development of targeted immunocytokines is hampered by the high potency of cytokines, which nullifies the effect of the targeting antibody and leads to toxicity through indiscriminate activation of all cytokine-response immune cell subsets (55–57). To circumvent the issue of potency, recent efforts have focused on reducing cytokine-receptor affinity through directed mutagenesis of the cytokine (58, 59), but cytokine modification may alter functional activity and also raises concerns about immunogenicity.
Here, we describe a novel use of immunocytokines to ‘shield’ a cytokine from non-specifically engaging immune cells and instead target it preferentially to TReg cells based on surface receptor expression levels. Our approach relies entirely on antibody engineering, thus obviating the need for cytokine manipulation and keeping both cytokine-receptor affinity and cytokine activity intact. Furthermore, our strategy completely eliminates off-target effects by fully sequestering the cytokine rather than simply lowering its receptor interaction affinity. Although the allosteric receptor-antibody exchange mechanism we describe is specific to the IL-2/JES6–1 system, the structure-based design principles we used to engineer an effective single-agent cytokine-antibody fusion can be extended to other ligand-antibody interactions for exclusive targeting of soluble factors to specific cell subsets of interest.
Our engineering workflow offered new insight into the relationship between antibody-cytokine affinity and signaling activity in the context of allosteric exchange. Previous studies have elucidated correlations between cytokine-receptor affinity and signaling activity (60–65). A systematic study of the interplay between cytokine-receptor complex stability and membrane-proximal signaling for the IL-13/IL-13Rα1/IL-4Rα complex revealed that cytokine activity correlated directly with cytokine-receptor affinity only outside of a ‘buffering region,’ an affinity regime within 100-fold (in either direction) of the wild-type interaction affinity wherein cytokine activity was insensitive to affinity changes (65). The complexities of activity-affinity relationships in other systems led us to speculate that the antibody-cytokine affinity in the IL-2/JES6–1 single-agent fusion could also exhibit non-linear behavior. Furthermore, since the exchange mechanism depends on the relative strengths of cytokine interaction between the IL-2Rα receptor subunit and the JES6–1 antibody, we would expect the activity of the immunocytokine on various cell subsets to depend strongly on binding parameters.
We predicted biphasic behavior of the IL-2RαHigh cell (i.e. TReg cell) activity bias of our engineered JES6–1 mutants with respect to their IL-2 affinities: at very low affinities the antibody would constitutively dissociate, resulting in unbiased activation of all IL-2-responsive cells, and at very high affinities, the antibody would never dissociate, obstructing activity on all cell subsets (Supplemental Fig. 1c). By modulating the affinity of the IL-2/antibody interaction over nearly two orders of magnitude (Fig. 1b, Table I), we aimed to probe the window of antibody-cytokine affinity ‘tunability’ for optimization of preferential TReg cell expansion. As illustrated in Fig. 3e, we indeed observed a biphasic activity curve as IL-2 affinity was varied, although the tuning range was found to be surprisingly narrow. JES6–1 mutants with up to a 2.2-fold decrease in IL-2 affinity compared to the parent antibody exhibited improved TReg selectivity, but no improvements were observed for mutants with IL-2 affinities that were reduced by 10-fold or more (Fig. 3e, Table II).
Other factors such as structural considerations also apparently contribute to the activity of IC mutants. For instance, the E60A and H100A mutants have similar affinities for IL-2, but diverge significantly in their activation potencies (Fig. 3e). The E60A mutation is in the VH domain whereas the H100A mutation is in the VL domain, suggesting that the location of the interface disruption with respect to chain affects IC activity. Consistent with this observation, VL mutants exhibit uniformly stronger signaling activity on IL-2Rα+ cells than do VH mutants. This phenomenon could also be impacted by the topology of the fusion itself, as the greater proximity to tethered IL-2 for VL compared to VH may render the VL interface more robust against affinity disruption. The dramatic (>10-fold) affinity losses observed with 3/4 VH mutants compared with only 1/4 VL mutants support the influence of topological factors on IC mutant activity (Table II). Further complicating the affinity-activity relationship is the lack of correlation between biased signal activation in vitro and selective cell subset expansion in vivo. Although the S34A+Y101A and Y41A+Y101A (JY3) IC mutants behaved similarly with respect to STAT5 signaling on IL-2Rα+ and IL-2Rα− cells, there was a clear discrepancy in their in vivo promotion of IL-2RαHigh versus IL-2RαLow cell growth (Fig. 3d and Supplemental Fig. 2b).
From a therapeutic development standpoint, the IC format has clear advantages over mixed complex administration as it eliminates dosing ratio considerations and concerns about the free cytokine inducing off-target effects and toxicities or undergoing rapid clearance from the bloodstream (43, 46). However, we unexpectedly found that our engineered IC elicited greater IL-2RαHigh cell expansion in an adoptive T cell transfer model (Fig. 4) and prevented DSS-induced colitis more effectively than the mixed complex (Fig. 5), even though the two formats induced similar TReg to effector cell expansion ratios (Figs. 4d and Supplemental Figs. 2c, 2e, and 3). One possibility for the superior phenotypic behavior of the engineered IC could be that it is positioned more optimally on the biphasic TReg to effector cell activity curve based on its altered antibody-cytokine affinity (Supplemental Fig. 1c). Alternatively, the more extensive IL-2Rα upregulation induced by JY3 IC versus the mixed complex (Supplemental Figs. 2d and 2f) may present an advantage for the immunocytokine by fueling the transcriptional feedback loop that perpetuates IL-2 signaling (17). Regardless of rationale, the enhanced behavior of JY3 IC over the mixed complex provides an immediately useful reagent for expanding TReg cells to combat autoimmune disease, and the structure-guided engineering strategy we used to develop this construct will inform the design of other mechanism-driven therapeutic immunocytokines. With a growing number of anti-cytokine antibodies in development, including those against IL-2, −4, −6, −7, and −15 (66), our novel approach can be readily extended to a broad range of cytokine-receptor systems for disease-relevant applications.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Garcia, Bluestone, Kovar, and Pande laboratories for helpful advice and discussions and A. Velasco and D. Waghray for technical assistance.
This work was supported by the US National Institutes of Health (R01 AI51321 to K.C.G.), the Mathers Fund, the Ludwig Foundation, the Parker Institute for Cancer Immunotherapy, and the Czech Science Foundation (Grants 13–12885S and 18–12973S to M.K.) The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health. K.C.G. is an investigator of the Howard Hughes Medical Institute, J.B.S. is the recipient of a Leukemia & Lymphoma Society Career Development Program fellowship, and A.P. is a recipient of a National Science Foundation Graduate Research fellowship.
Footnotes
COMPETING FINANCIAL INTERESTS
Provisional patents concerning the technology described in this work have been filed. V.S.P. is a consultant and SAB member of Schrodinger, LLC and Globavir, sits on the Board of Directors of Apeel Inc, Freenome Inc, Omada Health, Patient Ping, and Rigetti Computing, and is a General Partner at Andreessen Horowitz.
ADDITIONAL INFORMATION
Correspondence and requests for materials should be addressed to K.C.G. Supplemental information includes four supplemental figures and one supplemental video.
REFERENCES
- 1.Malek TR 2008. The Biology of Interleukin-2. Annu. Rev. Immunol 26: 453–479. [DOI] [PubMed] [Google Scholar]
- 2.Boyman O, and Sprent J. 2012. The role of interleukin-2 during homeostasis and activation of the immune system. Nat. Rev. Immunol 12: 180–190. [DOI] [PubMed] [Google Scholar]
- 3.Wang X, Rickert M, and Garcia KC. 2005. Structure of the quaternary complex of interleukin-2 with its alpha, beta, and gammac receptors. Science 310: 1159–1163. [DOI] [PubMed] [Google Scholar]
- 4.Spangler JB, Moraga I, Mendoza JL, and Garcia KC. 2015. Insights into Cytokine–Receptor Interactions from Cytokine Engineering. Annu. Rev. Immunol 33: 139–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stroud RM, and Wells JA. 2004. Mechanistic Diversity of Cytokine Receptor Signaling Across Cell Membranes. Sci STKE 2004: re7–re7. [DOI] [PubMed] [Google Scholar]
- 6.Murray PJ 2007. The JAK-STAT signaling pathway: input and output integration. J. Immunol. Baltim. Md 1950. 178: 2623–2629. [DOI] [PubMed] [Google Scholar]
- 7.Shevach EM 2012. Application of IL-2 therapy to target T regulatory cell function. Trends Immunol 33: 626–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng G, Yu A, and Malek TR. 2011. T-cell tolerance and the multi-functional role of IL-2R signaling in T-regulatory cells. Immunol. Rev 241: 63–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klatzmann D, and Abbas AK. 2015. The promise of low-dose interleukin-2 therapy for autoimmune and inflammatory diseases. Nat. Rev. Immunol 15: 283–294. [DOI] [PubMed] [Google Scholar]
- 10.Saadoun D, Rosenzwajg M, Joly F, Six A, Carrat F, Thibault V, Sene D, Cacoub P, and Klatzmann D. 2011. Regulatory T-Cell Responses to Low-Dose Interleukin-2 in HCV-Induced Vasculitis. N. Engl. J. Med 365: 2067–2077. [DOI] [PubMed] [Google Scholar]
- 11.Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea EPI, Armand P, Cutler C, Ho VT, Treister NS, Bienfang DC, Prasad S, Tzachanis D, Joyce RM, Avigan DE, Antin JH, Ritz J, and Soiffer RJ. 2011. Interleukin-2 and Regulatory T Cells in Graft-versus-Host Disease. N. Engl. J. Med 365: 2055–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boyman O, Kovar M, Rubinstein MP, Surh CD, and Sprent J. 2006. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science 311: 1924–1927. [DOI] [PubMed] [Google Scholar]
- 13.Boyman O, Surh CD, and Sprent J. 2006. Potential use of IL-2/anti-IL-2 antibody immune complexes for the treatment of cancer and autoimmune disease. Expert Opin. Biol. Ther 6: 1323–1331. [DOI] [PubMed] [Google Scholar]
- 14.Tang Q, Adams JY, Penaranda C, Melli K, Piaggio E, Sgouroudis E, Piccirillo CA, Salomon BL, and Bluestone JA. 2008. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity 28: 687–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu R, Zhou Q, La Cava A, Campagnolo DI, Van Kaer L, and Shi F-D. 2010. Expansion of regulatory T cells via IL-2/anti-IL-2 mAb complexes suppresses experimental myasthenia. Eur. J. Immunol 40: 1577–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grinberg-Bleyer Y, Baeyens A, You S, Elhage R, Fourcade G, Gregoire S, Cagnard N, Carpentier W, Tang Q, Bluestone J, Chatenoud L, Klatzmann D, Salomon BL, and Piaggio E. 2010. IL-2 reverses established type 1 diabetes in NOD mice by a local effect on pancreatic regulatory T cells. J. Exp. Med 207: 1871–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spangler JB, Tomala J, Luca VC, Jude KM, Dong S, Ring AM, Votavova P, Pepper M, Kovar M, and Garcia KC. 2015. Antibodies to Interleukin-2 Elicit Selective T Cell Subset Potentiation through Distinct Conformational Mechanisms. Immunity 42: 815–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Webster KE, Walters S, Kohler RE, Mrkvan T, Boyman O, Surh CD, Grey ST, and Sprent J. 2009. In vivo expansion of T reg cells with IL-2–mAb complexes: induction of resistance to EAE and long-term acceptance of islet allografts without immunosuppression. J. Exp. Med 206: 751–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park Y-H, Koo S-K, Kim Y, Kim H-M, Joe I-Y, Park C-S, Kim S-C, Han D-J, and Lim D-G. 2010. Effect of in vitroexpanded CD4+CD25+Foxp3+ regulatory T cell therapy combined with lymphodepletion in murine skin allotransplantation. Clin. Immunol 135: 43–54. [DOI] [PubMed] [Google Scholar]
- 20.Tomala J, Kovarova J, Kabesova M, Votavova P, Chmelova H, Dvorakova B, Rihova B, and Kovar M. 2013. Chimera of IL-2 Linked to Light Chain of anti-IL-2 mAb Mimics IL-2/anti-IL-2 mAb Complexes Both Structurally and Functionally. ACS Chem. Biol 8: 871–876. [DOI] [PubMed] [Google Scholar]
- 21.Dukkipati A, Park HH, Waghray D, Fischer S, and Garcia KC. 2008. BacMam system for high-level expression of recombinant soluble and membrane glycoproteins for structural studies. Protein Expr. Purif 62: 160–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yodoi J, Teshigawara K, Nikaido T, Fukui K, Noma T, Honjo T, Takigawa M, Sasaki M, Minato N, and Tsudo M. 1985. TCGF (IL 2)-receptor inducing factor(s). I. Regulation of IL 2 receptor on a natural killer-like cell line (YT cells). J. Immunol. Baltim. Md 1950 134: 1623–1630. [PubMed] [Google Scholar]
- 23.Kuziel WA, Ju G, Grdina TA, and Greene WC. 1993. Unexpected effects of the IL-2 receptor alpha subunit on high affinity IL-2 receptor assembly and function detected with a mutant IL-2 analog. J. Immunol 150: 3357–3365. [PubMed] [Google Scholar]
- 24.Ring AM, Lin J-X, Feng D, Mitra S, Rickert M, Bowman GR, Pande VS, Li P, Moraga I, Spolski R, Ozkan E, Leonard WJ, and Garcia KC. 2012. Mechanistic and structural insight into the functional dichotomy between IL-2 and IL-15. Nat. Immunol 13: 1187–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salafsky JS 2006. Detection of protein conformational change by optical second-harmonic generation. J. Chem. Phys 125: 074701. [DOI] [PubMed] [Google Scholar]
- 26.Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, and Lindahl E. 2015. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2: 19–25. [Google Scholar]
- 27.Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, and Shaw DE. 2010. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 78: 1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, and Klein ML. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 79: 926–935. [Google Scholar]
- 29.Šali A, and Blundell TL. 1993. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol 234: 779–815. [DOI] [PubMed] [Google Scholar]
- 30.Bussi G, Donadio D, and Parrinello M. 2007. Canonical sampling through velocity rescaling. J. Chem. Phys 126: 014101. [DOI] [PubMed] [Google Scholar]
- 31.Parrinello M, and Rahman A. 1981. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys 52: 7182–7190. [Google Scholar]
- 32.Hess B, Bekker H, Berendsen HJC, and Fraaije JGEM. 1997. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem 18: 1463–1472. [Google Scholar]
- 33.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, and Pedersen LG. 1995. A smooth particle mesh Ewald method. J. Chem. Phys 103: 8577–8593. [Google Scholar]
- 34.Schwantes CR, and Pande VS. 2013. Improvements in Markov State Model Construction Reveal Many Non-Native Interactions in the Folding of NTL9. J. Chem. Theory Comput 9: 2000–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beauchamp KA, Bowman GR, Lane TJ, Maibaum L, Haque IS, and Pande VS. 2011. MSMBuilder2: Modeling Conformational Dynamics on the Picosecond to Millisecond Scale. J. Chem. Theory Comput 7: 3412–3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McGibbon RT, Beauchamp KA, Harrigan MP, Klein C, Swails JM, Hernández CX, Schwantes CR, Wang L-P, Lane TJ, and Pande VS. 2015. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J 109: 1528–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cooper HS, Murthy SN, Shah RS, and Sedergran DJ. 1993. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab. Investig. J. Tech. Methods Pathol 69: 238–249. [PubMed] [Google Scholar]
- 38.Salafsky JS 2007. Second-harmonic generation for studying structural motion of biological molecules in real time and space. Phys. Chem. Chem. Phys. PCCP 9: 5704–5711. [DOI] [PubMed] [Google Scholar]
- 39.Moree B, Connell K, Mortensen RB, Liu CT, Benkovic SJ, and Salafsky J. 2015. Protein Conformational Changes Are Detected and Resolved Site Specifically by Second-Harmonic Generation. Biophys. J 109: 806–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thanos CD, Randal M, and Wells JA. 2003. Potent Small-Molecule Binding to a Dynamic Hot Spot on IL-2. J. Am. Chem. Soc 125: 15280–15281. [DOI] [PubMed] [Google Scholar]
- 41.Thanos CD, DeLano WL, and Wells JA. 2006. Hot-spot mimicry of a cytokine receptor by a small molecule. Proc. Natl. Acad. Sci 103: 15422–15427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Levin AM, Bates DL, Ring AM, Krieg C, Lin JT, Su L, Moraga I, Raeber ME, Bowman GR, Novick P, Pande VS, Fathman CG, Boyman O, and Garcia KC. 2012. Exploiting a natural conformational switch to engineer an interleukin-2 “superkine”. Nature 484: 529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pasche N, and Neri D. 2012. Immunocytokines: a novel class of potent armed antibodies. Drug Discov. Today 17: 583–590. [DOI] [PubMed] [Google Scholar]
- 44.Lotze MT, Matory YL, Ettinghausen SE, Rayner AA, Sharrow SO, Seipp CA, Custer MC, and Rosenberg SA. 1985. In vivo administration of purified human interleukin 2. II. Half life, immunologic effects, and expansion of peripheral lymphoid cells in vivo with recombinant IL 2. J. Immunol 135: 2865–2875. [PubMed] [Google Scholar]
- 45.Kontermann RE 2011. Strategies for extended serum half-life of protein therapeutics. Curr. Opin. Biotechnol 22: 868–876. [DOI] [PubMed] [Google Scholar]
- 46.Vazquez-Lombardi R, Roome B, and Christ D. 2013. Molecular Engineering of Therapeutic Cytokines. Antibodies 2: 426–451. [Google Scholar]
- 47.Finkelman FD, Madden KB, Morris SC, Holmes JM, Boiani N, Katona IM, and Maliszewski CR. 1993. Anti-cytokine antibodies as carrier proteins. Prolongation of in vivo effects of exogenous cytokines by injection of cytokine-anti-cytokine antibody complexes. J. Immunol 151: 1235–1244. [PubMed] [Google Scholar]
- 48.Roopenian DC, and Akilesh S. 2007. FcRn: the neonatal Fc receptor comes of age. Nat. Rev. Immunol 7: 715–725. [DOI] [PubMed] [Google Scholar]
- 49.Becker JC, Pancook JD, Gillies SD, Furukawa K, and Reisfeld RA. 1996. T cell-mediated eradication of murine metastatic melanoma induced by targeted interleukin 2 therapy. J. Exp. Med 183: 2361–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Halin C, Rondini S, Nilsson F, Berndt A, Kosmehl H, Zardi L, and Neri D. 2002. Enhancement of the antitumor activity of interleukin-12 by targeted delivery to neovasculature. Nat. Biotechnol 20: 264. [DOI] [PubMed] [Google Scholar]
- 51.Gillies SD, Lan Y, Williams S, Carr F, Forman S, Raubitschek A, and Lo K-M. 2005. An anti-CD20–IL-2 immunocytokine is highly efficacious in a SCID mouse model of established human B lymphoma. Blood 105: 3972–3978. [DOI] [PubMed] [Google Scholar]
- 52.Schrama D, Reisfeld RA, and Becker JC. 2006. Antibody targeted drugs as cancer therapeutics. Nat. Rev. Drug Discov 5: 147. [DOI] [PubMed] [Google Scholar]
- 53.Schliemann C, Palumbo A, Zuberbühler K, Villa A, Kaspar M, Trachsel E, Klapper W, Menssen HD, and Neri D. 2009. Complete eradication of human B-cell lymphoma xenografts using rituximab in combination with the immunocytokine L19-IL2. Blood 113: 2275–2283. [DOI] [PubMed] [Google Scholar]
- 54.Schilbach K, Alkhaled M, Welker C, Eckert F, Blank G, Ziegler H, Sterk M, Müller F, Sonntag K, Wieder T, Braumüller H, Schmitt J, Eyrich M, Schleicher S, Seitz C, Erbacher A, Pichler BJ, Müller H, Tighe R, Lim A, Gillies SD, Strittmatter W, Röcken M, and Handgretinger R. 2015. Cancer-targeted IL-12 controls human rhabdomyosarcoma by senescence induction and myogenic differentiation. OncoImmunology 4: e1014760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shusterman S, London WB, Gillies SD, Hank JA, Voss SD, Seeger RC, Reynolds CP, Kimball J, Albertini MR, Wagner B, Gan J, Eickhoff J, DeSantes KB, Cohn SL, Hecht T, Gadbaw B, Reisfeld RA, Maris JM, and Sondel PM. 2010. Antitumor Activity of Hu14.18-IL2 in Patients With Relapsed/Refractory Neuroblastoma: A Children’s Oncology Group (COG) Phase II Study. J. Clin. Oncol 28: 4969–4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, Smith M, Anderson B, Villablanca JG, Matthay KK, Shimada H, Grupp SA, Seeger R, Reynolds CP, Buxton A, Reisfeld RA, Gillies SD, Cohn SL, Maris JM, and Sondel PM. 2010. Anti-GD2 Antibody with GM-CSF, Interleukin-2, and Isotretinoin for Neuroblastoma. N. Engl. J. Med 363: 1324–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tzeng A, Kwan BH, Opel CF, Navaratna T, and Wittrup KD. 2015. Antigen specificity can be irrelevant to immunocytokine efficacy and biodistribution. Proc. Natl. Acad. Sci 112: 3320–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gillies SD, Lan Y, Hettmann T, Brunkhorst B, Sun Y, Mueller SO, and Lo K-M. 2011. A Low-Toxicity IL-2–Based Immunocytokine Retains Antitumor Activity Despite Its High Degree of IL-2 Receptor Selectivity. Clin. Cancer Res 17: 3673–3685. [DOI] [PubMed] [Google Scholar]
- 59.Garcin G, Paul F, Staufenbiel M, Bordat Y, der Heyden JV, Wilmes S, Cartron G, Apparailly F, Koker SD, Piehler J, Tavernier J, and Uzé G. 2014. High efficiency cell-specific targeting of cytokine activity. Nat. Commun 5: 3016. [DOI] [PubMed] [Google Scholar]
- 60.Pearce Kenneth H., Cunningham BC, Fuh G, Teeri T, and Wells JA. 1999. Growth Hormone Binding Affinity for Its Receptor Surpasses the Requirements for Cellular Activity. Biochemistry (Mosc.) 38: 81–89. [DOI] [PubMed] [Google Scholar]
- 61.Junttila IS, Creusot RJ, Moraga I, Bates DL, Wong MT, Alonso MN, Suhoski MM, Lupardus P, Meier-Schellersheim M, Engleman EG, Utz PJ, Fathman CG, Paul WE, and Garcia KC. 2012. Redirecting cell-type specific cytokine responses with engineered interleukin-4 superkines. Nat. Chem. Biol 8: 990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Piehler J, Thomas C, Garcia KC, and Schreiber G. 2012. Structural and dynamic determinants of type I interferon receptor assembly and their functional interpretation. Immunol. Rev 250: 317–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Walter MR 2014. The molecular basis of IL-10 function: from receptor structure to the onset of signaling. Curr. Top. Microbiol. Immunol 380: 191–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moraga I, Spangler J, Mendoza JL, and Garcia KC. 2014. Multifarious determinants of cytokine receptor signaling specificity. Adv. Immunol 121: 1–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Moraga I, Richter D, Wilmes S, Winkelmann H, Jude K, Thomas C, Suhoski MM, Engleman EG, Piehler J, and Garcia KC. 2015. Instructive roles for cytokine-receptor binding parameters in determining signaling and functional potency. Sci. Signal 8: ra114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mostböck S 2009. Cytokine/Antibody complexes: an emerging class of immunostimulants. Curr. Pharm. Des 15: 809–825. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.